

Abstract
We previously established a method to generate myeloid cells with a proliferative capability from pluripotent stem cells and designated them iPS‐ML. Human iPS‐ML cells share features with physiological macrophages including the capability to infiltrate into cancer tissues. We observed therapeutic effects of human iPS‐ML cells expressing interferon β (iPS‐ML/interferon (IFN)‐β) in xenograft cancer models. However, assessment of host immune system‐mediated therapeutic and adverse effects of this therapy is impossible by xenograft models. We currently evaluated the therapeutic effects of a mouse equivalent of human iPS‐ML/IFN, a mouse embryonic stem (ES) cell‐derived myeloid cell line producing IFN (ES‐ML/IFN). The ES‐MLs producing IFN‐β (β‐ML) and IFN‐γ (γ‐ML) and originating from E14 ES cells derived from the 129 mouse strain (H‐2b) were generated, and the MHC (H‐2K b , D b , and I‐A b) genes of the ES‐ML/IFN were disrupted using the clustered regularly interspaced short palindromic repeats (CRISPR)/CAS9 method. We used the ES‐ML/IFN to treat allogeneic BALB/c mice (H‐2d) transplanted with Colon26 cancer cells. Treatment with β‐ML but not with γ‐ML cells repressed the growth of colon cancer in the peritoneal cavity and liver. The transferred ES‐ML/IFN infiltrated into cancer tissues and enhanced infiltration of T cells into cancer tissues. ES‐ML/IFN therapy increased the number of immune cells in the lymphoid organs. Sensitization of both cancer antigen‐specific CD8+ T cells and natural killer (NK) cells were enhanced by the therapy, and CD8+ T cells were essential for the therapeutic effect, implying that donor MHC‐deficient β‐ML exhibited a therapeutic effect through the activation of host immune cells derived from allogeneic recipient mice. The results suggested the usefulness of HLA‐deficient human iPS‐ML/IFN‐β cells for therapy of HLA‐mismatched allogeneic cancer patients.
Keywords: embryonic stem cell, immunotherapy, interferon, macrophage, MHC
1. INTRODUCTION
Interferon (IFN)‐α, ‐β, and ‐γ exert direct anticancer effects via growth inhibition or death induction of cancer cells.1, 2, 3, 4, 5, 6, 7 In addition, activation of immune cells and inhibition of cancer angiogenesis by IFNs is known.8, 9, 10, 11 However, the clinical use of IFNs as anticancer agents is limited. The restricted clinical merit of IFNs is due to their rapid inactivation and inefficient tissue penetration after intravenous injection.12 In addition, systemic toxicity precludes high‐dose intravenous administration.13, 14, 15, 16 Drug delivery systems (DDSs) that make IFNs act specifically on cancer tissues may significantly improve the therapeutic benefits of IFNs.
Infiltration of macrophages is observed in various cancer tissues.17, 18, 19, 20, 21, 22, 23, 24 This infiltration depends on their expression of receptors for many kinds of leukocyte‐attracting factors such as chemokines, complement proteins, and various damage‐associated molecular pattern molecules (DAMPs).25, 26 We hypothesized that macrophages producing IFNs may be useful as DDS for cancer therapy.
We previously established a method to induce the proliferation of human induced pluripotent stem (iPS) cell‐derived myeloid cells to generate iPS cell‐derived myeloid/macrophage cell lines (iPS‐ML).27, 28, 29 Similar to physiological macrophages, iPS‐ML cells expressed various receptors related to tissue infiltration. When human iPS‐ML cells were injected into severe combined immunodeficiency (SCID) mice bearing cancers (xenograft cancer models), iPS‐ML cells accumulated and infiltrated into the cancer tissues, indicating a cancer‐directed migration capacity of iPS‐ML. iPS‐ML transduced with an expression vector for IFN‐β (iPS‐ML/IFN‐β) exhibited a therapeutic effect in several xenograft cancer models.30, 31
However, from experiments with xenograft cancer models, we could not evaluate the indirect therapeutic effects of iPS‐ML/IFN mediated by the host immune system. In addition, IFN toxicity could not be assessed by xenograft experiments because of the species differences in IFN‐β and IFN‐γ between humans and mice. To examine both the beneficial and adverse effects of iPS‐ML/IFN therapy, experiments using syngeneic cancer models with immunocompetent mice must be necessary. Because a iPS‐ML/IFN derived from mouse iPS cells has not yet been developed in our laboratory, in the current study we utilized 129Sv mouse ES cell‐derived myeloid cell lines producing IFNs (ES‐ML/IFN) as donor cells to treat mice in an allogeneic and cancer‐bearing mouse model, assuming the therapy with human iPS‐ML/IFN cells. In the current study, we disrupted the genes encoding H‐2Kb, Db, and I‐Aβb molecules in the 129Sv mouse (H‐2b)‐derived ES‐ML/IFN using the clustered regularly interspaced short palindromic repeats (CRISPR) method. We transferred these cells into cancer‐bearing allogeneic BALB/c mice (H‐2d) to investigate whether the MHC‐deficient ES‐ML/IFN can be used for cancer therapy in MHC‐mismatched recipients.
iPS‐ML cells can be expanded using a simple suspension culture procedure, and an automated cell culture system is applicable to provide sufficient quantities of cells required for clinical use. We presumed that we could use human leukocyte antigen (HLA)‐A/B/DR‐deficient iPS‐ML/IFN cells for treatment of any cancer patient irrespective of their HLA type, by avoiding acute rejection. If this concept is proven, mass production of cellular pharmaceuticals for anticancer therapy can be realized.
2. MATERIALS AND METHODS
2.1. Cell lines
Colon26, colon cancer cells, BALB‐MC, breast cancer cells, and SP2/0, myeloma cells derived from the BALB/c mouse strain were provided from RIKEN, JCRB Cell Bank, and the European Collection of Authenticated Cell Cultures (ECACC), respectively.
2.2. Generation of mouse ES cell‐derived myeloid cells producing IFNs
E14 ES cells derived from the 129Sv mouse strain were seeded onto a feeder cell layer of OP9 mouse bone marrow stromal cells. On d 5 or 6, cells were harvested, reseeded onto fresh OP9 cell layers, and cultured in the presence of granulocyte‐macrophage colony stimulating factor (GM‐CSF) (1000 U/mL). On d 13 and 14, myeloid lineage cells were recovered,32 and transduced with a lentiviral expression vector CSIIEF33 for cMYC to establish the ES‐ML line.34 Subsequently, ES‐ML cells were transduced with expression vectors for mouse GM‐CSF and M‐CSF. To generate ES‐ML lines producing high amounts of IFNs, genes encoding for the components of type I and type II IFN receptors were disrupted using zinc finger nucleases (Sigma CKOZFND33655 for ifnar2 and CKOZFND33659 for ifngr1) before introduction of expression vectors for mouse IFN‐β and IFN‐γ, respectively.
