

Abstract
Aberrant activation of the MET/hepatocyte growth factor (HGF) receptor participates in the malignant behavior of cancer cells, such as invasion‐metastasis and resistance to molecular targeted drugs. Many mutations in the MET extracellular region have been reported, but their significance is largely unknown. Here, we report the dysregulation of mutant MET originally found in a lung cancer patient with Val370 to Asp370 (V370D) replacement located in the extracellular SEMA domain. MET‐knockout cells were prepared and reconstituted with WT‐MET or V370D‐MET. HGF stimulation induced MET dimerization and biological responses in cells reconstituted with WT‐MET, but HGF did not induce MET dimerization and failed to induce biological responses in V370D‐MET cells. The V370D mutation abrogated HGF‐dependent drug resistance of lung cancer cells to epidermal growth factor receptor‐tyrosine kinase inhibitors (EGFR‐TKI). Compared with WT‐MET cells, V370D‐MET cells showed different activation patterns in receptor tyrosine kinases upon exposure to survival/growth‐stressed conditions. Surface plasmon resonance analysis indicated that affinity between the extracellular region of V370D‐MET and HGF was reduced compared with that for WT‐MET. Further analysis of the association between V370D‐MET and the separate domains of HGF indicated that the SP domain of HGF was unchanged, but its association with the NK4 domain of HGF was mostly lost in V370D‐MET. These results indicate that the V370D mutation in the MET receptor impairs the functional association with HGF and is therefore a loss‐of‐function mutation. This mutation may change the dependence of cancer cell growth/survival on signaling molecules, which may promote cancer cell characteristics under certain conditions.
Keywords: affinity, hepatocyte growth factor, loss of function, MET receptor, missense mutation
1. INTRODUCTION
Activation of MET receptor tyrosine kinase (RTK) plays a well‐defined role as a driver of oncogenesis and progression to malignant tumors.1, 2 The MET receptor is biosynthesized as a single‐chain precursor of ~190 kDa, which undergoes cleavage by furin during transport to the cell membrane.3 The resulting mature form is composed of a covalently linked 50‐kDa α‐chain and a membrane‐spanning 145‐kDa β‐chain.4 The extracellular domain (ECD) of the MET receptor consists of SEMA, PSI, and IPT1‐4 domains, and the intracellular region contains juxtamembrane and tyrosine kinase (TK) domains.5 The ligand for MET is hepatocyte growth factor (HGF), which is composed of NK4 (N‐terminal and four kringle domains, also known as the α‐chain) and SP (also known as the β‐chain) domains linked by a disulfide bridge.6
Activation of the HGF/MET pathway results in pleiotropic biological activities including on cell proliferation, migration, survival, and morphogenesis.6, 7 Aberrant activation of MET by ligand‐dependent or ligand‐independent mechanisms is associated with cancer development, metastasis, and drug resistance.2, 6, 8 Germline and somatic mutations of MET receptor were found in many malignancies.9, 10 Mutations in the MET tyrosine kinase domain (H1094Y/R, H1106D, M1131T, V1188L, L1195V, D1228H/N, Y1230C/D/H, M1250T/I) affect kinase activation and are likely to be gain‐of‐function mutations.6, 10, 11 Deletion of the juxtamembrane domain of MET caused by exon 14 skipping resulted in decreased MET degradation and prolonged downstream signaling activation.12 Two missense mutations in the MET juxtamembrane domain in small‐cell lung cancer patients increased tyrosine phosphorylation of cellular proteins.13 Thus, signal abnormalities and the significance of mutations in the intracellular domains of MET have been defined. In contrast, approximately 20 different mutations in the MET extracellular domains were found in cancer patients; however, except for a few cases, their significance and underlying mechanisms remain unknown.6, 11, 14
N375S is a missense mutation in the MET SEMA domain found in 13% of East Asian lung tumor patients. Studies based on HGF binding, molecular modeling, and apoptosis induced by MET inhibitors indicated that the N375S mutation conferred mild resistance to MET inhibition.15 Recently, the N375K mutation was identified in four siblings affected by epidermal growth factor receptor (EGFR)‐mutant lung adenocarcinomas.16 Thus, N375 is a “hotspot” for mutations and may play an important role in the normal functions of MET receptor. Here, we focus on the V370D missense mutation originally found in lung adenocarcinoma,11, 14 because V370 is located in the same α‐helix‐367‐375 structure as the hotspot N375. We obtained evidence that V370D‐MET is a loss‐of‐function mutation.
2. MATERIALS AND METHODS
2.1. Cells, reagents, and recombinant proteins
CHO‐K1 cell line was obtained from ATCC and cultured in Ham's F12 medium (FUJIFILM Wako Pure Chemical, Osaka, Japan) supplemented with 5% FBS (Sigma‐Aldrich, St Louis, MO, USA). Human lung adenocarcinoma PC‐9 cells, kindly provided by Prof. Seiji Yano (Kanazawa University, Japan), were cultured in RPMI‐1640 medium (FUJIFILM Wako Pure Chemical) supplemented with 10% FBS. HGF was prepared from the conditioned medium of CHO cells stably expressing human HGF. NK4 was kindly provided by Kringle Pharma (Ibaraki, Osaka, Japan). PHA665752 was purchased from Selleck Biotech (Tokyo, Japan). Anti‐pMET (Y1234/1235), anti‐MET, anti‐pAKT (S473), and anti‐GAPDH antibodies were purchased from Cell Signaling Technology Japan (Tokyo, Japan). For the expression of MET extracellular domains (MET‐ECD‐Fc‐His or MET‐ECD‐His), expression constructs were transiently transfected into Expi293F cells using the ExpiFectamine 293 Transfection Kit (ThermoFisher Scientific, Waltham, MA, USA). Secreted proteins were purified using cOmplete His‐Tag Purification Resin (Sigma‐Aldrich).
