

Abstract
Humans are exposed to large numbers of environmental chemicals, some of which potentially interfere with the endocrine system. The identification of potential endocrine disrupting chemicals (EDCs) has gained increasing priority in the assessment of environmental hazards. The U.S. Environmental Protection Agency (U.S. EPA) has developed the Endocrine Disruptor Screening Program (EDSP) which aims to prioritize and screen potential EDCs. The Toxicity Forecaster (ToxCast) program has generated data using in vitro high-throughput screening (HTS) assays measuring activity of chemicals at multiple points along the androgen receptor (AR) activity pathway. In the present study, using a large and diverse data set of 1667 chemicals provided by the U.S. EPA from the combined ToxCast AR assays in the framework of the Collaborative Modeling Project for Androgen Receptor Activity (CoMPARA). Two models were built using ADMET Predictor™; one is based on Artificial Neural Networks (ANNs) technology and the other uses a Support Vector Machine (SVM) algorithm; one model is a Decision Tree (DT) developed in R; and two models make use of differently combined sets of structural alerts (SAs) automatically extracted by SARpy. We used two strategies to integrate predictions from single models; one is based on a majority vote approach and the other on prediction convergence. These strategies led to enhanced statistical performance in most cases. Moreover, the majority vote approach improved prediction coverage when one or more single models were not able to provide any estimations. This study integrates multiple in silico approaches as a virtual screening tool for use in risk assessment of endocrine disrupting chemicals.
Keywords: endocrine disrupting chemicals, high-throughput screening, androgen receptor, in silico, Artificial Neural Networks, Support Vector Machine, Decision Tree
1. Introduction
Endocrine disrupting chemicals (EDCs) are natural or manufactured substances that potentially cause adverse health effects to humans and/or ecological species by disrupting their endogenous hormone systems (Mansouri et al., 2016). EDCs can interfere with endocrine pathways including estrogenic, androgenic, thyroid pathways, but also metabolism, fat storage, bone development and the immune system (Bergman et al., 2013). EDCs affect endocrine functions through different underlying mechanisms, i.e. inhibiting the biosynthesis or metabolism of endogenous ligands, mimicking or interfering with natural hormones and altering their mechanisms of action at the receptor level (Li and Gramatica, 2010, Mansouri et al., 2016). EDCs cover a wide range of chemically diverse substances (e.g. organochlorines, organophosphates, carbamates, pyrethroids) with broad application fields (e.g. pesticides, insecticides, herbicides) (Ruiz et al., 2017).
Nevertheless, many environmental chemicals have not yet been tested for their endocrine disrupting potential, in particular, their ability to interact with the estrogen and androgen receptors (ER and AR). To narrow this gap, the US EPA has been developing high-throughput in vitro methods to test for ER and AR activity (Dix et al., 2007, Judson et al., 2015, Kleinstreuer et al., 2017) as part of the ToxCast and Tox21 programs. (Kavlock et al. 2012, Thomas et al. 2013, Tice et al. 2013, Richard et al. 2016).
Within the EDSP framework, the EPA launched the Collaborative Estrogen Receptor Activity Prediction Project (CERAPP) with the intent to demonstrate the effectiveness of predictive computational models in prioritizing thousands of chemicals for ER activity. Within CERAPP, a number of computational models for predicting ER activity were developed and validated in collaboration between 17 groups in the U.S. and Europe (Mansouri et al., 2016). The resulting models, based on combined ToxCast in vitro HTS assays data, (Judson et al 2015) were integrated into a single consensus model. The present work describes one contribution to a similar consortium effort led by the EPA: Collaborative Modeling Project for Androgen Receptor Activity (CoMPARA), involving 25 international groups, to screen chemicals for their potential AR activity (Mansouri et al., 2018).
There is a wide and increasing collection of software and literature models for predicting ER and AR activity (Roncaglioni et al., 2008; Li and Gramatica, 2010; Lo Piparo and Worth, 2010; Ruiz et al., 2017; Trisciuzzi et al., 2015). Recently, an integrated strategy combining in silico methods and computational systems biology has been proposed to explore endocrine-disrupting chemical binding with nuclear hormone receptors (Ruiz et al., 2017).
This study aimed to develop predictive classification models for predicting AR binding activity on a large and diverse data set of 1667 chemicals provided by the U.S. EPA from the combined ToxCast AR assays (Kleinstreuer et al., 2017). Five models were built based on different algorithms: Artificial Neural Networks (ANNs), Support Vector Machine (SVM), Decision Tree (DT) and fragment-based. Hence, predictions of single models were combined based on a majority vote approach and considering only convergent predictions. We have presented the use of in silico modeling to provide confident predictions using ToxCast AR assays and the ability to use it in risk assessment of endocrine disrupting chemicals.
2. Materials and methods
2.1. Data Sets
2.1.1. Provided training set
1689 curated chemical structures with AR experimental activity were provided by the EPA’s National Center for Computational Toxicology as a training set to develop the in silico models. Experimental data were derived from a collection of 11 in vitro HTS assays exploring multiple points in the AR pathway including three receptor binding, two cofactor recruitment, one RNA transcription, three agonist-mode protein production and two antagonist-mode protein production. A chemical was considered as a binder if it was either an active agonist or antagonist. For more information about the ToxCast assays and how they were combined see Kleinstreuer et al., 2017. The mathematical model proposed by Judson et al. (2015) was used to integrate the in vitro data and calculate an area under the curve (AUC) score, ranging from 0 to 1, being approximately proportional to the consensus AC50 (activity concentration at half-maximal response) value across the active assays (Judson et al., 2015, Kleinstreuer et al., 2017). A given chemical was considered active if its AUC value was ≥0.1. AUC values between 0.001 and 0.1 indicate very weak potential activity and were considered inconclusive (Judson et al., 2015. Values below 0.001 were considered inactive and set to zero (Kleinstreuer et al., 2017). The training set provided by the AR data constitutes a very diverse set of molecular structures, chemical classes, physicochemical and structural properties. This results in a large domain of applicability (DoA). From the initial list of 1,689, we removed a duplicate structure and twenty-one chemicals identified by the EPA as potentially false negatives based on a bootstrapping study (Watt and Judson, 2018), obtaining a final list of 1667 compounds. AR experimental activities refer to the impact of the compounds on the AR pathway (active = 198 and inactive = 1469). Because these data contain an unbalanced ratio of active and inactive chemicals, applying appropriate evaluation metrics for model generation, such as balanced accuracy and Matthews correlation coefficient are best taken into consideration, as well as to try different sampling techniques and models to evaluate what works best (Section 2.3–2.5).