2.3. Disruption of genes encoding for H‐2K, D, and I‐Aβ in the ES‐ML/IFN
The E14 ES cell‐derived ES‐ML/IFN were transduced with a CAS9 nuclease expression vector together with guide RNA (gRNA) expression vectors by lentivirus vectors. The target sequence of gRNA for H‐2K b and H‐2D b was GAATCCGAGATATGAGCCGC and that for I‐Aβb was GGCTGCTGTGGTGGTGCTGA. After transduction of CRISPR/CAS9 vectors, the ES‐ML/IFN deficient for expression of H‐2K, H‐2D, and I‐A were purified using anti‐H‐2Kb, H‐2Db, and I‐Ab antibodies and a magnetic microbead‐based cell purification system (Miltenyi Biotec). The absence of specific MHC molecules was verified by flow cytometric analysis.
2.4. Analysis of sensitivity of Colon26 cells to IFN‐β and IFN‐γ
Colon26 cells expressing firefly luciferase (Colon26/Luc)34 were cultured in the presence of recombinant mouse (rm) IFN‐β (10 ng/mL) and/or rmIFN‐γ (10 ng/mL). After 3 d, luciferase substrate (Steadylite plus, Perkin‐Elmer) was added, and luminescence activity was quantified using a microplate reader (TriStar; Berthold Technologies). To detect apoptosis, the cells were stained with annexin V‐fluorescein isothiocyanate (FITC). To assess live cells based on nicotinamide adenine dinucleotide (NADH) quantification using the water‐soluble disulfonated tetrazolium salt, the Cell Counting Kit‐8 (CCK‐8; Dojindo Molecular Technologies) was used. To analyze proliferative activity, cells were centrifuged at 270 g for 6 min, fixed with cold 70% ethanol, incubated for 1 h at −20°C, and stained with an anti‐Ki67 antibody (clone REA183; Miltenyi Biotec).
2.5. Analysis of in vivo anticancer effects of the ES‐ML/IFN
Mouse experiments were performed under the approval of the Animal Research Committee of Kumamoto University (Approval number: A 27‐069). Next, 6−8‐wk‐old BALB/c mice were purchased from Kyudo CO., Ltd., CLEA Japan, Inc., and Japan SLC, and housed under specific pathogen‐free conditions at the Center for Animal Resources and Development (CARD, Kumamoto University).
Colon26/Luc cells (4 × 106 cells/mouse) were injected intraperitoneally (ip) into BALB/c mice. In another model, Colon26/Luc cells (5 × 105 cells/mouse) were injected into the liver using a 29G needle and syringe under laparotomy. Mice anesthetized by inhalation of isoflurane were injected ip with 2.5 mg luciferin and subjected to imaging analysis using an in vivo imaging system (NightOWL II; Berthold Technologies). Cancer‐bearing mice were treated by injection ip of ES‐ML cells that were producing IFN‐β (β‐ML) or IFN‐γ (γ‐ML).
2.6. Analysis of ES‐ML/IFN infiltration into cancer tissues
Green fluorescent protein (GFP)‐expressing Colon26 cells (Colon26/GFP, 2 × 106 cells/mouse) were injected ip into mice. After 3 d, β‐ML (1.6 × 107) and γ‐ML cells (4 × 106) were labeled with a red fluorescent linker PKH26 (Sigma) and injected ip. The next day, mice were euthanized, and the locations of Colon26/GFP cells and the PKH26‐labeled ES‐ML/IFN were detected macroscopically by fluorescence at excitation wavelengths of 475 and 500 nm and emission filters of 520 and 600 nm, respectively, using the NightOWL II. For microscopic examination, β‐ML and γ‐ML labeled with PKH26 were injected ip at 10 d after inoculation of Colon26/GFP cells. The next day, cancer tissues were isolated, fixed in 4% paraformaldehyde/phosphate‐buffered saline, and embedded in Tissue‐TEK OCT (Sakura Fine Technical). Tissue sections of 4‐6‐μm thickness were prepared using a cryostat and analyzed by fluorescence microscopy (Axio Observer Z1; Carl Zeiss).
2.7. Analysis of CD4+ cells and CD8+ cells infiltration into cancer tissues
Cancer nodules on the mesentery were resected from mice and frozen sections (4‐μm thickness) were prepared, fixed with cold acetone, incubated with 1% bovine serum albumin/TBS‐T with NaN3 for blocking, and stained with anti‐CD4 (GK1.5) and anti‐CD8 (53‐6.72) monoclonal antibodies. The sections were treated with a horseradish peroxidase‐conjugated anti‐rat secondary antibody (Nichirei). Diaminobenzidine (DAB; Nichirei) was used to visualize antibody reactions. Nuclei were counterstained with hematoxylin.
2.8. Flow cytometric analysis
The following monoclonal antibodies were used: FcR‐blocking antibody (anti‐mouse CD16/CD32, clone 2.4G2; BD Pharmingen), FITC, PE, PerCP, or PerCP/Cy5.5‐conjugated anti‐CD335 (clone 29A1.4; BioLegend), anti‐CD49b (clone HMα2; BioLegend), anti‐CD11b (clone M1/70; BioLegend), anti‐CD4 (clone RM4‐5; eBioscience), anti‐CD8a (clone 53‐6.7; eBioscience), anti‐CD45.2 (clone 104; BioLegend), anti‐IFN‐γ (clone XMG1.2; BioLegend), Rat IgG2a,k (BD Pharmingen), Rat IgG2b, k control (BD Pharmingen), Rat IgG1k isotype‐matched control Abs (BioLegend), and PE/Cy7‐conjugated Armenian hamster IgG (BioLegend) Abs.
Cells were labeled at 4°C with specific antibodies. For intracellular cytokine analysis, cells were resuspended in RPMI‐1640 medium supplemented with 20% FBS, and phorbol 12‐myristate 13‐acetate (50 ng/mL), ionomycin (500 ng/mL), and brefeldin A (10 μg/mL; all from Sigma) were added. After incubation for 4 h, the cells were washed, stained for surface molecule markers, and fixed and permeabilized using IntraPrep reagent (Beckman Coulter), followed by intracellular staining of IFN‐γ. The stained cells were analyzed using a FACSCalibur (BD Biosciences).
2.9. Life span of β‐ML in peritoneal dissemination model
Colon26 cells (4 × 106 cells/mouse) were injected ip into BALB/c mice. At 2 d later, β‐ML labeled with a red fluorescent linker PKH26 (Sigma) were injected ip (3 × 106 cells/mouse). Peritoneal lavage fluid was collected at 6, 48 and 96 h after injection of PKH26‐labeled β‐ML. The proportion of PKH26‐labeled β‐ML was analyzed by flow cytometric analysis.
2.10. Evaluation of the anticancer effect of CD8+ T cells in vitro
Colon26/Luc cells (4 × 106 cells/mouse) were injected ip to establish a peritoneal dissemination model. Mice were divided into treatment (administration of both β‐ML and γ‐ML) and control (no treatment) groups. Treatment group mice were administrated twice from d 4 with a mixture of β‐ML and γ‐ML. CD8+ T cells were isolated from mesenteric lymph nodes (MLNs) and para‐aortic lymph nodes (PANs) using magnetic beads (CD8 MicroBeads for mouse; Miltenyi Biotec). The CD8+ T cells (1 × 104 cells/well) were cultured with Colon26/Luc cells (5 × 103 cells/well). Luciferase substrate solution was added (50 μL/well) after 3 d of culture, and luminescence was measured.
2.11. Enzyme‐linked ImmunoSpot (ELISPOT) analysis of IFN‐γ production
Spleens and MLNs were resected from cancer‐bearing mice and dissociated mechanically to prepare cell suspensions. The prepared cells (1 × 105/well) were cultured with Colon26 or BALB‐MC cells (5 × 104/well). Then the number of cells producing IFN‐γ was measured by ELISPOT assay. In some experiments, the cells were cultured in the presence of a murine leukemia virus‐derived gp70 peptide (SPSYVYHQF) instead of Colon26 cells. In other assays, IFN‐γ production by purified CD8+ T cells was measured. The frequencies of IFN‐γ‐producing cells in natural killer (NK) cells isolated with an NK Cell Isolation Kit II (Miltenyi Biotec) were also analyzed by ELISPOT assay using syngeneic SP2/0 cells as NK target cells.
2.12. Depletion of CD4+ and CD8+ T cells in vivo
InVivoMab anti‐mouse CD4 (clone GK1.5; Bio X Cell) and IgG from rat serum (Sigma) were used as an isotype‐matched control antibody. To prepare the anti‐CD8 monoclonal antibody, hybridoma cells (clone 3.155; 1 × 106/mouse) were injected ip into ICR nude mice and ascites were harvested. Ascites were treated with Ascites Conditioning Reagent (Thermo Scientific), desalted with a Scientific™ Zeba™ Spin Desalting Colum (Thermo Scientific), and purified using a Melon Gel IgG Purification Kit (Thermo Scientific). In T cell‐depletion experiments, Colon26/Luc cells (1 × 106 cells/mouse) were injected ip into mice at d 0. Each antibody (50 μg/mouse) was injected ip at d −3, −1, and 6, and depletion of CD4+ and CD8+ T cells was checked by flow cytometry at d 3 and d 6 in blood samples.
2.13. Statistical analysis
When comparing two experimental groups, Student's t test or Mann‐Whitney U test was used. In some experiments, Tukey‐Kramer post‐hoc test after one‐way analysis of variance (ANOVA) was used to perform multiple comparisons. For ELISPOT assay, spot numbers were analyzed using the Poisson regression model. To evaluate the longitudinal fold change of luminescence counts, the mixed model was employed. The log‐rank test was used to analyze mouse survival and given as Kaplan‐Meier plots. These analyses were performed using GraphPad Prism, R version 3.3.1 and SAS Version 9.4 software. A P‐value < .05 was considered as significant. The log‐rank test was used to analyze mouse survival using GraphPad Prism software. A P‐value < .05 was considered as significant.
3. RESULTS
3.1. Pro‐apoptotic and growth‐inhibitory effects of rmIFN‐β and rmIFN‐γ on Colon26 cells
We examined the effect of rmIFN proteins on cultured Colon26 cells. Colon26/Luc cells were cultured for 3 d in the presence of rmIFN‐β, rmIFN‐γ, or both rmIFN‐β and rmIFN‐γ (denoted as IFN‐β+γ). After culture, the number of living Colon26/Luc cells was significantly lower in the wells supplemented with either IFN‐β or IFN‐γ in comparison with the wells of Colon26/Luc cells alone, indicating a decrease in the number of live cells due to the effects of rmIFNs (Figure 1A). The addition of IFN‐β was more effective than addition of IFN‐γ, and simultaneous addition of IFN‐β+γ was even more effective. The number of viable cells was also measured based on quantification of intracellular NADH. The results of this experiment also indicated that both IFN‐β and IFN‐γ reduced the number of live cells (Figure 1B).
Figure 1.
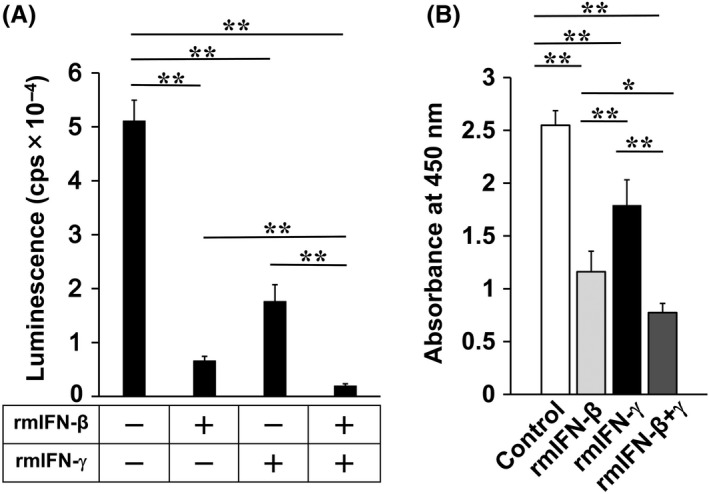
Sensitivity of Colon26 cells to rmIFN‐β and rmIFN‐γ. A, Colon26/Luc cells (5 × 103 cells/well) were co‐cultured with or without recombinant mouse (rm) IFN‐β (10 ng/mL), rmIFN‐γ (10 ng/mL), or rmIFN‐β + rmIFN‐γ (10 ng/mL each) for 3 d. The number of live Colon26/Luc cells was determined by measuring luciferase activity. B, Numbers of viable Colon26 cells after rmIFN treatments were analyzed based on quantification of NADH. P‐values were calculated using Tukey‐Kramer test (*P < .05, **P < .01)
The decreased number of live cells could be accounted for by induction of cell death or by growth inhibition. Induction of apoptosis by rmIFNs was examined by staining cells with annexin V‐FITC (Figure S1A). When Colon26 cells were cultured alone, apoptotic cells made up <1%. Addition of IFN‐β and IFN‐γ increased the frequencies of apoptotic cells to 4.9% and 9%, respectively, and addition of IFN‐β+γ increased the frequency of apoptotic cells to 50.2%. The frequency of proliferating cells was examined by Ki67 staining. IFN‐β, IFN‐γ, and IFN‐β+γ decreased Ki67‐positive cells from 12.4% (cultured alone) to 6.14%, 2.62%, and 2.81%, respectively (Figure S1B). Collectively, both rmIFN‐β and rmIFN‐γ had apoptosis‐inducing and growth‐inhibitory effects on Colon26 cells.
3.2. Disruption of the genes encoding for H‐2K, D, and I‐Aβ molecules in the ES‐ML/IFN
We disrupted the genes encoding H‐2Kb, Db, and I‐Aβb molecules in the ES‐ML/IFN. We transduced β‐ML with a mixture of lentiviral gRNA expression vectors along with a CAS9 nuclease expression vector. Subsequently, cells deficient in the expression of H‐2K, D, and I‐A were purified using anti‐MHC antibodies and magnetic beads. The absence of these MHC molecules in the resulting cells was verified by flow cytometric analysis (Figure S2). We also established MHC‐deficient γ‐ML cells using the same methods (data not shown). In all of the following in vivo experiments, the MHC‐deficient ES‐ML/IFN were transferred into allogeneic and cancer‐bearing BALB/c mice (H‐2d) for evaluation of their therapeutic effects.
3.3. Therapeutic effect of ES‐ML/IFN against peritoneally disseminated colon cancer
To evaluate the anticancer therapeutic effect of the ES‐ML/IFN, we injected ip the ES‐ML/IFN into cancer‐bearing mice. The half‐life of β‐ML administrated into the peritoneal cavity of the allogeneic and cancer‐bearing BALB/c mice is estimated to be c. 28 h (Figure S3), therefore the ES‐ML/IFN was administrated twice a week for 2‐4 wk to obtain anticancer effects. We determined 3 × 106 and 7.5 × 105 cells per injection per mouse as the tolerated doses of β‐ML and γ‐ML, respectively, because repetitive injections of these numbers of the ES‐ML/IFN did not cause apparent adverse effects in the mice even if β‐ML and γ‐ML were simultaneously injected.
To establish a model of peritoneal dissemination of colon cancer, we injected ip Colon26/Luc cells and monitored cancer growth by bioluminescence analysis. Mice engrafted with Colon26/Luc cells on d 2 were divided into therapy and no therapy (control) groups. Mice in the therapy group were treated by injection ip of β‐ML twice a week for 4 wk from d 2. Cancer growth was inhibited by treatment with β‐ML (Figure 2A,B). In addition, survival of the cancer‐bearing mice was extended by the therapy (Figure 2C), suggesting that the therapeutic benefit of the administration of β‐ML surpassed the harmful effects.
Figure 2.
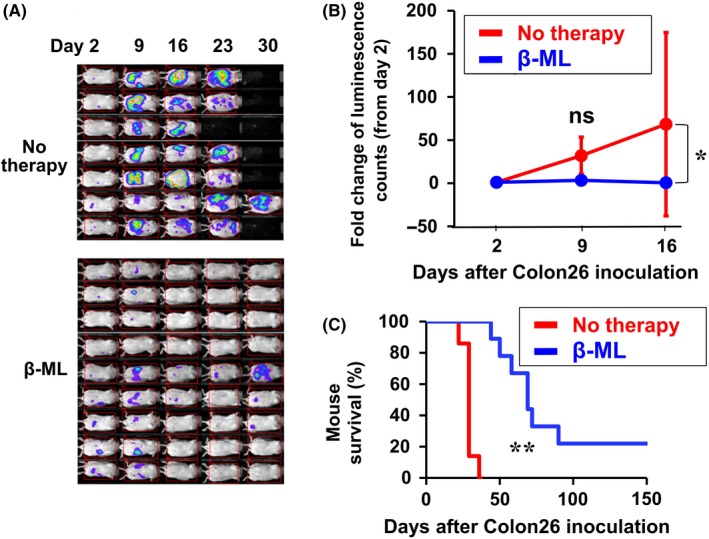
Therapeutic effect of β‐ML against peritoneally disseminated colon cancer. Colon26/Luc cells were injected ip. The mice were divided into no therapy (n = 7) and β‐ML therapy (n = 9) groups. The mice in the β‐ML therapy group were injected ip with β‐ML (3 × 106) from d 2, twice each week for 4 wk. A, Luminescence images are shown. B, Cancer growth of the two groups is indicated as the fold change of luminescence counts in comparison with counts observed on d 2. The linear mixed model was performed to compare the data of the no therapy and β‐ML groups, and the difference between the two groups is statistically significant (*P < .05). C, Kaplan‐Meier survival curves for β‐ML‐treated and no therapy groups are shown. The difference between the two groups is statistically significant (**P < .01, log‐rank test)
3.4. Therapeutic effect of the ES‐ML/IFN against colon cancer injected into liver
We injected Colon26/Luc cells into the left lobe of mouse liver. After 4 d, colonization of cancer cells was verified by an in vivo bioluminescence assay, and cancer‐positive mice were divided into control and treatment groups. Therapy with β‐ML inhibited cancer growth (Figure 3A,B) and extended the survival of mice (Figure 3C). Conversely, therapy with γ‐ML was not effective (Figure S4). Simultaneous injections of β‐ML and γ‐ML were also effective (Figure S5).
Figure 3.
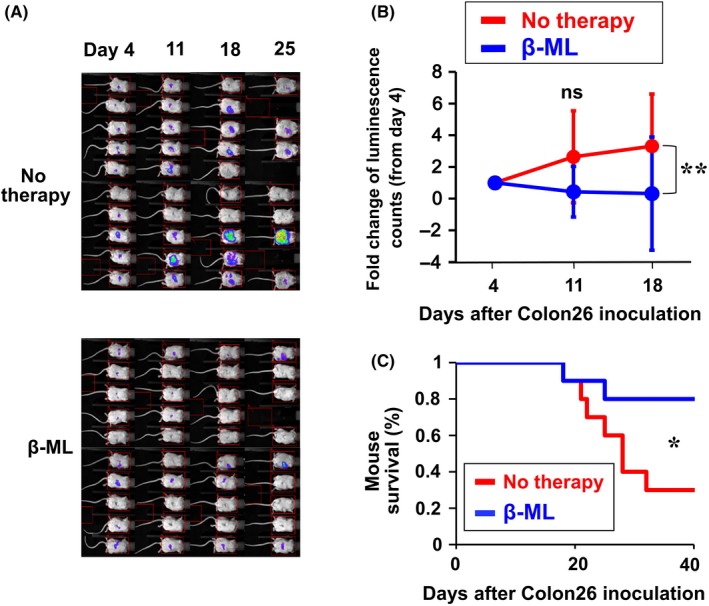
Therapeutic effect of β‐ML against colon cancer in liver. Colon26/Luc cells (5 × 105/mouse) were inoculated into the left liver lobe on d 0. On d 4, mice were divided into no therapy (n = 10) and β‐ML therapy (n = 10) groups. The mice in the therapy group were injected ip with β‐ML (3 × 106) from d 4, twice each week for 4 wk. A, Luminescence images of the mice are shown. B, Cancer growth is indicated as the fold change of luminescence counts in comparison with counts observed on d 4. The mean ± SD of the mouse groups are shown. The difference between the two groups was statistically significant (**P < .01, linear mixed model). C, Kaplan‐Meier survival curves are shown. The difference between the two groups was statistically significant (*P < .05, log‐rank test)
3.5. Infiltration of the ES‐ML/IFN into cancer tissues
We next examined infiltration of β‐ML and γ‐ML into cancer tissues. Colon26/GFP cells were injected ip into mice. After 3 d, β‐ML and γ‐ML labeled with red fluorescent dye PKH26 were injected ip. On the next day, the mice were euthanized, and the locations of cancer cells and the ES‐ML/IFN were examined macroscopically. The locations of cancer lesions formed by Colon26/GFP cells as well as the ES‐ML/IFN are shown in Figure S6A,B. Co‐localization of the ES‐ML/IFN with Colon26/GFP cells indicated accumulation of the ES‐ML/IFN at the cancer lesion site.