2.2. MET‐knockout cells
Cas 9 protein (1 μg; ThermoFisher Scientific) was added to 10 μL resuspension buffer (Buffer R) provided in the Neon Transfection System 10 μL Kit (ThermoFisher Scientific), followed by adding 250 ng gRNA. The mixture was incubated for 5 minutes at room temperature. CHO‐K1 cells were washed once in PBS and resuspended at 5 × 106 cells/mL in Buffer R containing the Cas9/gRNA complexes. Cells were electroporated under the following conditions: pulse voltage, 1560 V; pulse width, 5 ms; and pulse number, 10. The following gRNA was used: 5′‐AATGCCAGGTGACAGCACGGTGG‐3′. Human Met CRISPR/Cas9 knockout plasmids and homology‐directed repair plasmids were purchased from Santa Cruz Biotechnology (sc‐400101; Dallas, TX, USA). These vectors were transfected into PC‐9 cells with Lipofectamine LTX and Plus reagents (ThermoFisher Scientific). Stable transfectants were selected with 0.5 μg/mL puromycin and single‐cell clones were isolated. Gene editing was confirmed by site‐specific genomic PCR using KOD‐FX Neo (TOYOBO, Osaka, Japan) and Sanger sequencing using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA) with primers for human MET exon 3 (forward: 5′‐GTGTAGTGTCCTAAGGTCAG‐3′, reverse: 5′‐CAAGCACAGTGGAATGGACA‐3′) or hamster MET exon 1 (forward: 5′‐TCAGGGGTTGAACGTCTCTC‐3′, reverse: 5′‐ATGTAGTTTGTGGCTCCGAGG‐3′) coding regions.
2.3. Plasmids
Human MET cDNA (NP_000236.2) was inserted into the pCAGGS plasmid,17 and a flag‐tag sequence (GACTACAAGGACGATGACGACAAG) was connected to the 3′ end of it. For pCAGGS‐hMET‐ECD‐His, cDNA fragments were amplified by PCR from pCAGGS‐hMET‐Flag using the following primer sets: HindIII‐hMET‐Fo and PacI‐hMET‐ECD‐Re. After ligation with the synthesized DNA fragment PacI‐GGGS‐7His‐STOP‐NotI, the HindIII‐hMET‐ECD‐GGGS‐His‐STOP‐NotI DNA fragment was cloned into HindIII‐NotI sites of pCAGGS‐hMET‐Flag. For pCAGGS‐hMET‐ECD‐Fc‐His, a cDNA library was prepared from pEF‐Fc18transfected EHMES‐1 cells. Human IgG fragment crystallizable region (Fc) cDNA was amplified by PCR using the following primers from the cDNA library: PacI‐Fc‐Fo and KpnI‐Fc‐Re. After ligation with the synthesized DNA fragment KpnI‐GGGS‐7His‐Stop‐NotI, PacI‐Fc‐GGGS‐His‐NotI DNA was cloned into PacI‐NotI sites of pCAGGS‐hMET‐ECD‐His. For mutation‐containing pCAGGS‐hMET‐ECD‐His plasmids, mutagenesis PCR was carried out using WT pCAGGS‐hMET‐ECD‐His as a template for V370D primers. Detailed information on the primers and synthesized DNA sequences is presented in Table 1. For mutation‐containing pCAGGS‐hMET‐ECD‐Fc‐His and pCAGGS‐hMET‐Flag plasmids, cDNA fragments containing mutation sites were purified on agarose gel after digesting mutation‐containing pCAGGS‐hMET‐ECD‐His by HindIII and MfeI, and were cloned into HindIII‐MfeI sites of pCAGGS‐hMET‐ECD‐Fc‐His and pCAGGS‐hMET‐Flag, respectively.
Table 1.
Primers and DNA sequences used for plasmid construction
| Name | Sequence |
|---|---|
| HindIII‐hMET‐Fo | 5′‐TCGAGCTCAAGCTTACCATGAAG‐3′ |
| PacI‐hMET‐ECD‐Re | 5′‐GTCACAATTTAATTAATGTGAAATTCTGATCTGGTTGAACTAT‐3′ |
| PacI‐GGGS‐7His‐STOP‐NotI‐Fo | 5′‐TTAATTAATGGTGGAGGCGGTTCACATCACCACCATCATCACCATTGAGCGGCCGC‐3′ |
| PacI‐GGGS‐7His‐STOP‐NotI‐Re | 5′‐GCGGCCGCTCAATGGTGATGATGGTGGTGATGTGAACCGCCTCCACCATTAATTAA‐3′ |
| PacI‐Fc‐Fo | 5′‐CACAATTTAATTAATCACACATGCCCACCGTGC‐3′ |
| KpnI‐Fc‐Re | 5′‐AATCGGTACCTTTACCCGGGGACAGGGA‐3′ |
| KpnI‐GGGS‐7His‐Stop‐NotI‐Fo | 5′‐CGGTGGAGGCGGTTCACATCACCACCATCATCACCATTGAGC‐3′ |
| KpnI‐GGGS‐7His‐Stop‐NotI‐Re | 5′‐GGCCGCTCAATGGTGATGATGGTGGTGATGTGAACCGCCTCCACCGGTAC‐3′ |
| V370D primers | Forward: 5′‐AAATATGACAACGACTTCTTCAACAAG‐3′ |
| Reverse: 5′‐GTCGTTGTCATATTTGATAGGGAATGC‐3′ |
2.4. Stable transfection
pCAGGS‐hMET‐Flag (WT or V370D mutant) and hygromycin‐resistant plasmid were cotransfected in a 10:1 ratio into CHO‐METKO or PC‐9 METKO cells in 60‐mm dishes using Lipofectamine LTX with Plus transfection reagent (ThermoFisher Scientific) according to the manufacturer's instructions. After 24 hours, cells were selected by culturing in growth medium supplemented with Hygromycin B (for CHO METKO cells, 300 μg/mL; for PC‐9 METKO cells, 100 μg/mL) for 10 days. Stably expressing cells were obtained through limiting dilution and confirmed by western blotting using anti‐MET antibody.
2.5. Western blot and RTK array analysis
Cells were seeded at 3 × 105 per well in 12‐well plates and cultured overnight. After serum starvation for 4 hours, cells were stimulated with HGF for 10 minutes, followed by washing with ice‐cold PBS, lysis in 100 μL of 1 × SDS‐PAGE Laemmli sample buffer (FUJIFILM Wako Pure Chemical), and ultrasonification (Sonics & Materials Inc., Newtown, CT, USA). Cell lysates were analyzed by SDS‐PAGE with a 10% polyacrylamide gel and electroblotted onto a PVDF membrane (Bio‐Rad, Hercules, CA, USA). The membrane was treated with primary antibodies (1:1000), followed by HRP‐conjugated secondary antibodies (Dako, Carpinteria, CA, USA) (1:2000). Chemiluminescence was visualized and quantitated using ImmunoStar LD (FUJIFILM Wako Pure Chemical). For RTK array analysis, cell lysates containing 800 μg protein were analyzed using Proteome Profiler Mouse Phospho‐RTK Array Kit (R&D Systems, Minneapolis, MN, USA), in accordance with the manufacturer's instructions.
2.6. Biological assays
For cell proliferation assays, cells were seeded in growth medium at 1000 per well in 96‐well plates and cultured overnight. Cells were washed once with PBS and cultured with Ham's F12 medium supplemented with 2% FBS for 48 hours. For gefitinib resistance assays, cells were seeded in 96‐well plates at 3000 per well and cultured overnight. Cells were incubated with or without gefitinib (FUJIFILM Wako Pure Chemical) and HGF for 72 hours. To measure the sensitivity to PHA665752, cells were seeded in 96‐well plates at 3000 per well. Viable cell numbers were determined using Cell Titer 96 Aqueous One solution cell proliferation assay kit (Promega, Madison, WI, USA). For scattering assays, cells were seeded in growth medium at 1000 per well in 96‐well plates and cultured overnight. Cells were washed with PBS and cultured with Ham's F12 medium supplemented with 2% FBS for 48 hours. Migration assays were carried out using Transwell chambers (6.5‐mm diameter with 8‐μm pores; Corning, Corning, NY, USA). Cells (2 × 104) were seeded into the upper insert and cultured for 24 hours. They were fixed with 4% paraformaldehyde in PBS and cells attached to the lower side of the insert were stained with 0.2% crystal violet in 20% methanol.
2.7. Receptor dimerization
Receptor dimerization on living cells in culture was done by chemical crosslinking, as described elsewhere.19 Briefly, cells (2 × 106) were seeded in 60‐mm dishes and cultured overnight. After washing twice with ice‐cold Ham's F12 medium supplemented with 5% FBS, cells were stimulated with HGF in Ham's F12 supplemented with 5% FBS for 60 minutes at 4°C. They were then washed with ice‐cold PBS three times and treated with 1 mmol/L bis(sulfosuccinimidyl)suberate (BS3; ThermoFisher Scientific) in PBS for 60 minutes at 4°C. Unreactive BS3 was quenched with 50 mmol/L Tris (pH 8.0) and 150 mmol/L NaCl for 15 minutes at 4°C. MET receptors enriched by immunoprecipitation from cell lysate were subjected to SDS‐PAGE and western blot using anti‐MET antibody (25H2; Cell Signaling Technology Japan), followed by HRP‐conjugated antimouse IgG (Dako), and then visualized by chemiluminescent reaction using ImmunoStar LD (FUJIFILM Wako Pure Chemical).
2.8. Flow cytometry
Cells were stained with FITC‐conjugated anti‐MET antibody (eBioclone 97; ThermoFisher Scientific) in PBS supplemented with 2% FBS for 30 minutes at 4°C. After washing, analysis was carried out using FACSCanto II (Becton Dickinson, Franklin Lakes, NJ, USA).
2.9. Cell surface biotinylation
Cells (1.5 × 106) were seeded in 60‐mm dishes and cultured overnight. After washing with ice‐cold PBS three times, they were incubated with 0.8 mg/mL Sulfo‐NHS‐SS‐Biotin (ThermoFisher Scientific) in PBS at 4°C for 1 hour. Unreactive Sulfo‐NHS‐SS‐Biotin was quenched with 50 mmol/L Tris (pH 8.0) and 150 mmol/L NaCl for 10 minutes at 4°C. Cells were washed with ice‐cold PBS and solubilized in lysis buffer [40 mmol/L Tris‐HCl (pH 8.0), 1% Triton X‐100, 1% NP‐40, 10% glycerol, 0.15 M NaCl, 2 mmol/L EDTA, 1 mmol/L phenylmethanesulfonyl fluoride, 1 × cOmplete Protease Inhibitor Cocktail, Nacalai Tesque, Kyoto, Japan]. Cell lysates were passed through a 27‐G needle five times and centrifuged for 15 minutes at 15 000×g. Supernatants were incubated with 50 μL streptavidin‐coupled magnetic beads (Dynabeads M280 Streptavidin; ThermoFisher Scientific) for 1 hour in a rotating apparatus at room temperature. The beads were washed three times with lysis buffer and treated with SDS‐PAGE Laemmli sample buffer supplemented with 5% mercaptoethanol. Samples were then subjected to western blot.
2.10. Size‐exclusion chromatography analysis
An ÄKTApurifier System (GE Healthcare Life Sciences, Uppsala, Sweden) equipped with a Superdex 200 Increase 10/300 GL column (GE Healthcare Life Sciences) was used. Eighty micrograms of MET‐ECD‐His, 40 μg HGF, or a mixture of both was incubated in PBS with 0.1 mmol/L sucrose octasulfate sodium salt (Toronto Research Chemicals, Toronto, Ontario, Canada) for 10 minutes at room temperature, and then subjected to size‐exclusion chromatography. Samples were eluted with PBS containing 0.1 mmol/L sucrose octasulfate sodium salt at a flow rate of 0.5 mL/min at 4°C. Fractions of 0.3 mL were collected and analyzed using absorbance at 280 nm. Eluted fractions were subjected to SDS‐PAGE using a 5%‐20% polyacrylamide gradient gel and stained using a Rapid CBB Stain Kit (Nacalai Tesque, Kyoto, Japan).