2.1.2. Prediction set
The Endocrine Disruptor Knowledgebase ( EDKB) androgen receptor (AR) binding data set (Fang et al., 2003) was downloaded as an SDF (https://www.fda.gov/ScienceResearch/BioinformaticsTools/EndocrineDisruptorKnowledgebase/ucm136086.htm), and used to evaluate the predictive performance of our classification models and consensus models (see sections 2.4 Consensus modeling approaches, 2.5 In silico modeling evaluations). The logarithm of the AR relative binding affinity (RBA) values were reported for 202 chemicals. This value represent the strength of the binding to the AR receptor using Metribolone as a reference for the activity (logRBA = 2). First, we explored the congruence between the dataset formerly used by Fang et al. reporting quantitative data and our training set categorical data for the 102 compounds common to both datasets. We observed, as depicted in Fig. 1, that those compounds with very low activity values in Fang et al. (logRBA < −3) or inactive (arbitrary set to −5 and −10 by the authors) were mostly inactive also in our dataset (91% of concordance). Similarly, compounds with relatively higher logRBA values (logRBA > −1) were mostly active in our training set as well (93% of concordance). In the remaining central area, the behaviour of the two datasets is quite promiscuous, so we excluded the compounds falling in this range of values from our evaluation since the two series of data are not in agreement in this range. In total we included in the prediction set 35 compounds in the negative class (chemicals with logRBA < −3 and not included already in the training set) and 15 compounds in the positive class (chemicals with logRBA > −1 and not included already in the training set). Compounds with logRBA between −3 and −1 were excluded from the validation set.
Figure 1.
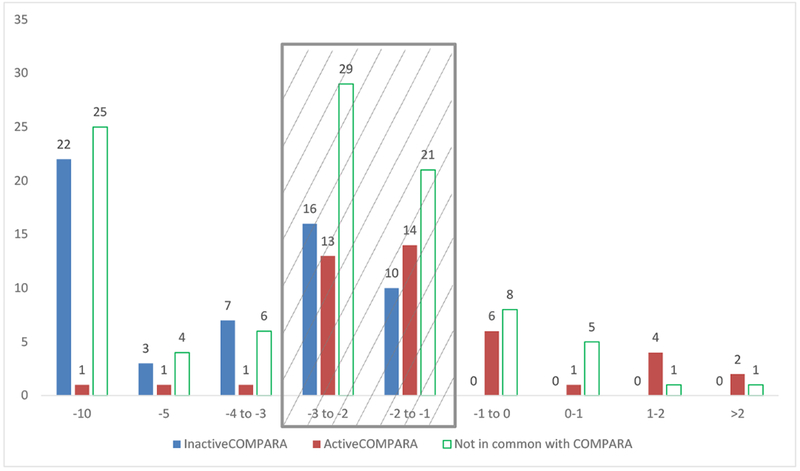
Prediction set activity values compared with the training set (TS) data. The training set classification values were compared to EDKB logRBA quantitative values to identify suitable chemicals as test set. The blue and red bars indicate the prevalence of training set negative or positive compounds respectively in the different ranges of EDKB logRBA values. While in the central area (with gray shadow) the behaviour is quite promiscuous. Good agreement is observed between negative TS compounds and quantitative logRBA data < −3, and between positive TS compounds and quantitative logRBA data > −1. Therefore, we considered as a prediction set those compounds not already included in the TS (white bars), but outside the gray area. All compounds with a quantitative logRBA < −3 were considered as negatives (35 in total) while those with a logRBA > −1 were considered as positives in the prediction set (15 compounds in total).
2.2. Software
In this study, the ADMET Modeler module of ADMET Predictor™ 8.0 (Simulations Plus Inc., 2015) was used to construct ANN and SVM classification QSAR models (section 2.3.1). R software (R Core Team, 2015) was used to build the DT model and SARpy for the automatic extraction of Structural Alerts (SAs) (Ferrari et al., 2013). The Recursive PARTitioning (rpart) module included in R software (Therneau and Atkinson, 2015) allows building CART (Classification and Regression Trees) models using a two-stage procedure (Section 2.3.2.).
SARpy (SAR in python) is a freely available software in the VEGA HUB platform (https://www.vegahub.eu/), which automatically generates Structural Alerts (SAs) without any a priori knowledge, exclusively based on their predictive performance on a training set used as input. Each SA is associated with a Likelihood Ratio (LR) value which is the likelihood that a given prediction would be expected for a compound with the target SA compared to the likelihood that the same prediction would be expected for a compound without the target SA. The LR ranges from 1 to infinity (∞). The higher the value is, the higher the predictive accuracy for a specific fragment will be (Ferrari et al., 2013) (Section 2.3.3).
2.3. Development and validation of classification models
2.3.1. Construction and validation of artificial neural networks (ANNs) and support vector machine (SVM) classification models using the ADMET Predictor/Modeler.
Data input and calculation/selection of descriptors: ADMET Predictor accepts data in which toxicities are linked to 2D molecular structures in a standard file and calculates more than 350 molecular and atomic-level descriptors. In the present investigation, 299 descriptors were selected after discarding non-numerical descriptors. At this stage, ADMET Modeler, a module of ADMET Predictor, is utilized to complete the next six steps in order to develop and validate ANN or SVM classification classification models.
Test and training set data distribution: Kohonen Self-Organizing Map (Kohonen, 1982) as implemented in ADMET Predictor was used to cluster the data sets based on their descriptors and perform the splits (Simulation Plus Inc., 2015). The Kohonen map is a square two-dimensional grid consisting of a number of cells. The default size of the Kohonen map is the largest square that will fit within the number of data points in the data set.
Selection of best descriptors: To select best descriptors, the method of sensitivity analysis (ways to sort descriptors by their relevant information content) was the truncated linear analysis (TLA) implemented in ADMET Predictor. TLA is a multiple linear regression model is built with the auto-scaled descriptors and the absolute value of each coefficient is used as a measure of the respective descriptor’s influence on the target property. The strategy used by this descriptor selection method is as follows: only those descriptors that have acceptable variance (non-zero), adequate representation in the data set (most influence on activity), and low correlation (close to zero) with other descriptors are retained for model building. Furthermore, the best models use the smallest number of descriptors to maximize the applicability domain covered by the model as well as reduce the chance of overtraining.
Split of training pool into training and verification sets: The training pool data is randomly divided into two separate sets: a training and a verification set. The training and verification sets contain, respectively, about two thirds and one third of the training set data. The initial assignments are then randomly shuffled for each fold of the ensemble. The verification set is used for early stopping to help prevent ANN overtraining.
Predictive algorithm selection: We used the artificial neural network ensemble (ANNE) technique to construct in silico models (Simulation Plus Inc., 2015). Artificial neural networks (ANNs) establish nonlinear correlation between molecular and/or structural descriptors of chemicals and their biological/toxicological activities. ADMET Modeler uses one hidden layer of neurons between the inputs and the outputs. The program utilizes ANNEs, which average the output of collections of ANNs. The prediction from an ensemble is the voting based on predicted values from each separately trained ANN, all of which use the same architecture (Simulations Plus Inc., 2015). We also used support vector machine (SVM) implemented in ADMET Predictor. SVM is an outgrowth of kernel methods. In such methods, the data is transformed with a kernel function (e.g., a radial basis function) and it is in this transformed space (“kernel space”) that the model is built. Care is taken in the construction of the kernel that it has a sufficiently high dimensionality that an underlying linear relationship can be found within it. A subset of transformed data points - the set of “support vectors” - is then used to specify a hyperplane that effectively serves as a linear model within this space.
Development of ANN and SVM models: The model-building progress can be observed in the ‘Ensemble Statistics’ Monitor of the program in which each ensemble displays the combined results of 33 best artificial neural networks (sub-models) using an averaging method. The combined results include the statistics of three data partitions: training set, verification set, and test set. The test set statistics of each ensemble are used to assess their external validity. The test set is a group of compounds that are set aside before training begins and is not involved in the training in any way. Once a model has been fully trained, the test set is used to assess the model’s predictive performance. This provides an objective estimate of the overall predictive accuracy of each ensemble that the program generates.
Selection of the best ANN and SVM classification models: Several statistical parameters (see details in Section 2.4) such as, sensitivity, specificity, false positive and negative rates, Youden index (Youden, 1950), Matthews’s correlation coefficient, and balanced accuracy were used to identify the best ANN classification models. For classification models, the fraction of incorrect predictions is call False Rate. The false rate is given by (FN + FP)/(TP + FN + TN + FP), where “T” and “F” indicate true and false predictions, respectively, and “P” and “N” indicate positive and negative predictions, respectively.
Applicability domain (AD): ADMET Predictor indicates whether a given compound is within the AD of each of the models. For each descriptor, xi (i = 1, …, …,N), its minimum (ximin) and maximum (ximax) values are determined over the training set of X. A new compound C is said to be within the AD of X if the value for each relevant descriptor ci (i = 1, …, …,N) calculated for C is within the corresponding interval [ximin, ximax] with tolerance equal to 10% of the interval length. Chemicals outside the AD and containing disjoint structures, metals and inorganic elements (e.g., Si, As, Se, Te, At) gave warning messages and were flagged as “unusable”, so they were considered not predicted and excluded from statistical analyses. For the CoMPARA data set, all chemicals containing the element Si were removed.
In the present study, a set of optimal experimental conditions for the ADMET Modeler were identified and used in the development of the classifications models. An artificial neural network is a connected set of mathematical functions. For each descriptor (or input), a coefficient (also called a “weight”) multiplies the value of the descriptor and that product is sent to a neuron (also “unit” or “node”). The neuron sums the inputs and subtracts a threshold weight as a bias term. The minimal coefficient of variation (CV) is the minimum % CV expressed as 100 x (standard deviation/mean) for a descriptor to be included for model building. Minimum representation is the minimum number of non-zero values for a descriptor which must be present in the raw data for that descriptor to be retained for model building. Maximum absolute correlation is the maximum allowed correlation between any two or more descriptors selected as independent variables to the model.
For the development of the ANN models:
1. A minimum test set of 10% of the chemicals was selected using the Kohonen method, 2) Seventy-five optimal descriptors were selected from 340 descriptors using sensitivity analyses (truncated linear analysis (TLA)), 3) Three ANN classification conditions were considered: (1) network neurons: minimum = 1, maximum = 20, step = 1; (2) network descriptors: minimum = 2, maximum = 200, step = 2; (3) network training: (a) number of Monte Carlo tries for starting points = 1 and (b) skip network architecture if the number of adjustable weights exceeds 75% of the data set size, 4) A binary classification was performed using Refine threshold conditions.
Similarly, for the SVM classification, models were identified and used the following set of conditions: 1) A minimum test set of 10% of the chemicals was selected using Kohonen method (Kohonen, 1982), 2) The minimum input CV = 1, minimum represent = 4, maximum input correlation = 0.98 using TLA sensitivity analyses, 3) For the SVM classifications, these conditions were considered: (1) descriptors: minimum = 5, maximum = 70, step = 5; (2) Ensemble settings (a) model per ensemble = 33 and (b) model to train = 4 0.
2.3.2. Construction and validation of Decision Tree (DT) model using the rpart R package
Data input and calculation/selection of descriptors: the full set of 2D descriptors was calculated using DRAGON 7 (v. 7.0.8, Kode srl, 2017); the descriptors have been pruned by discarding those with constant values, those with at least one missing value, and applying a filter to remove those correlated to another descriptor with a pairwise correlation greater than 0.9; the final pool of descriptors consisted of 1095 2D descriptors, such as topological indices, connectivity indices, P_VSA-like descriptors, functional group counts, constitutional descriptors, physico-chemical properties.
Test set and training set distribution: The dataset was randomly split into training (80%) and test sets (20%) keeping the same distribution of experimental activities using KNIME Rule-based Row Splitter and Partitioning nodes (Berthold et al., 2008).
Selection of the best descriptors (inputs): A stepwise variable selection using a linear discriminant analysis (LDA) and a bootstrap technique (based on a balanced resampling) for validation was performed to select the best variables, using an in-house code implemented in R.
- Predictive algorithm and model development: We used a decision tree (DT) algorithm, implemented in the “rpart” R package, applied to the selected descriptors. The algorithm works as follows:
- First, the variable which best splits the data into two groups is found. The data is separated, and then this process is applied independently to each sub-group, and so either on recursively until the subgroups reach a minimum size or until no improvement can be made. The initial split changes depending on the prior probabilities (prior) which are proportional to the data counts by default. We assumed “prior = 0.5, 0.5” in the rpart function, to balance the original bias in the training set between the size of the two classes.
- The second stage of the procedure consists in the use of a cross-validation procedure to trim back and simplify the full tree to avoid its overfitting.
Selection of the best DT model: The best model was selected based on the minimum cross-validated error, which included five variables.
The applicability domain (AD) of the DT is assessed using the bounding-box approach which consists of checking of the descriptors range from the training set, i.e., new samples to be predicted are considered out of AD if one or more input descriptors show values outside the range of the training set descriptors (Sahigara et al., 2012).
2.3.3. Development and validation of SARpy models
We used SARpy to develop two models based on two “definitions” and combinations of structural alerts. We extracted two rule sets and then combined them in two different models. First, we selected only one class as target (the active class, i.e., binders). In this way, the software extracts fragments related to the active class only and therefore all compounds that do not match any fragment are labelled as negative (i.e., non-binders). By manually increasing the minimum accepted threshold for LR on the training set, fewer rules are retained (only those having better performance) so fewer compounds will be labelled as positive.
Then, we extracted fragments by analyzing both classes together. In this way, the software extracts specific fragments for the active and inactive class. The class is assigned to a target chemical based on the matched fragments; if no fragment is identified, the compound is not predicted. In this case, also, using a manual setting, a few thresholds for LR were tested. By increasing the LR, fewer rules are kept for either the active or inactive class (only those having better performance), so the model tends to be more precise but with a larger proportion of unpredicted compounds.
Both rule sets were extracted using the entire dataset (N = 1667) without train/test splitting and tested on the non-overlapping part of the EDKB dataset.
Finally, two approaches were used to derive rules with SARpy:
SAs for the two classes: the best model developed on both classes (that usually results in a large number of unpredicted compounds) resulted in a list of 17 SAs and 110 SAs for active and for inactive compounds respectively;
SAs for the Active class only: the best model on the active compounds only resulted in a list of 22 SAs for active compound.
Two different strategies to integrate these rule sets was drawn up, by combining the two best models described above. The first combined model applies the two models in cascade assigns inactivity label to a given chemical if it was ether matched by an inactive SA in the two class’s model or not matched by the latter rule set (SARpy combined model 1). The second combined model (SARpy combined model 2) labels as active chemicals those which are positive in at least one of the two rule sets, regardless the inactive label that could be assigned in the two class model (conservative approach).
2.4. Consensus modeling approaches
The consensus approach combines the predictions of several (Q)SAR models to maximize their strengths, minimize their weaknesses, and increase their global prediction accuracy (Ruiz et al., 2017). Several androgen receptor in silico models were selected, based on different molecular descriptors and statistical and modeling techniques (ANNs, SVM, DT, SA extraction). They were merged into two types of consensus approaches. In the first (consensus I), all model’s predictions had to agree on the predicted class. In the second (consensus II), based on the majority agreement approach, the compound was classified according to the most frequently predicted class. Both consensus approaches ignored models that did not provide classifications for specific chemicals.
2.5. In silico modeling evaluations
A common way to evaluate the performance of classification models is to use a confusion matrix. Such a matrix was used to show the number of correct and incorrect predictions made by each classification model compared with the actual (experimental value) in the data. Several statistical parameters were used to evaluate the predictive power and reliability of the classification models, such as:
-
Sensitivity: proportion of actual positive cases correctly identified.
Sensitivity=TP(TP+FN)
-
Specificity: proportion of actual negative cases correctly identified.
Specificity=TN(TN+FP)
Balanced accuracy (BA): average of sensitivity and specificity.
Matthews correlation coefficient (MCC): a coefficient of +1 represents a perfect prediction, 0 an average random prediction, and −1 an inverse prediction (Manganelli et al., 2018).
Youden’s J statistic (also called Youden’s index): sum between specificity and sensitivity minus 1. It is a single statistic summarizing the performance of a dichotomous diagnostic test. It ranges from −1 to 1, and has a zero value for when there is the same proportion of positive results for groups with and without the disease, i.e., the test is useless. A value of 1 indicates that there are no false positives or false negatives, i.e., the test is perfect.
MCC, BA, and Youden’s index are generally regarded as balanced measures, which can be used even for classes of very different sizes (Powers, 2011)
3. Results and discussion
Here we compare the performance and predictive ability of the classification models on the CoMPARA data set. This included an initial list of 1689 chemicals structures, then duplicates and chemicals identified by the EPA as potentially false negatives based on a bootstrapping study (Watt and Judson, 2018) were removed, obtaining a final list of 1667 compounds (active = 198 and inactive = 1469).
Table 1 lists the model performance for the SVM, ANN and DT models on the training and test sets, which were used in their building and validation processes. The SVM model showed the lowest sensitivity compared to ANN and DT models. Thus, it predicts a larger percent of false negatives. Confusion matrices for the SVM and ANN classification models developed are shown in Fig. 2, Fig. 3, respectively. As previously explained, SARpy models were built using the full data set of 1667 chemicals, as reported in Table 2. It allows having a larger chemical diversity to automatically extract statistically significant fragments, to increase the applicability domain of the model and the prediction coverage, since the dataset is highly unbalanced and the ruleset extraction does not requires too much optimization procedures.
Table 1.
ANNs, SVM and DT classification model selection methods and statistical performance. For ANN and SVM classification models Kohonen Self-Organizing Map was used to select the test set, while for the DT developed in R, a random splitting in training and test set was performed. To select best descriptors of ADMET models, the method of sensitivity analysis (SA) was the truncated linear analysis (TLA) implemented in ADMET Predictor. For DT, a stepwise variable selection using a linear discriminant analysis (LDA) and a bootstrap technique (based on a balanced resampling) for validation was performed to select the best variables.
| Models | Test set selection method | SA method | No. D | Sensitivity (%) (Training/test set) |
Specificity (%) (Training/test set) |
Balance Accuracy (%) (Training/test set) |
MCC (%) (Training/test set) |
Youden (%) (Training/test set) |
False rate (%) (Training/test set) |
|---|---|---|---|---|---|---|---|---|---|
| ANN | Kohonen | TLA | 75 | 91/71 | 92/98 | 92/85 | 68/76 | 82/69 | 9/6 |
| SVM | Kohonen | TLA | 45 | 66/51 | 100/100 | 83/76 | 77/69 | 66/51 | 4/7 |
| DT | Random | Stepwise | 5 | 86/79 | 81/80 | 83/80 | 49/43 | 67/60 | 18/20 |
Figure 2.
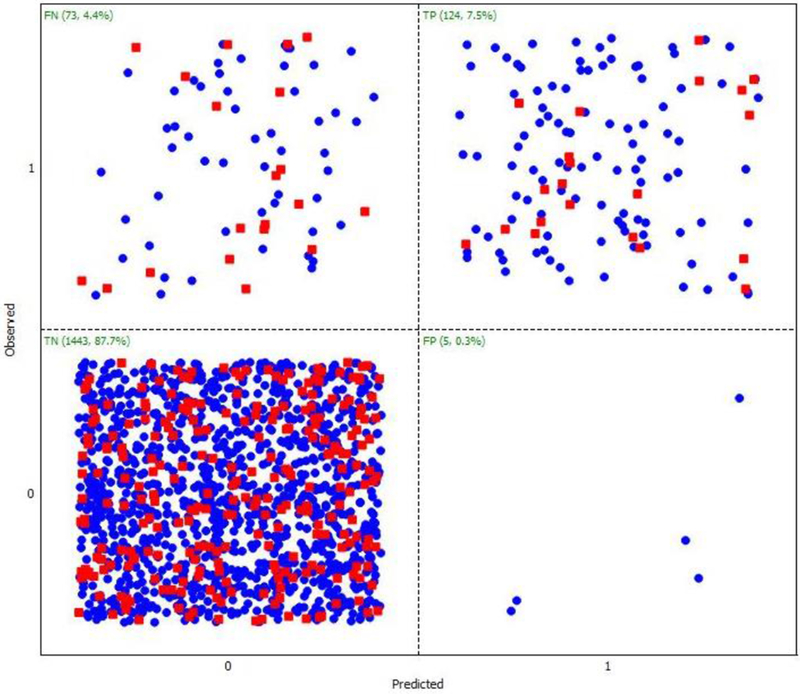
Statistical performance results for the Support Vector Machine (SVM) model using the ADMET Predictor.
Figure 3.
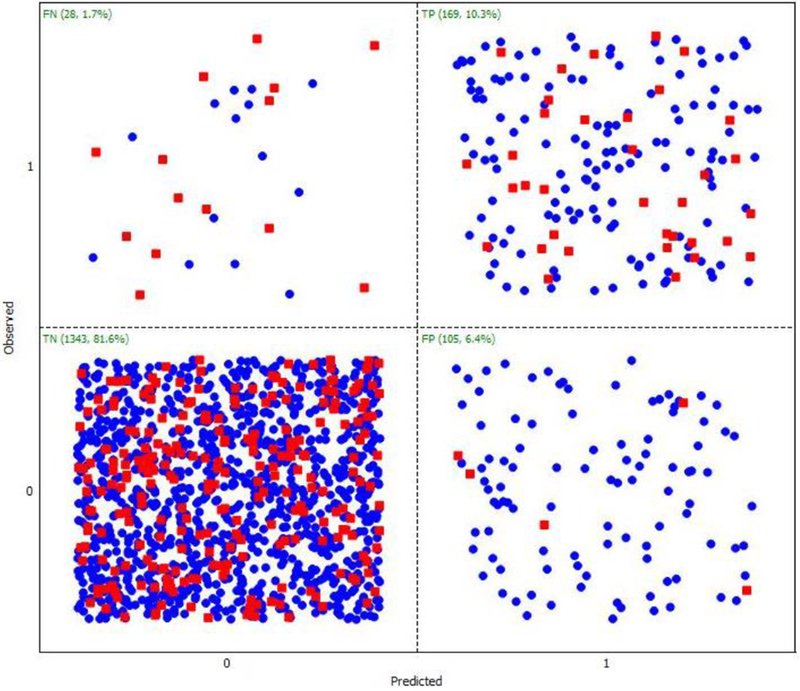
Statistical performance results for the Artificial Neural Networks (ANNs) model using the ADMET Predictor.
Table 2.
Statistical results of individual classification models and the two consensus models on the training set of 1667 chemicals. True Positives (TP) indicates the number of correctly classified positive chemicals, False Negatives (FN) indicates the number of misclassified positive chemicals, True Negative (TN) is the number of correctly classified positive chemicals, False positive (FP) refers to the misclassified experimentally negative chemicals. False rate is the fraction of incorrect predictions. Statistical parameters are the same as reported in Table 1.
| SVM | ANN | DT | SARpy1 | SARpy2 | Consensus I | Consensus II | |
|---|---|---|---|---|---|---|---|
| TP | 128 | 174 | 167 | 154 | 154 | 106 | 165 |
| FN | 68 | 22 | 31 | 44 | 44 | 11 | 32 |
| FP | 9 | 111 | 281 | 59 | 141 | 7 | 52 |
| TN | 1436 | 1334 | 1188 | 1410 | 1328 | 1113 | 1418 |
| Total | 1641 | 1641 | 1667 | 1667 | 1667 | 1237 | 1667 |
| accuracy | 0.95 | 0.92 | 0.81 | 0.94 | 0.89 | 0.99 | 0.95 |
| specificity | 0.99 | 0.92 | 0.81 | 0.96 | 0.90 | 0.99 | 0.96 |
| sensitivity | 0.65 | 0.89 | 0.84 | 0.78 | 0.78 | 0.91 | 0.84 |
| MCC | 0.76 | 0.69 | 0.48 | 0.71 | 0.58 | 0.91 | 0.77 |
| BA | 0.82 | 0.91 | 0.83 | 0.87 | 0.84 | 0.95 | 0.90 |
| False rate | 0.05 | 0.08 | 0.19 | 0.06 | 0.11 | 0.01 | 0.05 |
| Youden | 0.65 | 0.81 | 0.65 | 0.74 | 0.68 | 0.90 | 0.80 |
Table 2 and Table 3 show statistical results of the consensus models compared to the individual classifications models on overall data set of 1,667 chemicals (Table 2) and on the external set of 50 chemicals respectively (Table 3).
Table 3:
Statistical results of individual classification models and the two consensus models on the external set of 50 chemicals True Positives (TP) indicates the number of correctly classified positive chemicals, False Negatives (FN) indicates the number of misclassified positive chemicals, True Negative (TN) is the number of correctly classified negative chemicals, False positive (FP) refers to the misclassified experimentally negative chemicals. False rate is the fraction of incorrect predictions. Statistical parameters are the same as reported in Table 1.
| SVM | ANN | DT | SARpy1 | SARpy2 | Consensus I | Consensus II | |
|---|---|---|---|---|---|---|---|
| TP | 8 | 15 | 15 | 14 | 15 | 7 | 15 |
| FN | 7 | 0 | 0 | 1 | 0 | 0 | 0 |
| FP | 8 | 11 | 18 | 12 | 16 | 6 | 11 |
| TN | 25 | 22 | 17 | 23 | 19 | 13 | 24 |
| Total | 48 | 48 | 50 | 50 | 50 | 26 | 50 |
| accuracy | 0.69 | 0.77 | 0.64 | 0.74 | 0.68 | 0.77 | 0.78 |
| specificity | 0.76 | 0.67 | 0.49 | 0.66 | 0.54 | 0.68 | 0.69 |
| sensitivity | 0.53 | 1.00 | 1.00 | 0.93 | 1.00 | 1.00 | 1.00 |
| MCC | 0.29 | 0.62 | 0.47 | 0.54 | 0.51 | 0.61 | 0.63 |
| BA | 0.65 | 0.83 | 0.74 | 0.80 | 0.77 | 0.84 | 0.84 |
| False rate | 0.31 | 0.23 | 0.36 | 0.26 | 0.32 | 0.23 | 0.22 |
| Youden | 0.29 | 0.67 | 0.49 | 0.59 | 0.54 | 0.68 | 0.69 |
The consensus approach, which uses the unanimity criterion, clearly shows very high performance on the training set. This consensus model shows the highest values for all the considered parameters. All statistics were greater than 0.90 (with accuracy and specificity equal to 0.99), and false rate equal to 1%. However, the number of chemicals predicted with this model is the lowest: models gave convergent predictions for only 1,237 out of 1,667 chemicals, corresponding to 73% of data set.
The consensus model II shows results are not significantly different from those obtained with the individual models. It shows the second highest accuracy (0.95) together with SVM, and MCC (0.77) values, after the consensus I, but for the other statistical parameters there are other models with better values. ANN performed better in terms of sensitivity (0.89) and balanced accuracy (0.91). Prediction coverage for the ADMET Predictor models (ANN and SVM) was lower than for consensus II.
SVM gave the highest specificity (0.99), but the lowest sensitivity (0.65) and Youden index (0.65). The DT model had the lowest but still good or acceptable values of most statistical parameters. Nonetheless, it gave the second highest sensitivity (0.84) and a BA (0.83) close to the BA value of SVM (0.82). The SARpy model 1 performs better than SARpy 2 for all statistical parameters except for sensitivity, which is equal to 0.78 in both the models. SARpy 1 has very high accuracy and specificity, 0.94 and 0.96 respectively.
However, the comparison of statistical performance should be undertaken considering that the SVM, DT and ANN models were built after splitting the 1,667 chemicals into different sets for training and external validation; conversely, for SARpy models the whole CoMPARA data set was used as training set.
A quite different picture appears from the results on the EDKB prediction set (Table 3). This set is quite small (50 compounds), in contrast to the CoMPARA data set. Since this data set was part of the exercise carried out by the tens of laboratories participating to the CoMPARA exercise, we preferred to adhere to the common scheme of the exercise and use the CoMPARA set of compounds to build (and validate) the single models. Furthermore, the source of the data is different, and this may cause inhomogeneous values. Figure 1 shows there are a few CoMPARA compounds that are active but have low RBA values (log RBA<−3). Table 3 shows that consensus II appears to be the slightly better than consensus I, which also only predicts half of the data set. The ANN and SVM models are not able to predict two chemicals containing Si, which are out of their scope. Conversely, all 50 chemicals are matched by SARpy rules indicating they are within the scope of SARpy models. Also, they fall in the applicability domain of the DT model based on their descriptors range. ANN has statistics comparable to consensus II but it is unable to predict two chemicals containing Si. SARpy 1 follows it. SVM shows the lowest values for many statistical parameters (e.g., MCC and Youden equal to 0.29). Due to the low number of active compounds, the number of false negatives is quite low or null, and thus sensitivity values are high for all models except for SVM. The results of this and the other consensus models are affected by the results of the individual models. If we compare the results of the individual models on the training and test sets (as from Table 2, Table 3), we notice that ANN provides stable results. Compared to the statistical values obtained on the training set, the ANN and consensus II models are still satisfactory.
The other individual models show larger reductions of the performance going from training to external testing, which may indicate overtraining.
We notice that the MCC values are still quite good for all models, except for SVM. Indeed, the values of about 0.5 or higher as in the present case demonstrate that the models are predictive.
If we summarize the results from the training and test sets, in the present case the consensus between different models prove to be superior to the individual models in terms of statistics and prediction coverage. This has been reported also in other cases (Manganelli et al., 2016; Manganelli et al., 2018); DT has the lowest specificity (0.49). DT has a major advantage: since it is based on a very limited subset of descriptors (five), it guarantees easy interpretability of the model. The same reasoning can be extended to SARpy models, which are based on structural motifs, which can be involved in the activation of AR.
A deeper analysis of chemicals which were misclassified by all models (consensus I) in the two sets allowed an analysis of prediction errors related to specific chemical classes.
Misclassified chemicals in the training set included seven false positives and eleven false negatives with prominently different chemical structures. The false negatives showed a variety of chemical classes and could not be grouped based on their structure.
Misclassified chemicals in the training set included two false positives predictions for p, p’-DDT and methoxychlor (Table 4) which share the (2,2,2,-trichloro-1-phenylethyl)benzene moiety. Both are organochlorine chiral pesticide chemicals that undergo enantioselective biotransformation in vivo. p,p’-DDE is an important metabolite of p,p’-DDT, which is known to exhibit weak anti-androgenic properties (ATSDR 2002). The isomer o,p’-DDT is estrogenic. On the other hand, metabolites of methoxychlor, mono-demethylated and di-demethylated methoxychlor derivatives, are weakly estrogenic (Aoyama and Chapin 2014). Their dehydochlorinated product also exhibits estrogenicity. Similarly to p, p’-DDE and methoxychlor, the 4,4′-methylenebis(2,6-di-t-butylphenol) and phenol red (Table 4) share the common feature of two benzene rings connected by a carbon (diphenylmethane), on the other hand, SR125047 has two benzene rings joined directly, these chemicals show a similar hydrophobic core and terminal polar group. These four chemicals share similar structural features to well known androgen receptor binders such as bisphenol A derivatives, DES, PCBs and organochlorinated pesticides. Several authors have studied the structural requirements for chemical-receptor activity and showed the complexity involved in determining the mechanism of action of endocrine-active chemicals that simultaneously act as agonists or antagonists through one or more hormone receptors (De Coster et al., 2012; Minf et al., 2011; Vandenberg et al., 2012). Assessment of the in silico prediction of potential EDCs metabolites from a qualitative standpoint can substantially help in analysing the available metabolism data. Fast and reliable in silico predictions could accelerate the in vitro/in vivo characterization of EDCs metabolites. In silico tools could be used to explore or develop analysis of the potential endocrine disrupting effects of chemicals, the potential interference with the metabolism of endogenous hormones, and the prediction of metabolism of chemicals by phase I and II enzymes. One of the most frequently cited limitations of in vitro assays without metabolic capacity concerns the qualitative and quantitative deficiencies in the metabolism of test chemicals. The enantiomeric abundance of the DDT and methoxychlor metabolites depends on the relative catalytic activity of hepatic CYP450 isoforms. Reported evidence suggests that consideration of gastrointestinal microbiota metabolism of EDCs is important to fully evaluate the toxicity of environmental contaminants (Velmurugan et al., 2017).
Table 4:
Consensus I model misclassified seven chemicals in the training set as false positive. These chemicals were experimentally negative but were predicted as positive by all the in silico models.
| CAS | Name | Structure |
|---|---|---|
| NOCAS_47342 | SR125047 |  |
| 52-01-7 | Spironolactone | 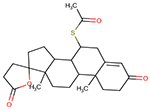 |
| 72-43-5 | Methoxychlor | 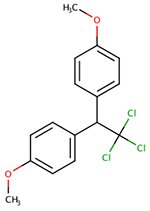 |
| 50-29-3 | p,p’-DDT | 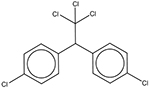 |
| 1434-54-4 | Pregnenolone carbonitrile | 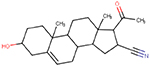 |
| 118-82-1 | 4,4′-Methylenebis(2,6-di-t-butylphenol) |  |
| 143-74-8 |
Phenol red |
 |
Other misclassified chemicals in the training sets as false positive were spironolactone, a steroid-based diuretic that acts by antagonism of aldosterone, and pregnenolone carbonitrile, a steroidal anti-glucocorticoid and pregnane X receptor agonist (Table 4). These chemicals share a steroid nucleus core consisting of three cyclohexane and one cyclopentane, as the androgens such as testosterone and dihydrotestosterone. In addition, the six false positives in the external sets: cholesterol, aldosterone, beta-sitosterol, pregnenolone, prednisolone, and ICI 164384 (Table 5) shared a steroid nucleus. These evidences suggested the models might encounter problems when predicting this chemical class. The misclassification may also be due to the different experimental assay used for EDBK compared to CoMPARA data. The mechanism of action of androgenicity is a complex process. A series of in vitro assays have been developed for the detection of potential androgens along the pathway of this mechanism. Most of these assays fall into one of three categories: a) AR competitive binding assays that measure the binding affinity of a chemical for AR; b) reporter gene assays that measure AR binding-dependent transcriptional and translational activity; and c) cell proliferation assays that measure the increase in cell number of target cells during the exponential phase of proliferation. Thus, these assays function at different levels of biologic complexity.
Table 5:
Consensus I model misclassified six chemicals in the external set as false positive. These chemicals were experimentally negative but were predicted as positive by all the in silico models.
| CAS | Name | Structure |
|---|---|---|
| 57-88-5 | Cholesterol | |
| 52-39-1 | Aldosterone |  |
| 83-46-5 | beta-Sitosterol |  |
| 145-13-1 | Pregnenolone |  |
| 50-24-8 | Prednisolone |  |
| 98007-99-9 | ICI 164384 |  |
ToxCast dataset experimentally tested 1855 chemicals as part of their 11 assays addressing androgenic activity at different pathway levels, then these were combined by a mathematical model to yield a unique AUC score for AR activity (Judson et al., 2015, Kleinstreuer et al., 2017). For the CoMPARA efforts, an initial list of 1689 chemicals was generated, then duplicate structures and twenty-one chemicals identified by the EPA as potentially false negatives based on a bootstrapping study (Watt and Judson, 2018) were removed, obtaining a final list of 1667 compounds. To evaluate our classification models, we applied them to predict the LogRBA for the FDA database (Fang et al., 2003) which limits our analysis to binding effects due to data availability. The two databases therefore, in their definition (AR overall activity and AR binding), are somehow different but still show some kind of agreement (that could be due to the relative importance given to the binding process in the weighting of all the assays). The CoMPARA data from the AR pathway model were used as the training set for binding activity (positive/negative). This data relates more to overall AR pathway activity, which includes binding but also covers all of the other downstream key events. Therefore, it is entirely possible that compounds that were missed by the binding assay but hit by all the other assays in the pathway were included in the training set as positives. We observed something similar when analysing CERAPP data addressing estrogenic pathway with 17 assays: a previously developed model for receptor binding affinity implemented in the VEGA platform was giving acceptable results also when used to predict this type of more complex output. A possible solution to rectify this issue may be the refinement of SA identified by SARpy related to this moiety, to make them more specific and able to discriminate within the steroid-based structures. It is also important to establish differences between moieties such as two aromatic rings that share a pair of carbon atoms (fused) versus benzene rings joined directly or benzene rings connected by carbon, ketone, or oxygen.
Due to the large number of EDCs and the limited understanding of their dose-response relationship for risk assessment, further research addressing these data gaps are required. This can be achieved through strategic laboratory and experimental studies. Consequently, the data generated will be used for the development of 2D, 3D QSAR models, docking and homology models that are required to have an appropriate range of similar structures and toxicity endpoints. Computational modeling and experimental approaches complement each other and help develop useful tools for risk assessment of these chemicals and other emerging chemicals that lack toxicity data. In silico models could be further evaluated, demonstrated and, if needed, refined using in vitro data that is being generated through high throughput and/or genomic studies. These in silico models could be used in the prioritization, hazard identification and dose–response assessment process of the 4-step risk assessment of environmental chemicals that lack appropriate data. In silico models could play a critical role in toxicology and public health as scientists move forward with modernizing and streamlining the risk assessment process and applying the findings of such risk assessments to public health
4. Conclusions.
The possibility to predict toxicity properties of chemical substances is increasing, due to the fruitful exploitation of collections of experimental data. In the present study, we used a large collection of data related to androgen receptor binding activity. A number of models have been developed, using different algorithms such as ANN, SVM, DT and structural alerts. These in silico models were internally and externally validated, and the analyses presented above support the transparency, robustness and reliability of the androgen binding predictions. Therefore, chemicals can be assessed for their potential androgenic toxicity. Useful results have been obtained with some variations among the models. The use of consensus approaches, through the unanimity or majority vote strategy, proved to be superior on the individual models for most statistical parameters and in terms of prediction coverage in the majority vote-based approach. The consensus II approach shows slightly better results than the best single in silico model for the external set.
There is a need to prioritize testing resources due to the large number of chemicals in commerce that may not impart endocrine toxicity. The appropriateness of the use of a single method or approach alone to address a complex toxicological endpoint has been evaluated against the use of multiple or integrated approaches as a weight of evidence. This study integrates in silico models and in vitro androgen binding tests as alternative methods of choice to evaluate the safety of data-poor chemicals in commerce.
Footnotes
Publisher's Disclaimer: Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the views or policies of the U.S. Environmental Protection Agency. The findings and conclusions in this abstract have not been formally disseminated by [the Centers for Disease Control and Prevention/the Agency for Toxic Substances and Disease Registry] and should not be construed to represent any agency determination or policy.
References
- Aoyama H, Chapin RE. 2014. Reproductive toxicities of methoxychlor based on estrogenic properties of the compound and its estrogenic metabolite, hydroxyphenyltrichloroethane. Vitam Horm 94:193–210. [DOI] [PubMed] [Google Scholar]
- ATSDR, 2002 ATSDR. Agency for toxic substances and disease registry 2002. Toxicological profile for DDT, DDE, and DDD. [PubMed] [Google Scholar]
- Bergman A, Heindel JJ, Kasten T, Kidd KA, Jobling S, Neira M, Zoeller RT, Becher G, Bjerregaard P, Bornman R and others. 2013. The impact of endocrine disruption: a consensus statement on the state of the science. Environ Health Perspect 121(4):A104–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold MR et al. (2008) KNIME: The Konstanz Information Miner In: Preisach C, Burkhardt H, Schmidt-Thieme L, Decker R (eds) Data Analysis, Machine Learning and Applications. Studies in Classification, Data Analysis, and Knowledge Organization. Springer, Berlin, Heidelberg [Google Scholar]
- De Coster S, van Larebeke N. 2012. Endocrine-disrupting chemicals: associated disorders and mechanisms of action. J Environ Public Health 2012:713696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix DJ, Houck KA, Martin MT, Richard AM, Setzer RW, Kavlock RJ. 2007. The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol Sci 95(1):5–12. [DOI] [PubMed] [Google Scholar]
- Fang H, Tong W, Branham WS, Moland CL, Dial SL, Hong H, Xie Q, Perkins R, Owens W, Sheehan DM. 2003. Study of 202 natural, synthetic, and environmental chemicals for binding to the androgen receptor. Chem Res Toxicol 16(10):1338–58. [DOI] [PubMed] [Google Scholar]
- Ferrari T, Cattaneo D, Gini G, Golbamaki Bakhtyari N, Manganaro A, Benfenati E. 2013. Automatic knowledge extraction from chemical structures: the case of mutagenicity prediction. SAR QSAR Environ Res 24(5):365–83. [DOI] [PubMed] [Google Scholar]
- Judson RS, Magpantay FM, Chickarmane V, Haskell C, Tania N, Taylor J, Xia M, Huang R, Rotroff DM, Filer DL and others. 2015. Integrated Model of Chemical Perturbations of a Biological Pathway Using 18 In Vitro High-Throughput Screening Assays for the Estrogen Receptor. Toxicol Sci 148(1):137–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavlock R, Chandler K, Houck K, Hunter S, Judson R, Kleinstreuer N, Knudsen T, Martin M, Padilla S, Reif D and others. 2012. Update on EPA’s ToxCast program: providing high throughput decision support tools for chemical risk management. Chem Res Toxicol 25(7):1287–302. [DOI] [PubMed] [Google Scholar]
- Kleinstreuer NC, Ceger P, Watt ED, Martin M, Houck K, Browne P, Thomas RS, Casey WM, Dix DJ, Allen D and others. 2017. Development and Validation of a Computational Model for Androgen Receptor Activity. Chem Res Toxicol 30(4):946–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kode srl, Dragon (software for molecular descriptor calculation) version 7.0.8, 2017, https://chm.kode-solutions.net
- Kohonen T (1982) Self-Organized Formation of Topologically Correct Feature Maps. Biol Cybern 43:59–69. [Google Scholar]
- Li J, Gramatica P. 2010. Classification and virtual screening of androgen receptor antagonists. J Chem Inf Model 50(5):861–74. [DOI] [PubMed] [Google Scholar]
- Piparo Lo, Elena and Andrew Worth. “Review of QSAR models and software tools for predicting developmental and reproductive toxicity.” JRC Scientific and Technical Reports EUR 24522 (2010) [Google Scholar]
- Long JS (1997). Regression Models for Categorical and Limited Dependent Variables. Thousand Oaks, CA: Sage Publications. [Google Scholar]
- Manganelli S, Benfenati E, Manganaro A, Kulkarni S, Barton-Maclaren TS, Honma M. 2016. New Quantitative Structure-Activity Relationship Models Improve Predictability of Ames Mutagenicity for Aromatic Azo Compounds. Toxicol Sci 153(2):316–26. [DOI] [PubMed] [Google Scholar]
- Manganelli S, Schilter B, Benfenati E, Manganaro A, Lo Piparo E. 2017. Integrated strategy for mutagenicity prediction applied to food contact chemicals. ALTEX. [DOI] [PubMed] [Google Scholar]
- Mansouri K, Abdelaziz A, Rybacka A, Roncaglioni A, Tropsha A, Varnek A, Zakharov A, Worth A, Richard AM, Grulke CM and others. 2016. CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ Health Perspect 124(7):1023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mnif W, Hassine AI, Bouaziz A, Bartegi A, Thomas O, Roig B. 2011. Effect of endocrine disruptor pesticides: a review. Int J Environ Res Public Health 8(6):2265–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2015). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: URL https://www.R-project.org/ [Google Scholar]
- Richard AM, Judson RS, Houck KA, Grulke CM, Volarath P, Thillainadarajah I, Yang C, Rathman J, Martin MT, Wambaugh JF and others. 2016. ToxCast Chemical Landscape: Paving the Road to 21st Century Toxicology. Chem Res Toxicol 29(8):1225–51. [DOI] [PubMed] [Google Scholar]
- Roncaglioni A, Piclin N, Pintore M, Benfenati E. 2008. Binary classification models for endocrine disrupter effects mediated through the estrogen receptor. SAR QSAR Environ Res 19(7–8):697–733. [DOI] [PubMed] [Google Scholar]
- Ruiz P, Sack A, Wampole M, Bobst S, Vracko M. 2017. Integration of in silico methods and computational systems biology to explore endocrine-disrupting chemical binding with nuclear hormone receptors. Chemosphere 178:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahigara F, Mansouri K, Ballabio D, Mauri A, Consonni V, Todeschini R. 2012. Comparison of different approaches to define the applicability domain of QSAR models. Molecules 17(5):4791–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simulations Plus Inc. (2015), Available: http://www.simulations-plus.com/ [accessed 21 April 2017].
- Sonnenschein C, Soto AM. 1998. An updated review of environmental estrogen and androgen mimics and antagonists. J Steroid Biochem Mol Biol 65(1–6):143–50. [DOI] [PubMed] [Google Scholar]
- Therneau Terry M. and Atkinson Elizabeth J. An introduction to recursive partitioning using the RPART routines. Rochester: Mayo Foundation; (2015). [Google Scholar]
- Tice RR, Austin CP, Kavlock RJ, Bucher JR. 2013. Improving the human hazard characterization of chemicals: a Tox21 update. Environ Health Perspect 121(7):756–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trisciuzzi D, Alberga D, Mansouri K, Judson R, Cellamare S, Catto M, Carotti A, Benfenati E, Novellino E, Mangiatordi GF and others. 2015. Docking-based classification models for exploratory toxicology studies on high-quality estrogenic experimental data. Future Med Chem 7(14):1921–36. [DOI] [PubMed] [Google Scholar]
- Trisciuzzi D, Alberga D, Mansouri K, Judson R, Cellamare S, Catto M, Carotti A, Benfenati E, Novellino E, Mangiatordi GF and others. 2015. Docking-based classification models for exploratory toxicology studies on high-quality estrogenic experimental data. Future Med Chem 7(14):1921–36. [DOI] [PubMed] [Google Scholar]
- Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR Jr., Lee DH, Shioda T, Soto AM, vom Saal FS, Welshons WV and others. 2012. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev 33(3):378–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youden WJ. 1950. Index for rating diagnostic tests. Cancer 3(1):32–5. [DOI] [PubMed] [Google Scholar]
- Zupan J; Gasteiger J Neural Networks in Chemistry and Drug Design, 2nd ed; Wiley-VCH: Weinheim, Germany, 1999; p 380. [Google Scholar]