For microscopic analysis, PKH26‐labeled β‐ML and γ‐ML were injected at 10 d after injection of cancer cells. The next day, cancer nodules were isolated, and frozen sections were prepared. Histological analysis indicated infiltration of the ES‐ML/IFN into the cancer capsule and inside cancer tissues (Figure S6C,D).
3.6. Enhanced infiltration of CD4+ and CD8+ cells into cancer tissues by treatment with the ES‐ML/IFN
We examined cancer‐infiltrating cells by immunohistochemical analysis. Colon26/Luc cells were injected ip, and mice were treated with β‐ML and γ‐ML twice a week for 2 wk. Then, we isolated peritoneal cancer tissues and subjected them to immunostaining to detect CD4+ cells and CD8+ cells. Higher numbers of both CD4+ cells and CD8+ cells were observed in cancer tissues of treated mice in comparison with those of untreated mice (Figure 4A,B).
Figure 4.
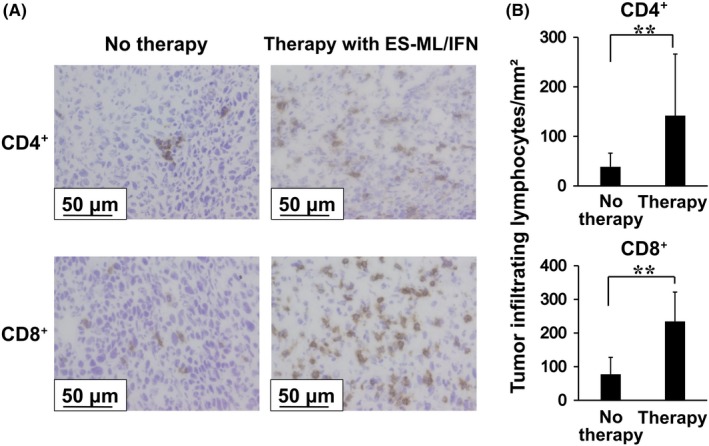
Enhanced infiltration of CD4+ cells and CD8+ cells into cancer tissues in mice treated with the ES‐ML/IFN. Colon26 cells (4 × 106/mouse) were injected ip into BALB/c mice. Mice were treated from d 4, twice each week for 2 wk with both β‐ML (3 × 106) and γ‐ML (7.5 × 105) (n = 9) or untreated (n = 9). Then, peritoneal cancer tissues were resected and subjected to immunohistochemical analyses. A, Tissue sections were stained with anti‐CD4 and anti‐CD8 antibodies, and reactions were visualized with DAB. Scale bars represent 50 μm. B, The numbers of CD4+ cells and CD8+ cells in the cancer tissues were significantly increased in the treated mice in comparison with mice without treatment. The difference between values from the no therapy and therapy groups was evaluated using Student's t test (**P < .01)
3.7. Increased numbers of immune cells in the spleen and lymph nodes of the ES‐ML/IFN‐treated cancer‐bearing mice
Next, we analyzed the influence of ES‐ML/IFN treatment on the number of immune cells in the lymphoid organs. Peritoneal cancer‐bearing mice were injected with β‐ML and γ‐ML twice a week and sacrificed after four times injection, and the numbers of immune cells in the lymph nodes and spleen were examined.
The total cell number of spleen and PANs was increased by the treatment (Figure 5). In spleen, NK cells, myeloid cells, and CD4+ cells were increased, while Th1 cells (CD4+ and IFN‐γ+), CD8+ cells, and effector CD8+ T cells (CD8+ and IFN‐γ+) were not increased. Conversely, all these subpopulations were increased in PANs, and the magnitude of increase of the cell numbers was much higher in PANs than in the spleen. ES‐ML/IFN treatment also increased the numbers of CD4+ cells and CD8+ cells in both mesenteric (draining) and inguinal (non‐draining) lymph nodes (Figure S7). The magnitude of increase of T cells in the draining lymph nodes was higher than those in the non‐draining lymph nodes. Collectively, injection ip of the ES‐ML/IFN into cancer‐bearing mice increased immune cells in lymphoid organs, and the increase was more marked in draining lymph nodes than in spleen or non‐draining lymph nodes.
Figure 5.
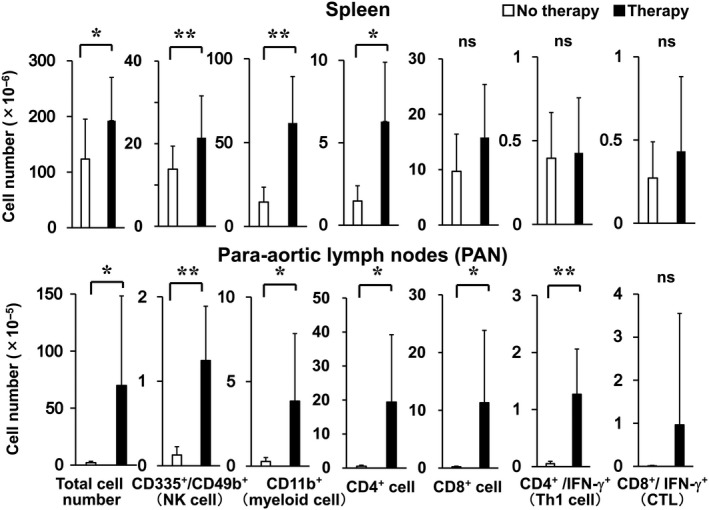
Increased numbers of immune cells in the spleen and para‐aortic lymph nodes (PANs) of cancer‐bearing mice treated with the ES‐ML/IFN. Mice with peritoneally disseminated Colon26 cells were treated with the ES‐ML/IFN as described in Figure 4 or untreated (no therapy). The numbers of immune cells in the spleen and PANs of mice were counted at d 17. Data for the no therapy group are indicated by white bars, and those of the therapy group (n = 12) are indicated by black bars. The difference between values of no therapy and therapy groups was evaluated using Student's t test (*P < .05, **P < .01)
3.8. Anticancer activity of CD8+ T cells isolated from cancer‐draining lymph nodes of the ES‐ML/IFN‐treated mice
We next examined the anticancer activity of CD8+ T cells in cancer‐bearing mice treated with the ES‐ML/IFN. Mice bearing intraperitoneal cancer were injected ip with β‐ML and γ‐ML twice from d 4. At d 11, we isolated CD8+ T cells from MLNs and PANs. PANs of untreated mice were small, and CD8+ T cells sufficient for this assay could not be prepared from PANs.