2.11. Surface plasmon resonance analysis
Binding constants of HGF, NK4, and SP to the MET receptor extracellular domains were determined by surface plasmon resonance (SPR) analysis using Biacore 3000 (GE Healthcare UK Ltd, Little Chalfont, UK). To detect the binding constants of HGF and MET receptor extracellular domains by SPR assay, running buffer composed of 10 mmol/L Tris‐HCl (pH 7.5), 150 mmol/L NaCl, and 0.05% (v/v) Tween 20 was used. Biotinylated HGF was immobilized on the streptavidin‐immobilized SPR sensor chip at a surface density of 500 resonance units (RU). Binding abilities were tested by injecting varying concentrations of MET‐ECD‐Fc‐His (WT or V370D) and quantifying them by the multiple cycle titration kinetic method. To determine the binding constants of NK4 and SP to the MET extracellular domain, the running buffer was composed of 10 mmol/L Tris‐HCl (pH 7.5), 300 mmol/L NaCl, and 0.05% (v/v) Tween 20. Biotin‐conjugated antihuman IgG F(c) antibody (Rockland Immunochemicals, Rockland Township, PA, USA) was captured on a streptavidin‐immobilized SPR sensor chip at a surface density of 3000 RU. MET‐ECD‐Fc‐His protein was immobilized to the sensor chip at a density of 500‐600 RU. Binding abilities were tested by injecting varying concentrations of NK4 and SP, and binding kinetics was quantified by the multiple cycle titration kinetic method.
2.12. Statistical analysis
Data were analyzed by one‐way ANOVA followed by Tukey's multiple comparison tests using GraphPad Prism 7. P‐values <.05 were considered statistically significant.
3. RESULTS
3.1. MET‐V370D is inefficiently cleaved and largely loses its response to HGF
Outlines of the structures of MET and HGF, and the position of the V370D mutation are shown in Figure 1A. Val370 is located in the α‐helix (amino acids 367‐375, shown in red in Figure 1B) in the MET‐SEMA domain, which does not participate in interactions with the HGF‐SP domain (Figure 1B). To analyze changes in the biochemical and functional characteristics of MET upon V370D mutation, we prepared endogenous MET‐knockout CHO cells (CHO‐METKO) (Figure 2A), and the CHO‐METKO cell clone was further manipulated to stably express WT human MET (CHO‐WT) or V370D mutant MET (CHO‐V370D) (Figure 2B). SDS‐PAGE under reducing conditions and western blotting analysis showed that the proportion of uncleaved MET relative to cleaved MET was higher in CHO‐V370D cells than in CHO‐WT cells (Figure 2B), indicating inefficient cleavage associated with the V370D mutation. When CHO‐WT cells were stimulated with HGF, MET phosphorylation (p‐MET) at Y1234/1235 and downstream p‐AKT at S473 were increased, indicating that wild‐type human MET was activated by HGF and that MET activation was linked to endogenous signaling molecules. In contrast, p‐MET in CHO‐V370D cells was marginal even at 100 ng/mL HGF (Figure 2B). Consistent with this, a marginal increase in p‐AKT by 100 ng/mL HGF was detected in CHO‐V370D cells (Figure 2B), indicating that HGF scarcely activates V370D mutant MET.
Figure 1.
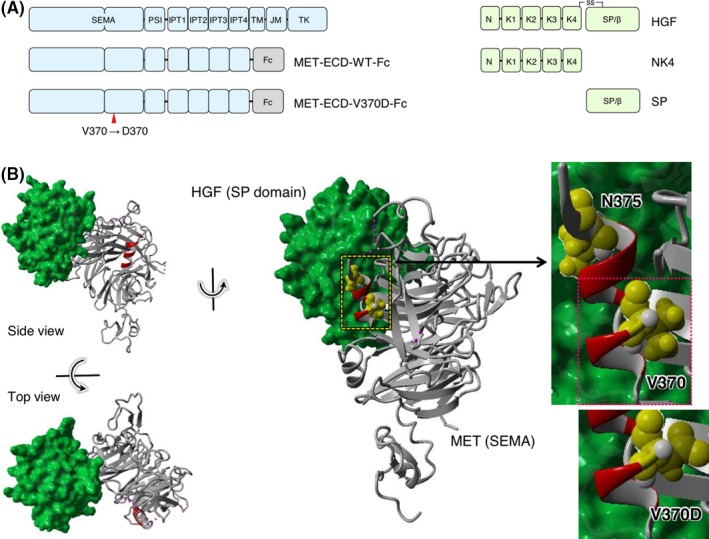
Structures of hepatocyte growth factor (HGF) and the MET receptor. A, Domain architecture of MET, HGF, and their recombinant domains. B, Complex structure composed of the SEMA domain of MET and the SP domain of HGF (PDB number: 1SHY)
Figure 2.
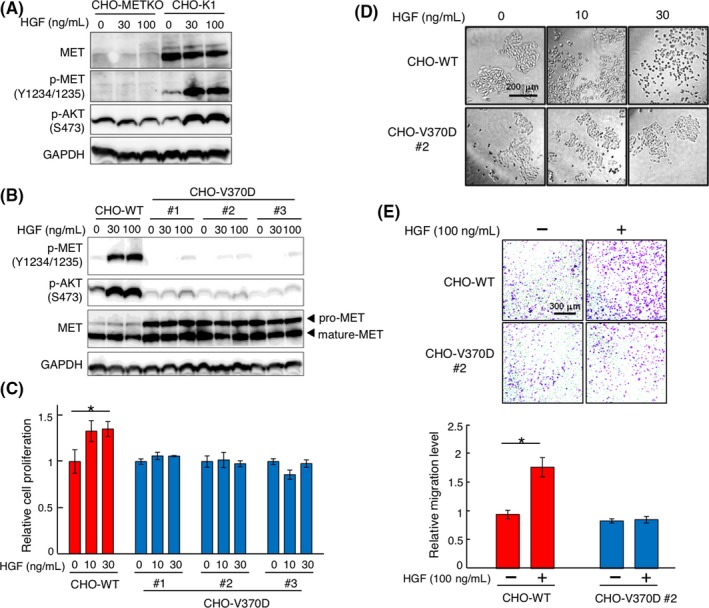
Hepatocyte growth factor (HGF)‐induced responses in CHO‐WT and CHO‐V370D cells. A, Activation of MET and AKT in CHO‐K1 and CHO‐K1 MET knockout (CHO‐METKO) cells. Cells were stimulated with HGF for 10 min. MET, p‐MET, p‐AKT, and GAPDH were detected by western blotting. B, Expression and activation of MET‐WT and MET‐V370D in CHO‐METKO cells and response to HGF. Cells were stimulated with HGF for 10 min. MET, p‐MET, p‐AKT, and GAPDH were detected by western blotting. C, Cell proliferation induced by HGF. Cells were cultured with or without HGF for 48 h. Each value indicates the mean ± SE of triplicate measurements. Asterisk indicates P < .05. D, HGF‐induced cell scattering. Cells were cultured with or without HGF for 48 hours, representative images of which are shown. E, HGF‐induced cell migration. Cells were seeded on Transwell membranes and cultured for 24 h. Each value indicates the mean ± SE of triplicate measurements. Asterisk indicates P < .05. Representative images are shown
Because HGF‐induced biological activities depend on MET receptor activation and subsequent signaling, we examined HGF‐induced cell proliferation, scattering and migration of CHO‐V370D cells. HGF significantly facilitated the growth of CHO‐WT cells but not CHO‐V370D cells (Figure 2C). HGF strongly induced the scattering and migration of CHO‐WT cells but not CHO‐V370D cells (Figure 2D,E). These results suggest that the V370D mutation is a loss‐of‐function mutation regarding HGF‐induced activation of the MET receptor.
3.2. V370D abrogates HGF‐dependent EGFR‐TKI resistance in PC‐9 tumor cells
Involvement of the HGF‐MET receptor pathway in resistance against EGFR‐TKI was well characterized in previous studies.20, 21, 22 To test whether the V370D mutation affects EGFR‐TKI resistance, we prepared PC‐9 human lung adenocarcinoma cells deficient in endogenous MET receptor (PC‐9 METKO) (Figure 3A). These cells were manipulated to stably express human wild‐type MET (PC‐9 WT) or V370D mutant MET (PC‐9 V370D) at similar levels (Figure 3B). In PC‐9 METKO cells, MET was undetectable and HGF failed to induce p‐MET and p‐AKT, whereas HGF increased p‐MET and p‐AKT levels in PC‐9 WT cells (Figure 3A,B). In contrast, basal phosphorylation levels of p‐MET and p‐AKT were different related to clonal variation (#1 and #2); however, p‐MET and p‐AKT levels were unchanged or marginally increased by HGF in PC‐9 V370D cells (Figure 3B).
Figure 3.
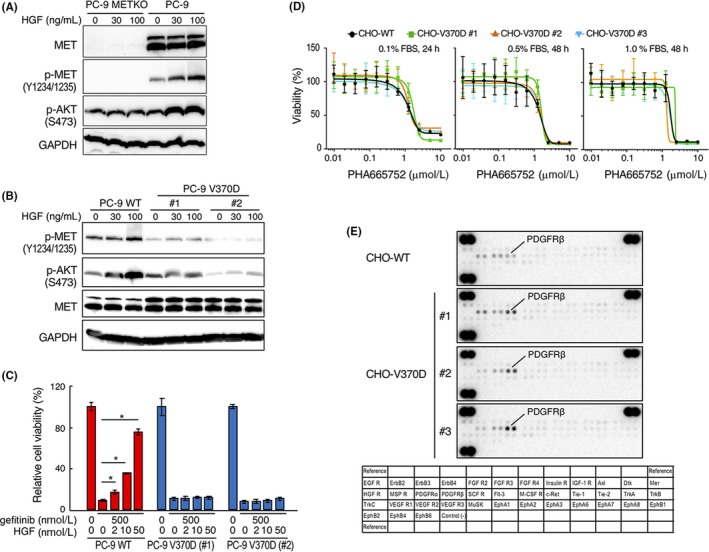
Distinct responses mediated by WT and V370D mutant MET. A, Expression and tyrosine phosphorylation of MET receptor in PC‐9 MET receptor‐knockout (PC‐9 METKO) cells. Cells were stimulated with HGF for 10 min. MET, p‐MET, p‐AKT, and GAPDH were detected by western blotting. B, Expression and activation of MET‐WT and MET‐V370D and response to HGF stimulation. Cells were stimulated with HGF for 10 min. MET, p‐MET, p‐AKT, and GAPDH were detected by western blotting. C, Cell viability after treatment with gefitinib and hepatocyte growth factor (HGF) in PC‐9 WT and PC‐9 V370D cells. Cells were treated with or without gefitinib or HGF for 3 d. Data represent mean ± SE (n = 3). *P < .05. D, Effect of MET kinase inhibitor on viability of CHO‐WT and CHO‐V370D cells. Cells were cultured in the absence or presence of PHA665752 in medium supplemented with different FBS concentrations for 24 or 48 h. Data represent mean ± SD (n = 4). E, Change in phosphorylation of RTK in CHO‐WT cells and CHO‐V370D cells (#1‐#3, independent clones) cultured under survival/growth‐stressed conditions. The cells were cultured in medium supplemented with 0.01% FBS and 20 ng/mL HGF for 10 d
We next examined the resistance characteristics of PC‐9 lung cancer cells to EGFR‐TKI gefitinib. In PC‐9 WT cells, cell growth was inhibited by gefitinib, but HGF rescued cell growth dose‐dependently even in the presence of gefitinib (Figure 3C). PC‐9 V370D growth was inhibited by gefitinib; however, HGF did not rescue cell growth inhibited by gefitinib. Taken together, these results indicate that HGF hardly activates V370D‐MET, which is associated with impaired biological responses to HGF.
Because N375S mutant MET conferred mild resistance to MET‐TKI,15 we tested the sensitivity of V370D‐MET to MET‐TKI (Figure 3D). Sensitivity to PHA665752 MET‐TKI was not changed between wild‐type MET and V370D mutant MET. To further address the potential involvement of V370D mutant MET in regulating cell characteristics, CHO cells expressing wild‐type or V370D‐MET were cultured under survival/growth‐stressed conditions induced by low‐level FBS in the presence of HGF and the change in phosphorylation of RTK was analyzed (Figure 3E). RTK array analysis indicated that the phosphorylation level of platelet‐derived growth factor receptor‐β (PDGFRβ) was higher in CHO‐V370D cells than in CHO‐WT cells. This implies that V370D‐MET may influence the availability or activation status of RTK under certain conditions.
3.3. MET‐V370D is normally located on the cell surface but is not dimerized by HGF
The impaired activation of V370D‐MET by HGF suggested a defect in the biochemical processes of MET activation. Because receptor dimerization is an essential step for the activation of RTK, we performed chemical cross‐linking to analyze MET dimerization (Figure 4A). CHO‐WT and CHO‐370D cells cultured in the absence or presence of HGF were treated with a chemical cross‐linker and subjected to SDS‐PAGE and western blotting. Addition of HGF induced detectable dimerization of MET receptors in CHO‐WT cells but not in CHO‐V370D cells (Figure 4A). This indicated that HGF cannot efficiently induce the dimerization of MET‐V370D and is incapable of efficiently activating MET.
Figure 4.
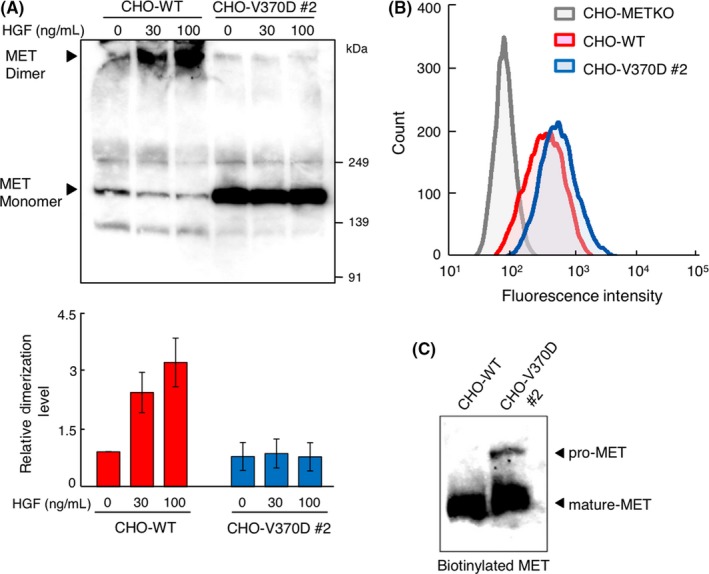
MET receptor dimerization and cell surface localization of V370D‐MET. A, MET receptor dimerization induced by hepatocyte growth factor (HGF). CHO‐WT and CHO‐V370D cells were stimulated with HGF followed by treatment with a cross‐linker bis(sulfosuccinimidyl)suberate (BS3). MET dimerization was analyzed by immunoprecipitation and western blotting using an anti‐MET antibody. Relative MET dimerization level was calculated by the band intensity in western blots. Each value indicates the mean ± SE of triplicate measurements. Representative image is shown. B, Cell surface expression of MET receptor. Cells were stained with FITC‐conjugated anti‐MET antibody and cell surface expression of MET was analyzed by flow cytometry. C, Cell surface MET receptor detected by biotinylation assay. Biotinylated cell surface proteins were pulled down, and cell surface MET was detected by western blotting using an anti‐MET antibody
To examine whether MET‐V370D was located on the cell membrane, we analyzed the cell surface expression of MET in CHO‐WT and CHO‐V370D cells by flow cytometry. Cell surface MET receptors were mostly undetectable in CHO‐METKO cells, but were successfully detected in CHO‐WT and CHO‐V370D cells (Figure 4B). Using another approach, the localization of MET was tested by the biotinylation of cell surface proteins labeled with NHS‐biotin, solubilized, pulled down using streptavidin beads, and subjected to western blotting with anti‐MET antibody. The levels of biotinylated MET were comparable between CHO‐WT and CHO‐V370D cells (Figure 4C). Thus, MET‐V370D is translocated to the cell membrane comparably to MET‐WT.
3.4. V370D mutation decreases MET‐HGF binding ability by blocking NK4 binding
To further investigate the mechanism underlying how the V370D mutation affects HGF‐induced MET receptor dimerization, the binding properties of MET‐WT and MET‐V370D to HGF were analyzed. Extracellular domains of MET‐WT (MET‐ECD‐WT) and MET‐V370D (MET‐ECD‐V370D) were prepared and their associations were analyzed by size‐exclusion chromatography and SDS‐PAGE (Figure 5). The elution profile of a mixture of HGF and MET‐ECD‐WT indicated that MET‐ECD‐WT was eluted with HGF as a major peak and the position of the major peak was shifted to earlier fractions than those of individual MET‐ECD‐WT and HGF (Figure 5, left). Protein staining after SDS‐PAGE indicated that the major peak was composed of MET‐ECD‐WT and HGF. This indicates that MET‐ECD‐WT associates with HGF. In contrast, for mixtures of MET‐ECD‐V370D and HGF, the elution profile indicated that these proteins formed two peaks, the positions of which were similar to those of individual MET‐ECD‐V370D and HGF (Figure 5, right). Protein staining of these peaks indicated a marginal band for HGF that was present in earlier and major fractions for MET‐ECD‐V370D, but MET‐ECD‐V370D and HGF were eluted separately. Therefore, MET‐ECD‐V370D has a low association with HGF in solution.
Figure 5.
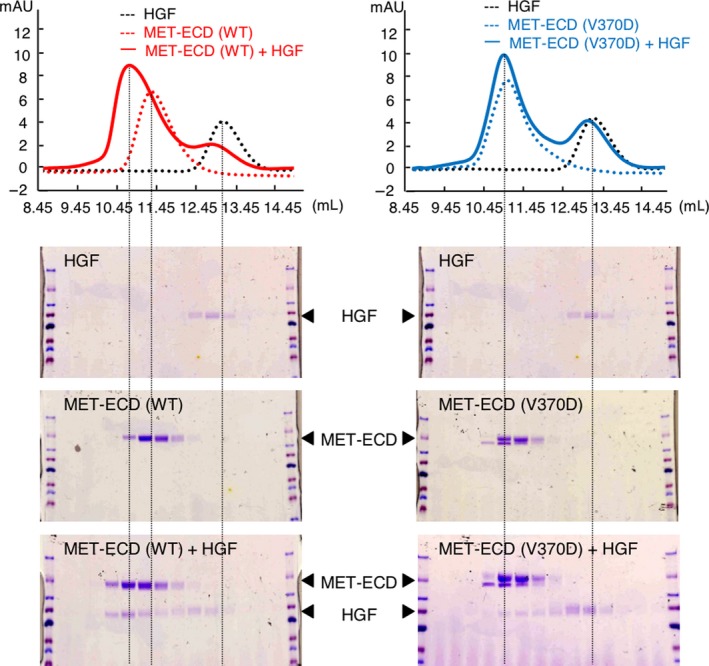
Binding of hepatocyte growth factor (HGF) to MET‐WT and MET‐V370D as evaluated by size‐exclusion chromatography. HGF and MET‐ECD‐His (WT or V370D) were subjected to size‐exclusion chromatography alone or as a mixture. Eluted fractions were analyzed by SDS‐PAGE and protein staining with Coomassie Brilliant Blue
To further evaluate the association between MET‐ECD and HGF, association kinetics was analyzed by SPR (Figure 6A). MET‐ECD‐Fc‐WT and MET‐ECD‐Fc‐V370D were prepared. Concentration‐dependent association kinetics of MET‐ECD‐Fc‐WT to immobilized HGF was calculated and showed high‐affinity association with a dissociation constant (K D) of 10 nmol/L. Analysis of the association kinetics between MET‐ECD‐Fc‐V370D and HGF showed a KD value of 45 nmol/L, indicating a decrease in the affinity to HGF compared with MET‐ECD‐Fc‐WT. As previous studies reported that HGF bound to MET receptor by using two independent binding interfaces located in NK4 and SP,23, 24 NK4 and SP were prepared and their association to MET‐ECD‐Fc (WT or V370D) was evaluated by SPR analysis. The SP domain of HGF bound to MET‐ECD‐Fc‐WT and MET‐ECD‐Fc‐V370D equally with K D values of 858 nmol/L and 914 nmol/L, respectively (Figure 6B). NK4 bound to MET‐ECD‐Fc‐WT with a K D value of 7 nmol/L. However, NK4 showed low binding affinity to MET‐ECD‐Fc‐V370D and the K D value could not be calculated (Figure 6C). Taken together, these results demonstrate that the V370D mutation in MET impairs association with the NK4 domain in HGF, which decreases its association with HGF.
Figure 6.
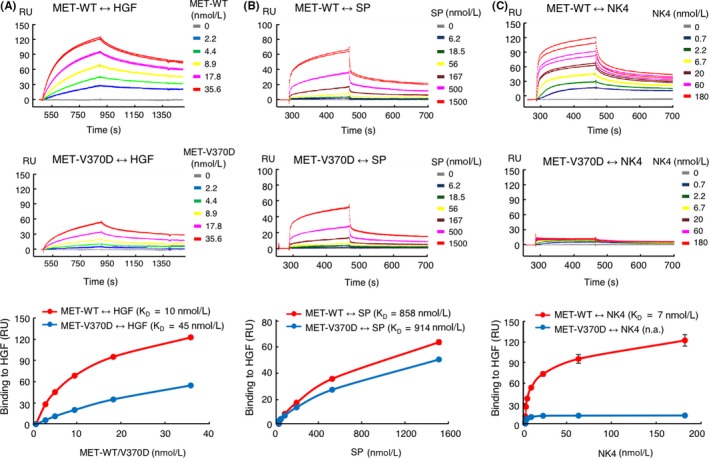
Binding of hepatocyte growth factor (HGF), SP, and NK4 to MET‐WT and MET‐V370D. Binding kinetics of HGF (A), SP (B), and NK4 (C) to MET‐WT or MET‐V370D was measured by surface plasmon resonance (SPR) analysis. In (A), biotinylated HGF was immobilized on a sensor chip and binding of MET‐ECD‐Fc (WT or V370D) was measured (n = 2). In (B) and (C), MET‐ECD‐Fc‐His (WT or V370D) was immobilized on a sensor chip and binding of SP or NK4 was measured (n = 2)
4. DISCUSSION
Biochemical analysis of separately prepared NK4 and SP indicated that NK4 binds to MET but does not activate MET; however, MET activation and MET‐dependent biological activities were reconstituted by combining NK4 and SP.23 Crystallographic analysis indicated that Thr124–Asp128 and Asp190–Phe192 in the SEMA domain of MET provide a binding interface to the SP domain of two‐chain HGF.25 Taking these findings together, HGF has two MET‐binding interfaces individually within NK4 and SP. The functional binding of these interfaces to MET is required for efficient activation of MET in a physiological context. Substantial loss of binding between the NK4 domain and mutant V370D‐MET indicates why it is a loss‐of‐function mutation.
Because the crystallographic structure of the association between the NK4 domain of HGF and MET has not been obtained, it cannot be explained why replacing Val370 with Asp370 prevents binding to HGF. Because Val370 is not located in the face that interacts with the SP domain (Figure 1B),25 Val370 might influence interactions with the NK4 domain directly or indirectly. Val370 is located in the α‐helix (amino acids 367‐375) and extends to the hydrophobic core of the SEMA domain.25 The replacement of Val to Asp changes the chemical characteristics from a hydrophobic to a negatively charged side chain. This change might induce unstable interactions of the α‐helix with NK4/HGF or structural changes that affect α‐helix orientation. In this context, a missense mutation of Asn375 located in α‐helix‐367‐375 is consistently found in different types of malignant tumors including lung cancer.11, 14 Asn375 to Lys375 replacement in MET reduced the affinity to HGF.16 Taking these findings together, α‐helix‐367‐375 may play a role in the association with HGF; thus, a change in orientation and/or position of α‐helix‐367‐375 might affect interactions between MET and HGF.
The N375K missense mutation in MET was identified by whole‐exome sequencing as the most likely causative mutation found in siblings affected by lung adenocarcinoma with EGFR mutation.16 Functional analysis of Asn375 to Lys375 replacement indicated that the association of HGF with Lys375‐MET was reduced and biological responses to HGF in cells expressing this mutant MET were decreased compared with those for wild‐type Asn375‐MET,16 indicating that N375K is a partial loss‐of‐function mutation of the MET receptor. MET with mutation located in the TK domain is constitutively active or susceptible to activation, and such gain‐of‐function mutations in RTK play a constitutive role in oncogenic alterations of cells. How loss‐of‐function mutations are associated with progression to malignant diseases cannot be explained. However, a recent study indicated that an inactive Braf mutation augmented MAPK signaling through the compensatory regulation of intracellular signaling, which promoted lung adenocarcinoma.26 An inactivating mutation in a signaling molecule may provide intrinsic pressure to replace signal activation. We found a difference in activation status in RTK between the cells expressing wild‐type and V370D‐MET under survival/growth‐stressed conditions. Although we cannot rule out the possibility that V370D mutations in lung adenocarcinoma patients14 are passenger mutations that do not substantially participate in the oncogenic process, the V370D mutation may change the dependence on other RTK and/or downstream signaling molecules, which may allow the expansion of cancer cells with V370D mutant MET under certain conditions.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
ACKNOWLEDGMENTS
This work was supported in part by the Project for Cancer Research and Therapeutic Evolution (P‐CREATE) from the Japan Agency for Medical Research and Development (AMED) to K.M., the Medical Research Fund of Takeda Science Foundation, the Mitani Foundation for Research and Development, JSPS Grant‐in‐Aid for Scientific Research (C) (16K08544) to K.S., and a Grant‐in‐Aid for Young Scientists (18K15306) to H.S. We thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Miao W, Sakai K, Sato H, et al. Impaired ligand‐dependent MET activation caused by an extracellular SEMA domain missense mutation in lung cancer. Cancer Sci. 2019;110:3340‐3349. 10.1111/cas.14142
REFERENCES
- 1. Park M, Dean M, Kaul K, Braun MJ, Gonda MA, Vande Woude G. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth‐factor receptors. Proc Natl Acad Sci USA. 1987;84:6379‐6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Comoglio PM, Trusolino L, Boccaccio C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat Rev Cancer. 2018;18:341‐358. [DOI] [PubMed] [Google Scholar]
- 3. Komada M, Hatsuzawa K, Shibamoto S, Ito F, Nakayama K, Kitamura N. Proteolytic processing of the hepatocyte growth factor/scatter factor receptor by furin. FEBS Lett. 1993;328:25‐29. [DOI] [PubMed] [Google Scholar]
- 4. Tempest PR, Stratton MR, Cooper CS. Structure of the met protein and variation of met protein kinase activity among human tumour cell lines. Br J Cancer. 1988;58:3‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gherardi E, Youles ME, Miguel RN, et al. Functional map and domain structure of MET, the product of the c‐met protooncogene and receptor for hepatocyte growth factor/scatter factor. Proc Natl Acad Sci. 2003;100:12039‐12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matsumoto K, Umitsu M, De Silva DM, Roy A, Bottaro DP. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. 2017;108:296‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Organ SL, Tsao MS. An overview of the c‐MET signaling pathway. Ther Adv Med Oncol. 2011;3:S7‐S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gherardi E, Birchmeier W, Birchmeier C, Vande Woude GF. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89‐103. [DOI] [PubMed] [Google Scholar]
- 9. Schmidt L, Duh FM, Chen F, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto‐oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68‐73. [DOI] [PubMed] [Google Scholar]
- 10. Petrini I. Biology of MET: a double life between normal tissue repair and tumor progression. Annal Transl Med. 2015;3:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sattler M, Reddy MM, Hasina R, Gangadhar T, Salgia R. The role of the c‐Met pathway in lung cancer and the potential for targeted therapy. Ther Adv Med Oncol. 2011;3:171‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kong‐Beltran M, Seshagiri S, Zha J, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006;66:283‐289. [DOI] [PubMed] [Google Scholar]
- 13. Ma PC, Kijima T, Maulik G, et al. c‐MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63:6272‐6281. [PubMed] [Google Scholar]
- 14. Ma PC, Tretiakova MS, MacKinnon AC, et al. Expression and mutational analysis of MET in human solid cancers. Genes Chromosom Cancer. 2008;47:1025‐1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krishnaswamy S, Kanteti R, Duke‐Cohan JS, et al. Ethnic Differences and Functional Analysis of MET Mutations in Lung Cancer. Clin Cancer Res. 2009;15:5714‐5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tode N, Kikuchi T, Sakakibara T, et al. Exome sequencing deciphers a germline MET mutation in familial epidermal growth factor receptor‐mutant lung cancer. Cancer Sci. 2017;108:1263‐1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Niwa H, Yamamura K, Miyazaki J. Efficient selection for high‐expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193‐199. [DOI] [PubMed] [Google Scholar]
- 18. Suda T, Nagata S. Purification and characterization of the fas‐ligand that induces apoptosis. J Exp Med. 1994;179:873‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ito K, Sakai K, Suzuki Y, et al. Artificial human Met agonists based on macrocycle scaffolds. Nat Commun. 2015;6:6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039‐1043. [DOI] [PubMed] [Google Scholar]
- 21. Yano S, Wang W, Li Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor‐activating mutations. Cancer Res. 2008;68:9479‐9487. [DOI] [PubMed] [Google Scholar]
- 22. Nanjo S, Arai S, Wang W, et al. MET copy number gain is associated with gefitinib resistance in leptomeningeal carcinomatosis of EGFR‐mutant Lung Cancer. Mol Cancer Ther. 2017;16:506‐515. [DOI] [PubMed] [Google Scholar]
- 23. Matsumoto K, Kataoka H, Date K, Nakamura T. Cooperative interaction between α‐ and β‐chains of hepatocyte growth factor on c‐Met receptor confers ligand‐induced receptor tyrosine phosphorylation and multiple biological responses. J Biol Chem. 1998;273:22913‐22920. [DOI] [PubMed] [Google Scholar]
- 24. Tolbert WD, Daugherty‐Holtrop J, Gherardi E, Vande Woude G, Xu HE. Structural basis for agonism and antagonism of hepatocyte growth factor. Proc Natl Acad Sci U S A. 2010;107:13264‐13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stamos J, Lazarus RA, Yao X, Kirchhofer D, Wiesmann C. Crystal structure of the HGF β‐chain in complex with the Sema domain of the Met receptor. EMBO J. 2004;23:2325‐2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nieto P, Ambrogio C, Esteban‐Burgos L, et al. A Braf kinase‐inactive mutant induces lung adenocarcinoma. Nature. 2017;548:239‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]