

Abstract
We retrospectively investigated the impact of the tumor microenvironment (TME) on the efficacy of epidermal growth factor receptor (EGFR)‐tyrosine kinase inhibitors (TKIs) as first‐line treatment in 70 patients with advanced EGFR‐mutant non‐small cell lung cancer and who were seen at Osaka City University Hospital (Osaka, Japan) between August 2013 and December 2017. Using immunohistochemical staining with 28‐8 and D7U8C Abs, the tumor proportion score was assessed for programmed cell death‐1 ligand‐1 (PD‐L1), as high (50% or more) or low (less than 50%), and ligand‐2 (PD‐L2) expression, respectively. The extent of CD8+ tumor‐infiltrating lymphocytes was evaluated on a scale of 0‐3, with 0‐1 as low and 2‐3 as high. The TME of the 52 evaluable pretreatment specimens was categorized into 4 subtypes, according to the respective PD‐L1 tumor proportion and CD8+ scores, as follows: (a) high/high (13.5%, n = 7); (b) low/low (42.3%, n = 22); (c) high/low (17.3%, n = 9); and (d) low/high (26.9%, n = 14). Expression of PD‐L2 was significantly the highest in type 1 (57.1% vs 4.5% vs 11.1% vs 7.1%, respectively; P = .0090). Response rate was significantly the lowest in type 1 (14.3% vs 81.8% vs 66.7% vs 78.6%, respectively; P = .0085). Progression‐free survival was the shortest in type 1 and the longest in type 4 (median, 2.4 vs 11.3 vs 8.4 vs 17.5 months, respectively; P = .00000077). The efficacy of EGFR‐TKIs differed according to the TME, and the phenotype with high PD‐L1 and CD8+ expression might be the subset that would poorly benefit from such treatment.
Keywords: epidermal growth factor receptor tyrosine kinase inhibitor, non‐small cell lung cancer, programmed cell death‐1 ligand‐1, programmed cell death‐1 ligand‐2, tumor microenvironment
Abbreviations
- DCR
disease control rate
- EGFR
epidermal growth factor receptor
- IHC
immunohistochemistry
- NSCLC
non‐small cell lung cancer
- PD‐1
programmed cell death‐1
- PD‐L1/2
programmed cell death‐1 ligand‐1/2
- PFS
progression‐free survival
- preT790M
pretreatment T790M
- RR
response rate
- TIL
tumor‐infiltrating lymphocyte
- TKI
tyrosine kinase inhibitor
- TME
tumor microenvironment
- TPS
tumor proportion score
1. INTRODUCTION
Lung cancer is the most common cause of cancer death worldwide.1 The discovery of driver oncogenes and the development of molecular‐targeting agents have placed NSCLC management into a new era of personalized medicine. Mutations in the EGFR gene are one of the most common driver oncogenes in NSCLC.2, 3 Epidermal growth factor receptor‐TKIs provide dramatic antitumor activity in advanced NSCLC patients with activating EGFR mutations. Clinical studies have confirmed EGFR‐activating mutations in exon 19 and exon 21 as the main predictor of clinical outcomes with TKI therapy for NSCLC; this has led to a paradigm shift on the use of TKI as a standard first‐line treatment for NSCLC patients with EGFR‐activating mutations.4, 5, 6, 7, 8
Following molecular‐targeted therapy, immunotherapy led a second paradigm shift for the treatment of NSCLC. Compared with conventional chemotherapy, immune checkpoint inhibitors that target PD‐1 or its ligand PD‐L1 have shown promising antitumor efficacy in patients with advanced NSCLC; in fact, nivolumab, pembrolizumab, and atezolizumab have been approved for the treatment of advanced NSCLC.9, 10, 11, 12 Recently, the first‐line treatment of patients with advanced NSCLC has been further developed; pembrolizumab monotherapy for a PD‐L1 TPS of 1% or higher and combination therapies with platinum doublet and pembrolizumab or atezolizumab, regardless of PD‐L1 expression, have become standard first‐line treatment options.13, 14, 15, 16, 17
However, patients with EGFR mutations from the KEYNOTE‐024, KEYNOTE‐042, and KEYNOTE‐189 trials and TKI‐naive EGFR‐mutant patients from the IMpower150 trial were excluded.13, 14, 15, 17 In fact, the preferential use of EGFR‐TKIs or immunotherapy as first‐line treatment for patients with EGFR‐activating mutations had not been strictly clear, because there had been no clinical trials that directly compared these therapies. Therefore, the correlation of EGFR mutation positivity and PD‐L1 expression and its impact on anti‐PD‐1/PD‐L1 therapies had been intensively investigated.
A retrospective study that investigated the frequencies of each PD‐L1 expression level in NSCLC harboring driver oncogenes revealed the existence of a subset of EGFR‐mutated NSCLC patients with high PD‐L1 expression,18 suggesting that the relationship between EGFR mutation positivity and PD‐L1 expression might not be mutually exclusive. However, subgroup analyses of the clinical trials and a meta‐analysis suggested that immune checkpoint inhibitors were less effective in patients with EGFR‐mutant NSCLC than in those with EGFR WT NSCLC.10, 11, 19 Another retrospective study found that NSCLC harboring EGFR mutations was associated with low overall response rate to PD‐1/PD‐L1 inhibitors and that low expression of both PD‐L1 and CD8+ TILs within the TME might underlie this unfavorable clinical response.20 In other words, the prediction of PD‐1/PD‐L1 inhibitor efficacy for NSCLC patients harboring EGFR mutations might need the assessment of both PD‐L1 expression in tumor cells and the TME, including CD8+ TILs.
Moreover, the relationship between the efficacy of EGFR‐TKI itself and the TME remains elusive. If the subset of TME type that is expected to benefit from immunotherapy is correlated with a certain subset that would poorly benefit from EGFR‐TKI treatment, there might be room for further investigation on the preferential of use of immunotherapy, even for EGFR‐mutant NSCLC patients. Therefore, we hypothesized that the efficacy of EGFR‐TKIs differed according to the TME, based on the tumor expression of PD‐L1 and CD8+ TIL. In this study, we aimed to investigate the association between TME and EGFR‐TKI efficacy, as well as the impact of such association.
2. MATERIALS AND METHODS
2.1. Patients
We had previously reported the association between PFS after first‐line conventional EGFR‐TKIs and the abundance of preT790M in EGFR‐TKI‐naive EGFR‐mutated NSCLC specimens that were assessed by droplet digital PCR.21 In the present retrospective study, the cohort used in that previous study was analyzed. The eligible patients who were screened comprised those with newly histologically proven locally advanced or metastatic lung adenocarcinoma, those with documented EGFR‐activating mutation (exon 19 deletion or L858R mutation), and those treated with first‐line EGFR‐TKIs at the Osaka City University Hospital (Osaka, Japan) between August 2013 and December 2017. This study was approved by the institutional review board of Osaka City University Hospital. All patients provided written informed consent before tissue collection. The study was carried out in accordance with the Declaration of Helsinki.
2.2. Data collection
The medical records were reviewed, and data regarding treatment history were extracted. The data were updated as of December 31, 2018, and the responses were assessed according to RECIST version 1.1.22 Progression‐free survival was measured from treatment initiation to clinical or radiographic progression or death from any cause. In this study, switching of EGFR‐TKI due to adverse events was considered as continuation of EGFR‐TKI treatment. In such cases, PFS was defined from the initiation of the first‐line EGFR‐TKI treatment to disease progression or death from any cause during the subsequent EGFR‐TKI treatment following the switch. Patients without documented clinical or radiographic disease progression and those who were still alive on the date of the last follow‐up were censored.
2.3. Immunohistochemistry
Consecutive 5‐μm‐thick sections from the formalin‐fixed paraffin‐embedded tumor specimens were analyzed by IHC staining at N Lab (Nagasaki, Japan). The histological samples that contained 100 or more tumor cells were included as the subjects of analysis. The samples were assessed or scored independently by 2 pathologists; in cases of disagreement, the slides were re‐examined and the 2 pathologists reached a consensus.
Immunohistochemistry for PD‐L1 was carried out using the PD‐L1 IHC 28‐8 pharmDx assay (Agilent Technologies), with Autostainer Link 48 (Agilent Technologies), in accordance with the manufacturer's instructions. Positivity for PD‐L1 was defined as membranous staining of tumor cells at any intensity. The PD‐L1 TPS was calculated as the percentage of the area with at least 100 viable tumor cells with PD‐L1 positivity. We classified patients as having test results that were strongly positive (TPS 50% or higher), weakly positive (TPS 1%‐49%), or negative (TPS less than 1%) (Figure S1).
The expression of PD‐L2 was analyzed by IHC staining using rabbit mAb (dilution 1:100, clone D7U8C; Cell Signaling Technology). For PD‑L2, the semiquantitative H‑score was calculated by multiplying the membranous and/or cytoplasmic intensity score (0, absent; 1, weak; 2, moderate; or 3, strong) with the percentage of stained cells. Specimens with an H‑score of 5 or higher were defined as PD‑L2‐positive, based on a previous study (Figure S2).23
The presence of CD8+ TILs in the NSCLC specimens was assessed with IHC staining, using mouse mAb (dilution 1:400, clone C8/144B; Agilent Technologies) on tumor cells and in tumor stroma where invasion of tumor cells was observed. For larger samples, the TILs expressing CD8 were counted in 4 random 1‐mm diameter samples on the whole tumor specimen, including the center and invasive margin of the tumor, in order to calculate the density of TILs.24 For smaller samples, density was evaluated using the count of CD8+ TILs in all tumor areas. The CD8+ TILs were semiquantitatively evaluated on a scale of 0 to 3, based on the extent of positive lymphocytes infiltrating within the tumor cells. Each score was defined on the basis of the fraction of tumor cells that contained CD8+ TILs on top: score 0, none or rare; score 1, less than 5%; score 2, 5% or more and less than 25%; and score 3, 25% or more, based on a previous study (Figure S3).20
We defined PD‐L1 expression with TPS 50% or more as PD‐L1 high and that with TPS less than 50% as low. A CD8+ TIL score of 2‐3 was defined as CD8+ TIL high and a CD8+ TIL score of 0‐1 as CD8+ TIL low. Consequently, based on PD‐L1 expression and CD8+ TILs, the TME was categorized into 4 subtypes: PD‐L1 high/CD8+ TIL high (type 1), PD‐L1 low/CD8+ TIL low (type 2), PD‐L1 high/CD8+ TIL low (type 3), and PD‐L1 low/CD8+ TIL high (type 4) (Figure 1).
Figure 1.

Representative images of the 4 tumor microenvironment subtypes of programmed cell death‐1 ligand‐1 (PD‐L1) and CD8+ immunostaining in non‐small cell lung cancer tissue. Scale bar = 100 μm. TIL, tumor‐infiltrating lymphocyte
2.4. Statistical analysis
Fisher's exact test was carried out for comparison of categorical data. The Kaplan‐Meier method was used to estimate the survival curves for PFS. Log‐rank tests were used to compare the survival curves among the TME subtypes. All P values were based on a 2‐sided hypothesis, and a P value less than .05 was considered to indicate statistical significance.
3. RESULTS
3.1. Patient characteristics
A total of 70 patients were eligible for this study. In our previous study, only 66 patients we assessed for preT790M status, because genome DNA extraction from the pretreatment samples was not successful in 4 patients. For the current study, among these 66 patients, we excluded 9 patients who had samples that had fewer than 100 viable tumor cells and 5 patients in whom the CD8+ TILs could not be assessed in the cell block. Therefore, 52 patients were finally analyzed in this study (Figure S4).
Among the 52 patients, the PD‐L1 TPS was 50% or more in 16 (30.8%) patients, 1%‐49% in 18 (34.6%) patients, and less than 1% in 18 (34.6%) patients. The proportions of each TME subtype were 13.5% (n = 7, type 1), 42.3% (n = 22, type 2), 17.3% (n = 9, type 3), and 26.9% (n = 14, type 4). The baseline patient characteristics according to the TME subtypes are shown in Table 1. Among the TME subtypes, there were no significant differences in age, gender, smoking status, stage, performance status, or EGFR status, but there was variability in the preT790M detection rate. In the current study cohort, only 7 (13.5%) patients had positive PD‐L2 expression. According to the TME subtype, positivity of PD‐L2 expression was significantly the highest for type 1 (57.1%) than for types 2, 3, or 4 (4.5%, 11.1%, and 7.1%, respectively, P = .0090).
Table 1.
Baseline characteristics of patients with non‐small cell lung cancer according to tumor microenvironment subtypes (N = 52)
| Characteristics | Number of patients, n (%) | P value | |||
|---|---|---|---|---|---|
| Type 1 (n = 7, 13.5%) | Type 2 (n = 22, 42.3%) | Type 3 (n = 9, 17.3%) | Type 4 (n = 14, 26.9%) | ||
| Age, years; median (range) | 67 (38‐88) | 71 (50‐82) | 71 (48‐77) | 74.5 (65‐81) | .1500 |
| <70 | 4 (57.1) | 10 (45.5) | 4 (44.4) | 2 (14.3) | |
| ≥70 | 3 (42.9) | 12 (54.5) | 5 (55.6) | 12 (85.7) | |
| Sex | |||||
| Male | 2 (28.6) | 7 (31.8) | 4 (44.4) | 4 (28.6) | .9000 |
| Female | 5 (71.4) | 15 (68.2) | 5 (55.6) | 10 (71.4) | |
| Smoking | |||||
| Smoker | 1 (14.3) | 8 (36.4) | 3 (33.3) | 3 (21.4) | .6700 |
| Never smoker | 6 (85.7) | 14 (63.6) | 6 (66.7) | 11 (78.6) | |
| Stage | |||||
| III | 1 (14.3) | 3 (13.6) | 0 (0.0) | 1 (7.1) | .7700 |
| IV | 6 (85.7) | 19 (86.4) | 9 (100.0) | 13 (92.9) | |
| ECOG PS | |||||
| 0‐1 | 4 (57.1) | 19 (86.4) | 8 (88.9) | 13 (92.9) | .2300 |
| ≥2 | 3 (42.9) | 3 (13.6) | 1 (11.1) | 1 (7.1) | |
| EGFR mutation status | |||||
| Exon19 deletion | 1 (14.3) | 12 (54.5) | 6 (66.7) | 5 (35.7) | .1400 |
| Exon21 L858R | 6 (85.7) | 10 (45.5) | 3 (33.3) | 9 (64.3) | |
| PreT790M | |||||
| Positive | 4 (57.1) | 8 (36.4) | 7 (77.8) | 3 (21.4) | .0430 |
| Negative | 3 (42.9) | 14 (63.6) | 2 (22.2) | 11 (78.6) | |
| First‐line EGFR‐TKI | |||||
| Gefitinib | 4 (57.1) | 8 (36.4) | 3 (33.3) | 5 (35.7) | .5800 |
| Erlotinib | 2 (28.6) | 3 (13.6) | 3 (33.3) | 4 (28.6) | |
| Afatinib | 1 (14.3) | 1 (4.5) | 0 (0.0) | 0 (0.0) | |
| Gefitinib/erlotiniba | 0 (0.0) | 9 (40.9) | 3 (33.3) | 4 (28.6) | |
| Afatinib/gefitiniba | 0 (0.0) | 1 (4.5) | 0 (0.0) | 0 (0.0) | |
| Erlotinib/afatiniba | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (7.1) | |
| PD‐L2 expression | |||||
| Positive | 4 (57.1) | 1 (4.5) | 1 (11.1) | 1 (7.1) | .0090 |
| Negative | 3 (42.9) | 21 (95.5) | 8 (88.9) | 13 (92.9) | |
In this study, switch of epidermal growth factor receptor‐ tyrosine kinase inhibitor (EGFR‐TKI) due to an adverse event was considered as continuation of EGFR‐TKI therapy.
PD‐L2, programmed cell death‐1 ligand‐2; PreT790M, pretreatment T790M; PS, performance status; TME, tumor microenvironment; Type 1, PD‐L1 high/CD8+ tumor‐infiltrating lymphocyte (TIL) high; Type 2, PD‐L1 low/CD8+ TIL low; Type 3, PD‐L1 high/CD8+ TIL low; Type 4, PD‐L1 low/CD8+ TIL high.
3.2. Efficacy of EGFR‐TKIs based on TME subtypes
In the current study cohort, the overall RR and disease control rate (DCR) after first‐line EGFR‐TKIs were 69.2% and 88.5%, respectively. According to the PD‐L1 expression, both RR and DCR were significantly worse with PD‐L1 high expression than with low PD‐L1 expression (RR, 43.8% vs 80.6%, P = .020; DCR, 62.5% vs 100.0%, P = .00039). According to the TME subtype, both RR and DCR were significantly the worst in type 1 than in types 2, 3, or 4 (RR, 14.3% vs 81.8% vs 66.7% vs 78.6%, respectively, P = .0085; DCR, 42.9% vs 100.0% vs 77.8% vs 100.0%, respectively, P = .00016) (Figure 2A). The details of the RRs are shown in Figure 2B.
Figure 2.
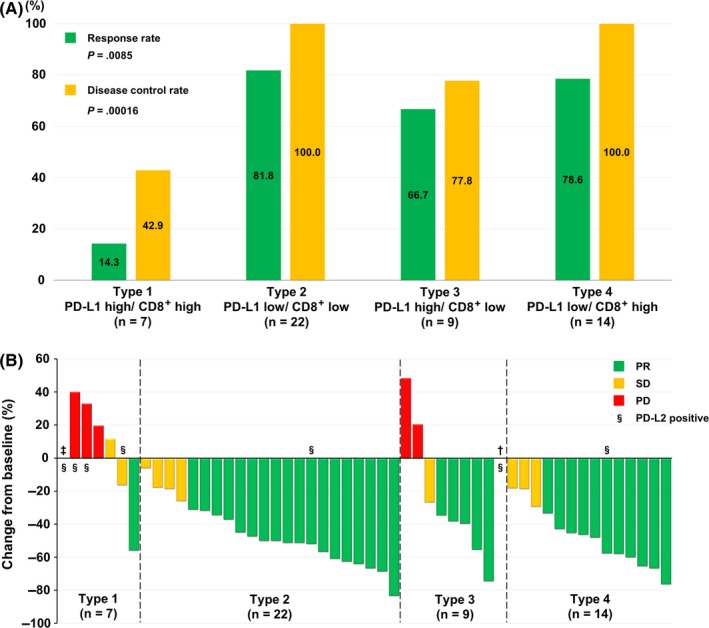
Association between response to epidermal growth factor receptor‐tyrosine kinase inhibitors (EGFR‐TKIs) and the tumor microenvironment (TME) in non‐small cell lung cancer. A, Comparison of the response and disease control rates for first‐line EGFR‐TKIs, according to the TME subtypes. B, Waterfall plot of the best percentage change from baseline in the cumulative longest tumor diameters. †In the type 3 group, 1 patient had a nonmeasurable lesion (malignant pleural effusion) at the initiation of first‐line EGFR‐TKI; apparent reduction of malignant pleural effusion was confirmed after the initiation of therapy. ‡ In the type 1 group, the tumor was not measurable in 1 patient who had disease progression without response. §A case with programmed cell death‐1 ligand‐2 (PD‐L2) expression. PD, progressive disease; PR, partial response; SD, stable disease
The PFS after first‐line EGFR‐TKIs was significantly shorter in patients with high PD‐L1 expression than in those with low PD‐L1 expression (median, 5.9 vs 13.2 months, P = .0059) (Figure 3A). According to the TME subtype, the PFS was the shortest in type 1 and the longest in type 4 (median: 2.4 vs 11.3 vs 8.4 vs 17.5 months, respectively, P = .00000077) (Figure 3B). In a PD‐L1 high setting, PFS was significantly shorter in patients with high CD8+ TIL (type 1) than in those with low CD8+ TIL (type 3) (P = .014). In a PD‐L1 low setting, PFS was significantly longer in patients with high CD8+ TIL (type 4) than in those with low CD8+ TIL (type 2) (P = .037) (Figure S5). In a CD8+ TIL high setting, PFS was significantly shorter in patients with high PD‐L1 (type 1) than in those with low PD‐L1 (type 4) (P = .00000056). In a CD8+ TIL low setting, there was no significant difference in the PFS between PD‐L1 high (type 3) and PD‐L1 low (type 2) patients (P = .77) (Figure S6). The details of the clinical course until disease progression are shown in Figure 3C.
Figure 3.
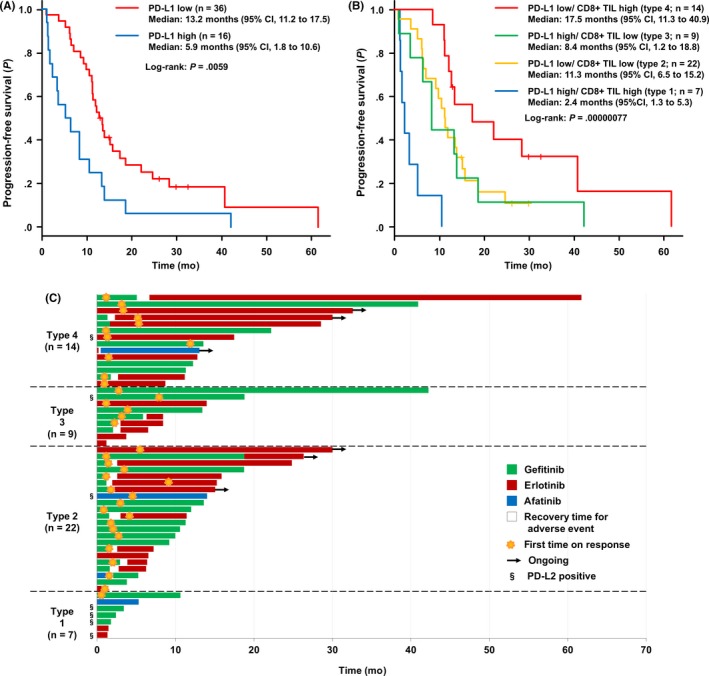
Impact of programmed cell death‐1 ligand‐1 (PD‐L1) expression status and the tumor microenvironment on progression‐free survival (PFS) in patients with non‐small cell lung cancer after first‐line epidermal growth factor receptor‐ tyrosine kinase inhibitors (EGFR‐TKIs). A, Kaplan‐Meier curves for PFS in EGFR‐mutated NSCLC patients treated with first‐line EGFR‐TKIs, according to PD‐L1 expression. B, Comparison of the Kaplan‐Meier curves for PFS after first‐line EGFR‐TKIs, according to the tumor microenvironment subtype (Type 1‐4). Plus signs denote censoring. C, Swimmer plot shows duration of first‐line EGFR‐TKIs treatment for patients on this study. CI, confidence interval; TIL, tumor‐infiltrating lymphocyte
3.3. Changes in TME after acquired resistance to EGFR‐TKIs
Of the current study cohort, 26 patients underwent rebiopsy to confirm the emergence of T790M after acquired resistance to EGFR‐TKIs. However, only 7 of these patients had available paired biopsy samples for the evaluation of PD‐L1/L2 TPS and CD8+ TILs. The clinical characteristics of these 7 patients and the changes in TME before and after developing acquired resistance to initial EGFR‐TKI therapy are summarized in Table 2. The results indicated that EGFR‐TKI treatment altered not only PD‐L1 expression status but also the CD8+ TILs and PD‐L2 expression status; accordingly, the TME phenotype shifted, although the regulatory mechanisms are unknown.
Table 2.
Changes in tumor microenvironment (TME) before and after developing acquired resistance to epidermal growth factor receptor‐tyrosine kinase inhibitor (EGFR‐TKI) therapy among patients with non‐small cell lung carcinoma and available paired tissues (N = 7)
| Patient | EGFR | Smoking | Pre | Rebiopsy | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Biopsy site Procedure |
PD‐L1 TPS | CD8+ score | PD‐L2 | TME subtype |
First‐line TKI Best response |
Rebiopsy site Procedure |
T790M | PD‐L1 TPS | CD8+ score | PD‐L2 | TME subtype | Response to osimertinib | |||
| 1 | L858R | No |
Primary lesion TBB |
<1% | 1 | − | Type 2 |
G/E PR |
Dissemination to PL Small resection |
+ | 3% | 2 | + | Type 4 | PR |
| 2 | L858R | No |
Primary lesion TBB |
0% | 2 | − | Type 4 |
G PR |
Primary lesion TBB |
− | 1% | 1 | − | Type 2 | − |
| 3 | Del19 | No |
Primary lesion TBB |
60% | 1 | − | Type 3 |
G/E PR |
Bone metastasis Resection |
+ | 30% | 0 | − | Type 2 | NE |
| 4 | Del19 | Yes |
Lymph node Needle biopsy |
60% | 1 | − | Type 3 |
G PR |
Primary lesion Resection |
− | 100% | 3 | − | Type 1 | − |
| 5 | L858R | No |
Primary lesion CT‐GLB |
0% | 2 | − | Type 4 |
E PR |
Primary lesion CT‐GLB |
+ | 100% | 3 | + | Type 1 | SD |
| 6 | L858R | No |
Primary lesion TBB |
30% | 2 | + | Type 4 |
E PR |
Primary lesion TBB |
+ | 8% | 1 | − | Type 2 | PR |
| 7 | L858R | No |
Primary lesion TBB |
70% | 1 | − | Type 3 |
E PR |
Primary lesion TBB |
− | 20% | 0 | − | Type 2 | − |
CT‐GLB, computed tomography‐guided percutaneous lung biopsy; E, erlotinib; G, gefitinib; NE, not evaluable; PD‐L1/2, programmed cell death‐1 ligand‐1/2; PL, pleura; PR, partial response; SD, stable disease; TBB, transbronchial biopsy; TPS, tumor proportion score.
3.3.1. Representative case
A 75‐year‐old male never‐smoker patient with stage IV lung adenocarcinoma harboring EGFR L858R mutation was given erlotinib as first‐line treatment, which confirmed partial response after 1.6 months. At baseline, IHC revealed a high proportion of CD8+ TILs (2+), without PD‐L1 or PD‐L2 expression. After disease progression, rebiopsy revealed EGFR T790M mutation, and the TME changed to type 1 (PD‐L1 TPS of 100% and CD8+ TIL of score 3), with positive PD‐L2 expression (80%). Osimertinib was started as a second‐line treatment, but, to our surprise, the response was not confirmed until 1.1 months after commencing treatment: however, the long‐term effect was not clear because osimertinib was discontinued due to an adverse event (Figures 4, S7, and S8).
Figure 4.
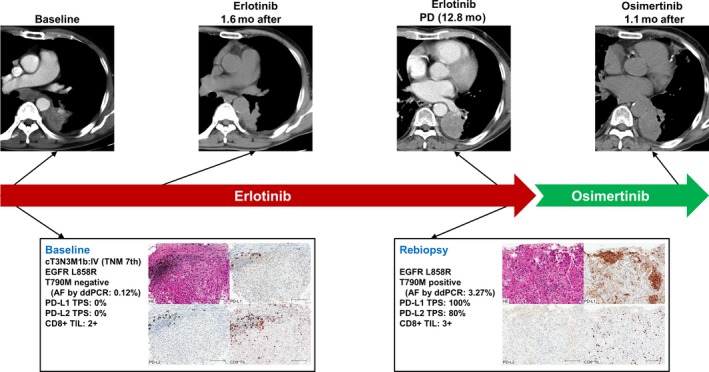
Representative case of non‐small cell lung cancer with altered tumor microenvironment and response to epidermal growth factor receptor‐tyrosine kinase inhibitor EGFR‐TKI) before and after developing acquired resistance to the initial EGFR‐TKI therapy. AF, allele frequency; ddPCR, droplet digital PCR; PD, progressive disease; PD‐L1/2, programmed cell death‐1 ligand‐1/2; TIL, tumor‐infiltrating lymphocyte; TPS, tumor proportion score
4. DISCUSSION
The present study indicated the existence of several subtypes of EGFR‐mutated NSCLC, according to the TME, based on tumor expression of PD‐L1 and CD8+ TILs. Our results suggested that differences in the TME could potentially impact treatment, and even the efficacy of EGFR‐TKIs.
A previous report proposed the stratification of the TME into 4 different types, based on the presence or absence of TIL and PD‐L1 expression, in order to predict the patients who will respond to anti‐PD‐1/PD‐L1 therapies. These include: type I, adaptive immune resistance (PD‐L1+/CD8+); type II, immune ignorance (PD‐L1−/CD8−); type III, intrinsic induction (PD‐L1+/CD8−); and type IV, immune tolerance (PD‐L1−/CD8+).25 Type I tumors are supposed to have adaptive PD‐L1 expression that is induced by preexisting intratumor T cells; therefore, these tumors are most likely to benefit from anti‐PD‐1/PD‐L1 blockade. A retrospective study by Gainor et al found that only 2.1% of patients had EGFR‐TKI‐naive tumors with concurrent PD‐L1 expression of 50% or more and high CD8+ TILs. They discussed that NSCLC harboring EGFR mutations was associated with low overall RR to PD‐1/PD‐L1 inhibitors and that low expression of both PD‐L1 and CD8+ TILs within the TME might underlie this unfavorable clinical response.20 Several studies have investigated the proportions of the 4 TME subtypes in NSCLC in larger cohorts. Velcheti et al analyzed the expression of PD‐L1 using 5H1 Ab and reported that the proportions of each subtype in 2 independent NSCLC cohorts (n = 302 and 155, respectively) were 12.9%‐23.9% for PD‐L1 high/TIL high, 41.3%‐47.4% for PD‐L1 low/TIL low, 11.9%‐12.3% for PD‐L1 high/TIL low, and 22.6%‐27.8% for PD‐L1 low/TIL high.26 Liu et al used the SP142 Ab to analyze PD‐L1 expression in a cohort of EGFR‐mutated or ALK‐rearranged NSCLC cases (n = 342) and reported that the proportions of each subtype were 5.0% for PD‐L1+/CD8+, 63.5% for PD‐L1−/CD8−, 11.1% for PD‐L1+/CD8−, and 20.5% for PD‐L1−/CD8+; whereas those in the EGFR and ALK WT NSCLC cohort (n = 339) were 14.2%, 50.3%, 7.0%, and 28.5%, respectively.27 In general, among the NSCLC tumors, type II seemed to be present in a majority and type I had been relatively present in a minority, although this tendency had the potential to become stronger in an EGFR‐mutant population.27 Our results showed a similar trend with those in previous studies, further revealing the prevalence of each TME subtype in EGFR‐mutated NSCLC.
Expression of another PD‐1 ligand, PD‐L2, in tumor cells is less prevalent than PD‑L1; however, PD‐L2 expression had been reported in a subset of some tumors, including NSCLC,28, 29 and has been cited as an independent predictor of poor overall survival in lung adenocarcinoma.30 Moreover, several studies reported subsets of PD‐L1+ patients who responded poorly to anti‐PD‐1 axis therapies, whereas some PD‐L1− patients have shown favorable responses;31, 32 these results suggested residual molecular interactions between PD‐1 and PD‐L2, and blockade of this pathway could be important to achieve treatment response.29 Although the expression of PD‐L2 in EGFR‐mutated NSCLC has been less studied, positive PD‐L2 expression has been reported in 13%‐20% of NSCLC, regardless of EGFR status.23, 29 In this study, a positive PD‐L2 expression was not associated with any clinicopathological characteristics, but it was significantly high in type 1, among the TME subtypes. Recent basic research showed that the expression of PD‐L2, as well as PD‐L1, in EGFR‐mutated or ALK‐rearranged NSCLC could be induced extrinsically by interferon‐γ and intrinsically by oncogenic signaling.33 The tendency of EGFR‐mutated NSCLC with high PD‐L1 expression to coexpress PD‐L2 through extrinsic or intrinsic mechanisms remains elusive in the context of basic research. However, the observation of highest PD‐L2 coexpression in type 1 tumors compared to other TME types could be reasonable evidence that type 1 tumors might benefit better from anti‐PD‐1 therapies than from anti‐PD‐L1 therapies in the clinical context.29, 34 Further clinical evaluation is warranted to support this hypothesis.
Expression of PD‐L1 has been shown to be related with clinical response to anti‐PD‐1 axis therapies in NSCLC.10, 11, 32 However, several recent reports showed that a relatively high PD‐L1 expression in EGFR‐mutated NSCLC was associated with worse response and PFS, even to EGFR‐TKI.18, 35 A retrospective study by Takashima et al36 reported that lower density of TILs and negative PD‐L1 expression correlated significantly with primary resistance to EGFR‐TKIs. However, in their study, not all the tumor samples analyzed by IHC staining were obtained in the advanced stage and the cases that used EGFR‐TKIs as second‐line or higher therapy were also included; therefore, their result might not have reflected the actual TME during the initial administration of EGFR‐TKIs. However, Su et al35 reported a high proportion of PD‐L1+/CD8+ cases among patients with de novo resistance to first‐line EGFR‐TKIs for advanced NSCLC; their results were similar to ours. These results, including our representative case, suggested that tumors with high expression of both PD‐L1 and CD8+ TIL, despite harboring EGFR mutations, are likely to benefit less from EGFR‐TKI therapies.
To our knowledge, our study was the first to examine whether PFS after first‐line EGFR‐TKIs in patients with advanced EGFR‐mutated NSCLC differed according to the PD‐L1 expression or TME subtype. Progression‐free survival was significantly shorter in patients with high PD‐L1 expression than in those with low PD‐L1 expression. High PD‐L1 expression had been reported by several recent studies to correlate with shorter PFS after EGFR‐TKI treatment in EGFR‐mutated NSCLC.18, 35 The results of our study were similar to those of the previous reports, in terms of this point. Moreover, the significantly shorter PFS in type 1 than in type 3 reflected the poor response in type 1. Progression‐free survival was significantly longer in type 4 than in type 2, although there was no difference in the response between these groups; PFS was not significantly different between types 2 and 3. These findings suggested the existence of subsets, among which the response or PFS of EGFR‐TKI differs, despite similar PD‐L1 expression status. In contrast, there might be subsets that do not differ much in their response to EGFR‐TKIs despite different PD‐L1 expression status; in these cases, the difference in EGFR‐TKI efficacy is likely to be associated with the difference in the TME, not in PD‐L1 expression. Both types 2 and 3 have been called “cold tumors,” which are characterized by a lack of T cell infiltration.25 This might explain the similar EGFR‐TKI efficacy between these groups. Moreover, in general, high CD8+ TIL has been associated with favorable clinical outcomes in NSCLC, including EGFR‐mutant cases.24, 37, 38 Therefore, the existence of CD8+ TILs might have some favorable impact on the longer duration of response to EGFR‐TKI therapy in type 4 than in type 2.
A phase II trial with small sample size evaluated the response to pembrolizumab of TKI‐naive patients with EGFR‐mutated advanced NSCLC and PD‐L1+ tumors did not have promising results.39 However, in previous clinical trials, the efficacy of immune checkpoint inhibitors had not been evaluated using the TME as a predictor. As previously mentioned, type 1 TME is a phenotype that had been considered to most likely benefit from anti‐PD‐1/PD‐L1 blockade. Actually, the response to anti‐PD‐1 inhibitors has been reported for several cases of EGFR‐mutated advanced NSCLC with both high PD‐L1 and CD8+ TILs.27, 35 Therefore, immunotherapy as a treatment option for such a selected subset, even EGFR‐mutant NSCLC patients, could leave room for further investigation in the context of personalized medicine.
There were several limitations in the present study. First, the retrospective and single‐center study design and the small number of patients make it difficult to draw definitive conclusions. Further investigation in a larger cohort is desirable. Second, there might have been biases in the clinical data, because a multivariate analysis could not be undertaken on a small number of patients. Third, the definition of PD‐L1 high expression was different from those in previous studies according to the Ab used for IHC or the cut‐off value; therefore, this difference in the evaluation method of PD‐L1 expression could have affected the results of different EGFR‐TKI efficacy according to the TME subtype. Fourth, the current study did not assess tumor mutation burden and its relationship with the TME. Fifth, the reason for the highest number of poor responders to EGFR‐TKI in type 1 cases was not investigated. Hepatocyte growth factor overexpression, mesenchymal‐epithelial transition factor (MET) gene or human epidermal growth factor receptor 2 (HER2) gene amplification, and others were reported as the mechanisms of primary resistance to EGFR‐TKI40, 41, 42, 43; these were not elucidated in this study. According to the study by Su et al,35 any specific relationship between the TME of cases that showed de novo resistance to EGFR‐TKIs and genetic profiles, such as EGFR T790M, MET amplification, ALK rearrangement, BIM deletion, and mutations of KRAS, PTEN, PIK3CA, HER2, were not observed. Further investigation is needed to reveal the association between a TME of high PD‐L1 and high CD8+ TILs itself and the undiscovered resistance mechanisms to EGFR‐TKI. Finally, in this study cohort, there were no type 1 TME cases treated with immunotherapy; further accumulation of cases is needed to explore and develop better treatment strategies for this phenotype.
In conclusion, differences in the TME based on PD‐L1 tumor expression and CD8+ TILs have potential impacts on the efficacy of EGFR‐TKI. A PD‐L1 high/CD8+ TIL high phenotype might be the subset that would poorly benefit from EGFR‐TKI treatment.
DISCLOSURE
H. Kaneda has received honoraria from AstraZeneca, Chugai Pharmaceutical, and Bristol‐Myers Squibb. S. Mitsuoka has received research funding from Ono Pharmaceutical. K. Asai has received honoraria from Boehringer Ingelheim Japan. N. Yamamoto has received honoraria from AstraZeneca, Chugai Pharmaceutical, Boehringer Ingelheim Japan, Ono Pharmaceutical, Bristol‐Myers Squibb, Taiho Pharmaceutical, and MSD as well as research funding from Chugai Pharmaceutical, Boehringer Ingelheim Japan, Ono Pharmaceutical, and Bristol‐Myers Squibb. T. Kawaguchi has received honoraria from Chugai Pharmaceutical and Boehringer Ingelheim Japan as well as research funding from AstraZeneca, Chugai Pharmaceutical, and Ono Pharmaceutical. Y. Koh has received honoraria from AstraZeneca, Chugai Pharmaceutical, Boehringer Ingelheim Japan, Ono Pharmaceutical, and Bristol‐Myers Squibb, as well as research funding from AstraZeneca, Boehringer Ingelheim Japan, Ono Pharmaceutical, and Bristol‐Myers Squibb. The other authors declare no potential conflicts of interest.
Supporting information
ACKNOWLEDGMENTS
This study was supported, in part, by AstraZeneca. We thank Emi Donoue of the research support platform of Osaka City University Graduate School of Medicine for the technical support.
Matsumoto Y, Sawa K, Fukui M, et al. Impact of tumor microenvironment on the efficacy of epidermal growth factor receptor‐tyrosine kinase inhibitors in patients with EGFR‐mutant non‐small cell lung cancer. Cancer Sci. 2019;110:3244–3254. 10.1111/cas.14156
Yasuhiro Koh and Tomoya Kawaguchi shared last authorship.
Funding information AstraZeneca
REFERENCES
- 1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359‐E386. [DOI] [PubMed] [Google Scholar]
- 2. Dearden S, Stevens J, Wu YL, Blowers D. Mutation incidence and coincidence in non small‐cell lung cancer: meta‐analyses by ethnicity and histology (mutMap). Ann Oncol. 2013;24:2371‐2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Midha A, Dearden S, McCormack R. EGFR mutation incidence in non‐small‐cell lung cancer of adenocarcinoma histology: a systematic review and global map by ethnicity (mutMapII). Am J Cancer Res. 2015;5:2892‐2911. [PMC free article] [PubMed] [Google Scholar]
- 4. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129‐2139. [DOI] [PubMed] [Google Scholar]
- 5. Han SW, Kim TY, Hwang PG, et al. Predictive and prognostic impact of epidermal growth factor receptor mutation in non‐small‐cell lung cancer patients treated with gefitinib. J Clin Oncol. 2005;23:2493‐2501. [DOI] [PubMed] [Google Scholar]
- 6. Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin‐paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947‐957. [DOI] [PubMed] [Google Scholar]
- 7. Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380‐2388. [DOI] [PubMed] [Google Scholar]
- 8. Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non‐small‐cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121‐128. [DOI] [PubMed] [Google Scholar]
- 9. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous‐cell non‐small‐cell lung cancer. N Engl J Med. 2015;373:123‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Borghaei H, Paz‐Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N Engl J Med. 2015;373:1627‐1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD‐L1‐positive, advanced non‐small‐cell lung cancer (KEYNOTE‐010): a randomised controlled trial. Lancet. 2016;387:1540‐1550. [DOI] [PubMed] [Google Scholar]
- 12. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non‐small‐cell lung cancer (OAK): a phase 3, open‐label, multicentre randomised controlled trial. Lancet. 2017;389:255‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reck M, Rodriguez‐Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med. 2016;375:1823‐1833. [DOI] [PubMed] [Google Scholar]
- 14. Mok TSK, Wu YL, Kudaba I, et al. Pembrolizumab versus chemotherapy for previously untreated, PD‐L1‐expressing, locally advanced or metastatic non‐small‐cell lung cancer (KEYNOTE‐042): a randomised, open‐label, controlled, phase 3 trial. Lancet. 2019;393:1819‐1830. [DOI] [PubMed] [Google Scholar]
- 15. Gandhi L, Rodriguez‐Abreu D, Gadgeel S, et al. Pembrolizumab plus chemotherapy in metastatic non‐small‐cell lung cancer. N Engl J Med. 2018;378:2078‐2092. [DOI] [PubMed] [Google Scholar]
- 16. Paz‐Ares L, Luft A, Vicente D, et al. Pembrolizumab plus chemotherapy for squamous non‐small‐cell lung cancer. N Engl J Med. 2018;379:2040‐2051. [DOI] [PubMed] [Google Scholar]
- 17. Socinski MA, Jotte RM, Cappuzzo F, et al. Atezolizumab for first‐line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378:2288‐2301. [DOI] [PubMed] [Google Scholar]
- 18. Yoneshima Y, Ijichi K, Anai S, et al. PD‐L1 expression in lung adenocarcinoma harboring EGFR mutations or ALK rearrangements. Lung Cancer. 2018;118:36‐40. [DOI] [PubMed] [Google Scholar]
- 19. Lee CK, Man J, Lord S, et al. Checkpoint inhibitors in metastatic EGFR‐mutated non‐small cell lung cancer‐a meta‐analysis. J Thorac Oncol. 2017;12:403‐407. [DOI] [PubMed] [Google Scholar]
- 20. Gainor JF, Shaw AT, Sequist LV, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD‐1 pathway blockade in non‐small cell lung cancer: a retrospective analysis. Clin Cancer Res. 2016;22:4585‐4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matsumoto Y, Sawa K, Oyanagi J, et al. Abstract 2613: predictive impact of low‐frequency pretreatment T790M mutation in patients with EGFR‐mutated non‐small cell lung cancer treated with EGFR tyrosine kinase inhibitors. Can Res. 2018;78:2613‐. [DOI] [PubMed] [Google Scholar]
- 22. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228‐247. [DOI] [PubMed] [Google Scholar]
- 23. Inoue Y, Yoshimura K, Mori K, et al. Clinical significance of PD‐L1 and PD‐L2 copy number gains in non‐small‐cell lung cancer. Oncotarget. 2016;7:32113‐32128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Obeid JM, Wages NA, Hu Y, Deacon DH, Slingluff CL Jr. Heterogeneity of CD8(+) tumor‐infiltrating lymphocytes in non‐small‐cell lung cancer: impact on patient prognostic assessments and comparison of quantification by different sampling strategies. Cancer Immunol Immunother. 2017;66:33‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T‐cell infiltration and PD‐L1. Cancer Res. 2015;75:2139‐2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Velcheti V, Schalper KA, Carvajal DE, et al. Programmed death ligand‐1 expression in non‐small cell lung cancer. Lab Invest. 2014;94:107‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu SY, Dong ZY, Wu SP, et al. Clinical relevance of PD‐L1 expression and CD8+ T cells infiltration in patients with EGFR‐mutated and ALK‐rearranged lung cancer. Lung Cancer. 2018;125:86‐92. [DOI] [PubMed] [Google Scholar]
- 28. Rozali EN, Hato SV, Robinson BW, Lake RA, Lesterhuis WJ. Programmed death ligand 2 in cancer‐induced immune suppression. Clin Dev Immunol. 2012;2012:656340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yearley JH, Gibson C, Yu N, et al. PD‐L2 expression in human tumors: relevance to anti‐PD‐1 therapy in cancer. Clin Cancer Res. 2017;23:3158‐3167. [DOI] [PubMed] [Google Scholar]
- 30. Zhang Y, Wang L, Li Y, et al. Protein expression of programmed death 1 ligand 1 and ligand 2 independently predict poor prognosis in surgically resected lung adenocarcinoma. Onco Targets Ther. 2014;7:567‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non‐small‐cell lung cancer. N Engl J Med. 2015;372:2018‐2028. [DOI] [PubMed] [Google Scholar]
- 33. Shibahara D, Tanaka K, Iwama E, et al. Intrinsic and extrinsic regulation of PD‐L2 expression in oncogene‐driven non‐small cell lung cancer. J Thorac Oncol. 2018;13:926‐937. [DOI] [PubMed] [Google Scholar]
- 34. Umezu D, Okada N, Sakoda Y, et al. Inhibitory functions of PD‐L1 and PD‐L2 in the regulation of anti‐tumor immunity in murine tumor microenvironment. Cancer Immunol Immunother. 2019;68:201‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Su S, Dong ZY, Xie Z, et al. Strong programmed death ligand 1 expression predicts poor response and de novo resistance to EGFR tyrosine kinase inhibitors among NSCLC patients with EGFR mutation. J Thorac Oncol. 2018;13:1668‐1675. [DOI] [PubMed] [Google Scholar]
- 36. Takashima Y, Sakakibara‐Konishi J, Hatanaka Y, et al. Clinicopathologic features and immune microenvironment of non‐small‐cell lung cancer with primary resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Lung Cancer. 2018;19:352‐359. e1. [DOI] [PubMed] [Google Scholar]
- 37. Al‐Shibli KI, Donnem T, Al‐Saad S, Persson M, Bremnes RM, Busund LT. Prognostic effect of epithelial and stromal lymphocyte infiltration in non‐small cell lung cancer. Clin Cancer Res. 2008;14:5220‐5227. [DOI] [PubMed] [Google Scholar]
- 38. Donnem T, Hald SM, Paulsen EE, et al. Stromal CD8+ T‐cell density‐a promising supplement to TNM staging in non‐small cell lung cancer. Clin Cancer Res. 2015;21:2635‐2643. [DOI] [PubMed] [Google Scholar]
- 39. Lisberg A, Cummings A, Goldman JW, et al. A phase II study of pembrolizumab in EGFR‐mutant, PD‐L1+ , tyrosine kinase inhibitor naive patients with advanced NSCLC. J Thorac Oncol. 2018;13:1138‐1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yano S, Yamada T, Takeuchi S, et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol. 2011;6:2011‐2017. [DOI] [PubMed] [Google Scholar]
- 41. Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gainor JF, Shaw AT. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J Clin Oncol. 2013;31:3987‐3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mazieres J, Peters S, Lepage B, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 2013;31:1997‐2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials