

What are MICAL proteins?
The MICALs are a family of actin regulatory oxidation-reduction (redox) enzymes that directly bind and disassemble actin filaments (F-actin). Three vertebrate MICAL genes, MICAL-1, MICAL-2 and MICAL-3, and one Drosophila Mical gene have been identified and are collectively called the MICALs. The MICALs are defined by a characteristic domain structure that includes an N-terminal redox domain, followed by other domains (Figure 1a). MICAL-like genes, which encode for proteins that are similar in domain organization to MICALs but lack the redox enzymatic portion, have also been found in mammals (MICAL-L1 and MICAL-L2 (also called JRAB)) and Drosophila (Mical-like). MICAL-likes are considered a distinct protein family and will not be covered below.
Figure 1. MICALs and their effects.
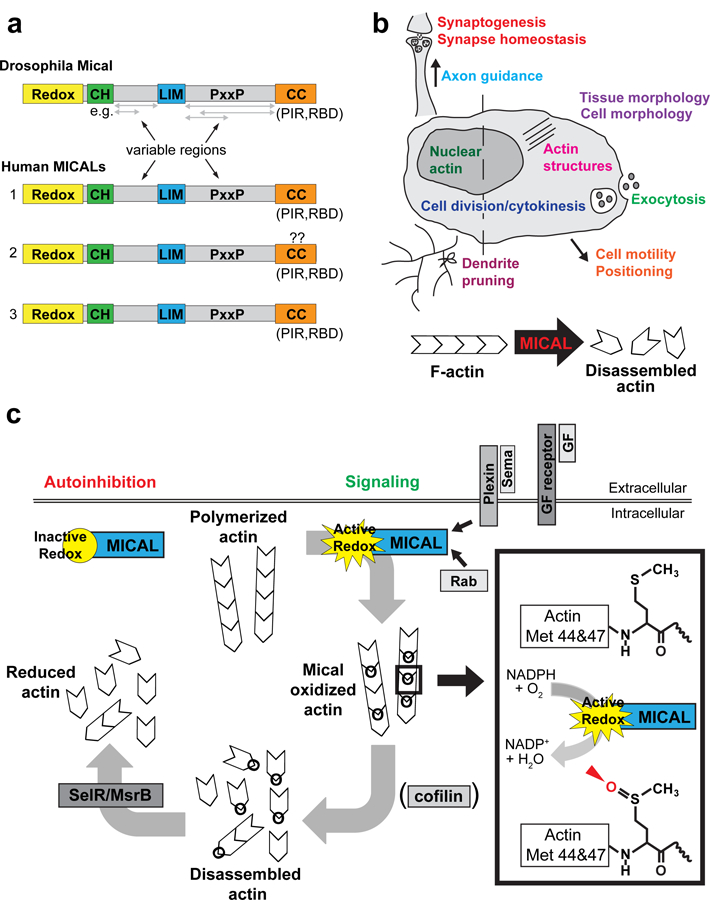
a) MICAL family proteins include Drosophila Mical and three vertebrate MICALs, MICAL-1, MICAL-2 and MICAL-3. All MICAL proteins consist of an N-terminal oxidation/reduction (redox) enzymatic domain, a calponin homology (CH) domain, a LIM domain, Src-homology 3 (SH3)-domain proline rich binding motifs (PxxP), and a C-terminal coiled coil domain (CC), which contains a Plexin interacting region (PIR) and a Rab binding domain (RBD). The regions between defined domains are denoted as “variable regions” because they differ in length for different MICALs and for the different protein isoforms of each MICAL. Question marks are placed over the CC domain for human MICAL-2 because many notated cDNAs for this protein do not include the CC domain, but the MICAL-2 genomic locus includes a CC coding region similar to MICAL-1 and MICAL-3. b) MICAL activity has been found to be involved in many cellular events (as listed) that contribute to both development and homeostasis in multiple cell types, including neurons. In many of these events, MICAL is known to destabilize and disassemble filamentous actin (F-actin). c) The current model for MICAL function is that in the absence of signaling, the redox activity of MICAL is autoinhibited and inactive. MICAL interacts with the Plexin receptor through its CC domain, and upon Sema/Plexin signaling, MICAL’s redox domain is activated. MICAL activity is also regulated through interactions with growth factor signaling and Rab family proteins. Active MICAL binds F-actin and uses NADPH as a coenzyme to specifically oxidize methionine (Met) 44 and 47 within the D-loop of actin (box). The D-loop mediates binding between actin monomers/filament subunits and MICAL’s targeted oxidation of Met 44 and 47 disrupts these interactions, resulting in F-actin disassembly. Cofilin is also known to accelerate the disassembly of MICAL-oxidized actin, and vice versa. MICAL-oxidized actin is reduced by the cellular reductase SelR/MsrB, creating a reversible system for redox regulation of actin dynamics.
Where are MICALs found?
As a group, MICAL family proteins are ubiquitously expressed, including in the developing and adult nervous system (neurons and glia), skeletal muscle, heart, fibroblasts, bone marrow, kidney, lung, thymus, liver, spleen, and testis. Intracellularly, MICALs are found in the cytoplasm (including closely associated with the plasma membrane), where they localize with actin-rich structures, as well as in the nucleus, where they associate with nuclear actin.
What do MICAL proteins do?
Using multiple different domains and protein interaction modules, MICALs interact with different proteins (see below) and much remains to be learned of these interactions and their role in the cell. However, MICALs are known to potently dismantle F-actin – directly binding F-actin and working via severing and rapid depolymerization to disassemble filaments (Figure 1b). These effects on actin occur in the cytoplasm, including within cellular extensions (such as axons), and in the nucleus.
Why is regulation of F-actin disassembly important?
Actin provides the structural underpinnings for dynamic cellular behaviors, such as morphological change, motility, cell division, and even gene transcription. To drive these cellular behaviors, actin forms polymers (F-actin) that are continually disassembled and assembled, reorganizing actin structures to fit the task at hand. Disassembly of existing F-actin – so as to prune F-actin networks and make them more malleable – is the first step toward initiating the cytoskeletal changes that are required for structural remodeling and diverse cellular behaviors.
How do MICALs disassemble actin filaments?
F-actin is made up of individual actin monomers/subunits bound to each other in the same end-to-end orientation to form a single-stranded polymer with a distinct polarity (Figure 1c). MICALs use a unique mechanism – enzymatic modification of individual actin subunits within F-actin – to disrupt filament stability. MICALs are classified as flavoprotein monooxygenase/hydroxylase enzymes. Such enzymes bind flavin adenine dinucleotide (FAD) and catalyze oxidation-reduction (redox) reactions using nicotinamide adenine dinucleotide phosphate (NADPH) as a co-enzyme. In these reactions, the substrate is oxidized while the co-enzyme NADPH is reduced. Studies with Drosophila Mical first revealed that F-actin is a Mical substrate. These studies showed that Mical selectively binds F-actin, and in the presence of NADPH, F-actin but not monomeric actin (G-actin), increases Mical’s enzymatic activity by more than 100-fold. Further analysis showed that Mical specifically and selectively oxidizes two methionine (Met) residues in actin, Met 44 and 47. Moreover, methionine has a unique oxidation pattern in that two stereoisomers (an R-isomer and an S-isomer) can be produced by oxidation – and MICALs stereo-specifically oxidize these two Met residues in the R-isomer conformation. Met 44 and 47 are situated within the D-loop of actin, which is located on the “pointed end” of actin and directly contacts the “barbed end” (opposite end) of the adjacent subunit within F-actin. Oxidation shifts the position of these Mets in the filament, altering the binding between actin subunits and causing filament instability and disassembly. The methionine residues modified by MICALs are present in all the different types of actin (e.g., cytoplasmic, nuclear, cardiac, neuronal, and muscle) and are conserved from invertebrates to humans. Importantly, all three human MICAL proteins also use the same mechanism to specifically oxidize and dismantle mammalian actins.
What makes the MICALs different from other redox proteins?
Enzymes that produce reactive oxygen species typically modify cysteine residues through the release of diffusible oxidants. These enzymes are generally nonspecific – modifying multiple protein targets and multiple cysteine residues on those targets. In contrast, MICALs are selective in their effects – they have a specific protein substrate that activates them (actin), they modify particular amino acid residues within that substrate (Met 44 and 47), and they stereospecifically modify substrate residues (Met R-isomer). MICALs are also preferentially activated by one form of their actin substrate, the polymerized form, further underscoring the specificity of these enzymes. These attributes, therefore, distinguish MICAL family enzymes from other physiologically-relevant redox enzymes characterized to date, such as NADPH oxidases and nitric oxide synthases, which have different mechanisms of activation and action, as well as broader specificity both with regards to protein substrates and the amino acids that are modified within those substrates.
Are any other proteins besides actin modified by MICAL’s enzymatic activity?
Currently, actin is the only known direct MICAL substrate (meaning that actin both activates and is modified by the catalytic activity of MICALs). However, the MICALs have also been found to release the diffusible oxidant H2O2 in conjunction with their reaction, so MICAL may also indirectly influence other molecules through the release of H2O2 (which, in contrast, is likely to modify cysteine and not specific Met residues).
How do MICALs differ from other actin disassembly factors?
Well-known actin disassembly proteins, such as gelsolin and actin depolymerizing factor (ADF)/cofilin, disassemble F-actin by binding to filaments and physically dissociating actin subunits through protein conformational changes. After actin monomers are liberated from the filament, they are recycled to build new F-actin. In contrast, because MICALs chemically modify actin to promote disassembly, MICAL activity does not replenish the pool of polymerization-competent actin monomers. Instead, MICAL-modified actin monomers do not polymerize efficiently.
Is MICAL modification of actin reversible?
Yes. MICALs are the first known enzymatic modifiers of methionine residues, and actin that has been oxidized by MICALs is reduced by a family of stereospecific cellular methionine sulfoxide reductases (called SelR in Drosophila and MsrB1, 2, and 3 in mammals) (Figure 1c). In vitro, SelR/MsrB activity can completely restore the polymerization capability of MICAL-oxidized actin. In vivo, SelR/MsrB counteracts Mical-mediated oxidation of actin to control cellular extension, remodeling, motility, axon guidance, synaptogenesis, and muscle morphology/function. Thus, MICALs and SelR/MsrBs function as a reversible system for controlling actin filament stability. Further, the stereospecific effects of MICALs/SelRs on selective methionine residues in actin provide a first demonstration that the stereospecific oxidation/de-oxidation of specific methionine residues (similar to the reversible phosphorylation of serine, threonine, and tyrosine residues) is a means to precisely modulate protein function.
Are MICALs always active in cells?
No, a series of different experiments indicate that MICALs are maintained in an autoinhibited state and that binding to specific effector proteins such as transmembrane receptors or small GTPases releases autoinhibition, allowing MICAL’s redox domain to become active (Figure 1c; see below).
When I think about redox, I think about energy production, metabolism, mitochondria, and the redox state of the cell. Is this how I should think of MICALs?
No, the MICALs appear to be locally positioned in the cell so they can be activated to exert their specific effects on actin and do not appear to globally affect the redox state of the cell.
Do MICALs function with other actin regulatory proteins – such as other actin disassembly factors?
Recent investigation has revealed that MICALs are capable of functioning with cofilin to promote rapid F-actin disassembly (Figure 1c). ADF/cofilin family actin disassembly factors are regarded as the major tool for F-actin dismantling in cells, but they are only weakly effective. Hence, ADF/cofilin-mediated F-actin severing cannot be the sole mechanism for many rapid F-actin disassembly events observed in cells. In vitro experiments with cofilin and Mical showed that Mical-mediated oxidation of actin enhances cofilin binding to actin filaments and dramatically boosts cofilin severing kinetics and F-actin disassembly. This Mical/cofilin synergy also occurs in vivo, where cell morphological and axon guidance phenotypes that depend on either Mical or cofilin are dramatically enhanced by the presence of both proteins. The expression patterns of ADF/cofilins and MICALs are widespread and similar, indicating that the coordinated activity of these two proteins may be a common mechanism for regulating F-actin stability.
What signaling pathways utilize MICALs?
Emerging data indicate that the MICALs, and the unusual redox manner in which they exert their cellular effects, are a component of classical signal transduction systems composed of ligand-receptor pairs, phosphorylation, dephosphorylation, GTPases, and classical cytoskeletal regulators (Figure 1c). MICALs are best known as effectors for Semaphorin/Plexin signaling. The Semaphorins (Semas), one of the largest families of extracellular guidance cues, signal through Plexin family transmembrane receptors to regulate numerous cellular effects in a number of different tissues. Sema/Plexin signaling is best associated with negative regulation of cellular morphology and motility – where it induces F-actin dismantling/collapse to promote cellular remodeling. Drosophila Mical was discovered in a screen for proteins that interact with Drosophila Plexin and parallel genetic studies indicated that Mical is required for axon guidance events that are controlled by Sema/Plexin. Once Mical was established as an actin disassembly factor, it became clear that MICALs provide a direct link between Sema/Plex signaling and F-actin collapse. Additional work has shown that MICALs are required for Sema/Plexin-mediated effects on axon and growth cone morphology in vitro and neuron migration and synaptic homeostasis in vivo. In addition, MICAL is required for F-actin rearrangements downstream of Semaphorin signaling in non-neuronal cells, such as vascular endothelial cells and podocytes (kidney cells).
In addition to functioning as an effector for the classically “inhibitory/repulsive” Sema/Plex signaling pathway, MICALs have been associated with growth factor signaling. MICAL-2 disassembles nuclear actin downstream of nerve growth factor (NGF) signaling to promote gene transcription through serum response factor (SRF). MICAL activity is also regulated by the Abl non-receptor protein tyrosine kinase, which is known to mediate the effects of different growth factors. Abl phosphorylates MICAL’s redox domain enhancing its enzymatic activity, and is required for MICAL-mediated cellular effects in response to Sema/Plex and growth factor signaling.
MICALs are also known to physically interact with Rab proteins, small GTPases that regulate vesicle trafficking. Rab6 and Rab8/MICAL-3 interactions control vesicle dynamics during exocytosis and cytokinesis through mechanisms that appear to be independent of MICAL-3’s enzymatic activity. On the other hand, MICAL-1’s enzymatic F-actin disassembly activity and Rab binding properties are utilized together during cytokinesis. In this case, Rab35 proteins recruit MICAL-1 to cell abscission sites where MICAL-1 disassembles actin to enable the terminal steps of cell division. Interactions between MICAL and other proteins, including among others, CasL, collapsin response mediator proteins (CRMPs), and the nuclear Dbf2-related (NDR) kinase, have also been identified.
What types of physiological processes do MICALs control?
Emerging genetic and knock-down data indicate that many actin-dependent events in numerous different cell types in vivo rely on MICAL-mediated actin disassembly (Figure 1b). MICALs are important for nervous system development and function, regulating neuronal cell migration, axon growth and guidance, growth cone vesicle dynamics, dendrite morphology, synapse development, synaptic homeostasis, and sensory organ formation. Outside of the nervous system, MICALs regulate cell viability, immunity, cytokinesis, and the morphology and function of skeletal muscle, the cardiovasculature, and kidney (podocyte) cells.
Are MICALs associated with human disease?
MICALs are increasingly being implicated in pathologies including different cancers, diabetic nephropathy, blood brain barrier dysfunction, muscular dystrophy, liver disease, susceptibility to infection, epilepsy, Alzheimer’s disease, aging, wide-ranging neurological disorders, skeletal abnormalities, and obesity. In addition, human patients with mutations in the actin residues oxidized by the MICALs have muscular and vascular system defects and accumulation of actin in muscles. Additional pathologies, such as neurodegeneration, aging, and deafness, are associated with defective SelR/MsrB function. Finally, the Redox susceptibility of actin’s Met-44 and Met-47 residues varies depending on actin’s conformation, ionic state, and polymerization condition, so it is possible that particular in vivo conditions and disease states may make these residues susceptible to abnormal redox regulation. In particular, the finding that MICALs target specific residues on actin and that oxidation of these specific residues induces robust disassembly of filaments, raises the possibility that certain pathologies and/or traumas allow oxidants abnormal access to actin where they nonspecifically work like MICALs (abnormally oxidizing Met-44 and Met-47 and pathologically disrupting F-actin-based cellular functions and behaviors).
Where can I find out more?
- 1.Terman JR, Mao T, Pasterkamp RJ, Yu HH, and Kolodkin AL (2002). MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell 109, 887–900. [DOI] [PubMed] [Google Scholar]
- 2.Beuchle D, Schwarz H, Langegger M, Koch I, and Aberle H (2007). Drosophila MICAL regulates myofilament organization and synaptic structure. Mech Dev 124, 390–406. [DOI] [PubMed] [Google Scholar]
- 3.Kirilly D, Gu Y, Huang Y, Wu Z, Bashirullah A, Low BC, Kolodkin AL, Wang H, and Yu F (2009). A genetic pathway composed of Sox14 and Mical governs severing of dendrites during pruning. Nat Neurosci 12, 1497–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hung RJ, Yazdani U, Yoon J, Wu H, Yang T, Gupta N, Huang Z, van Berkel WJ, and Terman JR (2010). Mical links semaphorins to F-actin disassembly. Nature 463, 823–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hung RJ, Pak CW, and Terman JR (2011). Direct redox regulation of F-actin assembly and disassembly by Mical. Science 334, 1710–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grigoriev I, Yu KL, Martinez-Sanchez E, Serra-Marques A, Smal I, Meijering E, Demmers J, Peranen J, Pasterkamp RJ, van der Sluijs P, et al. (2011). Rab6, Rab8, and MICAL3 Cooperate in Controlling Docking and Fusion of Exocytotic Carriers. Curr Biol 21, 967–974. [DOI] [PubMed] [Google Scholar]
- 7.Hung RJ, Spaeth CS, Yesilyurt HG, and Terman JR (2013). SelR reverses Mical-mediated oxidation of actin to regulate F-actin dynamics. Nat Cell Biol 15, 1445–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee BC, Peterfi Z, Hoffmann FW, Moore RE, Kaya A, Avanesov A, Tarrago L, Zhou Y, Weerapana E, Fomenko DE, et al. (2013). MsrB1 and MICALs Regulate Actin Assembly and Macrophage Function via Reversible Stereoselective Methionine Oxidation. Mol Cell 51, 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Battum EY, Gunput RA, Lemstra S, Groen EJ, Yu KL, Adolfs Y, Zhou Y, Hoogenraad CC, Yoshida Y, Schachner M, et al. (2014). The intracellular redox protein MICAL-1 regulates the development of hippocampal mossy fibre connections. Nat Commun 5, 4317. [DOI] [PubMed] [Google Scholar]
- 10.Lundquist MR, Storaska AJ, Liu TC, Larsen SD, Evans T, Neubig RR, and Jaffrey SR (2014). Redox Modification of Nuclear Actin by MICAL-2 Regulates SRF Signaling. Cell 156, 563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grintsevich EE, Yesilyurt HG, Rich SK, Hung RJ, Terman JR, and Reisler E (2016). F-actin dismantling through a redox-driven synergy between Mical and cofilin. Nat Cell Biol 18, 876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fremont S, Hammich H, Bai J, Wioland H, Klinkert K, Rocancourt M, Kikuti C, Stroebel D, Romet-Lemonne G, Pylypenko O, et al. (2017). Oxidation of F-actin controls the terminal steps of cytokinesis. Nat Commun 8, 14528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoon J, Kim SB, Ahmed G, Shay JW, and Terman JR (2017). Amplification of F-Actin Disassembly and Cellular Repulsion by Growth Factor Signaling. Dev Cell 42, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orr BO, Fetter RD, and Davis GW (2017). Retrograde semaphorin-plexin signalling drives homeostatic synaptic plasticity. Nature 550, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grintsevich EE, Ge P, Sawaya MR, Yesilyurt HG, Terman JR, Zhou ZH, and Reisler E (2017). Catastrophic disassembly of actin filaments via Mical-mediated oxidation. Nat Commun 8, 2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu H, Yesilyurt HG, Yoon J, and Terman JR (in press). The MICALs are a family of F-actin dismantling oxidoreductases conserved from Drosophila to humans. Scientific Reports [DOI] [PMC free article] [PubMed]