We co‐cultured the isolated CD8+ T cells with Colon26/Luc cells for 3 d and measured the number of live Colon26/Luc cells by luminescence activity. Cancer cells were significantly decreased by co‐culture with CD8+ T cells isolated from the ES‐ML/IFN‐treated mice (Figure S8A). Conversely, CD8+ T cells isolated from untreated mice did not exhibit such activity.
3.9. Activation of CD8+ T cells and NK cells in the cancer‐bearing mice treated with the ES‐ML/IFN
We evaluated the responsiveness of T cells from cancer‐bearing mice to Colon26 cells. Spleen and MLN cells were isolated from mice bearing intraperitoneal Colon26/Luc cancer cells and treated ip twice with β‐ML and γ‐ML or untreated. Isolated cells were co‐cultured with Colon26/Luc, and production of IFN‐γ by these cells was analyzed.
Whole spleen and MLN cells of untreated mice showed no or a very low response to the cancer cells (Figure 6A). Conversely, cells isolated from mice treated with β‐ML and γ‐ML secreted IFN‐γ in response to Colon26 cells, indicating that T cells reactive to cancer cells were primed in the cancer‐bearing mice by therapy with the ES‐ML/IFN.
Figure 6.
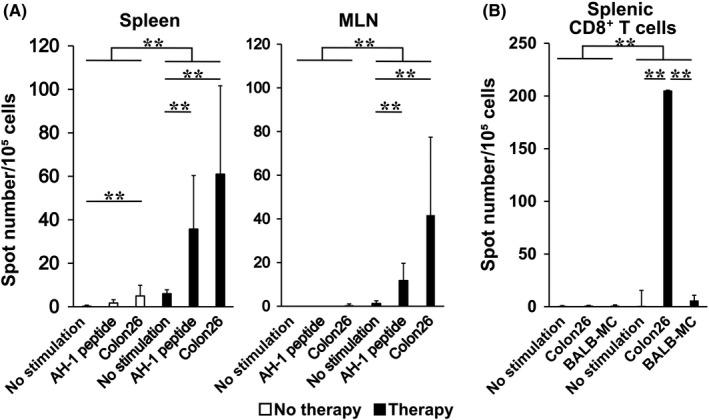
Priming of cancer cell‐reactive CD8+ T cells following treatment with the ES‐ML/IFN. A, Mice bearing peritoneally disseminated Colon26 cells were treated twice with both β‐ML (3 × 106) and γ‐ML (7.5 × 105) from d 4 or untreated and then euthanized. Whole spleen cells and mesenteric lymph node (MLN) cells were isolated at d 11 after cancer dissemination. The cells were cultured alone, with AH1 peptide, or with Colon26 cells; IFN‐γ secretion by the cells was measured by ELISPOT assay. Average spot numbers + SD are indicated. B, CD8+ T cells were purified from spleen cells and cultured alone, with Colon26 cells, or with BALB‐MC cells. To compare ELISPOT counts between the no therapy and therapy groups, a Poisson regression model, which is used to distinguish types of therapy for a covariate, was established (**P < .01)
Colon26 cells produce a murine leukemia virus gp70‐derived and H2‐Ld‐binding peptide AH‐1. T cells isolated from the treated mice also reacted to this peptide, further indicating the induction of cancer antigen‐specific activation of T cells following the therapy (Figure 6A). Purified CD8+ cells (Figure 6B), but not CD4+ cells (data not shown), secreted IFN‐γ when co‐cultured with Colon26 cells. The response of CD8+ T cells to BALB‐MC, a mammary cancer cell line derived from the BALB/c strain, was much lower than that to Colon26 cells. Therefore, priming of CD8+ T cells by ES‐ML/IFN therapy was specific to Colon26 cells (Figure 6B).
To compare β‐ML and γ‐ML for their activity to prime T cells, we injected cancer‐bearing mice with either β‐ML or γ‐ML and examined the reactivity of MLNs cells. β‐ML primed Colon26‐reactive T cells much more effectively than did γ‐ML (Figure S8B).
We also examined production of IFN‐γ by NK cells of treated and untreated mice. NK cells from both groups did not produce IFN‐γ when co‐cultured with Colon26/Luc (data not shown). However, significantly higher numbers of NK cells derived from the treated mice produced IFN‐γ upon co‐culture with syngeneic SP2/0 cells, compared with those cells from untreated mice, indicating activation of NK cells following the ES‐ML/IFN therapy (Figure S9).
3.10. Contribution of CD8+ T cells to therapeutic effect of the ES‐ML/IFN
To examine the contribution of CD8+ T cells to therapeutic effect of the ES‐ML/IFN, we conducted a depletion experiment with anti‐CD8 monoclonal antibody.
We injected anti‐CD8 monoclonal antibody ip at d −3, −1, and 6. Colon26/Luc cells were injected ip at d 0 to establish the peritoneal dissemination model and treated with β‐ML and γ‐ML from d 4, twice a week for 2 wk. The therapeutic effect was significantly impaired by depletion of CD8+ T cells (Figure 7). Conversely, the difference in cancer progression between CD4+ T cell‐depleted and non‐depleted mice was not statistically significant (Figure S10).
Figure 7.
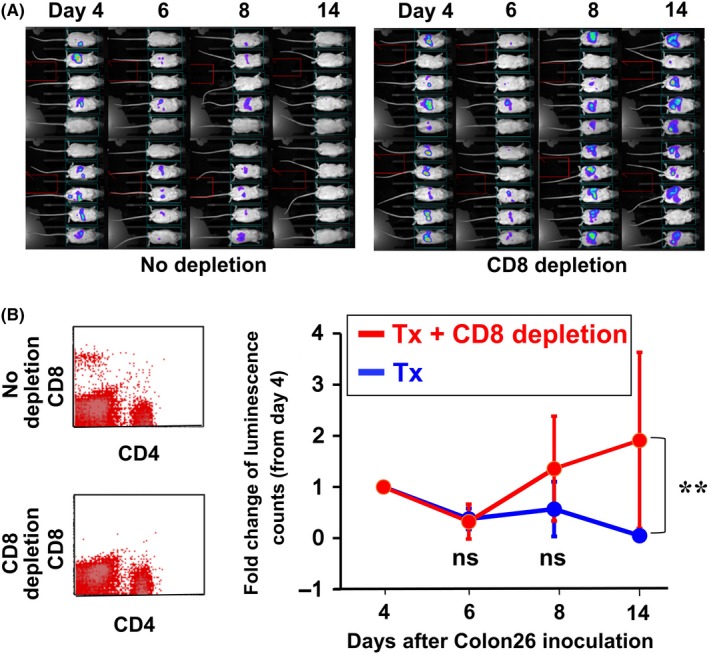
Contribution of CD8+ T cells to the therapeutic effect of the ES‐ML/IFN. An anti‐CD8 antibody was administrated ip at d −3, −1, and 6 (50 μg/injection/mouse). Colon26/Luc cells (1 × 106 cells/mouse) were injected ip on d 0, and mice were treated with the ES‐ML/IFN as described in Figure 4. Cancer growth was monitored by in vivo luminescence analysis from d 4. A, Imaging data of luciferase assay are shown. B, The presence of CD4+ and CD8+ T cells in peripheral blood was examined to confirm depletion of CD8+ T cells at d −1. C, Fold changes in luminescence counts of each mouse in comparison with counts observed on d 4. The difference between the two groups was statistically significant (**P < .01, linear mixed model)
4. DISCUSSION
We currently tested a strategy to use pluripotent stem cell‐derived myeloid cells as DDS to improve cancer‐tissue selectivity of the effect of IFN. Although administration of the allogeneic ES‐ML/IFN might have led to some individual differences in therapeutic efficacy as compared with the case with the syngeneic ES‐ML/IFN, a treatment with the tolerated dose of the ES‐ML/IFN suppressed the cancer growth and significantly prolonged the survival of the cancer‐bearing mice without causing apparent adverse effects. The most important reason why the allogenic BALB/c (H2d) recipients rather than 129Sv (H2b) syngeneic recipients were used in this study, was to assess the effectiveness of our therapeutic approach assuming a use for allogeneic and HLA‐deficient human iPS‐ML/IFN for HLA‐incompatible cancer patients. The results in the current study provide a rationale for clinical trials to evaluate the efficacy and safety of cancer therapy using human iPS‐ML/IFN.
Regarding the anticancer effect of IFNs, direct effect and indirect potency mediated through the activation of immune cells are conceivable. The direct anticancer effects of IFNs are exerted by inducing growth arrest or apoptosis of cancer cells as demonstrated by in vitro analysis (Figures 1 and S1). These effects were probably induced via the Jak‐Stat intracellular signaling pathway.8 It is known that p53 promotes apoptosis through extrinsic death receptor‐mediated and intrinsic mitochondrial pathways.35 p53 is activated functionally in response to diverse stress stimuli including activation of oncogenes, DNA damage, nutrient deprivation, or hypoxia.36 Conversely, IFN‐β is reported to enhance the expression of p53 protein, although IFN‐β signaling itself does not activate p53. Therefore, p53 and IFN‐β are important for the induction of apoptosis in cancer cells.37 Based on these issues, stress stimuli may be involved in the direct anticancer effects of IFNs via the ES‐ML/IFN in our system, especially under in vivo conditions.
An in vivo study also clarified the therapeutic effect of the ES‐ML/IFN including indirect potency. While sole treatment with γ‐ML did not exhibit any therapeutic effect against Colon26 cancer in vivo (Figure S4), β‐ML did show manifest therapeutic effects (Figures 2 and 3). Indeed, their sensitizing efficacy for cancer cell‐reactive CD8+ T cells was much more effective than that of γ‐ML (Figure S8B). The immune‐stimulating effect of β‐ML on host cells was likely to be essential to elicit the therapeutic effect in our in vivo models, because the depletion of CD8+ T cells in the mice canceled the therapeutic effect of β‐ML (Figure 7). It is conceivable that the effective induction of cancer‐specific cytotoxic T cells in the ES‐ML/IFN‐treated mice is mediated by host‐derived antigen‐presenting cells including dendritic cells and macrophages, which have been activated by type I interferons, because allogeneic ES‐ML itself should function as a drug delivery system for interferons but not antigen‐presenting cells in the tumor microenvironment.
In our preliminary results, the number of F4/80+ macrophages in cancer tissues tended to be increased by treatment with the ES‐ML/IFN (data not shown), although the difference between nontreated and treated groups was not statistically significant. Conversely, MHC‐class I expression in cancer tissue was significantly elevated in the ES‐ML/IFN‐treated mice (data not shown). These results suggest a possibility that the expression levels of upregulated MHC‐class I in both cancer cells and antigen‐presenting cells in the tumor microenvironment were induced by IFN‐γ secreted from immune cells, including cancer‐reactive cytotoxic T cells, that were activated by the allogeneic ES‐ML/IFN as demonstrated in Figures 4 and 6. However, the details of host immune cells‐mediated therapeutic effects of ES‐ML/IFN therapy remains to be elucidated.
Considering clinical application of iPS cell‐based anti‐cell therapy, use of iPS cells originated from the patient to be treated is not practical. Generation of clinical grade iPS cells for individual patients is very expensive. In addition, generation of iPS cells and differentiation of these cells to produce therapeutic cells may take several months and miss the timing to treat cancer patients. Therefore, use of pre‐established allogeneic iPS cell‐derived cells may be necessary. A major problem in cell therapy using allogeneic cells is rejection of transferred cells by the host immune system.
Recently, we generated HLA class I‐deficient human iPS cells and iPS‐ML using CRISPR technology. We observed that HLA‐deficient iPS‐ML avoided recognition by alloreactive T cells (unpublished data). The current study indicated that the MHC (K, D, and I‐A)‐deficient ES‐ML/IFN could exert a therapeutic effect in MHC‐mismatched allogeneic BALB/c mice. The results suggested that MHC‐deficient ES‐ML could avoid allogeneic MHC‐directed acute rejection by the host immune system.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
We thank Mitchell Arico from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript. This work was supported by a JSPS KAKENHI grant (grant no. 17H04272) and grants from AMED (grant nos. 16ck0106080h0003 and 17lm0203040h0001) to S. Senju and a JSPS Grants‐in‐Aid for Scientific Research on Innovative Areas (grant no. 16H 06498) to Y. Nishimura.
Umemoto S, Haruta M, Sakisaka M, et al. Cancer therapy with major histocompatibility complex‐deficient and interferon β‐producing myeloid cells derived from allogeneic embryonic stem cells. Cancer Sci. 2019;110:3027–3037. 10.1111/cas.14144
Contributor Information
Satoshi Umemoto, Email: satos0526-surg@outlook.jp.
Yasuharu Nishimura, Email: mxnishim@gpo.kumamoto-u.ac.jp.
REFERENCES
- 1. Shen J, Xiao Z, Zhao Q, et al. Anti‐cancer therapy with TNFalpha and IFNgamma: a comprehensive review. Cell Prolif. 2018;51:e12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sayers TJ, Wiltrout TA, McCormick K, Husted C, Wiltrout RH. Antitumor effects of α‐interferon and γ‐interferon on a murine renal cancer (Renca) in vitro and in vivo . Cancer Res. 1990;50:5414‐5420. [PubMed] [Google Scholar]
- 3. Chawla‐Sarkar M, Lindner DJ, Liu Y‐F, et al. Apoptosis and interferons: role of interferon‐stimulated genes as mediators of apoptosis. Apoptosis. 2003;8:237‐249. [DOI] [PubMed] [Google Scholar]
- 4. Cascinu S, Del Ferro E, Fedeli A, et al. Cytokinetic effects of interferon in colorectal cancer tumors: implications in the design of the interferon/5‐fluorouracil combinations. Cancer Res. 1993;53:5429‐5432. [PubMed] [Google Scholar]
- 5. Asmana Ningrum R. Human interferon alpha‐2b: a therapeutic protein for cancer treatment. Scientifica. 2014;2014:970315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Booy S, van Eijck CH, Dogan F, van Koetsveld PM, Hofland LJ. Influence of type‐I interferon receptor expression level on the response to type‐I interferons in human pancreatic cancer cells. J Cell Mol Med. 2014;18:492‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li P, Du Q, Cao Z, et al. Interferon‐gamma induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (HCC) cells through interferon‐regulatory factor‐1 (IRF‐1). Cancer Lett. 2012;314:213‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16:131‐144. [DOI] [PubMed] [Google Scholar]
- 9. Rizza P, Moretti F, Capone I, Belardelli F. Role of type I interferon in inducing a protective immune response: perspectives for clinical applications. Cytokine Growth Factor Rev. 2015;26:195‐201. [DOI] [PubMed] [Google Scholar]
- 10. Shou P, Chen Q, Jiang J, et al. Type I interferons exert anti‐tumor effect via reversing immunosuppression mediated by mesenchymal stromal cells. Oncogene. 2016;35:5953‐5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405‐414. [DOI] [PubMed] [Google Scholar]
- 12. Chiang J, Gloff CA, Yoshizawa CN, Williams GJ. Pharmacokinetics of recombinant human interferon‐beta ser in healthy volunteers and its effect on serum neopterin. Pharm Res. 1993;10:567‐572. [DOI] [PubMed] [Google Scholar]
- 13. Petrella T, Verma S, Spithoff K, Quirt I, McCready D. Adjuvant interferon therapy for patients at high risk for recurrent melanoma: an updated systematic review and practice guideline. Clin Oncol. 2012;24:413‐423. [DOI] [PubMed] [Google Scholar]
- 14. Quesada JR, Talpaz M, Rios A, Kurzrock R, Gutterman JU. Clinical toxicity of interferons in cancer patients: a review. J Clin Oncol. 1986;4:234‐243. [DOI] [PubMed] [Google Scholar]
- 15. Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon alfa‐2b adjuvant therapy of high‐risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 1996;14:7‐17. [DOI] [PubMed] [Google Scholar]
- 16. Belardelli F, Ferrantini M, Proietti E, Kirkwood JM. Interferon‐alpha in tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002;13:119‐134. [DOI] [PubMed] [Google Scholar]
- 17. Jedinak A, Dudhgaonkar S, Sliva D. Activated macrophages induce metastatic behavior of colon cancer cells. Immunobiology. 2010;215:242‐249. [DOI] [PubMed] [Google Scholar]
- 18. Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155‐1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605‐612. [DOI] [PubMed] [Google Scholar]
- 20. Caux C, Ramos RN, Prendergast GC, Bendriss‐Vermare N, Ménétrier‐Caux C. A milestone review on how macrophages affect tumor growth. Cancer Res. 2016;76:6439‐6442. [DOI] [PubMed] [Google Scholar]
- 21. Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor‐associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25:315‐322. [DOI] [PubMed] [Google Scholar]
- 22. Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer. 2017;117:1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Noy R, Pollard JW. Tumor‐associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Williams CB, Yeh ES, Soloff AC. Tumor‐associated macrophages: unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer. 2016;2:15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roussos ET, Condeelis JS, Patsialou A. Chemotaxis in cancer. Nat Rev Cancer. 2011;11:573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hernandez C, Huebener P, Schwabe RF. Damage‐associated molecular patterns in cancer: a double‐edged sword. Oncogene. 2016;35:5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haruta M, Tomita Y, Imamura Y, et al. Generation of a large number of functional dendritic cells from human monocytes expanded by forced expression of cMYC plus BMI1. Hum Immunol. 2013;74:1400‐1408. [DOI] [PubMed] [Google Scholar]
- 28. Senju S, Haruta M, Matsumura K, et al. Generation of dendritic cells and macrophages from human induced pluripotent stem cells aiming at cell therapy. Gene Ther. 2011;18:874‐883. [DOI] [PubMed] [Google Scholar]
- 29. Zhang R, Liu TY, Senju S, et al. Generation of mouse pluripotent stem cell‐derived proliferating myeloid cells as an unlimited source of functional antigen‐presenting cells. Cancer Immunol Res. 2015;3:668‐677. [DOI] [PubMed] [Google Scholar]
- 30. Koba C, Haruta M, Matsunaga Y, et al. Therapeutic effect of human iPS‐cell‐derived myeloid cells expressing IFN‐beta against peritoneally disseminated cancer in xenograft models. PLoS ONE. 2013;8:e67567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sakisaka M, Haruta M, Komohara Y, et al. Therapy of primary and metastatic liver cancer by human iPS cell‐derived myeloid cells producing interferon‐beta. J Hepatobiliary Pancreat Sci. 2017;24:109‐119. [DOI] [PubMed] [Google Scholar]
- 32. Senju S, Hirata S, Matsuyoshi H, et al. Generation and genetic modification of dendritic cells derived from mouse embryonic stem cells. Blood. 2003;101:3501‐3508. [DOI] [PubMed] [Google Scholar]
- 33. Miyoshi H, Blomer U, Takahashi M, Gage FH, Verma IM. Development of a self‐inactivating lentivirus vector. J Virol. 1998;72:8150‐8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haga E, Endo Y, Haruta M, et al. Therapy of peritoneally disseminated colon cancer by TAP‐deficient embryonic stem cell‐derived macrophages in allogeneic recipients. J Immunol. 2014;193:2024‐2033. [DOI] [PubMed] [Google Scholar]
- 35. Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22:9030‐9040. [DOI] [PubMed] [Google Scholar]
- 36. Aubrey BJ, Kelly GL, Janic A, et al. How does p53 induce apoptosis and how does this relate to p53‐mediated tumour suppression? Cell Death Differ. 2017;25:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takaoka A, Hayakawa S, Yanai H, et al. Integration of interferon‐α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424:516‐523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials