

Abstract
p97/VCP is an endoplasmic reticulum (ER)‐associated protein that belongs to the AAA (ATPases associated with diverse cellular activities) ATPase family. It has a variety of cellular functions including ER‐associated protein degradation, autophagy, and aggresome formation. Recent studies have shown emerging roles of p97/VCP and its potential as a therapeutic target in several cancer subtypes including multiple myeloma (MM). We conducted a cell‐based compound screen to exploit novel small compounds that have cytotoxic activity in myeloma cells. Among approximately 2000 compounds, OSSL_325096 showed relatively strong antiproliferative activity in MM cell lines (IC 50, 100‐500 nmol/L). OSSL_325096 induced apoptosis in myeloma cell lines, including a bortezomib‐resistant cell line and primary myeloma cells purified from patients. Accumulation of poly‐ubiquitinated proteins, PERK, CHOP, and IREα, was observed in MM cell lines treated with OSSL_325096, suggesting that it induces ER stress in MM cells. OSSL_325096 has a similar chemical structure to DBeQ, a known p97/VCP inhibitor. Knockdown of the gene encoding p97/VCP induced apoptosis in myeloma cells, accompanied by accumulation of poly‐ubiquitinated protein. IC 50 of OSSL_325096 to myeloma cell lines were found to be lower (0.1‐0.8 μmol/L) than those of DBeQ (2‐5 μmol/L). In silico protein–drug‐binding simulation suggested possible binding of OSSL_325096 to the ATP binding site in the D2 domain of p97/VCP. In cell‐free ATPase assays, OSSL_325096 showed dose‐dependent inhibition of p97/VCP ATPase activity. Finally, OSSL_325096 inhibited the growth of subcutaneous myeloma cell tumors in vivo. The present data suggest that OSSL_325096 exerts anti‐myeloma activity, at least in part through p97/VCP inhibition.
Keywords: apoptosis, ERAD, myeloma, OSSL_320596, p97/VCP
Abbreviations
- ERAD
ER‐associated protein degradation
- ER
endoplasmic reticulum
- iMiD
immunomodulatory drug
- MM
multiple myeloma
- PI
proteasome inhibitor
- UPS
ubiquitin‐proteasome system
1. INTRODUCTION
Multiple myeloma (MM) is a malignant plasma cell dyscrasia, typically developing in elderly patients.1, 2, 3, 4 It is characterized by monoclonal gammopathy and clonal plasma cell infiltration in bone marrow.5 There are currently several novel agents available, including PI, iMiD, histone deacetylase inhibitors, and monoclonal antibodies for CD38 and SLAMF7.6, 7, 8, 9, 10, 11, 12, 13 Although these novel drugs have improved the clinical outcome of MM patients,14, 15 they are not a curative option for myeloma patients.
Previous studies suggest there are many potent therapeutic target molecules or pathways in MM. As MM cells usually produce a large amount of monoclonal immunoglobulins, molecules involved in the UPS seem to be attractive targets for eradicating MM cells. MM cells show ER stress with a large amount of monoclonal immunoglobulins, and the UPS plays critical roles for the survival of MM cells.16, 17 Inhibition of the UPS results in an overload of ER stress, leading to apoptotic cell death in MM. For example, proteasome inhibitors are predicted to inhibit the degradation of such overloaded monoclonal immunoglobulins and induce excess ER stress in MM cells. However, acquired resistance to bortezomib is quite commonly observed in the clinic.18 The mechanisms by which MM cells acquire resistance to bortezomib or other PI are poorly understood.19, 20, 21, 22
p97/VCP is an ER‐associated protein that belongs to the hexameric AAA ATPase family.23, 24, 25, 26 It plays critical roles in the UPS and other intracellular signaling pathways by cooperating with many cofactors. The most well‐known and probably most critical role of p97/VCP in the UPS is to release poly‐ubiquitinated proteins from the ER membrane into the cytosol for proteasomal degradation. This pathway culminates in the form of the p97/VCP‐Ufd1‐Npl4 complex. p97/VCP consists of two ATPase domains, D1 and D2, and an N‐terminal domain. Of the two ATPase domains, D2 acts as the main ATPase, and produces the driving force. The D1 domain promotes hexamerization of p97/VCP,27 and the N‐terminal domain is essential for binding and stabilization of substrates. p97/VCP is also involved in numerous other biological processes including autophagy, mitophagy, DNA damage response, DNA replication, and many others.28 Recently, it has been shown that dysfunction of p97/VCP is responsible for several neuromuscular diseases. For example, mutations in VCP, the gene encoding p97/VCP, are thought to be one of the causes for inclusion body myopathy with Paget's disease of bone and frontotemporal dementia (IBMPFD).24, 28 A subset of mutations is also found in amyotrophic lateral sclerosis (ALS) cases,20 indicating this protein plays important roles in neurons.
Recently, some studies have shed light on p97/VCP as a potential target for cancer therapy including MM, and efforts to discover small molecule inhibitors against p97/VCP are producing notable results.29, 30 DBeQ, the first selective inhibitor discovered for p97/VCP, has a relatively strong inhibitory activity (IC50, 1‐2 μmol/L) against recombinant p97/VCP.31 DBeQ cooperates with bortezomib to induce apoptosis in MM cells.29
We screened approximately 2000 small molecule compounds to exploit novel small compounds that have antiproliferative activity against MM cell lines by reusing a compound library for discovering HIV capsid inhibitors in our laboratory. Among those compounds, OSSL_325096 showed a relatively strong activity against MM. In the present study, we show anti‐MM activity of OSSL_325096 through p97/VCP inhibition in an ATP‐competitive manner.
2. MATERIALS AND METHODS
2.1. Cell culture and clinical specimens
Human MM cell lines KMS12PE, KMS11, KMS11btz, KMM1, KHM11, U266, and RPMI‐8226 were cultured in RPMI 1640 medium containing 10% (vol/vol) FBS at 37°C in 5% CO2. HEK293T cells were cultured in DMEM medium containing 10% (vol/vol) FBS at 37°C in 5% CO2. Primary MM cells were collected from the pleural effusion or bone marrow of two patients with MM from the Department of Hematology, Kumamoto University School of Medicine, using CD138 MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany). PBMC were collected from the whole blood of a healthy donor. Patients and the healthy donor provided written informed consent which was approved by the Ethics Committee on the Use of Human Subjects (Kumamoto University Graduate School of Medicine).
2.2. Reagents and compounds
OSSL_325096 was first purchased from Princeton BioMolecular Research, Inc. (Monmouth Junction, NJ, USA). For evaluation of efficacy in vivo, we also chemically synthesized OSSL_325096 as described in Figure S1. In the procedure, we synthesized the (R)‐isomer and the (S)‐isomer of OSSL_325096. We first tested both for myeloma cell lines and used S‐type OSSL_325096 for the in vivo assay.
2.3. Cell viability analysis
In total, 5 × 104 cells per well were seeded into 96‐well plates. Cells were treated with DMSO or OSSL_325096 at varying concentrations. Cell viability was assessed at 96 hours using CCK‐8 Reagent (Dojindo Molecular Technologies, Kumamoto, Japan).
2.4. Flow cytometric analyses of apoptosis with annexin V/7‐AAD double staining
Cells treated with DMSO or OSSL_325096 for 48‐72 hours were washed with annexin V buffer, stained with annexin‐V and 7‐AAD (BioLegend, San Diego, CA, USA), and analyzed using a FACSVerse cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
2.5. Lentiviral constructs and stable cell lines
A doxycycline‐inducible shRNA‐expressing vector and lentivirus packaging plasmids were obtained through Addgene (Watertown, MA, USA); Tet‐pLKO‐puro was a gift from Dmitri Wiederschain (Addgene plasmid #21915, Immuno‐Oncology Research, Sanofi, Boston, MA, USA) and psPAX2 and pMD2.G were gifts from Didier Trono (Addgene plasmids #12260 and #12259, Laboratory of Virology and Genetics, Swiss Federal Institute of Technology in Lausanne, Lausanne, Switzerland). Each of four shRNA oligos targeting VCP (shVCP #1, #2, #4, #5) and one non‐targeting oligo (control shRNA) were cloned into Tet‐pLKO‐puro (Data S1). Lentiviruses were produced in HEK293T cells according to Addgene's protocol. Stable cell lines were generated by lentiviral infection. Condensed lentiviral solution was added to KMS11 and KMS12PE cells with 8 μg/mL polybrene (Sigma‐Aldrich, St Louis, MO, USA). Cells were cultured with 1 μg/mL puromycin (Wako Pure Chemical Corp., Osaka, Japan) from 48 hours after infection. For the induction of shRNAs, doxycycline (Sigma‐Aldrich) was added to a concentration of 1 μg/mL in the culture medium.
2.6. RNA extraction, cDNA synthesis and RT‐qPCR
RNA was extracted from myeloma cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and cDNA synthesis was carried out using ReverTra Ace (Toyobo, Osaka, Japan). Expression levels of VCP were analyzed using SYBR Premix Ex Taq II (Takara Bio, Kusatsu, Japan) (Data S1). Target gene expression levels were normalized against ACTB expression. Reactions were carried out using an Eco Real‐Time PCR system (Illumina, San Diego, CA, USA).
2.7. Protein preparation, SDS‐PAGE and western blotting
Antibodies against caspase‐3, CHOP, ubiquitinated proteins, and actin were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies against PERK, VCP, IRE1α, ATF4, ATF6, XBP1, and XBP1s antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Cell lysates were separated on NuPAGE Bis‐Tris precast gels (Invitrogen) and transferred to PVDF membranes using an iBlot Dry Blotting system (Invitrogen). The membranes were blocked with 5% non‐fat dry milk and incubated with the primary antibodies at 4°C overnight. Then the membranes were incubated with a HRP‐conjugated secondary antibody (Amersham Biosciences, Little Chalfont, UK). The antibody‐bound proteins were visualized using an ECL Prime kit (Amersham Biosciences).
2.8. In silico docking simulations between p97/VCP and compounds
Crystal structures of p97/VCP (PDB ID: 3CF1) were obtained from the RCSB Protein Data Bank (http://www.rcsb.org) for analysis. Hydrogen moieties were added to 2‐D structures of ATP or OSSL_325096, and each structure was energy‐minimized with the MMFF94x force field as implemented in MOE 2013.08 (Chemical Computing Group, Montreal, Canada). All docking simulations were carried out with LeadIT version 2.1.3 (BioSolveIT GmbH, St Augustin, Germany).
2.9. Expression and purification of recombinant p97/VCP
His‐tagged human VCP (hVCP) cDNA was amplified from KMS12PE cells by PCR and cloned into PCR‐linearized pET30a (+) (Merck Millipore, Burlington, MA, USA) using an In‐Fusion HD Cloning Kit (Takara Bio) (Data S1). The pET30a‐hVCP plasmid was transformed into Escherichia coli BL21 (Rosetta; Novagen, Madison, WI, USA) and transformed bacteria were precultured in LB medium containing kanamycin and chloramphenicol overnight at 37°C. Protein expression was induced with 1 mmol/L isopropyl‐beta‐d‐thiogalactopyranoside. His‐tagged hVCP was purified as previously described;31 >95% protein purity was confirmed by SDS‐PAGE.
2.10. ATPase activity assay
Recombinant p97/VCP was diluted in assay buffer (50 mmol/L Tris‐HCl [pH 8.0], 20 mmol/L MgCl, 1 mmol/L EDTA, 1 mmol/L DTT) to a final concentration of 0.5 μmol/L. Then, 72 μL of the mixture was dispensed into a 96‐well plate and 4 μL of compound stocks of various concentrations of OSSL_325096 or DMSO was added to each well. The plate was incubated for 10 minutes at room temperature. Then, 10 μL of 0.5 mmol/L ATP solution was added to each well and incubated for 30 minutes at room temperature. ATPase activity was quantified using a QuantiChrom ATPase/GTPase Assay Kit (BioAssay Systems, Hayward, CA, USA).
2.11. RNA sequencing and gene expression analysis
RNA was extracted and purified using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) and an RNeasy Mini Kit (Qiagen, Hilden, Germany). Primary RNA sequencing was carried out by Novogene (Beijing, China). Raw sequence data were mapped to hg19 and expression values were determined as Reads Per Kilobase of transcript, per Million mapped reads (RPKM) using Biomedical Genomics Workbench 5 software (Qiagen Bioinformatics, Hilden, Germany). Gene Expression Omnibus (GEO) accession number for the RNA‐seq data reported in this paper is GSE118320. Genes with maximum average RPKM of more than 50‐fold, and twofold up‐ or downregulation were evaluated for gene set enrichment analysis (GSEA), which was 2 on online public GSEA software published by the Broad Institute (http://software.broadinstitute.org/gsea/index.jsp) using a “hallmark” gene set.
2.12. Xenograft models
KMS12PE or KMS11 myeloma cell lines were injected s.c. into female Rag2 −/− Jak3 −/− BALB/c mice (5 × 106 cells/mouse). A total of 250 mg/mL DMSO stock solution of OSSL_325096 was diluted in 25% PEG to a final concentration of 10 mg/mL. When s.c. tumors became visible, mice were i.p. given vehicle (25% PEG containing 4% DMSO) or 50 mg/kg OSSL_325096 twice a week (every Monday and Thursday). Tumor volumes were calculated as follows: V (cm3) = length (cm) × width (cm)2 × π/6. All mice were killed at humane endpoints. Endpoints included food and water intake difficulties, moribund symptoms, weight loss (20% or more over several days), and marked increase in tumor size (10% or more of body weight). All animal experiments were approved by the Committee for Ethics in Animal Experimentation at Kumamoto University Graduate School of Medicine.
3. RESULTS
3.1. OSSL_325096, a potent p97/VCP inhibitor, suppresses cell proliferation and induces ER stress and apoptotic cell death in MM cell lines
We screened approximately 2000 small molecule compounds to identify those with antiproliferative activity in MM cell lines. OSSL_325096 was one such small molecule that inhibited MM cell proliferation (Figure 1A). It has a very similar structure to DBeQ, a known p97/VCP inhibitor.31 This structure is also found in several other previously reported p97/VCP inhibitors, including CB‐5083.32 This class of inhibitors is reported to inhibit p97/VCP activity by binding to the ATP‐binding site located in the D2 domain of p97/VCP in an ATP‐competitive and reversible manner. Ninety‐six hours of treatment with various concentrations of OSSL_325096 showed a dose‐dependent antiproliferative effect on all six MM cell lines tested, including one bortezomib‐resistant cell line, KMS11btz (IC50, 0.1‐0.5 μmol/L; Figure 1B).19 In contrast, OSSL_325096 at more than 10‐fold higher concentrations was required to reduce the viability of PBMC from a healthy donor (Figure 1B). Moreover, DBeQ was less toxic in these myeloma cell lines, and IC50 in myeloma cell lines were almost similar to that for PBMC (IC50, 1‐10 μmol/L; Figure 1B). As determined by 7‐AAD/annexin V staining, the percentages of pre‐apoptotic/dead cells in KMS12PE and KMS11 cells were increased by OSSL_325096 treatment compared with those treated with DMSO (Figure 2A,B).
Figure 1.
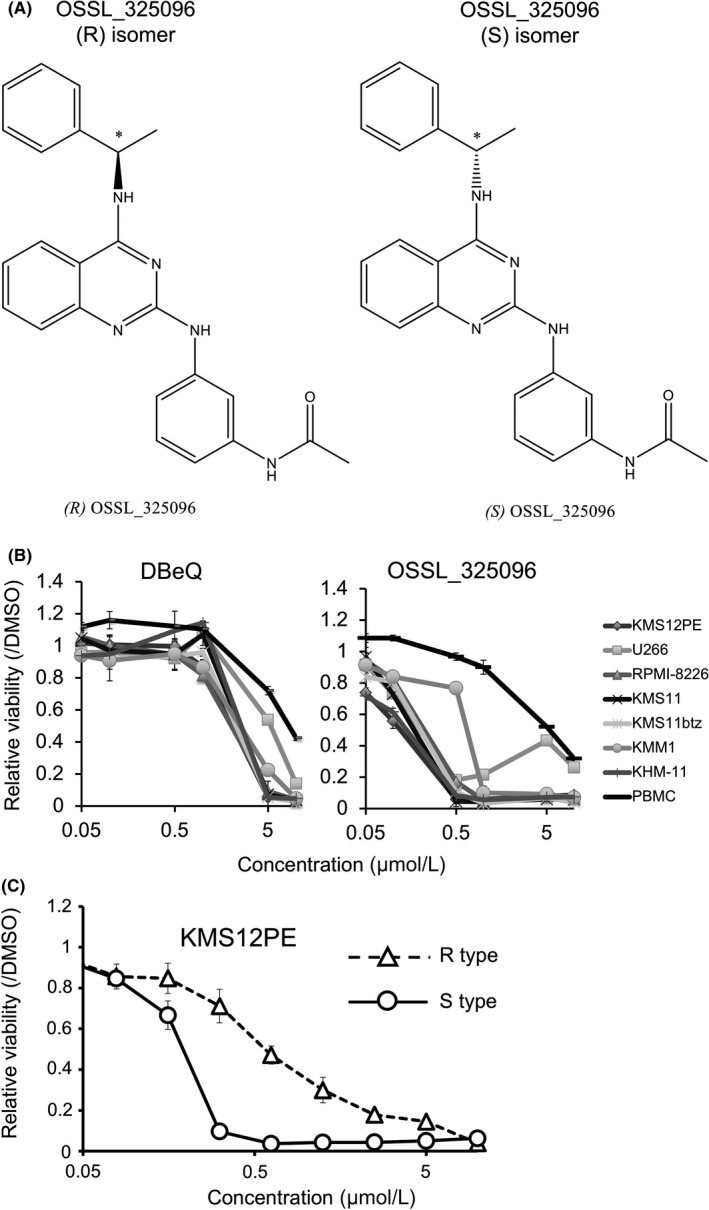
OSSL_325096 inhibits multiple myeloma (MM) cell proliferation. A, Chemical structure of OSSL_325096. (R)‐ and (S)‐OSSL_325096 are two stereoisomers (enantiomers) that differ in the configuration of the chiral carbon assigned by the asterisk. Such a simple difference in configuration resulted in a significant difference in bioactivity. B, Antiproliferative activity of OSSL_325096 and DBeQ in MM cell lines. Cells were treated with 0.1% DMSO, 0.05, 0.1, 0.5, 1, 5, or 10 μmol/L OSSL_325096 or DBeQ for 96 h and cell viability was evaluated by WST‐8 assay. Cell viability of DMSO‐treated samples served as a control. C, (S)‐OSSL_325096 showed higher antiproliferative activity than (R)‐OSSL_325096. KMS12PE cells were treated with 100, 25, 6.25, 1.56, or 0.39 μmol/L of R type or S type OSSL_325096 for 96 h and cell viability was evaluated by WST‐8 assay. Data represent the mean ± SD derived from three separate experiments
Figure 2.
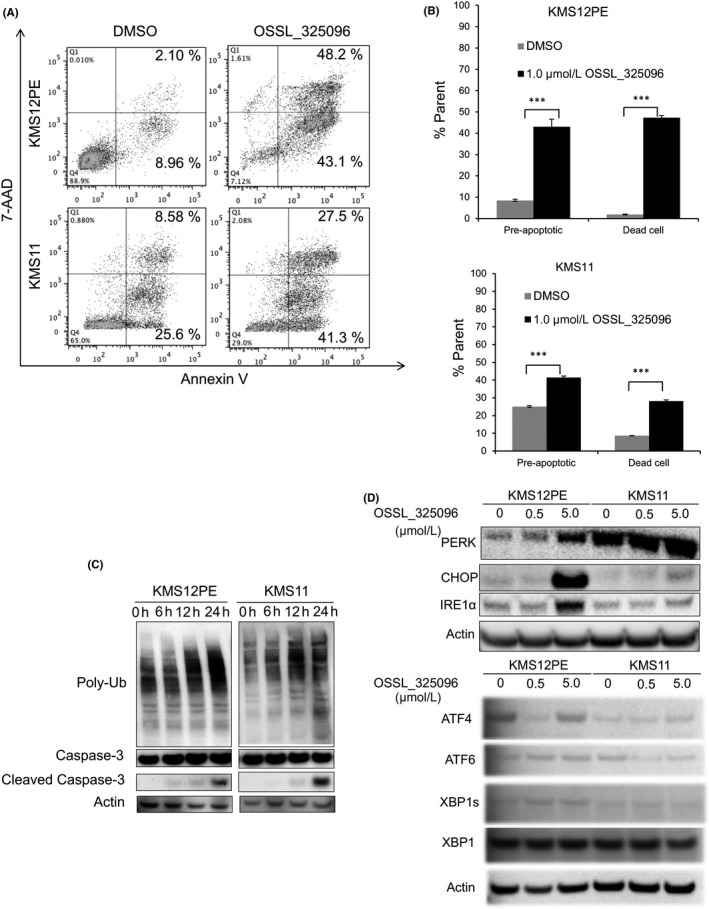
OSSL_325096 induces apoptosis in multiple myeloma (MM) cell lines. Cells were treated with DMSO or 1 μmol/L OSSL_325096 for 48 h. A, Dot plots and B, bar charts of cells stained with annexin V and 7AAD after 48‐h treatment. Pre‐apoptotic cells are defined as annexin V+ 7AAD − cells and dead cells are annexin V+ 7AAD + cells. Data represent the mean ± SD derived from three separate experiments. Significance values are indicated as ***P < .001 (Student's t‐test). C, Western blots of ubiquitin, non‐cleaved and cleaved caspase‐3, and actin at various time points in KMS12PE and KMS11 cells treated with OSSL_325096. Poly‐ubiquitinated proteins accumulated and apoptosis was induced in KMS12PE and KMS11 cells treated with 1 μmol/L OSSL_325096. D, Western blots of PERK, CHOP, IRE1α, ATF4, ATF6, XBP1 and XBP, and actin in OSSL_325096‐treated KMS12PE and KMS11 cells. Endoplasmic reticulum (ER) stress seems to be induced in KMS12PE and KMS11 cells treated with OSSL_325096
In addition, we evaluated markers of cellular ERAD and apoptosis by western blotting. Treatment with OSSL_325096 induced the accumulation of poly‐ubiquitinated proteins, expression of markers for ER‐stress, PERK, CHOP, and IREα, and cleavage of caspase‐3 (Figure 2C,D). This result indicates that OSSL_325096 induced ER‐stress and caspase‐3‐mediated apoptosis in MM cells and suggests it has antiproliferative activity and induces apoptotic cell death in vitro.
3.2. (S)‐OSSL_325096 has higher cytotoxic activity in KMS12PE cells than (R)‐OSSL_325096
To verify the difference between (R)‐ and (S)‐isomers of OSSL_325096, we chemically synthesized both isomers as described in Figure S1. We tested both isomers in KMS12PE myeloma cells and found that the (S)‐isomer has higher cytotoxic activity than the (R)‐isomer (Figure 1C). Therefore, we produced a large amount of (S)‐OSSL_325096 and used this preparation for further evaluation.
3.3. OSSL_325096 induces apoptosis in bortezomib‐resistant cells and primary MM cells
We also evaluated whether OSSL_325096 is effective in bortezomib‐resistant myeloma cells. KMS11btz is a previously reported bortezomib‐resistant cell line generated by continuous exposure to bortezomib in vitro.19 Treatment with 5 nmol/L bortezomib for 48 hours induced apoptotic cell death in the parental cell line KMS11 but not in KMS11btz cells (Figure 3A). In contrast, OSSL_325096 induced apoptotic cell death in both KMS11 and KMS11btz cells. The data indicate that OSSL_325096 was effective in bortezomib‐resistant MM cells. Next, we treated primary MM cells from two MM patients. The patients were heavily treated and refractory to most available anti‐myeloma agents, including lenalidomide and bortezomib. OSSL_325096 succeeded in inducing apoptosis in both primary myeloma cells (Figure 3B). In contrast, OSSL_325096 did not induce apoptosis in PBMC from a healthy donor. These data indicate that OSSL_325096 has antitumor activity in multiple drug‐resistant MM cells, including those resistant to lenalidomide and bortezomib, and is less toxic to normal cells.
Figure 3.
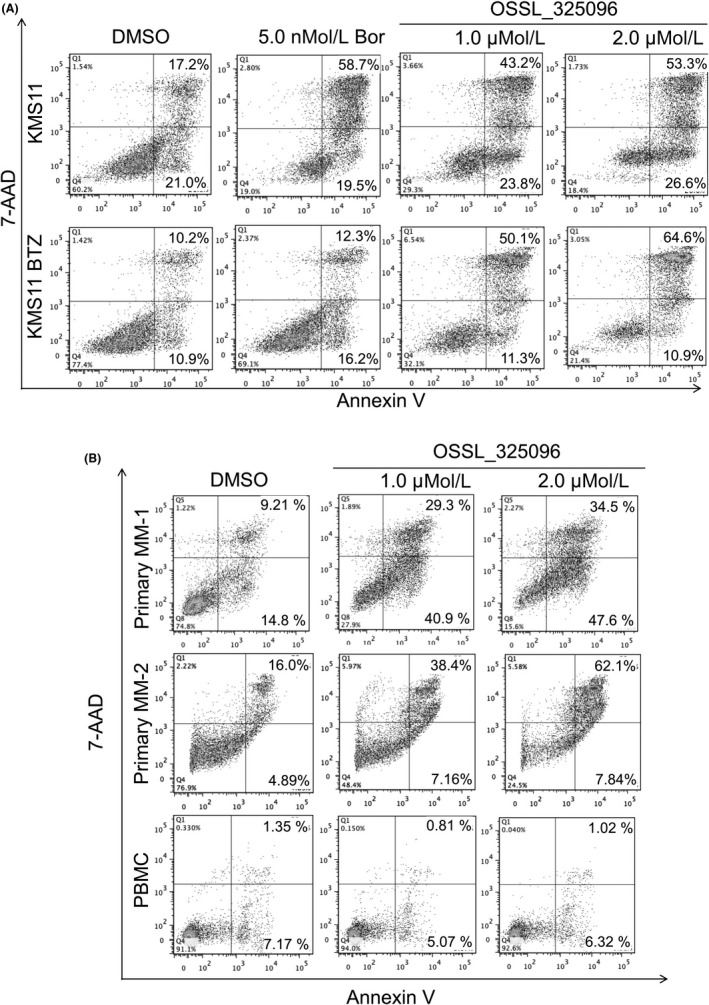
OSSL_325096 induces apoptosis in bortezomib‐resistant multiple myeloma (MM) cells and primary MM cells from two bortezomib‐refractory patients but not in PBMC from a healthy donor. Dot plots of cells treated with DMSO or 1 or 2 μmol/L OSSL_325096 with annexin V and 7‐AAD staining. A, KMS11 or KMS11/BTZ cells were treated with DMSO, 5 nmol/L bortezomib (Bor), or 1 μmol/L or 2 μmol/L OSSL_325096 for 48 h. B, Primary myeloma cells derived from two patients or PBMC from a healthy donor were treated with DMSO, 1 μmol/L, or 2 μmol/L OSSL_325096 for 48 h
3.4. OSSL_325096 inhibits ATPase activity of p97/VCP
As OSSL_325096 has a similar structure to a known p97/VCP inhibitor, DBeQ, we next investigated whether OSSL_325096 has inhibitory activity against p97/VCP. In silico flexible docking simulations were carried out using structural data of the catalytic domain active sites of p97/VCP. The 3‐D or 2‐D docking poses and total binding scores between p97/VCP and each compound are shown in Figure 4A and B. ATP, a natural ligand of p97/VCP, showed a high binding score of −32.8 KJ/mol. OSSL_325096 also had a favorable binding score that was lower than that of ATP. The data indicate that OSSL_325096 may have inhibitory activity against p97/VCP in myeloma cells, whereas it may not have complete inhibitory activity against p97/VCP to prevent survival of normal PBMC.
Figure 4.
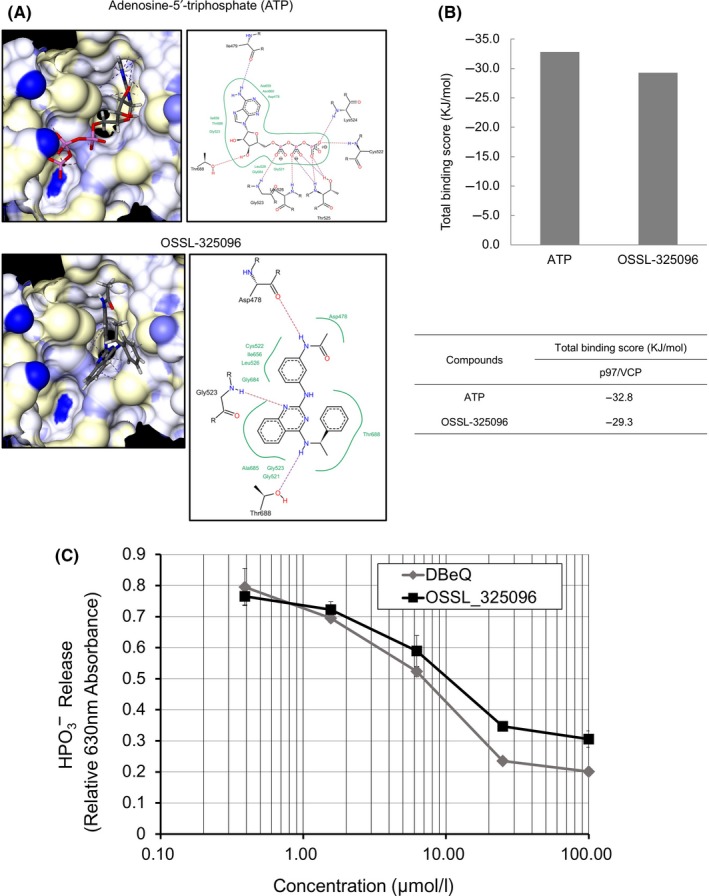
OSSL_325096 functions as an inhibitor of p97/VCP by inhibiting its ATPase activity in vitro. A, Docking simulation results of ATP or OSSL_325096 with the active site of the catalytic domain of p97/VCP (PDB ID: 3CF1). Hydrogen bond interactions between the molecular surface of p97/VCP and compounds are indicated by dotted lines. Hydrophobic interactions are represented as green curves in the 2‐D docking poses. All docking simulations were carried out with LeadIT version 2.1.3 (BioSolveIT GmbH, St Augustin, Germany). B, Summary of the total binding scores between p97/VCP and compounds are shown. Total binding scores were calculated as the sum of receptor‐ligand interactions. C, In vitro ATPase inhibition assay shows that OSSL_325096 suppressed ATPase activity of p97/VCP in a dose‐dependent way. Recombinant p97/VCP was incubated with DMSO or escalating doses of OSSL_325096 or DBeQ before addition of ATP. Degradation of ATP was quantified as HPO 3− release, presented as relative 630 nm absorbance. Data represent mean ± SD derived from three separate experiments
Next, using a cell‐free assay, we tested whether OSSL_325096 is a real inhibitor of p97/VCP. As previously reported, DBeQ and its derivatives inhibit p97/VCP function by binding to the D2 domain in an ATP‐competitive way.31 We used purified p97/VCP protein to carry out p97/VCP inhibition assays. OSSL_325096 inhibited hydrolysis of ATP in a dose‐dependent method (Figure 4C). The p97/VCP inhibitory activity of OSSL_325096 and DBeQ were very similar (IC50 10.25 ± 1.57 μmol/L vs 6.99 ± 0.50 μmol/L). However, the IC50 for cell proliferation inhibition of OSSL_325096 was much lower compared with that of DBeQ (Figure 1B). These data suggest that OSSL_325096 may also have other cytotoxic mechanisms in addition to VCP inhibition.
3.5. Knockdown of VCP induces accumulation of ERAD‐associated proteins and apoptosis in MM cell lines
For elucidation of the biological phenotype of MM cells by p97/VCP inhibition, we carried out knockdown of VCP in KMS12PE and KMS11 myeloma cell lines. Doxycycline‐inducible shRNAs for VCP were stably introduced in the MM cell lines (shVCP #1, #2, #4, #5). Knockdown efficacy was determined by RT‐qPCR. Of four shRNAs, shVCP #5 showed the highest knockdown efficacy in both KMS12PE and KMS11 cells (Figure 5A). VCP knockdown induced apoptosis in the majority of KMS12PE and KMS11 cells (Figure 5B‐D), and also induced accumulation of poly‐ubiquitinated proteins and cleavage of caspase‐3 in KMS12PE and KMS11 cells (Figure 5B). These data indicate that VCP knockdown induces ERAD‐associated protein accumulation, leading to apoptosis. Therefore, p97/VCP is a promising therapeutic target for MM.
Figure 5.
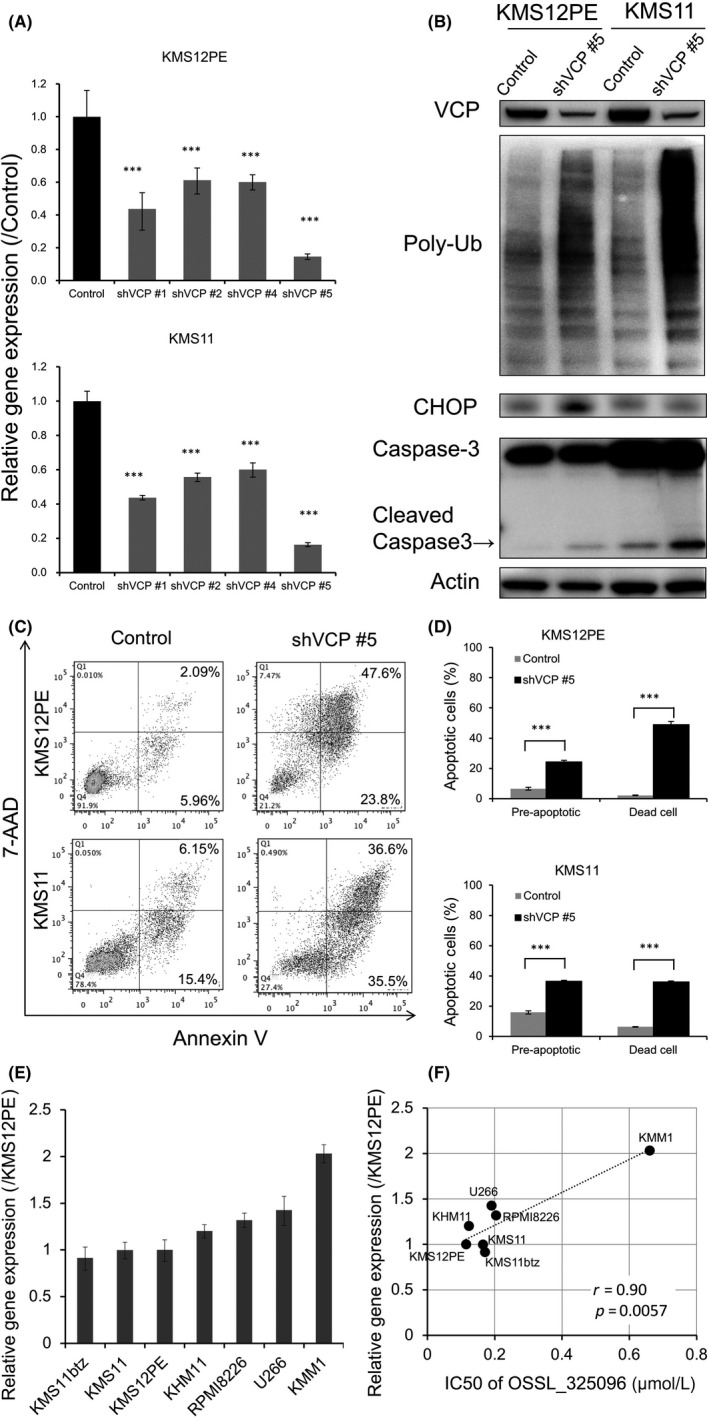
Knockdown of VCP induces accumulation of endoplasmic reticulum‐associated protein degradation (ERAD)‐associated proteins and apoptosis in multiple myeloma (MM) cell lines. shRNA‐mediated knockdown of VCP was carried out in MM cell lines. KMS12PE or KMS11 cells were stably transfected with tetracycline‐inducible shRNA plasmids, and shRNAs were induced by exposure of 1 μmol/L doxycycline for 48 h. Non‐targeting shRNA served as a control. A, Knockdown efficacies were determined by RT‐qPCR after shRNA induction in KMS12PE and KMS11 cells. shVCP#5 induced the strongest knockdown of VCP mRNA levels to <20% of that with control shRNA. Data represent mean ± SD derived from three separate experiments. B, Western blotting for VCP, poly‐ubiquitinated protein (Poly‐Ub), CHOP, and caspase‐3 in KMS12PE and KMS11 cells was carried out after shVCP#5 induction. Along with knockdown of p97/VCP protein, poly‐ubiquitinated protein accumulated and caspase‐3 was cleaved in KMS12PE and KMS11 cells. C, Dot plots and D, bar charts of MM cells stained with annexin V and 7AAD after shVCP#5 induction. VCP knockdown induced apoptosis in KMS12PE and KMS11 cells. Data represent the mean ± SD derived from three separate experiments. Significance values are indicated as ***P < .001 (Student's t‐test). E, Expression levels of VCP in myeloma cell lines. Real‐time PCR of VCP was carried out with myeloma cell lines. β‐Actin expression levels were used for standardization of VCP expression levels. F, Dot plot graph of VCP expression levels and IC 50 of OSSL_325096 in myeloma cell lines is shown. Significance values are indicated as P‐value
3.6. Expression levels of VCP are negatively correlated with sensitivities to OSSL_325096 in myeloma cell lines
Next, we evaluated whether VCP expression levels are correlated with the sensitivities to OSSL_325096 in myeloma cell lines. Expression level of p97/VCP varies between myeloma cell lines (Figure 5E). The cytotoxic IC50 of OSSL_325096 is higher in myeloma cell lines with high VCP expression and positively correlated with the expression levels of VCP in myeloma cell lines (Figure 5F). Therefore, the data indicated that VCP expression levels in myeloma cells are negatively correlated with sensitivities to OSSL_325096.
3.7. OSSL_325096 activates an apoptosis pathway and represses a cell cycle‐related pathway in MM cells
Next, we further analyzed the mechanisms of anti‐myeloma activity of OSSL_325096 using RNA sequencing. Whole transcriptomes of KMS12PE and KMS11 cells treated with DMSO or 1 μmol/L OSSL_325096 were quantified and gene expression levels in both cell lines were compared. GSEA of upregulated and downregulated genes in both KMS12PE and KMS11 following OSSL_325096 treatment was carried out. Significantly, the genes downregulated more than twofold were H1F0, RPS17L, and BCL2, and both H1F0 and BCL2 are involved in apoptosis (Figure 6A) (Table S1). In contrast, a total of 11 genes were upregulated more than twofold, including cyclin B1 (CCNB1), cell division cycle 20 homolog (CDC20), and heat shock proteins (HSPA8, HSPA1B). GSEA of the upregulated genes showed that overlapping pathways included “cell cycle”, “G2M checkpoint”, and “Genes involved in APC/C:Cdc20‐mediated degradation of Cyclin B” (Figure 6B) (Table S2). The data suggest that OSSL_325096 downregulates anti‐apoptotic proteins and upregulates heat shock‐related proteins and G2M checkpoint proteins that may result in cell cycle arrest.
Figure 6.
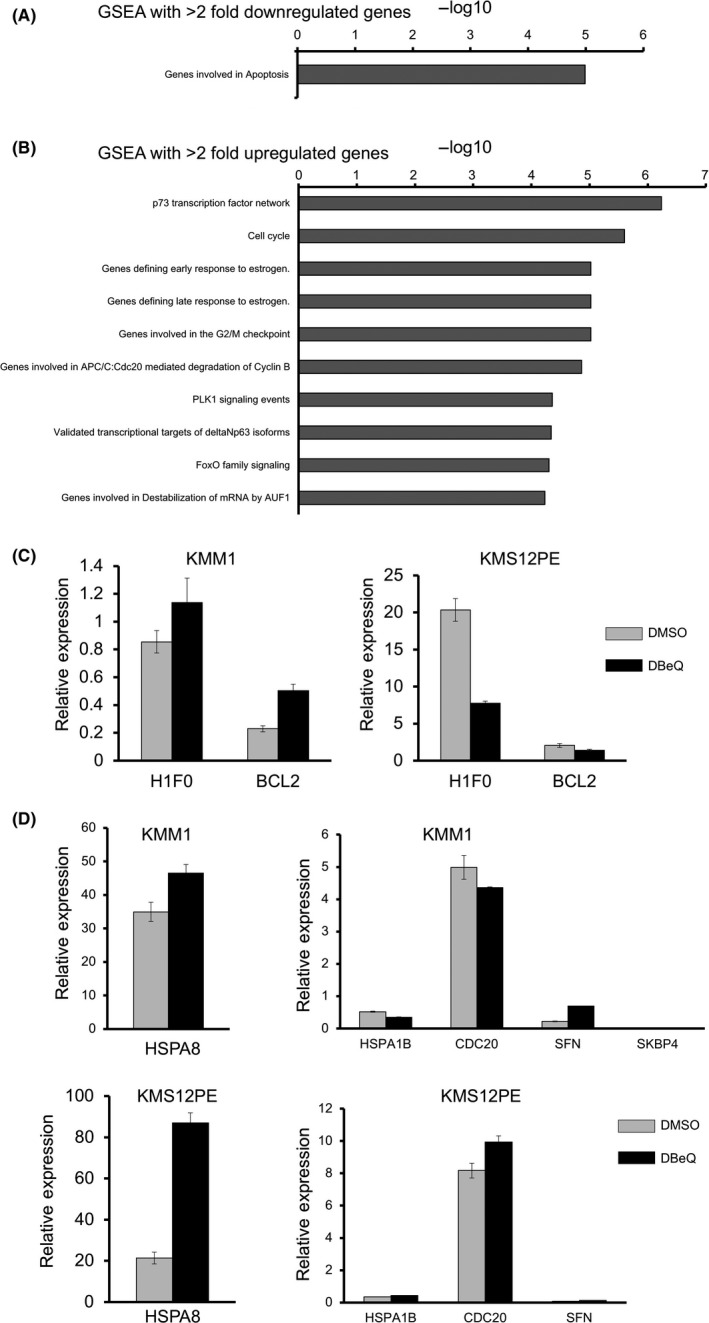
OSSL_325096 downregulated BCL2 and upregulated cell cycle and heat shock‐related proteins. Gene set enrichment analysis (GSEA) was carried out based on RNA‐seq data of KMS12PE and KMS11 cells treated with DMSO or 1 μmol/L OSSL_325096. GSEA with >2 fold downregulated genes (A), and GSEA with >2 fold upregulated genes (B), with their maximum average RPKM of more than 50 were carried out. C,D, Real‐time PCR analysis of H1F0 and bcl2 (C), and HSPA1B, HSPA8,CDC20,SFN, and SKBP4 (D) in KMM1 and KMS12PE myeloma cells treated with 10 μmol/L of DBeQ. Means ± SD are shown. RPKM, Reads Per Kilobase of transcript, per Million mapped reads
We also checked the expression levels of genes which were highly downregulated or upregulated by OSSL_325096 in myeloma cells treated with DBeQ by real‐time PCR. H1F0 and BCL2, which are downregulated by OSSL_325096, were downregulated only in KMS12PE but not in KMM1 cells (Figure 6C). Among the most upregulated genes by OSSL_325096, namely HSPA1B, HSPA8, CDC20, and SFN, only HSPA8 and SFN were upregulated in both KMM1 and KMS12PE cells (Figure 6D). Therefore, gene expression profiles are not completely similar between myeloma cells treated with OSSL_325096 and DBeQ, suggesting that OSSL_320596 may also have other molecular targets.
3.8. OSSL_325096 suppresses MM cell proliferation in vivo
Finally, we evaluated the efficacy of OSSL_325096 in MM cells in vivo. We used two MM cell lines, KMS11 and KMS12PE, to generate xenograft mice. The xenograft mice (eight mice for each cell line) were treated four times (twice a week for 2 weeks) with 50 mg/kg OSSL_325096 or vehicle. In both xenograft models, OSSL_325096 significantly inhibited MM tumor growth (Figure 7A). During the treatment, OSSL_325096‐treated mice had body weight loss (5%‐8% of original weight), but their body weights soon recovered after discontinuation of OSSL_325096 (Figure 7B). No mice died in the OSSL_325096‐treated group. These data indicate that OSSL_325096 had potent anti‐MM activity in vivo, and that the toxicity was reversible and not severe.
Figure 7.
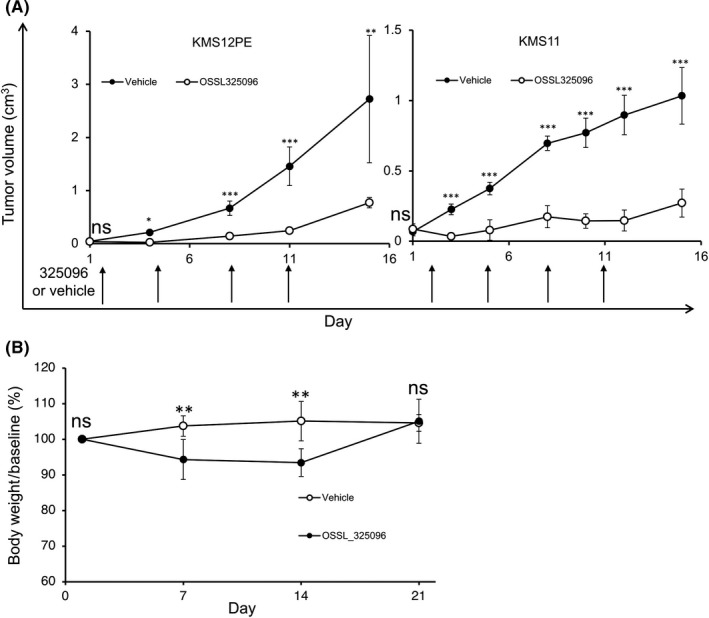
OSSL_325096 suppressed multiple myeloma (MM) cell proliferation in vivo without lethal toxicity. KMS12PE or KMS11 cells were s.c. transplanted into Rag2 −/− Jak3 −/− BALB/c mice and 50 mg/kg OSSL_325096 or vehicle was given to the mice i.p. after subcutaneous tumors became visible. A, Tumor volumes are presented as mean ± SD. Arrows indicate number of days of OSSL_325096 or vehicle dosage. B, Body weights of mice were monitored every week after giving OSSL_325096 or vehicle. Body weight at day 1 was regarded as baseline (set as 100%). Body weight is presented as mean ± SD. Significance values are indicated as *P < .05, **P < .01, ***P < .001 (Student's t‐test)
4. DISCUSSION
p97/VCP is a ubiquitous protein involved in many biological processes that are reported to be important for MM survival and it is thought to be a non‐redundant protein.28 p97/VCP is also involved in many essential cellular processes including protein degradation occurring through the ubiquitination‐proteasome degradation pathway. E1, E2, and E3 enzymes add ubiquitin to proteins for degradation, and E3 and p97/VCP interact and promote ERAD.23, 24, 25, 26 Ubiquitinated substrates are extracted and disassembled from the protein complex of p97/VCP and delivered to the proteasome. Therefore, p97/VCP inhibition may have a similar effect to proteasome inhibition. Indeed, PI such as bortezomib, carfilzomib, and ixazomib bind to and inhibit the proteasome and are effective for MM.6, 33, 34, 35, 36 In addition, IMiD, thalidomide, lenalidomide, and pomalidomide, bind to cereblon, a well‐known E3 ubiquitin ligase, and change substrate specificity, leading to degradation of IKZF1 and IKZF3.37, 38, 39 Therefore, IMiD are effective for MM because they target ubiquitination‐proteasome degradation. As p97/VCP is also involved in the ubiquitination‐proteasome degradation pathway, p97/VCP may be an ideal target for treatment of myeloma. Furthermore, p97/VCP inhibitor, OSSL_325096, may synergistically eradicate myeloma cells with PI and IMiD. However, it is possible that p97/VCP inhibition may interfere with the degradation of IKZF1 and IKZF3 by inhibiting disassociation from the p97/VCP protein complex for delivery to the proteasome. Further investigation is necessary to elucidate whether p97/VCP inhibitors have synergistic or cancellation effects with IMiD and PI.
Many novel agents that target specific pathways in MM cells have recently been developed and are improving the prognosis of MM patients. However, because of the genetic and molecular complexity of MM, no MM patients achieve a complete cure. Therefore, the development of other types of novel agents is necessary for MM patients. VCP inhibition is a promising strategy to treat myeloma patients. However, there remains some concern regarding the general toxicity of targeting p97/VCP for MM therapy, because p97/VCP is a ubiquitous protein and is involved in many essential cellular processes. In fact, embryonic knockout of Vcp in mice is lethal,40 and mutations of VCP induce several neurological diseases.24, 28 These observations indicate that complete or irreversible inhibition of p97/VCP can induce severe general or neurological toxicities. Thus, inhibition levels of p97/VCP for cancer therapy may need reversibility and less toxicity. The data from in silico docking simulations and ATPase activity assays indicate that OSSL_325096 acts as an ATP‐competitive and reversible inhibitor of p97/VCP. Furthermore, inhibition levels of OSSL_325096 against the ATP binding site in the D2 domain of p97/VCP were weaker than those of ATP, suggesting this compound may not block the function of p97/VCP completely. Treatment with OSSL_325096 induced slight weight loss in xenograft mice, which was reversible and not lethal. More detailed examination for dosage and administration is needed for further understanding of anti‐MM activity and the adverse effects of OSSL_325096 in vivo.
p97/VCP inhibition activity of OSSL_325096 is very similar to that of DBeQ (IC50 10.25 ± 1.57 μmol/L vs. 6.99 ± 0.50 μmol/L). In contrast, the cytotoxic IC50 of OSSL_325096 is much lower than that of DBeQ, suggesting that OSSL_325096 may have other molecular targets in myeloma cells. Indeed, downregulated and upregulated genes by OSSL_325096 and those by DBeQ are not consistent in myeloma cells. As mentioned above, p97/VCP is a multifunctional protein involved in many essential cellular processes. Therefore, to understand the effectiveness of OSSL_325096, identification of its other possible molecular targets that trigger off‐target effects is now under investigation.
We also examined the molecular mechanisms of myeloma cell death induced by OSSL_325096. RNA‐seq analysis of myeloma cells treated with OSSL_325096 shows that of three genes downregulated twofold, H1F0 and BCL2 are associated with apoptosis. It is reasonable that BCL2, which protects B cells from apoptosis, is downregulated in OSSL_325096‐treated MM cells. GSEA of genes upregulated twofold identified several pathways that were activated in OSSL_325096‐treated myeloma cells including p73, cell cycle, G2M checkpoint, CDC20‐mediated degradation of cyclin B, PLK, and FOXO pathways. These data are compatible with the fact that OSSL_325096 induces apoptosis in MM cells, whereas OSSL_325096 does not induce cell cycle arrest (data not shown).
It was recently reported that another p97/VCP inhibitor, CB‐5083, is highly effective in myeloma cells in vitro and in vivo. Indeed, CB‐5083 is already undergoing a Phase 1 trial in myeloma patients, although there is no report on efficacy in patients. Growth inhibition of 50% (GI50) of CB‐5083 is reportedly 428 nmol/L in KMS11 cells, whereas the IC50 of OSSL_325096 in KMS11btz cells is 100‐500 nmol/L, suggesting that there is not much difference in efficacy between the two compounds. The structures of both compounds are different, and therefore OSSL_325096 is also a good candidate as a new type of p97/VCP inhibitor. IC50 of OSSL_325096 (100‐500 nmol/L) for viability of KMS11btz cells is much lower than that for p97/VCP inhibition activity, so OSSL_325096 should have other target molecules for killing myeloma cells. Further investigation of the mechanisms of anti‐myeloma activities and pharmacokinetics of OSSL_325096 are necessary for clinical application. To find more effective and less toxic agents, we may also use OSSL_325096 as a lead compound and carry out structure modification of OSSL_325096 to identify more effective compounds.
DISCLOSURE
Y. Okuno has received research funding from Celgene. The remaining authors declare no conflicts of interest.
Supporting information
ACKNOWLEDGMENTS
We thank H. Nikki March, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript. This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (JSPS grant‐in‐aid number 26461427).
Nishimura N, Radwan MO, Amano M, et al. Novel p97/VCP inhibitor induces endoplasmic reticulum stress and apoptosis in both bortezomib‐sensitive and ‐resistant multiple myeloma cells. Cancer Sci. 2019;110:3275‐3287. 10.1111/cas.14154
REFERENCES
- 1. Ozaki S, Handa H, Saitoh T, et al. Trends of survival in patients with multiple myeloma in Japan: a multicenter retrospective collaborative study of the Japanese Society of Myeloma. Blood Cancer J. 2015;5:e349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kyle R, Rajkumar S. Multiple myeloma. Blood. 2008;111:2962‐2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364:1046‐1060. [DOI] [PubMed] [Google Scholar]
- 4. Raab M, Podar K, Breitkreutz I, Richardson P, Anderson K. Multiple myeloma. Lancet. 2009;374:324‐339. [DOI] [PubMed] [Google Scholar]
- 5. Singhal S, Mehta J, Desikan R, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341:1565‐1571. [DOI] [PubMed] [Google Scholar]
- 6. Hideshima T, Richardson P, Chauhan D, et al. The proteasome inhibitor PS‐341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Can Res. 2001;61:3071‐3076. [PubMed] [Google Scholar]
- 7. Hideshima T, Richardson P, Anderson KC. Novel therapeutic approaches for multiple myeloma. Immunol Rev. 2003;194:164‐176. [DOI] [PubMed] [Google Scholar]
- 8. Jain S, Diefenbach C, Zain J, O'Connor OA. Emerging role of carfilzomib in treatment of relapsed and refractory lymphoid neoplasms and multiple myeloma. Core Evid. 2011;6:43‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Richardson PG, Blood E, Mitsiades CS, et al. A randomized phase 2 study of lenalidomide therapy for patients with relapsed or relapsed and refractory multiple myeloma. Blood. 2006;108:3458‐3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in combination with low‐dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood. 2014;123:1826‐1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dimopoulos M, Spencer A, Attal M, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357:2123‐2132. [DOI] [PubMed] [Google Scholar]
- 12. Jakubowiak AJ, Benson DM, Bensinger W, et al. Phase I trial of anti‐CS1 monoclonal antibody elotuzumab in combination with bortezomib in the treatment of relapsed/refractory multiple myeloma. J Clin Oncol. 2012;30:1960‐1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N Engl J Med. 2015;373:1207‐1219. [DOI] [PubMed] [Google Scholar]
- 14. Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. 2014;28:1122‐1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chauhan D, Bianchi G, Anderson KC. Targeting the UPS as therapy in multiple myeloma. BMC Biochem. 2008;9(Suppl 1):S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lub S, Maes K, Menu E, De Bruyne E, Vanderkerken K, Van Valckenborgh E. Novel strategies to target the ubiquitin proteasome system in multiple myeloma. Oncotarget. 2016;7:6521‐6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leung‐Hagesteijn C, Erdmann N, Cheung G, et al. Xbp1s‐negative tumor B cells and pre‐plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell. 2013;24:289‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ri M, Iida S, Nakashima T, et al. Bortezomib‐resistant myeloma cell lines: a role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia. 2010;24:1506‐1512. [DOI] [PubMed] [Google Scholar]
- 20. Abdel Malek MA, Jagannathan S, Malek E, et al. Molecular chaperone GRP78 enhances aggresome delivery to autophagosomes to promote drug resistance in multiple myeloma. Oncotarget. 2015;6:3098‐3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alonso S, Hernandez D, Chang YT, et al. Hedgehog and retinoid signaling alters multiple myeloma microenvironment and generates bortezomib resistance. J Clin Investig. 2016;126:4460‐4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zaal EA, Wu W, Jansen G, Zweegman S, Cloos J, Berkers CR. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saffert P, Enenkel C, Wendler P. Structure and function of p97 and Pex1/6 type II AAA+ complexes. Front Mol Biosci. 2017;4:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xia D, Tang WK, Ye Y. Structure and function of the AAA+ ATPase p97/Cdc48p. Gene. 2016;583:64‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA‐ATPase in the ubiquitin system. Nat Cell Biol. 2012;14:117‐123. [DOI] [PubMed] [Google Scholar]
- 26. Meyer H, Weihl CC. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci. 2014;127:3877‐3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dantuma NP, Hoppe T. Growing sphere of influence: Cdc48/p97 orchestrates ubiquitin‐dependent extraction from chromatin. Trends Cell Biol. 2012;22:483‐491. [DOI] [PubMed] [Google Scholar]
- 28. Yamanaka K, Sasagawa Y, Ogura T. Recent advances in p97/VCP/Cdc48 cellular functions. Biochem Biophys Acta. 2012;1823:130‐137. [DOI] [PubMed] [Google Scholar]
- 29. Auner HW, Moody AM, Ward TH, et al. Combined inhibition of p97 and the proteasome causes lethal disruption of the secretory apparatus in multiple myeloma cells. PLoS ONE. 2013;8:e74415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Le Moigne R, Aftab BT, Djakovic S, et al. The p97 inhibitor CB‐5083 is a unique disrupter of protein homeostasis in models of multiple myeloma. Mol Cancer Ther. 2017;16:2375‐2386. [DOI] [PubMed] [Google Scholar]
- 31. Chou TF, Brown SJ, Minond D, et al. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin‐dependent and autophagic protein clearance pathways. Proc Natl Acad Sci USA. 2011;108:4834‐4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou HJ, Wang J, Yao B, et al. Discovery of a first‐in‐class, potent, selective, and orally bioavailable inhibitor of the p97 AAA ATPase (CB‐5083). J Med Chem. 2015;58:9480‐9497. [DOI] [PubMed] [Google Scholar]
- 33. San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359:906‐917. [DOI] [PubMed] [Google Scholar]
- 34. Harousseau JL, Palumbo A, Richardson PG, et al. Superior outcomes associated with complete response in newly diagnosed multiple myeloma patients treated with nonintensive therapy: analysis of the phase 3 VISTA study of bortezomib plus melphalan‐prednisone versus melphalan‐prednisone. Blood. 2010;116:3743‐3750. [DOI] [PubMed] [Google Scholar]
- 35. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372:142‐152. [DOI] [PubMed] [Google Scholar]
- 36. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;374:1621‐1634. [DOI] [PubMed] [Google Scholar]
- 37. Ito T, Ando H, Suzuki T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345‐1350. [DOI] [PubMed] [Google Scholar]
- 38. Kronke J, Udeshi ND, Narla A, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lu G, Middleton RE, Sun H, et al. The myeloma drug lenalidomide promotes the cereblon‐dependent destruction of Ikaros proteins. Science. 2014;343:305‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Muller JM, Deinhardt K, Rosewell I, Warren G, Shima DT. Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem Biophys Res Comm. 2007;354:459‐465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials