

Abstract
The molecular chaperone heat shock protein 90 (HSP90) has been used by cancer cells to facilitate the function of numerous oncoproteins, and it can be argued that cancer cells are ‘addicted’ to HSP90. However, although recent reports of the early clinical efficacy of HSP90 inhibitors are encouraging, the optimal use of HSP90-targeted therapeutics will depend on understanding the complexity of HSP90 regulation and the degree to which HSP90 participates in both neoplastic and normal cellular physiology.
Molecular chaperones help nascent polypeptides fold correctly and multimeric protein complexes assemble productively, while minimizing the danger of aggregation in the protein-rich intracellular environment. Heat shock protein 90 (HSP90) is an evolutionarily conserved molecular chaperone that participates in stabilizing and activating more than 200 proteins — referred to as HSP90 ‘clients’ — many of which are essential for constitutive cell signalling and adaptive responses to stress1,2. To accomplish this task, HSP90, the chaperone HSP70, and additional proteins termed co-chaperones form the dynamic complex known as the HSP90 chaperone machine3. Cancer cells use the HSP90 chaperone machinery to protect an array of mutated and overexpressed oncoproteins from misfolding and degradation. Therefore, HSP90 is recognized as a crucial facilitator of oncogene addiction and cancer cell survival4.
In the past 5 years, the complex nature of HSP90 regulation and the many ways in which it participates in cell physiology have been clarified. Considerable progress has been made in understanding the dynamic conformational flexibility of HSP90 and in recognizing the contribution made by post-translational modifications to the regulation of the HSP90 chaperone machine. HSP90 is now known to be a key mediator of cellular homeostasis5, a function that it accomplishes partly by facilitating numerous transient low-affinity protein-protein interactions6. Recent bioinformatic and proteomic analyses have uncovered several previously unrecognized roles for HSP90 in regulating cell physiology under normal and stressed conditions2,7,8, and the involvement of the chaperone in transcriptional regulation and chromatin remodelling, although previously appreciated9, is now being studied more extensively10,11.
Progress in the clinical evaluation of targeting HSP90 in cancer has also been evident. The first HSP90 inhibitor, 17-AAG (tanespimycin), entered clinical trials in 1999. In 2004, a second HSP90 inhibitor, 17-DMAG (alvespimycin), entered a first-in-human study. Owing to extensive efforts in rational drug design and discovery12,13, 13 HSP90 inhibitors are currently undergoing clinical evaluation in cancer patients, 10 of which have entered the clinic in the past 3 years14. Considerable progress has also been made in identifying optimal cancer indications and effective drug combinations15. This Review describes recent advances in our understanding of HSP90 regulation and function as they affect cancer biology and inform the use of HSP90 inhibitors for the treatment of cancer.
Conformational flexibility of HSP90
HSP90 is a member of a small superfamily of functionally unrelated proteins (that also comprises DNA gyrase, histidine kinase and the DNA mismatch repair protein MutL) that possess a unique ATP-binding pocket that is distinct from the ATP-binding cleft of protein kinases16. The conserved chaperone structure consists of three domains: an amino terminal region (N domain) that contains an ATP and drug-binding site and co-chaperone-interacting motifs; a middle (M) domain that provides docking sites for client proteins and co-chaperones, and which participates in forming the active ATPase; and a carboxy-terminal (C) domain that contains a dimerization motif, a second drug-binding region and interaction sites for other co-chaperones17–19. Dimerization of two HSP90 protomers through their C domains is necessary for chaperone function20. Although HSP90 is primarily a cytoplasmic protein, mammalian cells also express two compartmentally restricted HSP90 homologues (BOX 1). HSP90 is also secreted from and found on the surface of cancer cells (BOX 2).
Box 1 |.
Homologues of HSP90
The molecular chaperone heat shock protein 90 (HSP90) homologue glucose-regulated protein 94 (GRP94) is found in the endoplasmic reticulum (ER), whereas tumour necrosis factor receptor-associated protein 1 (TRAP1) is localized to mitochondria. Like other HSP90 proteins, both GRP94 and TRAP1 possess ATPase activity but both lack known co-chaperones155–157. Recent studies suggest that GRP94 is essential for the maturation and secretion of insulin-like growth factors, which are autocrine mitogens that have a key role in transformation. ATP binding and hydrolysis are essential for the chaperone activity of GRP94, and a comparison of the nucleotide-binding pocket of GRP94 with that of HSP90 suggests that GRP94-specific inhibitors can be designed155,158. In light of these recent findings, GRP94 should be evaluated as a bona fide anticancer target.
TRAP1 protects mitochondria from oxidative stress159,160. Intriguingly, TRAP1 expression is low in the mitochondria of normal tissues (with the exception of the brain and testis) but is markedly increased in tumour mitochondria161. TRAP1 silencing in prostate cancer cell lines caused apoptosis, as did its targeting with mitochondria-specific HSP90 inhibitors162. TRAP1 inhibition leads to the collapse of mitochondrial integrity, cytochrome c release, and caspase activation in several tumour cell lines and in several murine tumour models with little effect on non-transformed cells and minimal in vivo toxicity161. Like HSP90, TRAP1 is a phosphoprotein the phosphorylation of which by PTEN-induced putative kinase 1 is necessary to prevent oxidative stress-induced apoptosis 163.
Box 2 |.
Extracellular HSP90
Recent studies indicate that molecular chaperone heat shock protein 90 (HSP90) is not confined to the intracellular environment. Many types of cells express HSP90 on the cell surface and secrete HSP90 into the extracellular space164,165. Moreover, the level of HSP90 on the cell surface is higher in cancer cells than in normal cells and correlates with metastasic activity166. Blocking or neutralizing secreted HSP90 has been shown to inhibit cell motility in vitro and tumour metastasis in vivo167. Although the molecular mechanisms underlying HSP90 secretion are not fully understood, secretion is stimulated by environmental stresses and growth factors168,169, and is affected by post-translational modifications to the chaperone, including phosphorylation and acetylation57. HSP90 secretion may also be conformationally restricted and affected by nucleotide-dependent modulation of amino terminal (N) region and middle (M) domain interaction170.
HSP90 stimulates cell migration through its interaction with the cell surface receptor CD91, and this property is independent of its ability to bind and hydrolyse ATP168. In addition, HSP90 binds to the extracellular domain of HER2 (also known as ERBB2), and a neutralizing antibody against extracellular HSP90 attenuates heregulin-induced HER2 phosphorylation, downstream kinase signalling, rearrangement of the actin cytoskeleton and subsequent cell migration171. Specific inhibition of extracellular HSP90 does not affect cancer cell proliferation in vitro or tumour xenograft growth in vivo167. Therefore, in contrast to intracellular HSP90, the functions of which encompass but are not limited to the regulation of cell migration172, the function of extracellular HSP90 seems to be restricted to the regulation of cell motility and metastasis.
Various biophysical techniques have indicated that HSP90 cycles through several conformations that ultimately result in correctly orienting the N and M domains to form an active ATPase18,21–25 (FIG. 1; see Supplementary information S1 (movie)). HSP90 inhibitors currently under clinical evaluation all disrupt the chaperone cycle by replacing ATP in the N domain nucleotide-binding pocket16.
Figure 1 |. The HSP90 chaperone cycle.

Although molecular chaperone heat shock protein 90 (HSP90) samples multiple conformations in the absence of ATP or other factors, current models propose that ATP binding and hydrolysis, as well as a precisely sequenced interaction with an array of co-chaperones, subtly shift the conformational equilibrium, presumably by lowering the energy barrier between certain conformations, thus providing directionality to the HSP90 cycle23,26,27. ATP binding to the undimerized (open) amino terminal (N) domain of HSP90 promotes repositioning of a ‘lid’ segment (red) that leads to transient dimerization of the N domains. Subsequent structural rearrangements result in the ‘closed and twisted’ conformation of HSP90 that is committed to ATP hydrolysis. Binding of the co-chaperone activator of HSP90 ATPase 1 (AHA1) enhances the rate of ATP hydrolysis-dependent HSP90 cycling by increasing the rate of the conformational alterations that result in the acquisition of ATPase competence. The dashed arrow reflects the difficulty of HSP90 in achieving the ATPase-competent conformation in the absence of AHA1. The co-chaperones STIP1 (also known as p60H0P) and cell division cycle 37 homologue (CDC37), and N domain-binding HSP90 inhibitors, exert an opposite effect to that of AHA1 by preventing the initial structural changes necessary for N domain dimerization. Prostaglandin E synthase 3 (PTGES3; also known as p23) slows the ATPase cycle by stabilizing the closed conformation that is committed to ATP hydrolysis. C, carboxy-terminal domain; M, middle domain; P., inorganic phosphate.
Co-chaperones modulate HSP90 function
Co-chaperones of HSP90 have diverse effects on HSP90 function in eukaryotes (FIG. 2). Some co-chaperones, including activator of HSP90 ATPase 1 (AHA1), prostaglandin E synthase 3 (PTGES3; also known as p23), STIP1 (also known as p60H0P), and cell division cycle 37 homologue (CDC37), modulate the rate of the HSP90 cycle by affecting the conformational dynamics of the chaperone18,21,26–30 (FIG. 1). Co-chaperone-mediated modulation of the HSP90 cycle rate is an important means of regulating the dwell time of client proteins in the chaperone complex31.
Figure 2 |. Co-chaperones and post-translational modifications modulate HSP90 chaperone activity.

Numerous co-chaperones assist heat shock protein 90 (HSP90) in its chaperone activity. Co-chaperones modulate HSP90 ATPase activity and so determine the rate of chaperone cycling (for example, cell division cycle 37 homologue (CDC37), activator of HSP90 ATPase 1 (AHA1), p23 and STIP1 (also known as p60HOP)), recruit certain classes of client proteins to HSP90, and/or participate in chaperoning specific categories of clients (such as CDC37, STIP1,TAH1, PIH1, suppressor of G2 allele of SKP1 (SUGT1; also known as SGT1), FKBP51 and FKBP52) and post-translationally modify HSP90 itself, its co-chaperones and its client proteins (such as PP5 and CHIP). Post-translational modifications profoundly affect HSP90 function by affecting co-chaperone and client interaction, ATP binding and ATP hydrolysis. The three major regulatory post-translational modifications are phosphorylation, acetylation and S-nitrosylation. An updated list of HSP90-interacting co-chaperones and HSP90-dependent client proteins can be found at the Picard laboratory website (see Further information) maintained by the laboratory of D. Picard, University of Geneva, Switzerland.
Some co-chaperones are adaptors that deliver specific substrates to HSP90. For example, CDC37 delivers protein kinase clients, and STIP1 participates in delivering steroid hormone receptor clients to HSP90 (REFS 32–35). Steroid hormone receptor function is further modified by other co-chaperones, including FKBP51and FKBP52 (REF. 36). The co-chaperone suppressor of G2 allele of SKP1 (SUGT1; also known as SGT1) associates with the N domain of HSP90 to assemble the core kinetochore complex, which is the initial step in kinetochore assembly37. The co-chaperones TAH1 and PIH1 are required for HSP90 to chaperone the nascent forms of several ribonucleoproteins that participate in processes as diverse as ribosome synthesis, DNA replication, telomere maintenance and antioxidant defence38. In addition, these co-chaperones link HSP90 to the chromatin-remodelling factors RVB1 and RVB2, suggesting that HSP90 also participates in the epigenetic modulation of transcription10.
Other co-chaperones affect HSP90 function by catalysing reactions such as ubiquitin ligation (the E3 ubiquitin ligase CHIP) and dephosphorylation (the phosphatase PP5). CHIP-dependent ubiquitylation of HSP90 clients directs them to degradation in the proteasome, thus short circuiting additional cycles of chaperone-mediated refolding35. In cells, PP5-mediated dephosphorylation of CDC37 regulates the ability of HSP90 to chaperone the protein kinase RAF1, inhibits RAF1-dependent MAPK activation and sensitizes cells to HSP90 inhibition39.
Co-chaperone expression may also affect cancer cell sensitivity to HSP90 inhibitors.
The deletion of p23 in yeast causes hypersensitivity to the structurally dissimilar natural product inhibitors geldanamycin and radicicol (which is isolated from the soil-borne fungus Humicola fuscoatra), and overexpression of this co-chaperone, as is seen in cancer40, protects cells from these drugs29. Similarly, silencing of AHA1 or CDC37, which are also overexpressed in cancer33, sensitizes cancer cells to both geldanamycin and 17-AAG (the clinical drug derived from geldanamycin)41,42. Therefore, targeting these proteins, or their interaction with HSP90, might be therapeutically beneficial, especially when combined with HSP90 inhibitors42.
Post-translational modification of HSP90
Phosphorylation.
HSP90 is subject to several post-translational modifications that affect its chaperone function1,43 (FIG. 2). Early studies reported hyperphosphorylation of HSP90 in response to serine/threonine phosphatase inhibition, leading to reduced association with its client kinase p60v-src in NIH3T3 cells44. More recently, PP5 was shown to dephosphorylate HSP90 in vitro, and its deletion in yeast cells inhibited HSP90 function45. Together with the effect of PP5 on CDC37, discussed above, these data point to the importance of regulated phosphorylation and dephosphorylation for the proper functioning of the HSP90 chaperone machinery.
The phosphorylation status of several individual serine, threonine and tyrosine residues has been shown to uniquely affect HSP90 function. Vascular endothelial growth factor receptor 2 (VEGFR2)-associated HSP90 is phosphorylated on Tyr301 by SRC, and this is essential for VEGFR2-induced angiogenesis46. Similarly, phosphorylation of Ser226 and Ser255 in HSP90 regulates apoptosome formation by modulating the affinity of HSP90 for apoptotic peptidase activating factor 1 (APAF1) in non-transformed cells. Constitutively reduced phosphorylation of Ser226 and Ser255 in some leukaemias strengthens HSP90–APAF1 association, abrogates cytochrome c-induced apoptosome assembly and confers chemoresistance47.
HSP90 has been identified as a substrate of the kinases BRAF48 and casein kinase II (CK2)49. Although the effect of BRAF-mediated HSP90 phosphorylation is unknown, CK2-mediated phosphorylation is required for HSP90 to chaperone several kinases, including CK2 itself50. As BRAF and CK2 are HSP90 clients, it is possible that HSP90 phosphorylation by certain client kinases establishes a positive feedback loop ensuring their chaperone-dependent stabilization and activity.
The relationship between the tyrosine kinase WEE1 and HSP90 supports this hypothesis. WEE1 is an HSP90 client protein that regulates the G2/M transition in the cell cycle by phosphorylating cyclin-dependent kinase 1 (CDK1)51. WEE1 also directly phosphorylates a conserved tyrosine residue in the N domain of HSP90 (REFS 52,53). This positively influences the ability of HSP90 to chaperone a select group of client protein kinases with cancer relevance, including HER2 (also known as ERBB2), p60v-src, RAF1, CDK4 and WEE1 itself, but it negatively affects geldanamycin and 17-AAG binding to HSP90. WEE1 silencing or its pharmacological inhibition in cancer cells, sensitizes these cells to HSP90 inhibition. A more detailed understanding of HSP90 phosphorylation could provide new strategies to increase the efficacy of HSP90 inhibitors and retard or reverse the acquisition of resistance to these drugs.
Acetylation.
The effect of HSP90 acetylation on chaperone activity has been extensively examined. Acetylation of HSP90 has been observed in cells treated with his-tone deacetylase (HDAC) inhibitors or after silencing of HDAC6, and this correlates with the destabilization of several HSP90 client proteins54–57. At least 11 lysine residues in HSP90 have been found to be acetylated56,57 (PhosphoSitePlus® website; see Further information). HSP90 hyperacetylation that is induced by HDAC inhibition abrogates p23, client protein and ATP binding, and enhances the binding of N domain inhibitory drugs55,57. In some murine tumour models, especially leukaemias, the combination of HDAC and HSP90 inhibitors is synergistic57. However, antagonism has been observed in several murine solid tumour models, suggesting that the outcome of this drug combination might depend on tumour type, host microenvironment and other factors58.
S-nitrosylation.
S-nitrosylation is another post-translational modification of HSP90. Nitric oxide (NO)-mediated nitrosylation of Cys597 in the HSP90 C domain inhibits chaperone activity in endothelial cells59. Cys597 is in the middle of a conformational switch region in the HSP90 C domain that molecular modelling suggests is able to transmit structural information to the HSP90 N domain60. S-nitrosylation of Cys597 inhibits HSP90 ATPase activity61, confirming the hypothesis that environmentally generated structural cues propagate between spatially distant regions of HSP90 to affect chaperone function. Some of the observed anti-tumour activity of exogenously administered NO may result from its inhibition of HSP90 in cancer cells. Supporting this hypothesis, an earlier report described NO-mediated telomere shortening that was dependent on HSP90 inhibition62. These findings are consistent with the crucial role of HSP90 in maintaining telomerase activity63, and with the importance of telomere maintenance to the immortality of cancer cells.
HSP90 modulates nuclear events
Effects of HSP90 on transcription.
Approximately 3% of the intracellular HSP90 pool is found in the nucleus64, and the chaperone can regulate several nuclear events (FIG. 3). HSP90 regulates the activity of steroid hormone receptors (SHRs), including glucocorticoid, androgen and oestrogen receptors. Several SHRs are important cancer-related proteins and are validated anticancer molecular targets65. Because SHRs shuttle between the cytoplasm and the nucleus, and demonstrate distinct activities in each cellular compartment, regulation of both SHR subcellular distribution and transcriptional activity are key determinants of their function. The HSP90 machine has a complex but crucial role in integrating these processes by regulating SHR cellular location, protein stability, competency to bind ligand and transcriptional activity66.
Figure 3 |. HSP90 modulates nuclear events.

Four examples of the effect of heat shock protein 90 (HSP90) on nuclear events are shown. a | The androgen receptor (AR) exemplifies the importance of HSP90 in steroid hormone receptor (SHR) function. b | The importance of HSP90 in regulating heat shock transcription factor 1 (HSF1) activity is also shown. c | The effect of HSP90 on SMYD3-mediated histone methylation and its effect on the transcription of cancer-associated genes, such as NKX, WNT and C/EBP. d | An HSP90–BCL6 complex suppresses the transcription of several tumour suppressor genes and contributes to tumorigenesis. ATR, ataxia telangiectasia and Rad3-related; DHT, dihydrotestosterone; HRE, hormone response element; HSE, heat shock element; P, phosphorylation; PSA, prostate-specific antigen; RNA Pol II, RNA polymerase II.
HSP90 also regulates the activity of the heat shock transcription factor 1 (HSF1). In mammalian cells exposed to proteotoxic stress, or to an HSP90 inhibitor such as geldanamycin, HSF1 monomers dissociate from HSP90, undergo trimerization, nuclear translocation and activation, and subsequently upregulate the coordinate expression of heat shock proteins, including HSP70 (REF. 67). HSP90 is also required for the disassembly of transcriptionally active HSF1 trimers ein Xenopus laevis oocytes, and the inhibition of HSP90 by geldanamycin increases HSF1 trimer stability and prolongs the heat shock response, providing an additional mechanism for HSP90 inhibitor-mediated enhancement of HSF1 activity68. Beyond transcriptional regulation of the heat shock response, HSF1 regulates the expression of numerous other genes that are involved in cell survival under stressful conditions. Recently, HSF1 expression has been shown to be an important facilitator of oncogenesis69,70, and efforts are underway to identify HSF1 inhibitors71. In addition to their potential use as anticancer drugs, such agents may increase the efficacy of HSP90 inhibitors.
HSP90 affects other nuclear events in addition to its regulation of SHRs and HSF1 (REF. 10) (FIG. 3). BCL-6 is a transcriptional repressor of genes such as ataxia telangiectasia and Rad3-related (ATR) and TP53 and is oncogenic in diffuse large B cell lymphomas (DLBCLs)72,73. HSP90 binds to BCL-6, and this complex represses BCL-6 target genes74. The HSP90 inhibitor PUH71 rapidly reduces BCL-6 expression by decreasing BCL-6 protein stability. This derepresses BCL-6 target genes and causes apoptosis of DLBCL cells, suggesting that the HSP90–BCL-6 interaction is crucial for DLBCL cell survival74.
HSP90 inhibitors are also expected to have a deleterious impact on HSP90-dependent transcription factors that have tumour suppressor properties, such as interferon regulatory factor 1 (IRF1). Loss of IRF1 expression cooperates with Ras mutation to transform cells, and the deletion of IRF1 is associated with certain cancers75. HSP90 positively regulates IRF1, modulating both IRF1 protein turnover and transcriptional activity76. HSP90 inhibition promotes IRF1 degradation, in a manner similar to its effect on other HSP90 client proteins. IRF1 transcriptional activity is inhibited before its degradation, suggesting that HSP90 also affects IRF1 binding to DNA. Therefore, the cumulative effect of HSP90 inhibition on HSP90-regulated transcriptional events is multifactorial and will almost certainly depend on the duration of HSP90 inhibition and the cellular context in which it occurs.
Effects of HSP90 on chromatin.
The co-chaperones TAH1 and PIH1 link HSP90 to the chromatin remodelling factors RVB1 and RVB2 (REF. 10). In Drosophila melanogaster, the association of trithorax (Trx) (a member of the TrxG gene family that encodes proteins that participate in chromatin remodelling) with HSP90 is required for the maintenance of active chromatin at sites of gene expression11. MLL (the mammalian orthologue of D. melanogaster Trx) also depends on HSP90 (REF. 11). As MLL fusion proteins are strongly leukaemogenic77, targeting HSP90 may be a new approach to treating these cancers.
Another chromatin-modifying enzyme, the arginine methyltransferase PRMT5, has been identified as an HSP90 client, and HSP90 inhibition caused a decrease in PRMT5 expression in several cancer cell lines58. PRMT5-mediated methylation negatively regulates two tumour suppressor genes, ST7 and NM23 (REF. 78). HSP90 inhibitors might promote their derepression, although this remains to be examined.
The histone methyltransferase SMYD3 is over-expressed in several cancers, including hepatocellular carcinoma and colorectal cancer, and it may have an important role in carcinogenesis, as it promotes proliferation79. SMYD3 methylates Lys4 on histone H3 (H3-K4). In vitro, this activity is enhanced by HSP90, with which SMYD3 interacts in cells, and HSP90 inhibitor treatment suppresses SMYD3 activity in cancer cells79. The less characterized methyltransferase SMYD2 is also overexpressed in some cancers and has been implicated in malignant progression80. Like SMYD3, SMYD2 interacts with HSP90 and its activity is enhanced by HSP90 association81.
The effects of HSP90 on DNA mutation.
HSP90 facilitates the folding of DNA polymerase-η into an active conformation, and its activity is inhibited by 17-AAG, thus sensitizing cells to the cytotoxic effects of ultraviolet radiation82. A similar mechanism may contribute to the reported hypersensitivity of HSP90 inhibitor-treated cancer cells to other DNA-damaging agents. HSP90 inhibition might also retard DNA polymerase-η-mediated mutagenic events that allow cancer cells to acquire more malignant phenotypes82.
However, HSP90 inhibition may not always suppress DNA mutation. In D. melanogaster, HSP90 is required for the suppression of transposon activity in germ cells83. Transposons are suppressed by an RNA-silencing mechanism that is mediated by Piwi-interacting RNAs (piRNAs)84. Biogenesis of these small RNAs requires HSP90. Inactivating mutations of the chaperone, or HSP90 inhibitor treatment, reduces piRNA expression, enhances transposon mobility and results in de novo mutations83. Loss of PRMT5 activity causes decreased methylation of several piRNA-interacting proteins, and decreases piRNA expression in D. melanogaster germ cells85. The effect of HSP90 inhibitors (described above) on PRMT5 may contribute to their ability to abrogate piRNA-mediated regulation of transposon mobility. Identification of HSP90 as a suppressor of transposable element-induced mutations thus raises the possibility that, in contrast to their effect on DNA polymerase-η-generated mutations, HSP90 inhibitors might increase mutation frequency in certain contexts.
HSP90 inhibitor clinical trials
There are now 13 HSP90 inhibitors undergoing clinical evaluation (TABLE 1), and 23 active HSP90 inhibitor oncology trials14. Although there are currently no approved HSP90-targeted drugs, there has been considerable progress on several fronts, and potential routes to approval are becoming apparent. One important advance has been in the drugs themselves. 17-AAG is undergoing Phase III evaluation with an improved formulation that overcomes several toxicities that were common to earlier trials14. At the same time, several chemically distinct HSP90 inhibitors with improved properties, including oral biological availability, have recently entered the clinic or will soon undergo clinical evaluation. A second area of advance is in choosing the appropriate indication. Recent preclinical and disease-specific clinical studies have illuminated some key points to consider in the further development of HSP90 inhibitors.
Table 1 |.
HSP90 inhibitors in clinical trials
| Structure | Inhibitor | Phase | Route | Source |
|---|---|---|---|---|
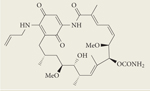 |
Tanespimycin (17-AAG) | II/III | Intravenous | BMS |
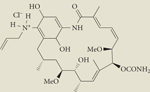 |
Retaspimycin hydrochloride (IPI-504) | II | Intravenous | Infinity |
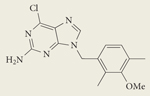 |
BIIB021 CNF2024 | II | Oral | Biogen Idec |
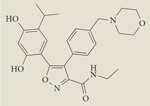 |
AUY922 | I/II | Intravenous | Novartis |
| Resorcinol derivative* | STA-9090 | I/II | Intravenous | Synta |
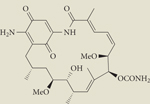 |
IPI-493 | I | Oral | Infinity |
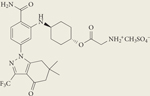 |
SNX-5422 mesylate | I | Oral | Pfizer, Inc. |
| Small molecule* | BIIB028 | I | Intravenous | Biogen Idec |
| Small molecule* | KW-2478 | I | Intravenous | Kyowa Hakko Kirin |
| Small molecule* | AT13387 | I | Oral or intravenous | Astex Therapeutics |
| Small molecule* | XL888 | I | Oral | Exelixis |
| Small molecule* | HSP990 | I | Oral | Novartis |
| Small molecule* | MPC-3100 | I | Oral | Myriad Pharmaceuticals |
| Nanoparticle albumin-bound 17-AAG | ABI-010 | I | Intravenous | Abraxis Bioscience |
HSP90, heat shock protein 90.
The structures are not reported.
HSP90 and prostate cancer: tumour microenvironment and target complexity may complicate clinical efficacy.
Initial preclinical observations suggested that castrate-resistant prostate cancer might respond favourably to HSP90 inhibitor therapy because the activity of several HSP90 client proteins, including androgen receptor (AR), remains crucial for disease progression. Nevertheless, IPI-504 (retaspimycin), a water-soluble stabilized dihydroquinone form of 17-AAG, was not efficacious as monotherapy in castrate-resistant prostate cancer in a Phase II trial. Although 1 of 4 patients without bony metastases had a prostate-specific antigen (PSA) decline of 48% from baseline after 9 cycles of treatment, 15 patients in the study had bony metastatic disease, and no PSA decline or objective responses were observed in this cohort86. Lack of clinical benefit was also reported in a Phase II trial of 17-AAG in patients with castrate-resistant prostate cancer87.
These findings are consistent with data in mice showing that experimental prostate cancer growth in bone is enhanced by 17-AAG as a consequence of localized transient activation of SRC signalling in the tumour microenvironment88. Furthermore, the cooperative interaction between AR and HSP27 facilitates AR transcriptional activity89. As HSP27 is induced subsequent to HSP90 inhibition, this may provide an additional mechanism for antagonizing the expected anticancer activity of HSP90 inhibitors in this setting.
Drug dose and schedule considerations.
Clinical investigators must also ascertain whether the chosen drug dose and schedule of drug delivery is sufficient to affect intratumoral HSP90 function long enough to influence the growth and/or viability of the cancer. For example, a Phase II trial of 17-AAG reported no objective clinical responses in patients with metastatic melanoma90. However, pharmacodynamic evaluation of tumour biopsy samples taken before and after drug treatment revealed minimal HSP90 inhibition. Therefore, even though preclinical data are promising for this indication91,92, the identification of a drug formulation and schedule capable of providing a more prolonged suppression of tumour HSP90 activity is necessary to determine whether significant clinical responses are attainable.
The importance of client driver protein sensitivity: HER2.
By contrast, promising results have been seen with HSP90 inhibitors in HER2-positive breast cancer, and they emphasize the value of choosing a cancer that is driven by an HSP90 client protein, such as HER2, that is exceptionally sensitive to HSP90 inhibition93. In a Phase II trial in patients with HER2-positive metastatic breast cancer and progressive disease following trastuzumab treatment, patients received standard weekly doses of trastuzumab together with 17-AAG. Using Response Evaluation Criteria In Solid Tumours (RECIST) criteria, investigators reported a response rate of 24%, and an overall clinical benefit (including stable disease) was seen in 57% of evaluable patients94. Given these results, there is interest in further evaluating 17-AAG and other HSP90 inhibitors in patients with HER2-positive cancer (NCT01081600 and NCT00526045). Preclinical studies suggest that HSP90 inhibitors may also have activity in cancers harbouring HER2 mutations and in triple-negative breast cancer95–97.
Multiple myeloma: is proteotoxic stress a predictor of HSP90 inhibitor efficacy?
HSP90 inhibitors have also shown promising activity in multiple myeloma. In Phase I and II clinical trials, 17-AAG sensitized tumours to the proteasome inhibitor bortezomib, and in some cases overcame bortezomib resistance98. As a result of HSP90 inhibition, many cellular proteins are targeted for degradation in the proteasome. When combined with a proteasome inhibitor, HSP90 inhibition can overload the protein degradation machinery and lead to apoptosis99. It is likely that the biology of multiple myeloma, a tumour that is highly sensitive to proteotoxic stress and strongly dependent on efficient proteasome function for maintenance of cell viability, may cause these tumours to be extremely sensitive to the dual inhibition of HSP90 and the proteasome100. Furthermore, clinical trial data suggest that HSP90 inhibitors may ameliorate bortezomib-induced neuropathy, potentially through the induction of HSP70, which has been shown to have a pro-survival function in neuronal cells101. In vitro, HSP90 inhibitors enhance natural killer (NK)-dependent recognition and lysis of myeloma cells, providing an additional rationale for the use of these agents in this indication102 (BOX 3). As the combination of 17-AAG and bortezomib has been associated with durable responses in heavily pretreated patients with multiple myeloma, this regimen was recently evaluated in a randomized Phase II/III clinical trial (NCT00514371), and additional HSP90 inhibitors are also being evaluated in this setting (NCT00708292 and NCT01063907).
Box 3 |.
HSP90 and modulation of tumour surveillance
The modulation of the host immune system is a promising therapeutic stratagem for treating cancer. Molecular chaperone heat shock protein 90 (HSP90) inhibition affects host immunity in several ways. HSP90 inhibitors promote the transcription of both major histocompatibility (MHC) class I chain-related A and B (MICA and MICB) genes in a panel of myeloma cell lines102. Transcriptional upregulation was dependent on heat shock transcription factor 1 (HSF1) binding to the promoter region of both genes. MICA and MICB are ligands for a receptor on natural killer (NK) cells important for tumour recognition and lysis. HSP90 inhibitors increase myeloma cell stimulation of NK cell degranulation, which was prevented with a blocking antibody to their MICA and MICB receptor. These data link tumour cell HSF1 to enhanced host NK cell activity, and suggest that the stimulation of tumour HSF1 activity by HSP90 inhibitors might enhance host tumour surveillance.
The receptor tyrosine kinase ephrin receptor A2 (EphA2) was recently identified as an HSP90 client172,173. EphA2 is abundantly expressed in a broad range of cancers and its expression correlates with poor clinical outcome. Tumour EphA2 is recognized as a self protein by the host and CD8+ T cells are poorly competent to recognize EphA2+ tumour cells in vitro and in vivo. This can be enhanced by pretreating CD8+ T cells with receptor agonists that promote proteasome-mediated degradation of the kinase and its upregulated expression in class I complexes at the cell surface174. This is also achieved by HSP90 inhibitors, suggesting that HSP90 inhibitors may significantly improve the anti-tumour activity of host CD8+ T cells.
Finally, HSP90 inhibitors may also be useful for selectively preventing graft-versus-host disease, by targeting alloreactive T cells in haematopoietic stem cell transplant recipients without impairing pathogen- or disease-specific immunity175.
HSP90 inhibitors and DNA damage.
HSP90 inhibitors may prove useful when combined with DNA damaging agents, because of their ability to abrogate S and G2/M cell cycle checkpoint controls by promoting the degradation of the client kinases CHK1 and WEE1 (REFS 103,104). This and the dependence of DNA polymerase-η on HSP90 (REF. 82) explain why HSP90 inhibitors sensitize cancer cells to DNA damage independently of p53 status103. A Phase I study of the topoisomerase inhibitor irinotecan and 17-AAG reported no partial responses, although tumour shrinkage was seen in six patients105. Importantly, the drug combination could be administered with acceptable toxicity at a dose and schedule that resulted in the loss of phospho-CHK1, abrogation of the G2/M checkpoint and cell death in tumour biopsy samples. Another recent Phase I study reported that the combination of 17-AAG and gemcitabine demonstrated clinical activity106, and these results have led to a Phase II evaluation of this drug combination in patients with advanced ovarian cancer (NCT00093496). Similarly, radiation produces DNA damage and radioresistance can occur as a result of cell cycle arrest. HSP90 inhibitors have been shown to provide p53-independent radiosensitization in preclinical models and clinical evaluation of this therapeutic combination is being considered107.
HSP90 inhibitors in leukaemia.
Extensive preclinical and preliminary clinical data also suggest a benefit of HSP90 inhibitors in acute myelogenous leukaemia (AML)108,109. FLT3, a tyrosine kinase and HSP90 client, is frequently mutated and constitutively active in a subpopulation of patients with AML and is generally a poor prognostic indicator in older patients110. However, FLT3 mutation may confer enhanced HSP90 dependence in this haematological malignancy.
Similarly, the dependence of chronic myelogenous leukaemia (CML) on BCR-ABL, a client that is readily degraded in response to HSP90 inhibition111, suggests that drug-resistant CML may be an appropriate indication for the use of HSP90 inhibitors, either alone or in conjunction with ABL tyrosine kinase inhibitors (TKIs)112. Indeed, BCR-ABL T315I, although cross-resistant to all first- and second-line ABL TKIs, and disproportionately represented in patients failing treatment with these agents113, remains sensitive to HSP90 inhibition114. A Phase I/II clinical trial is evaluating the efficacy of the synthetic HSP90 inhibitor STA-9090 (Synta Pharmaceuticals) in leukaemias, including AML and CML (NCT00964873).
Finally, ZAP70 is expressed in patients with aggressive chronic lymphocytic leukaemia (CLL). In CLL cells, ZAP70 is HSP90-dependent and ZAP70 expression makes these cells highly sensitive to HSP90 inhibition in vitro115. Preliminary clinical evaluation of a purine-based HSP90 inhibitor has suggested possible in vivo efficacy in CLL116.
HSP90 inhibitors and non-small-cell lung cancer: prevention of the oncogenic switch.
HSP90 inhibitors also may be effective in non-small-cell lung cancer (NSCLC). In a recent study, 44% of 29 NSCLC tumours had a deletion on chromosome 14 (14q32.2–33)117 that encompasses HSP90A, and this was correlated with a significant survival benefit. Stratification of these patients by tumour HSP90a expression also demonstrated a significant correlation between prolonged survival and HSP90 level, and this was validated in an additional 307 patients with NSCLC. It would be interesting to determine whether HSP90 expression is predictive of the response of patients with NSCLC to HSP90 inhibitors. Clinical evaluation of several HSP90 inhibitors in NSCLC is currently in progress (NCT01031225 and NCT00431015).
Although HSP90 expression in tumours was not evaluated, clinical data have shown efficacy of HSP90 inhibitors in combination with epidermal growth factor receptor (EGFR) TKIs in NSCLC, even in patients who have progressed on TKI therapy118. Preclinical data show that HSP90 inhibitors abrogate the oncogenic switch that allows cancer cells to signal through other receptor tyrosine kinases when one is blocked. Oncogenic switching is frequently induced as a resistance mechanism to TKIs, but most of the induced and/or mutated alternative kinases that have been identified, including HER2, BRAF, MET and ALK, are HSP90 clients that are sensitive to HSP90 inhibition119. Furthermore, alternative EGFR mutations that provide resistance to TKIs remain sensitive to HSP90 inhibitors95,120. For these reasons, HSP90 inhibitors may emerge as useful agents to combine with TKIs to combat oncogene switch-mediated development of drug resistance in NSCLC and other malignancies.
Pharmacodynamic assessment of HSP90 inhibition
Although in some cases tumour tissue has been available for pharmacodynamic analysis109,121, most pharmacodynamic assays to assess HSP90 inhibition in patients enrolled in Phase I trials have been carried out on peripheral blood mononuclear cells (PBMCs). As all HSP90 inhibitors currently under clinical evaluation interact with the N-terminal ATP-binding pocket and are predicted to cause client protein degradation, initial pharmacodynamic assays focused on the evaluation of client protein levels. However, the evaluation of client protein responses in PBMCs, although useful for establishing the biological activity of HSP90 inhibitors in early Phase clinical trials, has proved to be of minimal use in predicting either clinical activity or biological response to HSP90 inhibition in tumour tissue122,123. This is not surprising, as HSP90 inhibitors preferentially accumulate in tumour cells rather than normal cells124,125, and the sensitivity of HSP90 to inhibitors seems to be fundamentally different in tumour cells compared with normal cells126. Furthermore, those client proteins most sensitive to HSP90 inhibition (for example, mutated proteins and HER2) are preferentially expressed in tumour cells. HSP90 inhibitor-induced expression of PBMC HSP70, although useful in establishing biologically active drug dosing, has also not correlated with clinical response127. Preclinical data suggest that serum HSP70 levels might be a useful pharmacodynamic marker of drug response. A sensitive high-throughput enzyme-linked immunosorbent assay revealed that serum HSP70 increased several-fold in tumour xenograft-bearing mice, but not in non-tumour-bearing mice, following treatment with an HSP90 inhibitor, perhaps owing to tumour lysis128.
Given the clear need for alternative pharmacodynamic approaches to monitor the clinical efficacy of HSP90 inhibitors, several non-invasive functional imaging techniques are being explored. Positron emission tomography (PET) with [18F]fluorodeoxyglucose (FDG-PET) was incorporated into a Phase I trial of the HSP90 inhibitor IPI-504 in patients with metastatic gastrointestinal stromal tumours129. A reduced FDG-PET signal in tumours correlated with drug dose, and reactivation of tumour FDG uptake correlated with planned breaks in drug administration. Decreased FDG uptake returned on re-dosing with IPI-504. These findings suggest that, at least in highly glycolytic tumours, FDG-PET may provide a useful pharmacodynamic correlate of HSP90 inhibitor anti-tumour activity.
A recent study demonstrated HER2 PET to be a sensitive and robust pharmacodynamic assay that is able to monitor tumour HER2 expression in real time following systemic HSP90 inhibitor administration130. These results have been confirmed131–133, and clinical evaluation of 89Zr-trastuzumab as a PET imaging agent in patients treated with HSP90 inhibitor is underway (NCT01081600). Similar approaches can probably be developed to image other HSP90-dependent transmembrane kinases; clinical evaluation of 89Zr-bevacizumab to image VEGFR in HSP90 inhibitor-treated patients with breast cancer is in progress (NCT01081613).
17-AAG-dependent tumour metabolic changes have been observed in mouse xenograft models by monitoring total choline intensity with proton magnetic resonance imaging134,135. This non-invasive technique was sensitive enough to detect early tumour drug responses in treated animals and, when considered with data from an earlier study136, suggests that altered choline metabolism may be a useful biomarker of response to HSP90 inhibitors.
Developing resistance to HSP90 inhibitors
In preclinical studies, inherent resistance to 17-AAG has been observed in cells with low endogenous expression of NAD(P)H/quinone oxidoreductase I (NQO1)137,138, an enzyme that reduces the quinone moiety of 17-AAG to its dihydroquinone form, which has much improved HSP90-binding properties138. Mechanisms underlying the development of in vitro resistance to 17-AAG in glioblastoma cells are consistent with this, and include reduced NQO1 levels, sometimes accompanied by reduced NQO1 mRNA expression, and expression of an inactive NQO1 polymorphism139. Importantly, no cross-resistance to non-ansamycin HSP90 inhibitors was seen, and this mechanism is relevant only for drugs containing a quinone moiety.
HSF1-dependent HSP70 and HSP27 induction frequently occurs in response to HSP90 inhibitor treatment140. Increased expression of one or both of these proteins in normal tissues may protect them from some of the adverse effects of HSP90 inhibitors, but similar protection is likely to occur in tumour cells. Indeed, silencing of HSP70 and/or HSP27 dramatically increases cancer cell sensitivity to geldanamycin141,142. Efforts are underway to discover and validate pharmacological inhibitors of HSP70 (REFS 143,144), and an antisense inhibitor of HSP27 has been described145. However, the effect of their combination with HSP90 inhibitors remains to be explored.
Similar to geldanamycin, radicicol binds to the N-terminal ATP-binding pocket of HSP90 (REF. 146). Interestingly, a single point mutation in the N domain of H. fuscoatra HSP90 reduces its affinity for radicicol without affecting ATP binding147, demonstrating that the appearance of a drug resistance-conferring mutation with no effect on HSP90 function is possible. However, no similar mutation (or polymorphism) in cancer cell HSP90 has been reported.
The co-chaperones CDC37, AHA1 and p23 are highly expressed in cancer cells40 and their depletion has been shown to increase the efficacy of HSP90 inhibitors. Although the upregulation of one or more of these co-chaperones in response to drug treatment has not yet been reported, it is a possible outcome of prolonged drug pressure. Finally, as discussed above, additional post-translational mechanisms might mediate cancer cell resistance to HSP90 inhibitors52.
Alternative methods for targeting HSP90
Coumarin antibiotic HSP90 inhibitors.
Although all HSP90 inhibitors currently in the clinic recognize the ATP-binding pocket in the N domain of the chaperone, the HSP90 C domain can also be targeted by drugs, such as the coumarin antibiotics novobiocin, clorobiocin and coumermycin A1. However, the poor affinity of these compounds for HSP90, coupled with their higher affinity for type II topoisomerases, precluded their further evaluation as clinically useful HSP90 inhibitors17. Recently, important advances have been made in improving the affinity of this class of compounds for HSP90 and, at the same time, reducing their affinity for topoisomerase. One such compound, termed F-4, demonstrated superior efficacy to 17-AAG in inducing apoptosis in LNCaP and PC-3 prostate cancer cell lines148. Another novobiocin derivative, termed KU135 (which had an HSP90 binding Kd of 1–2 μM), was highly active in Jurkat T cell leukaemia lymphocytes, inhibiting cell proliferation and promoting apoptosis more potently than 17-AAG did149. Others have independently evaluated a set of novobiocin derivatives and have identified several promising compounds that have anti-proliferative activity in several cancer cell lines150. In X. laevis, novobiocin abrogated heat-induced HSF1 activation and also geldanamycin-stimulated HSF1 activity when the two drugs were used in combination68. Although not yet confirmed in cancer cells, these findings strongly support further medicinal chemistry development and the pre-clinical evaluation of these and other C domain HSP90 inhibitors in cancer.
InhibitingHSP90: co-chaperone interactions.
Although the N and C domains of HSP90 contain druggable motifs, identifying inhibitors the target primarily hydrophobic protein–protein interaction surfaces has proved to be much more difficult. However, recent molecular docking studies predict that the natural product celastrol, although not uniquely an HSP90 inhibitor, disrupts the interaction of HSP90 with its co-chaperone CDC37, and destabilizes several HSP90 client kinases151. NMR spectroscopy has identified a single amino acid in human CDC37 that is crucial for complex formation with HSP90, suggesting additional routes for the disruption of HSP90–CDC37 association152. Inhibitors that block the association of specific co-chaperones with HSP90 are a new paradigm for drugging the HSP90 chaperone machine.
Concluding remarks
For many years, the primary function of HSP90 was thought to be the stabilization of proteins and protein complexes in the cytoplasm. Much of the work on HSP90 for the past 20 years has focused on the identification of these HSP90 clients. The identification of clients that are crucial for the maintenance of each of the proposed hallmarks of cancer suggested that cancer itself might be an indication for an HSP90 inhibitor. Indeed, a recent in vitro study identified HSP90 as a suppressor of the mammalian pro-apoptotic protein inositol hexakisphosphate kinase 2 in cancer cells and suggested that HSP90 inhibition should be uniformly cytotoxic to cancer cells153. However, data in animal models and 10 years of HSP90 inhibitor clinical trials have shown that this is not the case, and such a simple approach to HSP90-targeted drug development might not be an optimally effective therapeutic strategy.
There are now ample data demonstrating uniquely important roles in cancer for cell surface HSP90, secreted HSP90, HSP90-like chaperones in the mitochondria and endoplasmic reticulum, and HSP90 in the nucleus. Furthermore, the two cytosolic isoforms of HSP90 (HSP90α and HSP90β) may have different roles in cancer because they are not functionally redundant in mammals, at least during embryonic development154. In the next 5–10 years, the discovery and validation of isoform- and cell location-specific inhibitors will enlarge the armamentarium of HSP90 inhibitor-based therapeutics.
Finally, global analyses have identified many new potential partner proteins in the HSP90 interactome, and in the past 5 years diverse roles for HSP90 beyond protein stabilization have been identified. Going forwards, the optimal development and application of HSP90-targeted therapeutics will depend on synthesizing information gained from a careful genetic analysis of primary and metastatic tumours with an appreciation of the unique environmental context in which the tumour is thriving at the expense of the host.
Supplementary Material
At a glance.
Heat shock protein 90 (HSP90) is a molecular chaperone of numerous oncoproteins. Therefore, cancer cells can be considered to be ‘addicted’ to this molecule.
HSP90 is also a mediator of cellular homeostasis. As such, it facilitates numerous transient low-affinity protein–protein interactions that have only recently been identified using bioinformatic and proteomic techniques.
Although primarily a cytoplasmic protein, HSP90 affects diverse nuclear processes, including transcription, chromatin remodelling and DNA damage-induced mutation.
HSP90 is a conformationally dynamic protein. ATP binding to the amino (N) domain and its subsequent hydrolysis by HSP90 drive a conformational cycle that is essential for chaperone activity.
In eukaryotes, co-chaperones and post-translational modifications regulate both client interactions with HSP90 and HSP90 ATPase activity.
Co-chaperones and post-translational modifications can also affect the efficacy of HSP90 inhibitors.
HSP90 inhibitors currently under clinical evaluation interact with the N domain ATP-binding pocket, prevent ATP binding, and stop the chaperone cycle, leading to client protein degradation.
Because of the HSP90 client repertoire, HSP90 inhibitors may combat oncogene switching, which is an important mechanism of tumour escape from tyrosine kinase inhibitors.
Derivatives of the coumarin antibiotic novobiocin represent an alternative strategy for inhibiting HSP90 by targeting a unique carboxy-terminal (C) domain.
Optimal development of HSP90-directed therapeutics will depend on synthesizing information gained from careful genetic analysis of primary and metastatic tumours with an understanding of the unique environmental context in which the tumour is thriving at the expense of the host.
Acknowledegements
The authors would like to thank K. Beebe, Y. S. Kim, and all members of the Neckers and Trepel laboratories for their helpful comments.
Glossary
- Co-chaperone
Protein that assists or alters the function of other chaperones
- Oncogene addiction
The hypothesis that tumours arising as a result of a particular oncogenic lesion are dependent on the continued expression of that oncogene
- Kinetochore
Specialized assembly of proteins that binds to a region of the chromosome called the centromere and is essential for chromosome segregation during eukaryotic cell division
- Apoptosome
A caspase-activating complex that is formed when cytochrome c is released from mitochondria. It initiates oligomerization of APAF1, which binds procaspase-9 and thereby initiates the caspase cascade that leads to programmed cell death
- S-nitrosylation
The covalent attachment of a nitrogen monoxide group to the thiol side chain of cysteine
- Proteotoxic stress
Protein damage caused by physical or chemical agents such as heat, heavy metals, hypoxia and some anticancer drugs
- DNA polymerase-η
A member of the DNA polymerase Y family, a group of low-fidelity DNA polymerases that can replicate through damaged DNA
- Transposon
Mobile genetic element that can insert in different positions in the genome and cause mutations
- Piwi-interacting RNA (piRNA)
A class of germline-specific small RNA molecule that suppresses transposon mobility by RNA silencing
- Castrate-resistant prostate cancer
Prostate cancer that no longer responds to androgen deprivation therapy
- Prostate-specific antigen (PSA)
A protein produced by the prostate that is increased in the blood of men with prostate cancer, benign prostatic hyperplasia, or infection and inflammation of the prostate
- Pharmacodynamic
The relationship between drug Concentration (pharmacokinetics) and its biological effects (what the drug does to the body)
- Trastuzumab
A humanized monoclonal antibody that binds HeR2 on tumour cells and prevents uncontrolled proliferation caused by aberrant HeR2 signalling
- RECIST
A set of published rules that define when cancer patients improve (‘respond’), stay the same (‘stable’) or worsen (‘progression’) during treatments
- Triple-negative breast Cancer
Breast cancer that lacks expression of oestrogen, progesterone and HeR2 receptors
- Neuropathy
Refers to any disease or injury affecting nerves or nerve cells
- Graft-versus-host disease
A common complication of allogeneic bone marrow transplantation in which functional immune cells in the transplanted marrow recognize the recipient as foreign and mount an immunological attack
- Alloreactive T cell
White blood cell that recognizes a complex composed of a major histocompatibility complex (MHC) molecule and a peptide in which the MHC or peptide are derived from a genetically different member of the same species
- Graft-versus-host disease
A common complication of allogeneic bone marrow transplantation in which functional immune cells in the transplanted marrow recognize the recipient as foreign and mount an immunological attack
- Alloreactive T cell
White blood cell that recognizes a complex composed of a major histocompatibility complex (MHC) molecule and a peptide in which the MHC or peptide are derived from a genetically different member of the same species
- FDG-PET
A radio-labelled imaging methodology for detecting cancers that relies on increased glucose uptake by the tumour — a characteristic of cancers and other pathologies
- Proton magnetic resonance
The resonance of protons to radiation in a magnetic field. Proton magnetic resonance spectra yield a great deal of information about molecular structure as most organic molecules contain hydrogen atoms that absorb energy of different wavelengths depending on their bonding environment
- Non-ansamycin HSP90 inhibitor
HSP90 inhibitor lacking the benzoquinone ansamycin backbone found in tanespimycin (17-AAG), alvespimycin (17-DMAG) and retaspimycin (IPI-504)
Footnotes
Competing interests statement
The authors declare no competing financial interests.
DATABASES
ClinicalTrials.gov: http://clinicaltrials.gov/
NCT00093496 | NCT00431015 | NCT00514371 |
NCT00526045 | NCT00708292 | NCT00964873 |
NCT01031225 | NCT01063907 | NCT01081600 |
National Cancer Institute Drug Dictionary:
http://www.cancer.gov/drugdictionary/bortezomib
UniProtKB: http://www.uniprot.org
FURTHER INFORMATION
Len Neckers’s homepage: http://ccr.cancer.gov/staff/staff.asp?profileid=5712
Jane Trepel’s homepage: http://ccr.cancer.gov/staff/staff.asp?profileid=7178
Guiseppe Giaccone’s homepage: http://ccr.cancer.gov/staff/staff.asp?profileid=12505
PhosphoSitePlus®: www.phosphosite.org/
Picard laboratory: http://www.picard.ch/downloads/downloads.htm
SUPPLEMENTARY INFORMATION
See online article: S1 (movie)
References
- 1.Wandinger SK, Richter K. & Buchner J. The Hsp90 chaperone machinery. J. Biol. Chem. 283, 18473–18477 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Zhao R. et al. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell 120, 715–727 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Pratt WB & Toft DO Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp. Biol. Med. (Maywood) 228, 111–133 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Whitesell L. & Lindquist SL HSP90 and the chaperoning of cancer. Nature Rev. Cancer 5, 761–772 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Dezwaan DC & Freeman BC HSP90: the Rosetta stone for cellular protein dynamics? Cell Cycle 7, 1006–1012 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Pratt WB, Morishima Y. & Osawa Y. The Hsp90 chaperone machinery regulates signaling by modulating ligand binding clefts. J. Biol. Chem. 283, 22885–22889 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McClellan AJ et al. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 131, 121–135 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Tsaytler PA, Krijgsveld J., Goerdayal SS, Rudiger S. & Egmond MR Novel Hsp90 partners discovered using complementary proteomic approaches. Cell Stress Chaperones 14, 629–638 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freeman BC & Yamamoto KR Disassembly of transcriptional regulatory complexes by molecular chaperones. Science 296, 2232–2235 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Zhao R. & Houry WA Hsp90: a chaperone for protein folding and gene regulation. Biochem. Cell Biol. 83, 703–710 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Tariq M., Nussbaumer U., Chen Y., Beisel C. & Paro R. Trithorax requires Hsp90 for maintenance of active chromatin at sites of gene expression. Proc. Natl Acad. Sci. USA 106, 1157–1162 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eccles SA et al. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 68, 2850–2860 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Chiosis G. & Tao H. Purine-scaffold Hsp90 inhibitors. IDrugs 9, 778–782 (2006). [PubMed] [Google Scholar]
- 14.Kim YS et al. Update on Hsp90 inhibitors in clinical trial. Curr. Top. Med. Chem. 9, 1479–1492 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Workman P., Burrows F., Neckers L. & Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann. N. Y. Acad. Sci. 1113, 202–216 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Pearl LH & Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem. 75, 271–294 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Donnelly A. & Blagg BS Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr. Med. Chem. 15, 2702–2717 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ali MM et al. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 440, 1013–1017 (2006).This paper describes the first crystal structure of a full-length HSP90–co-chaperone complex.
- 19.Prodromou C. & Pearl LH Structure and functional relationships of Hsp90. Curr. Cancer Drug Targets 3, 301–323 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Wayne N. & Bolon DN Dimerization of Hsp90 is required for in vivo function. Design and analysis of monomers and dimers. J. Biol. Chem. 282, 35386–35395 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Onuoha SC, Coulstock ET, Grossmann JG & Jackson SE Structural studies on the co-chaperone Hop and its complexes with Hsp90. J. Mol. Biol. 379, 732–744 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Vaughan CK et al. Structure of an Hsp90-Cdc37-Cdk4 complex. Mol. Cell 23, 697–707 (2006).This paper describes the first structure of an HSP90–co-chaperone–client protein complex.
- 23.Southworth DR & Agard DA Species-dependent ensembles of conserved conformational states define the Hsp90 chaperone ATPase cycle. Mol. Cell 32, 631–640 (2008).This paper reports the species-dependence of the conformational states sampled by HSP90.
- 24.McLaughlin SH, Ventouras LA, Lobbezoo B. & Jackson SE Independent ATPase activity of Hsp90 subunits creates a flexible assembly platform. J. Mol. Biol. 344, 813–826 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Meyer P. et al. Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 23, 1402–1410 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mickler M., Hessling M., Ratzke C., Buchner J. & Hugel T. The large conformational changes of Hsp90 are only weakly coupled to ATP hydrolysis. Nature Struct. Mol. Biol. 16, 281–286 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Hessling M., Richter K. & Buchner J. Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nature Struct. Mol. Biol. 16, 287–293 (2009).References 26 and 27 dissect the conformational intermediates of the HSP90 chaperone cycle.
- 28.Panaretou B. et al. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha 1. Mol. Cell 10, 1307–1318 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Forafonov F. et al. p23/Sba1p protects against Hsp90 inhibitors independently of its intrinsic chaperone activity. Mol. Cell. Biol. 28, 3446–3456 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Retzlaff M. et al. Asymmetric activation of the hsp90 dimer by its cochaperone aha1. Mol. Cell 37, 344–354 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Koulov AV et al. Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol. Biol. Cell 21, 871–884 (2010).This paper describes how AHA1 interaction with HSP90 affects client interaction with the HSP90 complex and chaperone efficiency.
- 32.Miyata Y. & Nishida E. Evaluating CK2 activity with the antibody specific for the CK2-phosphorylated form of a kinase-targeting cochaperone Cdc37. Mol. Cell. Biochem. 316, 127–134 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Smith JR & Workman P. Targeting CDC37: an alternative, kinase-directed strategy for disruption of oncogenic chaperoning. Cell Cycle 8, 362–372 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Echeverria PC et al. Nuclear import of the glucocorticoid receptor-hsp90 complex through the nuclear pore complex is mediated by its interaction with Nup62 and importin p. Mol. Cell. Biol. 29, 4788–4797 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pratt WB, Morishima Y., Murphy M. & Harrell M. Chaperoning of glucocorticoid receptors. Handb. Exp. Pharmacol. 172, 111–138 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Wochnik GM et al. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J. Biol. Chem. 280, 4609–4616 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Zhang M. et al. Structural and functional coupling of Hsp90- and Sgt1-centred multi-protein complexes. EMBO J. 27, 2789–2798 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boulon S. et al. The Hsp90 chaperone controls the biogenesis of L7Ae RNPs through conserved machinery. J. Cell Biol. 180, 579–595 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vaughan CK et al. Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol. Cell 31, 886–895 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McDowell CL, Bryan Sutton R. & Obermann WM Expression of Hsp90 chaperone [corrected] proteins in human tumor tissue. Int. J. Biol. Macromol. 45, 310–314 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Gray PJ Jr, Stevenson MA & Calderwood SK Targeting Cdc37 inhibits multiple signaling pathways and induces growth arrest in prostate cancer cells. Cancer Res. 67, 11942–11950 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Holmes JL, Sharp SY, Hobbs S. & Workman P. Silencing of HSP90 cochaperone AHA1 expression decreases client protein activation and increases cellular sensitivity to the HSP90 inhibitor 17-allylamino- 17-demethoxygeldanamycin. Cancer Res. 68, 1188–1197 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Scroggins BT & Neckers L. Post-translational modification of heat shock protein 90: impact on chaperone function. Expert Opin. Drug Discov. 2, 1403–1414 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Mimnaugh EG, Worland PJ, Whitesell L. & Neckers LM Possible role for serine/threonine phosphorylation in the regulation of the heteroprotein complex between the hsp90 stress protein and the pp60v-src tyrosine kinase. J. Biol. Chem. 270, 28654–28659 (1995). [DOI] [PubMed] [Google Scholar]
- 45.Wandinger SK, Suhre MH, Wegele H. & Buchner J. The phosphatase Ppt 1 is a dedicated regulator of the molecular chaperone Hsp90. EMBO J. 25, 367–376 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duval M., Le Boeuf F., Huot J. & Gratton JP Src-mediated phosphorylation of Hsp90 in response to vascular endothelial growth factor (VEGF) is required for VEGF receptor-2 signaling to endothelial NO synthase. Mol. Biol. Cell 18, 4659–4668 (2007).This paper reports the tyrosine phosphorylation of HSP90 by a client kinase.
- 47.Kurokawa M., Zhao C., Reya T. & Kornbluth S. Inhibition of apoptosome formation by suppression of Hsp90β phosphorylation in tyrosine kinase-induced leukemias. Mol. Cell. Biol. 28, 5494–5506 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Old WM et al. Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol. Cell 34, 115–131 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lees-Miller SP & Anderson CW Two human 90-kDa heat shock proteins are phosphorylated in vivo at conserved serines that are phosphorylated in vitro by casein kinase II. J. Biol. Chem. 264, 2431–2437 (1989). [PubMed] [Google Scholar]
- 50.Miyata Y. Protein kinase CK2 in health and disease: CK2: the kinase controlling the Hsp90 chaperone machinery. Cell. Mol. Life Sci. 66, 1840–1849 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harvey SL, Charlet A., Haas W., Gygi SP & Kellogg DR Cdk1-dependent regulation of the mitotic inhibitor Wee1. Cell 122, 407–420 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Mollapour M. et al. Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol. Cell 37, 333–343 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mollapour M., Tsutsumi S. & Neckers L. Hsp90 phosphorylation, Wee1 and the cell cycle. Cell Cycle 9, 1–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu X. et al. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J. Natl Cancer Inst. 94, 504–513 (2002). [DOI] [PubMed] [Google Scholar]
- 55.Kovacs JJ et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 18, 601–607 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Scroggins BT et al. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell 25, 151–159 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang Y. et al. Role of acetylation and extracellular location of heat shock protein 90α in tumor cell invasion. Cancer Res. 68, 4833–4842 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maloney A. et al. Gene and protein expression profiling of human ovarian cancer cells treated with the heat shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 67, 3239–3253 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Martinez-Ruiz A. et al. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc. Natl Acad. Sci. USA 102, 8525–8530 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morra G., Verkhivker G. & Colombo G. Modeling signal propagation mechanisms and ligand-based conformational dynamics of the Hsp90 molecular chaperone full-length dimer. PLoS Comput. Biol. 5, e1000323 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Retzlaff M. et al. Hsp90 is regulated by a switch point in the C-terminal domain. EMBO Rep. 10, 1147–1153 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Compton SA, Elmore LW, Haydu K., Jackson-Cook CK & Holt SE Induction of nitric oxide synthase-dependent telomere shortening after functional inhibition of Hsp90 in human tumor cells. Mol. Cell. Biol. 26, 1452–1462 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Toogun OA, Dezwaan DC & Freeman BC The hsp90 molecular chaperone modulates multiple telomerase activities. Mol. Cell. Biol. 28, 457–467 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Csermely P., Schnaider T., Soti C., Prohaszka Z. & Nardai G. The 90-kDa molecular chaperone family: structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 79, 129–168 (1998). [DOI] [PubMed] [Google Scholar]
- 65.Conzen SD Minireview: nuclear receptors and breast cancer. Mol. Endocrinol. 22, 2215–2228 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Echeverria PC & Picard D. Molecular chaperones, essential partners of steroid hormone receptors for activity and mobility. Biochim. Biophys. Acta 1803, 641–649 (2009). [DOI] [PubMed] [Google Scholar]
- 67.Zou J., Guo Y., Guettouche T., Smith DF & Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94, 471–480 (1998). [DOI] [PubMed] [Google Scholar]
- 68.Conde R., Belak ZR, Nair M., O’Carroll RF & Ovsenek N. Modulation of Hsf1 activity by novobiocin and geldanamycin. Biochem. Cell Biol. 87, 845–851 (2009). [DOI] [PubMed] [Google Scholar]
- 69.Dai C., Whitesell L., Rogers AB & Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130, 1005–1018 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Min JN, Huang L., Zimonjic DB, Moskophidis D. & Mivechi NF Selective suppression of lymphomas by functional loss of Hsf 1 in a p53-deficient mouse model for spontaneous tumors. Oncogene 26, 5086–5097 (2007).References 69 and 70 highlight the importance of HSF1 for carcinogenesis.
- 71.Au Q., Zhang Y., Barber JR, Ng SC & Zhang B. Identification of inhibitors of HSF1 functional activity by high-content target-based screening. J. Biomol. Screen. 14, 1165–1175 (2009). [DOI] [PubMed] [Google Scholar]
- 72.Ci W. et al. The BCL6 transcriptional program features repression of multiple oncogenes in primary B cells and is deregulated in DLBCL. Blood 113, 5536–5548 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cerchietti LC et al. A peptomimetic inhibitor of BCL6 with potent antilymphoma effects in vitro and in vivo. Blood 113, 3397–3405 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cerchietti LC et al. A purine scaffold Hsp90 inhibitor destabilizes BCL-6 and has specific antitumor activity in BCL-6-dependent B cell lymphomas. Nature Med. 15, 1369–1376 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Choo A. et al. The role of IRF1 and IRF2 transcription factors in leukaemogenesis. Curr. Gene Ther. 6, 543–550 (2006). [DOI] [PubMed] [Google Scholar]
- 76.Narayan V., Eckert M., Zylicz A., Zylicz M. & Ball KL Cooperative regulation of the interferon regulatory factor-1 tumor suppressor protein by core components of the molecular chaperone machinery. J. Biol. Chem. 284, 25889–25899 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bach C. & Slany RK Molecular pathology of mixed-lineage leukemia. Future Oncol. 5, 1271–1281 (2009). [DOI] [PubMed] [Google Scholar]
- 78.Pal S., Vishwanath SN, Erdjument-Bromage H., Tempst P. & Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 24, 9630–9645 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hamamoto R. et al. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nature Cell Biol. 6, 731–740 (2004).This paper reports that HSP90 inhibitor treatment suppresses SMYD3 activity in cancer cells.
- 80.Komatsu S. et al. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis 30, 1139–1146 (2009). [DOI] [PubMed] [Google Scholar]
- 81.Abu-Farha M. et al. The tale of two domains: proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol. Cell. Proteomics 7, 560–572 (2008). [DOI] [PubMed] [Google Scholar]
- 82.Sekimoto T. et al. The molecular chaperone Hsp90 regulates accumulation of DNA polymerase η at replication stalling sites in UV-irradiated cells. Mol. Cell 37, 79–89 (2010). [DOI] [PubMed] [Google Scholar]
- 83.Specchia V. et al. Hsp90 prevents phenotypic variation by suppressing the mutagenic activity of transposons. Nature 463, 662–665 (2010). [DOI] [PubMed] [Google Scholar]
- 84.Brennecke J. et al. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128, 1089–1103 (2007). [DOI] [PubMed] [Google Scholar]
- 85.Nishida KM et al. Functional involvement of Tudor and dPRMT5 in the piRNA processing pathway in Drosophila germlines. EMBO J. 28, 3820–3831 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oh WK et al. A single arm phase II trial of IPI-504 in patients with castration resistant prostate cancer (CRPC). Genitourinary Cancers Symp. Abstr. 219 (2009). [Google Scholar]
- 87.Heath EI et al. A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone- refractory metastatic prostate cancer. Clin. Cancer Res. 14, 7940–7946 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yano A. et al. Inhibition of Hsp90 activates osteoclast c-Src signaling and promotes growth of prostate carcinoma cells in bone. Proc. Natl Acad. Sci. USA 105, 15541–15546 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zoubeidi A. et al. Cooperative interactions between androgen receptor (AR) and heat-shock protein 27 facilitate AR transcriptional activity. Cancer Res. 67, 10455–10465 (2007). [DOI] [PubMed] [Google Scholar]
- 90.Solit DB et al. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin. Cancer Res. 14, 8302–8307 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Grbovic OM et al. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl Acad. Sci. USA 103, 657–662 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.da Rocha Dias S. et al. Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino- 17-demethoxygeldanamycin. Cancer Res. 65, 10686–10691 (2005). [DOI] [PubMed] [Google Scholar]
- 93.Mimnaugh EG, Chavany C. & Neckers L. Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J. Biol. Chem. 271, 22796–22801 (1996). [DOI] [PubMed] [Google Scholar]
- 94.Modi S. et al. Phase II trial of the Hsp90 inhibitor tanespimycin (Tan) + trastuzumab (T) in patients (pts) with HER2-positive metastatic breast cancer (MBC). J. Clin. Oncol. Abstr. 26, 1027 (2008). [Google Scholar]
- 95.Xu W. et al. Sensitivity of epidermal growth factor receptor and ErbB2 exon 20 insertion mutants to Hsp90 inhibition. Br. J. Cancer 97, 741–744 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chandarlapaty S. et al. Inhibitors of HSP90 block p95-HER2 signaling in Trastuzumab-resistant tumors and suppress their growth. Oncogene 29, 325–334 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Caldas-Lopes E. et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc. Natl Acad. Sci. USA 106, 8368–8373 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Richardson PG et al. Tanespimycin + bortezomib demonstrates safety, activity, and effective target inhibition in relapsed/refractory myeloma patients: updated results of a phase 1/2 study. 51st Am. Soc. Hematogy Annu. Meet. Abstr. (2009). [Google Scholar]
- 99.Mimnaugh EG, Xu W., Vos M., Yuan X. & Neckers L. Endoplasmic reticulum vacuolization and valosin-containing protein relocalization result from simultaneous hsp90 inhibition by geldanamycin and proteasome inhibition by velcade. Mol. Cancer Res. 4, 667–681 (2006). [DOI] [PubMed] [Google Scholar]
- 100.Mitsiades CS et al. Antimyeloma activity of heat shock protein-90 inhibition. Blood 107, 1092–1100 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Frebel K. & Wiese S. Signalling molecules essential for neuronal survival and differentiation. Biochem. Soc. Trans. 34, 1287–1290 (2006). [DOI] [PubMed] [Google Scholar]
- 102.Fionda C. et al. Heat shock protein-90 inhibitors increase MHC class I-related chain A and B ligand expression on multiple myeloma cells and their ability to trigger NK cell degranulation. J. Immunol. 183, 4385–4394 (2009).This paper shows that HSP90 inhibitors enhance NK-dependent recognition and lysis of myleoma cells in an HSF1-dependent manner.
- 103.Tse AN, Sheikh TN, Alan H., Chou TC & Schwartz GK 90-kDa heat shock protein inhibition abrogates the topoisomerase I poison-induced G2/M checkpoint in p53-null tumor cells by depleting Chk 1 and Wee1. Mol. Pharmacol. 75, 124–133 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Arlander SJ et al. Chaperoning checkpoint kinase 1 (Chk1), an Hsp90 client, with purified chaperones. J. Biol. Chem. 281, 2989–2998 (2006). [DOI] [PubMed] [Google Scholar]
- 105.Tse AN et al. A phase 1 dose-escalation study of irinotecan in combination with 17-allylamino-17-demethoxygeldanamycin in patients with solid tumors. Clin. Cancer Res. 14, 6704–6711 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hubbard J. et al. Phase I study of 17-allylamino-17 demethoxygeldanamycin, gemcitabine and/or cisplatin in patients with refractory solid tumors. Invest. New Drugs 15 January 2010. [epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hwang M., Moretti L. & Lu B. HSP90 inhibitors: multi-targeted antitumor effects and novel combinatorial therapeutic approaches in cancer therapy. Curr. Med. Chem. 16, 3081–3092 (2009). [DOI] [PubMed] [Google Scholar]
- 108.Reikvam H., Ersvaer E. & Bruserud O. Heat shock protein 90 - a potential target in the treatment of human acute myelogenous leukemia. Curr. Cancer Drug Targets 9, 761–776 (2009). [DOI] [PubMed] [Google Scholar]
- 109.Lancet JE et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022,17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia 24, 699–705 (2010). [DOI] [PubMed] [Google Scholar]
- 110.Weisberg E. et al. FLT3 inhibition and mechanisms of drug resistance in mutant FLT3-positive AML. Drug Resist. Updat. 12, 81–89 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shiotsu Y. et al. Novel oxime derivatives of radicicol induce erythroid differentiation associated with preferential G1 phase accumulation against chronic myelogenous leukemia cells through destabilization of Bcr-Abl with Hsp90 complex. Blood 96, 2284–2291 (2000). [PubMed] [Google Scholar]
- 112.Peng C., Li D. & Li S. Heat shock protein 90: a potential therapeutic target in leukemic progenitor and stem cells harboring mutant BCR-ABL resistant to kinase inhibitors. Cell Cycle 6, 2227–2231 (2007). [DOI] [PubMed] [Google Scholar]
- 113.O’Hare T., Eide CA & Deininger MW New Bcr-Abl inhibitors in chronic myeloid leukemia: keeping resistance in check. Expert Opin. Investig. Drugs 17, 865–878 (2008). [DOI] [PubMed] [Google Scholar]
- 114.Peng C. et al. Inhibition of heat shock protein 90 prolongs survival of mice with BCR-ABL-T315I-induced leukemia and suppresses leukemic stem cells. Blood 110, 678–685 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Castro JE et al. ZAP-70 is a novel conditional heat shock protein 90 (Hsp90) client: inhibition of Hsp90 leads to ZAP-70 degradation, apoptosis, and impaired signaling in chronic lymphocytic leukemia. Blood 106, 2506–2512 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Elfiky A. et al. BIIB021, an oral, synthetic non-ansamycin Hsp90 inhibitor: phase I experience. J. Clin. Oncol. Abstr. 26, 2503 (2008). [Google Scholar]
- 117.Gallegos Ruiz MI et al. Integration of gene dosage and gene expression in non-small cell lung cancer, identification of HSP90 as potential target. PLoS ONE 3, e0001722 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sequist LV et al. A phase II trial of IPI-504 (retaspimycin hydrochloride), a novel Hsp90 inhibitor, in patients with relapsed and/or refractory stage IIIb or stage IV non-small cell lung cancer (NSCLC) stratified by EGFR mutation status. J. Clin. Oncol. Abstr. 27, 8073 (2009). [Google Scholar]
- 119.Shimamura T. & Shapiro GI Heat shock protein 90 inhibition in lung cancer. J. Thorac. Oncol. 3, S152–S159 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shimamura T. et al. Hsp90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistance. Cancer Res. 68, 5827–5838 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Banerji U. et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J. Clin. Oncol. 23, 4152–4161 (2005). [DOI] [PubMed] [Google Scholar]
- 122.Grem JL et al. Phase I and pharmacologic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with solid tumors. J. Clin. Oncol. 23, 1885–1893 (2005). [DOI] [PubMed] [Google Scholar]
- 123.Ramanathan RK et al. Phase I pharmacokinetic and pharmacodynamic study of 17-dimethylaminoethyl-amino-17-demethoxygeldanamycin, an inhibitor of heat-shock protein 90, in patients with advanced solid tumors. J. Clin. Oncol. 28, 1520–1526 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Eiseman JL et al. Pharmacokinetics and pharmacodynamics of 17-demethoxy 17-[[(2-dimethylamino)ethyl]amino]geldanamycin (17DMAG, NSC 707545) in C.B-17 SCID mice bearing MDA-MB-231 human breast cancer xenografts. Cancer Chemother. Pharmacol. 55, 21–32 (2005). [DOI] [PubMed] [Google Scholar]
- 125.Vilenchik M. et al. Targeting wide-range oncogenic transformation via PU24FCl, a specific inhibitor of tumor Hsp90. Chem. Biol. 11, 787–797 (2004). [DOI] [PubMed] [Google Scholar]
- 126.Kamal A. et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425, 407–410 (2003). [DOI] [PubMed] [Google Scholar]
- 127.Kummar S. et al. Phase I trial of 17-dimethylamino-ethylamino-17-demethoxygeldanamycin (17-DMAG), a heat shock protein inhibitor, administered twice weekly in patients with advanced malignancies. Eur. J. Cancer 46, 340–347 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dakappagari N. et al. An investigation into the potential use of serum Hsp70 as a novel tumour biomarker for Hsp90 inhibitors. Biomarkers 15, 31–38 (2009). [DOI] [PubMed] [Google Scholar]
- 129.Demetri GD et al. Inhibition of the heat shock protein 90 (Hsp90) chaperone with the novel agent IPI-504 to overcome resistance to tyrosine kinase inhibitors (TKIs) in metastatic GIST: updated results of a phase I trial. J. Clin. Oncol. Abstr. 25, 10024 (2007). [Google Scholar]
- 130.Smith-Jones PM, Solit D., Afroze F., Rosen N. & Larson SM Early tumor response to Hsp90 therapy using HER2 PET: comparison with 18F-FDG PET. J. Nucl. Med. 47, 793–796 (2006). This paper describes a new non-invasive imaging approach to monitor anti-tumour HSP90 inhibitor activity in vivo. [PMC free article] [PubMed] [Google Scholar]
- 131.Oude Munnink TH et al. 89Zr-trastuzumab PET visualises HER2 downregulation by the HSP90 inhibitor NVP-AUY922 in a human tumour xenograft. Eur. J. Cancer 46, 678–684 (2009). [DOI] [PubMed] [Google Scholar]
- 132.Kramer-Marek G., Kiesewetter DO & Capala J. Changes in HER2 expression in breast cancer xenografts after therapy can be quantified using PET and 18F-labeled affibody molecules. J. Nucl. Med. 50, 1131–1139 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Holland JP et al. Measuring the pharmacodynamic effects of a novel Hsp90 inhibitor on HER2/neu expression in mice using Zr-DFO-trastuzumab. PLoS ONE 5, e8859 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Le HC et al. Proton MRS detects metabolic changes in hormone sensitive and resistant human prostate cancer models CWR22 and CWR22r. Magn. Reson. Med. 62, 1112–1119 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chung YL et al. Magnetic resonance spectroscopic pharmacodynamic markers of the heat shock protein 90 inhibitor 17-allylamino, 17-demethoxygeldanamycin (17AAG) in human colon cancer models. J. Natl Cancer Inst. 95, 1624–1633 (2003). [DOI] [PubMed] [Google Scholar]
- 136.Liu D. et al. Use of radiolabelled choline as a pharmacodynamic marker for the signal transduction inhibitor geldanamycin. Br. J. Cancer 87, 783–789 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kelland LR, Sharp SY, Rogers PM, Myers TG & Workman P. DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxy-geldanamycin, an inhibitor of heat shock protein 90. J. Natl Cancer Inst. 91, 1940–1949 (1999). [DOI] [PubMed] [Google Scholar]
- 138.Guo W. et al. Formation of 17-allylamino-demethoxygeldanamycin (17-AAG) hydroquinone by NAD(P)H:quinone oxidoreductase 1: role of 17-AAG hydroquinone in heat shock protein 90 inhibition. Cancer Res. 65, 10006–10015 (2005). [DOI] [PubMed] [Google Scholar]
- 139.Gaspar N. et al. Acquired resistance to 17-allylamino-17-demethoxygeldanamycin (17-AAG, tanespimycin) in glioblastoma cells. Cancer Res. 69, 1966–1975 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Erlichman C. Tanespimycin: the opportunities and challenges of targeting heat shock protein 90. Expert Opin. Investig. Drugs 18, 861–868 (2009). [DOI] [PubMed] [Google Scholar]
- 141.McCollum AK, Teneyck CJ, Sauer BM, Toft DO & Erlichman C. Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxy-geldanamycin through a glutathione-mediated mechanism. Cancer Res. 66, 10967–10975 (2006). [DOI] [PubMed] [Google Scholar]
- 142.Powers MV, Clarke PA & Workman P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell 14, 250–262 (2008). [DOI] [PubMed] [Google Scholar]
- 143.Evans CG, Chang L. & Gestwicki JE Heat shock protein 70 (Hsp70) as an emerging drug target. J. Med. Chem. 24 March 2010. (doi: 10.1021/jm100054f). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Powers MV et al. Targeting HSP70: the second potentially druggable heat shock protein and molecular chaperone? Cell Cycle 9, 1542–1550 (2010). [DOI] [PubMed] [Google Scholar]
- 145.Hadchity E. et al. Heat shock protein 27 as a new therapeutic target for radiation sensitization of head and neck squamous cell carcinoma. Mol. Ther. 17, 1387–1394 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Roe SM et al. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 42, 260–266 (1999). [DOI] [PubMed] [Google Scholar]
- 147.Prodromou C. et al. Structural basis of the radicicol resistance displayed by a fungal hsp90. ACS Chem. Biol. 4, 289–297 (2009). [DOI] [PubMed] [Google Scholar]
- 148.Matthews SB et al. Characterization of a novel novobiocin analogue as a putative C-terminal inhibitor of heat shock protein 90 in prostate cancer cells. Prostate 70, 27–36 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shelton SN et al. KU135, a novel novobiocin-derived C-terminal inhibitor of the 90-kDa heat shock protein, exerts potent antiproliferative effects in human leukemic cells. Mol. Pharmacol. 76, 1314–1322 (2009). References 148 and 149 describe new C-terminal HSP90 inhibitors with potent anticancer activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Radanyi C. et al. Antiproliferative and apoptotic activities of tosylcyclonovobiocic acids as potent heat shock protein 90 inhibitors in human cancer cells. Cancer Lett. 274, 88–94 (2009). [DOI] [PubMed] [Google Scholar]
- 151.Zhang T. et al. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 7, 162–170 (2008). [DOI] [PubMed] [Google Scholar]
- 152.Sreeramulu S., Gande SL, Gobel M. & Schwalbe H. Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew. Chem. Int. Ed. Engl. 48, 5853–5855 (2009). [DOI] [PubMed] [Google Scholar]
- 153.Chakraborty A. et al. HSP90 regulates cell survival via inositol hexakisphosphate kinase-2. Proc. Natl Acad. Sci. USA 105, 1134–1139 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Voss AK, Thomas T. & Gruss P. Mice lacking HSP90β fail to develop a placental labyrinth. Development 127, 1–11 (2000). [DOI] [PubMed] [Google Scholar]
- 155.Dollins DE, Warren JJ, Immormino RM & Gewirth DT Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol. Cell 28, 41–56 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Frey S., Leskovar A., Reinstein J. & Buchner J. The ATPase cycle of the endoplasmic chaperone Grp94. J. Biol. Chem. 282, 35612–35620 (2007). [DOI] [PubMed] [Google Scholar]
- 157.Leskovar A., Wegele H., Werbeck ND, Buchner J. & Reinstein J. The ATPase cycle of the mitochondrial Hsp90 analog Trapl. J. Biol. Chem. 283, 11677–11688 (2008). [DOI] [PubMed] [Google Scholar]
- 158.Immormino RM et al. Different poses for ligand and chaperone in inhibitor-bound Hsp90 and GRP94: implications for paralog-specific drug design. J. Mol. Biol. 388, 1033–1042 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Felts SJ et al. The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J. Biol. Chem. 275, 3305–3312 (2000). [DOI] [PubMed] [Google Scholar]
- 160.Hua G., Zhang Q. & Fan Z. Heat shock protein 75 (TRAP1) antagonizes reactive oxygen species generation and protects cells from granzyme M-mediated apoptosis. J. Biol. Chem. 282, 20553–20560 (2007). [DOI] [PubMed] [Google Scholar]
- 161.Kang BH et al. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J. Clin. Invest. 119, 454–464 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Leav I. et al. Cytoprotective mitochondrial chaperone TRAP-1 as a novel molecular target in localized and metastatic prostate cancer. Am. J. Pathol. 176, 393–401 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pridgeon JW, Olzmann JA, Chin LS & Li L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 5, e172 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sidera K. & Patsavoudi E. Extracellular HSP90: conquering the cell surface. Cell Cycle 7, 1564–1568 (2008). [DOI] [PubMed] [Google Scholar]
- 165.Eustace BK et al. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nature Cell Biol. 6, 507–514 (2004). This paper describes an important role for secreted HSP90 in cancer cell motility and invasion. [DOI] [PubMed] [Google Scholar]
- 166.Becker B. et al. Induction of Hsp90 protein expression in malignant melanomas and melanoma metastases. Exp. Dermatol. 13, 27–32 (2004). [DOI] [PubMed] [Google Scholar]
- 167.Tsutsumi S. et al. A small molecule cell-impermeant Hsp90 antagonist inhibits tumor cell motility and invasion. Oncogene 27, 2478–2487 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Cheng CF et al. Transforming growth factor alpha (TGFα)-stimulated secretion of HSP90alpha: using the receptor LRP-1/CD91 to promote human skin cell migration against a TGFβ-rich environment during wound healing. Mol. Cell. Biol. 28, 3344–3358 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Li W. et al. Extracellular heat shock protein-90α: linking hypoxia to skin cell motility and wound healing. EMBO J. 26, 1221–1233 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tsutsumi S. et al. Hsp90 charged-linker truncation reverses the functional consequences of weakened hydrophobic contacts in the N domain. Nature Struct. Mol. Biol. 16, 1141–1147 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sidera K., Gaitanou M., Stellas D., Matsas R. & Patsavoudi E. A critical role for HSP90 in cancer cell invasion involves interaction with the extracellular domain of HER-2. J. Biol. Chem. 283, 2031–2041 (2008). [DOI] [PubMed] [Google Scholar]
- 172.Annamalai B., Liu X., Gopal U. & Isaacs JS Hsp90 is an essential regulator of EphA2 receptor stability and signaling: implications for cancer cell migration and metastasis. Mol. Cancer Res. 7, 1021–1032 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kawabe M. et al. Heat shock protein 90 inhibitor 17-dimethylaminoethylamino- 17-demethoxygeldana-mycin enhances EphA2+ tumor cell recognition by specific CD8+ T cells. Cancer Res. 69, 6995–7003 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wesa AK et al. Enhancement in specific CD8+ T cell recognition of EphA2+ tumors in vitro and in vivo after treatment with ligand agonists. J. Immunol. 181, 7721–7727 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Stuehler C. et al. Selective depletion of alloreactive T cells by targeted therapy of heat shock protein 90: a novel strategy for control of graft-versus-host disease. Blood 114, 2829–2836 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.