

Abstract
PURPOSE
We developed a precision medicine program for patients with advanced cancer using integrative whole-exome sequencing and transcriptome analysis.
PATIENTS AND METHODS
Five hundred fifteen patients with locally advanced/metastatic solid tumors were prospectively enrolled, and paired tumor/normal sequencing was performed. Seven hundred fifty-nine tumors from 515 patients were evaluated.
RESULTS
Most frequent tumor types were prostate (19.4%), brain (16.5%), bladder (15.4%), and kidney cancer (9.2%). Most frequently altered genes were TP53 (33%), CDKN2A (11%), APC (10%), KTM2D (8%), PTEN (8%), and BRCA2 (8%). Pathogenic germline alterations were present in 10.7% of patients, most frequently CHEK2 (1.9%), BRCA1 (1.5%), BRCA2 (1.5%), and MSH6 (1.4%). Novel gene fusions were identified, including a RBM47-CDK12 fusion in a metastatic prostate cancer sample. The rate of clinically relevant alterations was 39% by whole-exome sequencing, which was improved by 16% by adding RNA sequencing. In patients with more than one sequenced tumor sample (n = 146), 84.62% of actionable mutations were concordant.
CONCLUSION
Integrative analysis may uncover informative alterations for an advanced pan-cancer patient population. These alterations are consistent in spatially and temporally heterogeneous samples.
INTRODUCTION
Genomic profiling is widely used in cancer care to identify actionable alterations for individual patients within the context of precision medicine (PM).1 However, only 2% to 11% of those patients with sequencing performed receive a genomically matched therapy, which may be a result of the availability and accessibility of clinical trials,2-6 patient factors and comorbidities, disease state or alternative options, or patient preference.5
Despite these drawbacks, PM studies continue to provide insights into the molecular underpinnings of cancer. We and others have shown that performing whole-exome sequencing (WES) and RNA sequencing is feasible in a clinical setting and may provide relevant information beyond targeted gene panels in certain settings.3,7,8 Sequencing matched tumor and normal (germline) DNA has the additional benefit of uncovering unforeseen hereditary conditions, including cancer risk mutations. In particular, germline DNA repair defects (DRDs) are more common than previously anticipated across adult advanced cancer populations.9-11 Robinson et al12 published their MET500 cohort with WES and RNA sequencing data from 500 patients with metastatic disease of varied tumor primary and biopsy sites. Our study, obtained from our own cohort of 759 samples from 515 patients with advanced and metastatic cancer, complements this data set as it adds WES data from approximately the same number of patients. Of importance, our data add needed information about tumor evolution, clonality, and tumor heterogeneity by analyzing multiple samples obtained from individual patients.
PATIENTS AND METHODS
Patients with locally advanced and metastatic cancer were prospectively enrolled in an institutional review board (IRB)–approved PM Trial (IRB No. 1305013903) with written informed consent. The consent process included explaining the risk and potential consequences of tumor biopsy, somatic and germline sequencing, as well as offering the opportunity to participate in a rapid autopsy program.13 Tumor DNA for WES was obtained from fresh frozen or formalin-fixed, paraffin-embedded tissue. Seventy image-guided biopsies from prostate cancer bone metastases using an optimized bone biopsy protocol14 were performed. Frozen or formalin-fixed, paraffin-embedded tissue slides were evaluated by study pathologists for diagnosis and tumor cell content. Germline DNA was obtained from blood samples (circulating mononuclear cells), buccal swabs, or benign tissue as described previously.3 Part of the data presented in this manuscript have already been published.3,13,15
CONTEXT
Key objective
We assessed whether developing a multidimensional precision oncology program is feasible and informative for patients with cancer with advanced disease.
Knowledge generated
We established a comprehensive clinical genomics program for this patient group. The rate of clinically relevant alterations across 515 patients with advanced solid tumors was 39% by whole-exome sequencing, which was improved by 16% by adding RNA sequencing. Multisample analysis of individual patients revealed a concordance rate of clinically significant alterations of 84.62%.
Relevance
Multidimensional genomic analysis is feasible and informative in a clinical setting and can improve clinical care for some patients.
WES was performed on each patient’s tumor/matched germline DNA pair using previously described protocols.3 We used a clinical-grade WES test—Exome Cancer Test Version 1—approved by New York State Department of Health (ID# 43032) and described in detail in Rennert et al.16 This approach allows for assessment of more than 21,000 genes through the development and implementation of computational approaches for tumor mutational burden and neoepitope analysis, as well as integration with other data, including RNA sequencing, to improve the identification of clinically relevant and actionable alterations. RNA sequencing was performed on a subset of cases with sufficient fresh frozen tissue available. For details of RNA sequencing data analysis, see the Data Supplement. To evaluate the concordance of tier 1 and tier 2 alterations between multiple samples from the same patient and to gain high-fidelity results, a cutoff of 20% for variant allele frequency was used. In addition, we developed patient-derived organoids from fresh tissues using previously described protocols,4,17 and we used cell lines to functionally validate outlier targetable alterations.
WES alterations were categorized on the basis of on their actionability and clinical or biologic relevance.3 Alterations in 49 actionable or clinically significant genes were reported within Category 1, alterations in 508 known cancer-associated genes within Category 2, and somatic alterations of unknown significance within Category 3.3 We developed an open-access, dynamic, Web-based PM knowledge base as an interactive online tool where variants are carefully interpreted in the context of tumor type.18
WES results were conveyed to the referring physician in the form of an Exome Cancer Test Version 1 report.3 Selected cases are presented at a regular, continuing medical education–accredited PM tumor board, which discusses sequencing results in the context of a patient’s history, available literature, and treatment options, including active clinical trials.
Pathogenic germline findings were reported to the referring physician if they occurred in any of the genes deemed reportable by the American College of Medical Genetics and Genomics (ACMG),19 and these patients were referred for genetic counseling and results were confirmed by targeted testing in a Clinical Laboratory Improvement Amendments–/Clinical Laboratory Evaluation Program–certified laboratory.
Study cohort demographic data were obtained through electronic health record search. Ethnicity was inferred through a computational analysis (EthSEQ; https://cran.r-project.org/package=EthSEQ) of germline single-nucleotide polymorphisms.20
RESULTS
Patient Characteristics
Between February 2013 and December 2016, 515 patients were prospectively enrolled (Fig 1 and Data Supplement). The majority of patients presented with metastatic (n = 319; 62%) or recurrent (n = 46; 8.9%) disease. Of patients, 149 (28.9%) had primary tumors available for study analysis. Disease status was unknown for one patient (0.2%). Median number of prior systemic therapies before tissue evaluation was three (range, zero to 14). The majority of patients were of European (n = 231; 44.9%) or Ashkenazi (n = 165; 32.1%) heritage (EthSEQ data; Data Supplement), compatible with the location of the Englander Institute for Precision Medicine (Upper East Side of Manhattan, New York, NY).21
FIG 1.
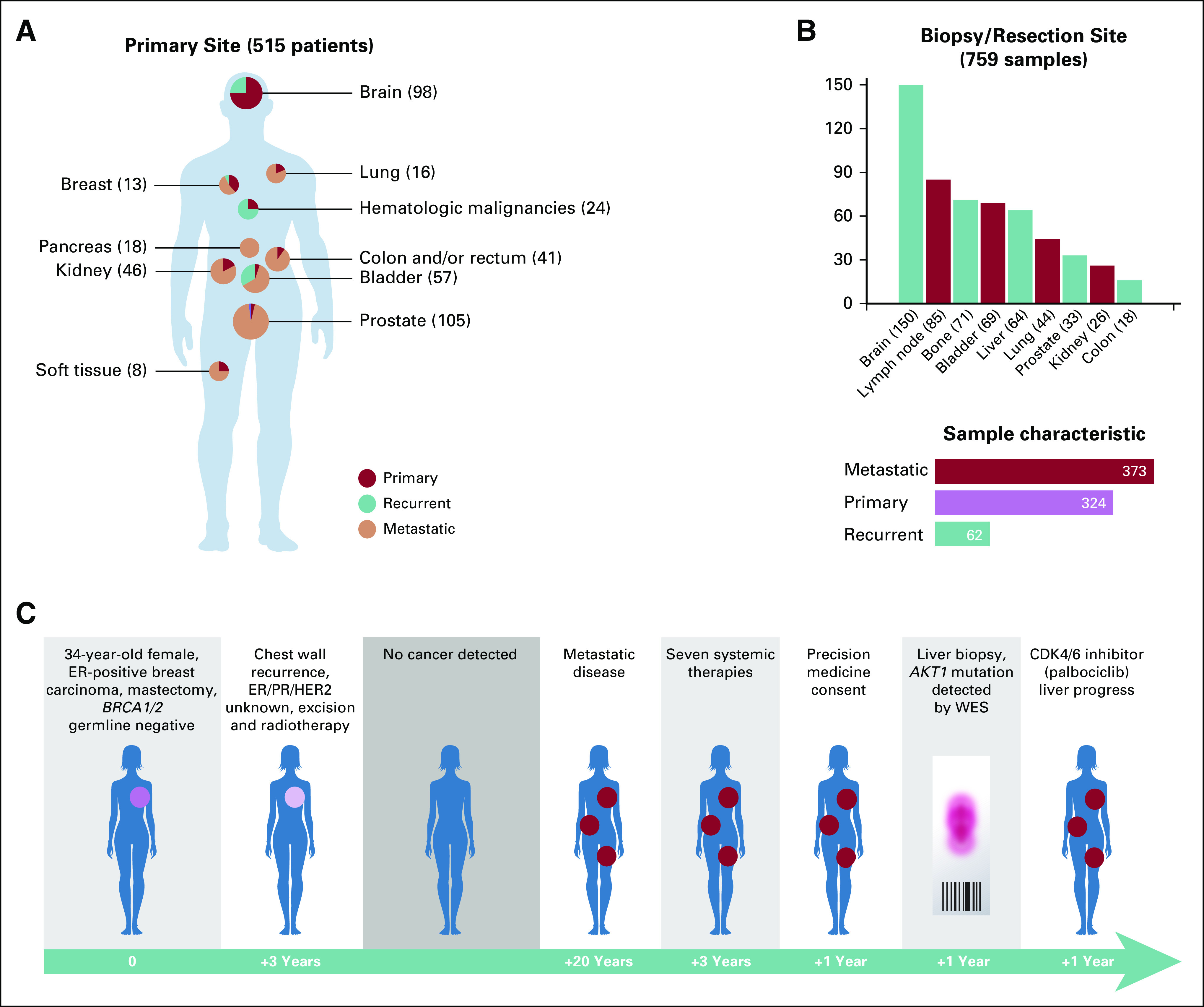
Selected primary tumor sites of 515 Englander Institute for Precision Medicine patients and patient status at time of enrollment. (A) The majority of patients with non-CNS tumors presented with metastatic disease. (B) Most common biopsy or resection sites and information, whether a sample was procured from a primary, metastatic, or recurrent site. (C) Clinical history of a female patient with breast cancer who participated in the precision medicine trial. An AKT1 mutation was discovered in a liver sample and she was treated with an AKT1 inhibitor, albeit without achieving disease remission. ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; PR, progesterone receptor; WES, whole-exome sequencing.
WES Results
Tier 1, 2, or 3 mutations were identified in 27.4%, 72.3%, and 95.1% of all samples, respectively. Tier 1, 2, or 3 somatic copy number alterations were found in 52.4%, 88.4%, and 100% of all samples, respectively. Figure 1C shows an example of a patient treated according to WES results. Mutation rate across tumor types ranged from zero to 37 mutations per megabase, with a mean of 1.2 mutations/Mb. The highest mutation rates were observed in tumors of the colon/rectum and bladder. Copy number burden (megabase altered by copy number variation) ranged from zero to 2,098 per sample (mean, 155.59 Mb). Higher somatic copy number alteration (SCNA) values were found in samples from endometrial and colorectal primary tumors. The most common somatic alterations were found in TP53 (33%), CDKN2A (11%), APC (10%), KMT2D (8%), PTEN (8%), and BRCA2 (7%; Fig 2).
FIG 2.

Landscape of genomic alterations in the Englander Institute for Precision Medicine cohort. The most common alterations are nonsynonymous TP53 mutations (Category 2). This alteration is found often in advanced prostate cancer, bladder cancer, and brain tumors. Amp, amplification; Del, deletion; Ins, insertion.
Multiple Samples
Three hundred eighty-two spatially and temporally heterogeneous samples from 146 patients underwent WES. Of these, 185 (48.4%) were metastatic, 153 (40.1%) were primary, and 44 (11.5%) were recurrent tumor samples. Most samples were paired primary and metastatic tumors, and concordance of alterations between primary and metastatic samples in clinically informative genes is shown in Figure 3. The primary tumor was sequenced at a different timepoint in 29 patients—for example, before and after systemic therapy. Employing a variant allele frequency cutoff of 20% for genomic alterations, multiple samples from 132 patients could be evaluated. Of Category 1 and 2 alterations, 84.62% and 85.75%, respectively, of mutations and SCNA variations were shared.
FIG 3.
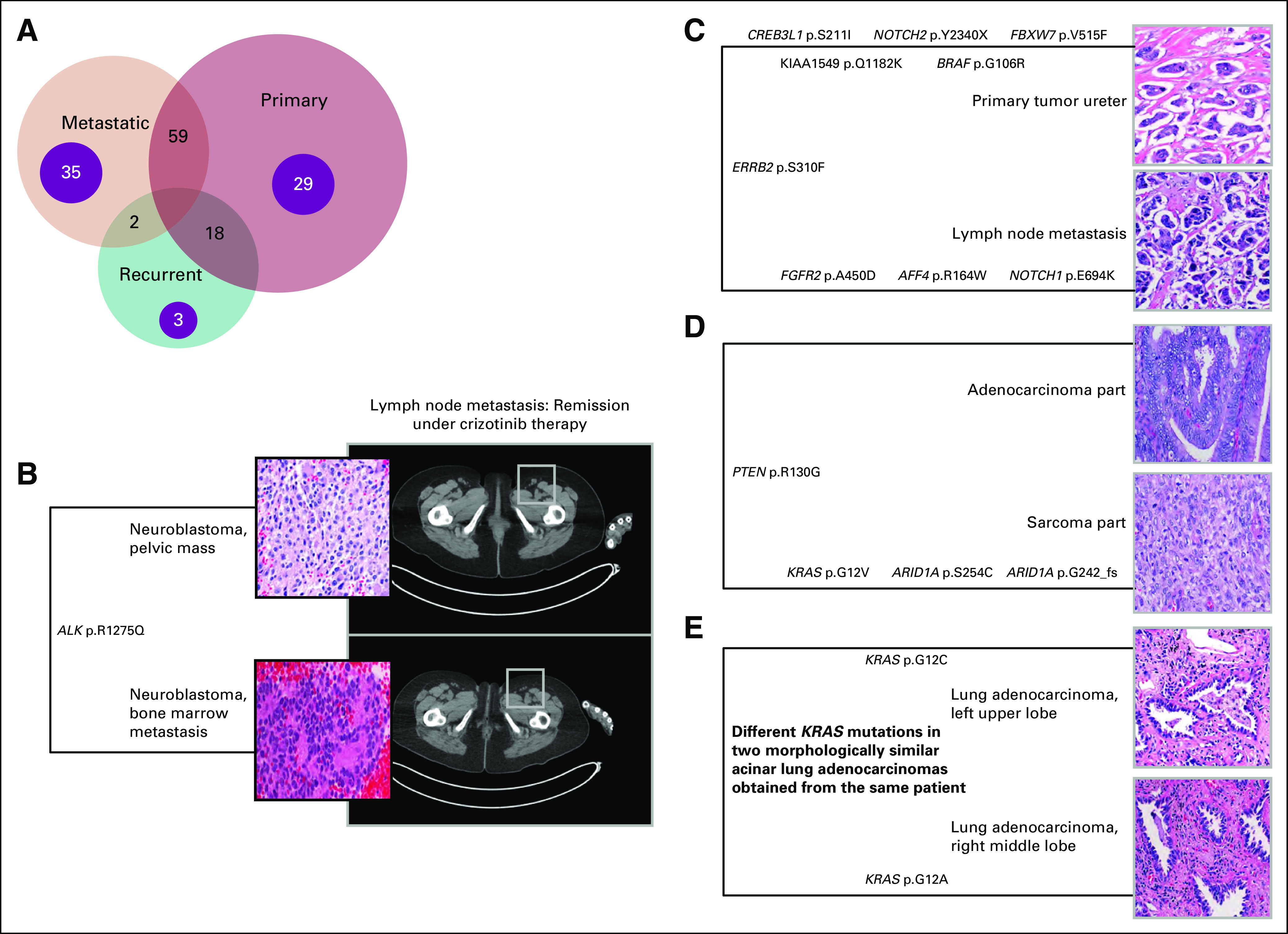
Multiple samples from same patients enrolled in the precision medicine trial at the Englander Institute for Precision Medicine (EIPM) and clinically relevant examples. (A) Venn Diagram showing the relationship of multiple tissue samples from individual patients. Multiple samples from 146 individuals in the EIPM cohort underwent whole-exome sequencing. Paired primary–metastatic tumor samples from 59 patients were the most frequent combination. Spatially and/or temporally heterogeneous metastases and primary tumors from 35 and 29 patients, respectively, also underwent sequencing. Primary tumors were usually sequenced at different timepoints—for example, tumor samples at initial diagnosis and subsequent residual tumor postneoadjuvant treatment. (B) Activating ALK mutation in both primary tumor and bone marrow metastasis; partial remission after crizotinib therapy. (C) Shared and potentially actionable mutation in the ERBB gene in both primary tumor and lymph node metastasis of an individual with micropapillary urothelial carcinoma. (D) Early divergent evolution in both histologically distinct epithelial and mesenchymal components in an individual with uterine carcinosarcoma. (E) Different KRAS mutations in two morphologically similar lung cancer samples from the same patient, thus confirming two separate primary tumors.
Germline Findings
WES of germline DNA was performed in all patients to filter single-nucleotide variants and SCNAs that are only present in the tumor (Fig 4). Sequencing germline DNA also serves as an internal quality control to determine whether both samples truly originate from the same patient.22 The ClinVar and ExAC databases are our main sources for germline variant evaluation.23,24 Variants are reported in an internal report using a five-tiered system as recommended by the ACMG and the Association for Molecular Pathology.19 Deleterious germline DRDs involving 12 genes were identified in 55 patients, comprising 10.7% of patients in this cohort (Data Supplement). The most frequently mutated genes were CHEK2 (11 patients), BRCA2 (nine patients), BRCA1 (nine patients), MSH6 (nine patients), and ATM (four patients; Fig 2). Twelve patients had additional loss of function in the other allele. Positive germline findings were confirmed by targeted sequencing for 53 patients. In addition, 44 additional likely pathogenic variants in 38 patients in MSH6 (14 cases); APC (seven cases); CHEK2 (five cases); POLE, PMS2, MSH2 BRCA1, and ATM (three cases each); and TP53, CDH1, and BRIP1 (one case each) were discovered in our cohort.
FIG 4.
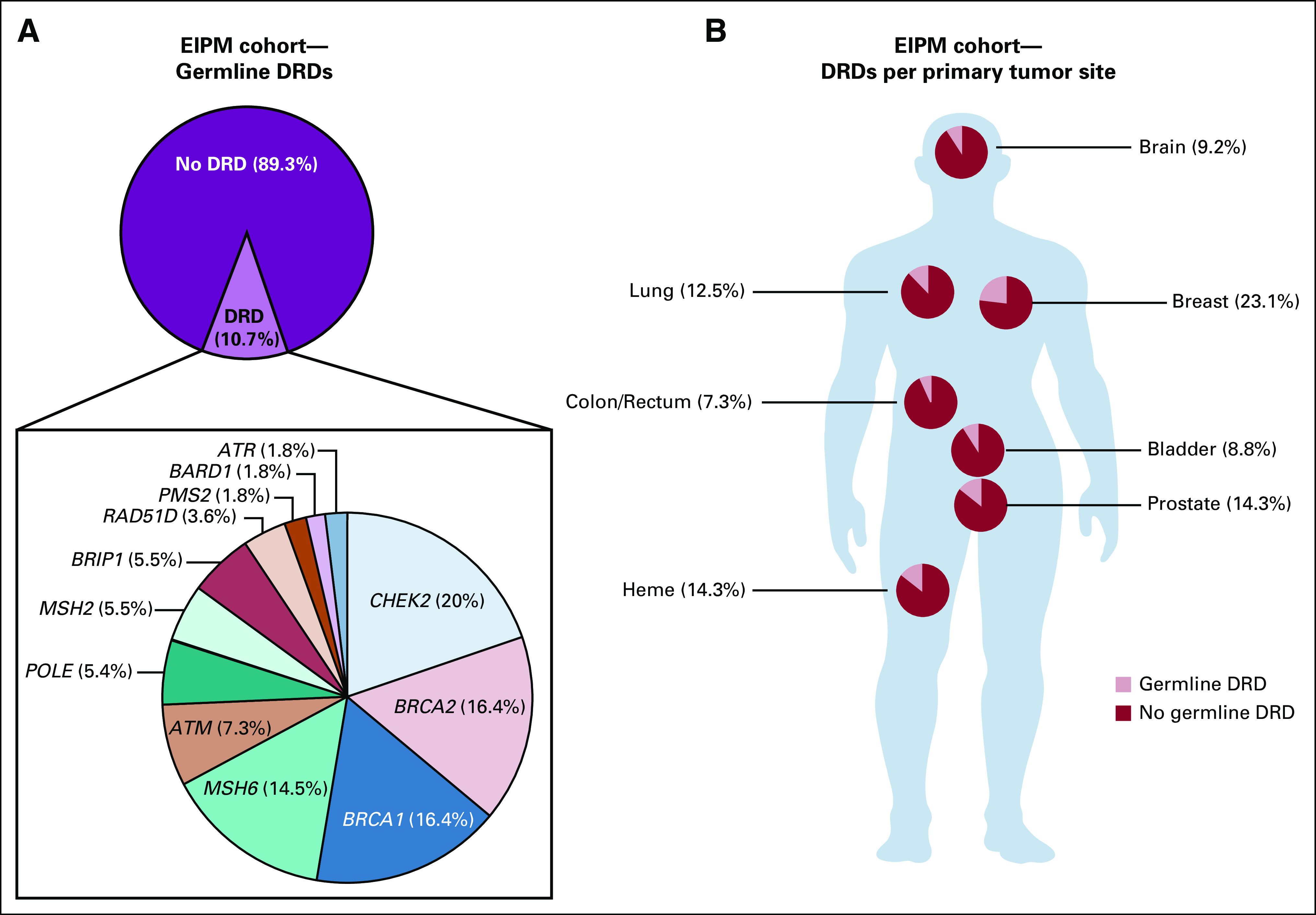
Germline DNA-repair defects (DRDs) in Englander Institute for Precision Medicine (EIPM) cohort. (A) The frequency of germline DRD alterations in our cohort is 10.7%. CHEK2, BRCA1/2, and MSH6 are the most frequently mutated genes. (B) High frequency of germline mutations in breast, prostate, and lung tumors is observed.
The prevalence of DRD in our cohort of patients with metastatic prostate cancer was 14.3%, with BRCA2 mutations being the most frequent variants (Data Supplement). Pathogenic germline DRDs were found in 9.2% of patients with primary brain tumors. The majority of these were in astrocytic neoplasms with WHO classification grades I to IV. We did not identify pathogenic mutations involving mismatch repair genes, which have been described in primary brain tumor patients, in particular as biallelic losses.25 CHEK2 was altered in four cases, including one medulloblastoma. The detected c.1100delC mutation has not been described in medulloblastoma patients to date.
DRDs involving homologous recombination genes may result in increased sensitivity to DNA-damaging agents, such as poly(APD-ribose) polymerase (PARP) inhibitors or platinum-based chemotherapy.26 Of the 55 patients with germline DRD defects, 12 had received platinum-based chemotherapy with follow-up available. Ten patients showed benefit in the form of stable disease or radiographic response.27 Sixteen patients (28.6%) with pathogenic germline defects succumbed to their disease. Median time from cancer diagnosis to death was 2 years.
RNA Sequencing
RNA sequencing was performed when sufficient fresh frozen tumor tissue was available after WES. RNA sequencing was performed successfully in 235 samples from 219 patients. We identified druggable outliers and potentially targetable gene fusions in 89 patients (17.3%). Of these 89 patients, 50 did not harbor a targetable genomic alteration identified by WES, resulting in an increase in the rate of actionable alteration detection of approximately 15%. We confirmed the drug sensitivity of select outlier genes using cell line experiments compared with randomly selected drugs (Fig 5).
FIG 5.
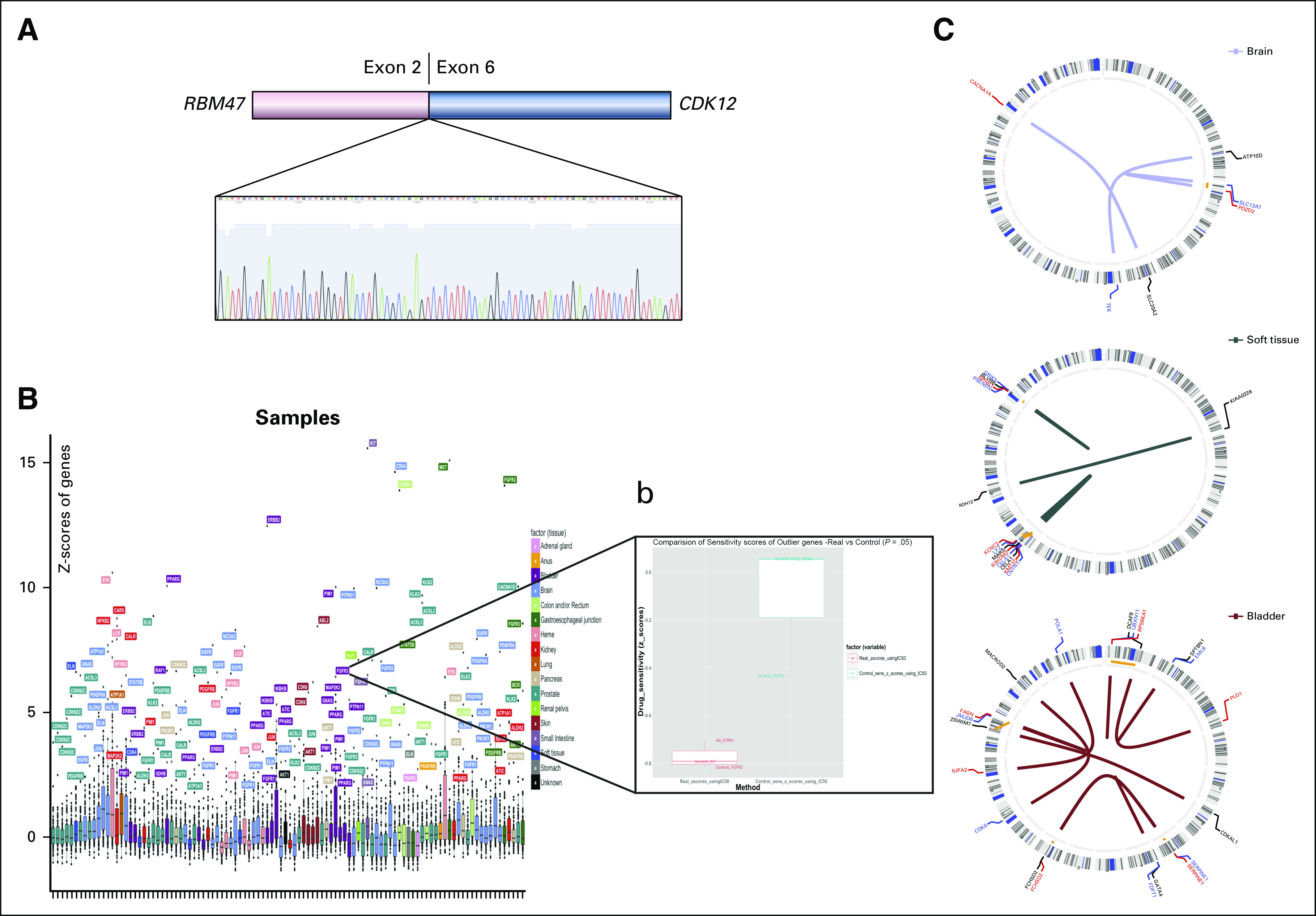
Detection of potential clinically relevant alterations through transcriptome analysis. (A) Novel RBM47-CDK12 gene fusion in a metastatic prostate cancer sample, detected by RNA sequencing and confirmed by polymerase chain reaction. (B) RNA sequencing outlier analysis and investigation of FGFR3 outlier in a bladder cancer cell line. (C) Circos plots of gene fusions identified in select cases (brain, soft tissue, bladder).
Nine novel fusions in a variety of cancer types were detected. A novel RBM47-CDK12 gene fusion was found in a prostate cancer bone metastasis, which was confirmed by reverse-transcription polymerase chain reaction and Sanger-Sequencing (Fig 5). An additional targetable NCOA4-RET fusion, which has been described in papillary thyroid cancer, non–small-cell lung cancer, and colorectal cancer, was found in a brain metastasis of a patient with unknown primary.28
Organoids
Part of the program was the development of patient-derived organoids from patient biopsies for high-throughput drug screening.4 Altogether 60 organoids were developed from 98 patients with an overall success rate of 61%. High-throughput drug screening was performed in a subset of patient-derived organoids as previously described.4
Case Studies
Germline results.
A pathogenic MSH6 mutation was detected in a 26-year-old patient with metastatic breast carcinoma who had undergone previous outside testing for BRCA1/2 germline mutations with a negative result. Although the association between mismatch repair mutations and breast cancer is not sufficiently established, these patients are at risk for secondary cancers, like colorectal or endometrial carcinoma.29 As testing was limited to BRCA1/2 in this case, this important germline finding in MSH6 was originally undetected. Other potentially significant variants in DRD genes, such as PALB2, would have been missed as well.30 This case underlines the importance of multigene panel or WES testing in patients not only with suspected hereditary cancer but also in those, especially young patients, with metastatic disease to detect underrecognized germline alterations.31
Tumor evolution and clonality.
Four illustrative cases are highlighted in Figure 3. We sequenced both the sarcoma and adenocarcinoma components from a primary uterine carcinosarcoma and discovered distinct molecular differences. Uterine carcinosarcomas are thought be of monoclonal origin.32 We detected a shared PTEN R130G missense mutation in both histologic subtypes. A deleterious effect has been predicted for this variant.33 In a previously published analysis of 13 uterine carcinosarcomas analyzed by targeted sequencing, eight cases demonstrated 100% identical mutations in both the carcinoma and sarcoma part.34 In contrast, we observed an early divergence with only the one PTEN mutation of 54 nonsynonymous shared mutations. Of note, a KRAS mutation was only observed in the sarcoma component.
In two samples of micropapillary urothelial carcinoma obtained from the primary tumor and a regional lymph node metastasis of one individual, a shared, potentially actionable mutation involving ERBB2 was detected.35
The problem of multifocal lung tumors and uncertainty regarding clinical management is not uncommon and has been addressed before.36,37 In one patient, two histologically similar lung acinar adenocarcinomas in different lobes were confirmed to be separate, synchronous primary tumors on the basis of two distinct driver mutations in the KRAS gene and other alterations observed by WES, thus resulting in two pT1 tumors rather than one pT4 tumor and eliminating the need to administer adjuvant chemotherapy.38 Whereas this alteration might also have been detected using a targeted gene panel, a background of different mutated genes in both samples that were detected by WES resulted in a stronger argument for two synchronous primary tumors. This case again demonstrates the importance of using next-generation sequencing to correctly identify synchronous primary tumors.39
Another patient with metastatic neuroblastoma was confirmed to have an activating ALK mutation (R1275Q) in both primary tumor and bone marrow metastasis. The patient has had stable disease for 13 months on therapy with sequential ALK inhibitors40 (Fig 3).
DISCUSSION
We have established a PM program for patients with advanced cancer using tumor/normal WES and integrative molecular profiling to detect genomic and other actionable alterations, improve clinical decision making, and study tumor evolution in a pan-cancer cohort. The patient population in our study is distinct from several reported studies in that our focus was on the evaluation of advanced tumors.41,42 The majority of patients (70.8%) presented with metastatic or recurrent disease at the time of enrollment.
Whereas publicly accessible molecular data for several tumor types are available, both for primary and metastatic cancers,43,44 these specimens are rarely matched—that is, obtained from the same patient.45 Our cohort of patients with locally advanced and metastatic cancer allowed for the collection of multiple matched, often primary and metastatic samples, resulting in a unique feature of this cohort: Genomic data from spatially and/or temporally heterogeneous, matched tissue samples were available in 146 patients. Our data contribute to the understanding of tumor evolution and heterogeneity. Of clinical importance is the finding that almost 85% of Category 1 alterations were shared between multiple samples from the same patient. These results are in concordance with published data in specific cancer types. In one study, when comparing actionable mutations between presurgery biopsies and resected specimen in patients with non–small-cell lung cancer, the concordance rate was found to be 79%.46 Similar findings have been reported for primary and recurrent breast cancer, with 86.6% of mutations and 85.5% of SCNAs being concordant.47 Our data confirm that it may reasonable to select the most accessible location or even archival material from the primary tumor for molecular analysis for certain cancer types.48,49
New research indicates that pathogenic germline mutations are more frequently found in patients with advanced cancer,9,12 and our data confirm this. Germline mutations that involve the DNA repair pathway, found in 10.7% of our patients, are predictive of response to PARP inhibitors and platinum-based therapy and have important familial implications for cancer screening.50 Family history was indicative of a heritable component in only one half of these patients. This discrepancy should be addressed to select patients and their families for genetic counseling.
WES provides the additional advantage of uncovering likely pathogenic mutations. According to the ACMG, a likely pathogenic variant is backed up by sufficient evidence to aid in clinical decision making.19 Our cohort is enriched for patients with metastatic prostate cancer, who have been reported to harbor DRDs in 11.8% of cases.9 This might explain, in addition to the high frequency of Ashkenazi Jewish ancestry, why we observe a higher rate of germline DRD compared with other pan-cancer cohorts (Data Supplement).10
Novel gene fusions were also discovered, including a RBM47-CDK12 gene fusion in a prostate cancer metastasis. This fusion might result in the loss of CDK12 activity, which has recently been described to delineate a distinct immunogenic subtype of metastatic castration-resistant prostate cancer.51 In our study, WES was prioritized over RNA sequencing whenever tissue availability was of concern. Biopsies in our study were usually performed for diagnostic purposes in the context of clinical care, hence the lower availability of fresh frozen tissue for additional RNA sequencing.
Targeted sequencing of cancer-related genes offers several advantages over WES, including deeper coverage, quicker turnaround time, lower cost, and fewer requirements for an elaborate computational pipeline. Here, we show that WES and RNA sequencing may provide an additional and complimentary layer of information in certain settings, particularly for patients with advanced cancer who experience progression after standard therapies. The highlighted cases illustrate the potential utility of WES for uncovering clinically informative somatic and germline alterations. WES also considers the rapidly expanding spectrum of actionable alterations, including alterations for which targeted treatment may not be available at the time of analysis, but for which clinical trials might be planned. Emergence of resistance pathways may also be identified.
Limitations of WES are lower coverage, higher cost, and slower turnaround time. In addition, whereas targeted next-generation sequencing testing of somatic DNA are sometimes ordered as part of clinical care, using germline DNA as normal control for WES necessitates securing informed consent.
Implementing -omic data into clinical care requires an interdisciplinary team. Genomic data and its interpretation in the context of tumor type and primary site must be easily accessible to enable clinicians to integrate the data into everyday patient care. Shared data platforms may also help to close the gap between research and clinical care. With the launch of the American Association of Cancer Research Project Genomics Evidence Neoplasia Information Exchange for sharing genomic and clinical data sets, there are now efforts toward developing a unified database to ultimately advance clinical care.44
One limitation of our study is the lack of uniformity of the cohort in terms of tumor type and therapy, which prevents us from making generalized statements about treatment response. Its strength lies in its ability to identify outliers and N-of-one cases. This data set also reports on a large number of matched temporally and spatially heterogeneous samples, highlighting the concordance of actionable alterations in different samples.
As other PM trials have shown, uncovering genomic alterations may not be sufficient in identifying actionable alterations. This study demonstrates how integrating genomics with transcriptomic and other potential readouts may ultimately improve actionability and contribute toward our growing understanding of the molecular underpinnings of cancer. Data for this study may be accessed through cBioportal (http://www.cbioportal.org).52
ACKNOWLEDGMENT
The authors acknowledge additional logistic and technical support provided by the Weill Cornell Biorepository facility (Maria T. Salpietro, Christopher Louie, Alyssa F. Polak), Gillian Hodes, Amin Yakubov, Victor Chan, Jyothi Manohar, Elyze C. Merzier, and Rebecca Meyer. Current primary affiliations are University of Bonn, Germany (V.S.); University of Bern, Switzerland (M.A.R.); Dana-Farber Cancer Institute, Boston, MA (H.B.).
Footnotes
Supported by the Englander Institute for Precision Medicine of Weill Cornell Medicine and New York Presbyterian, the Translational Research Program of the Department of Pathology and Laboratory Medicine at Weill Cornell Medicine. Funded by Department of Defense Grants No. PC121341 (H.B.) and PC160264 (D.R., H.B.), the Prostate Cancer Foundation (L.P., M.A.R., and H.B.), and National Cancer Institute SPORE in Prostate Cancer Grant No. P50-CA211024 (J.M.M., M.A.R., O.E., and H.B.).
M.A.R., O.E., and H.B. contributed equally to this work and share senior authorship.
AUTHOR CONTRIBUTIONS
Conception and design: Verena Sailer, Hanna Rennert, Rob Kim, Bishoy M. Faltas, Eleni Andreopoulou, Linda T. Vahdat, Douglas S. Scherr, Manish A. Shah, Wei Song, Juan Miguel Mosquera, Mark A. Rubin, Olivier Elemento, Himisha Beltran
Financial support: Rob Kim, Scott T. Tagawa, Mark A. Rubin, Olivier Elemento, Himisha Beltran
Administrative support: Verena Sailer, Chantal Pauli, Noah Greco, Rob Kim, Theresa Y. MacDonald, Eleni Andreopoulou, Olivier Elemento, Himisha Beltran
Provision of study materials or patients: Noah Greco, Rob Kim, Bishoy M. Faltas, Eleni Andreopoulou, Linda T. Vahdat, Douglas S. Scherr, Koen van Besien, Christopher E. Barbieri, Brian D. Robinson, Allyson J. Ocean, Manish A. Shah, David M. Nanus, Scott T. Tagawa, Juan Miguel Mosquera, Mark A. Rubin, Olivier Elemento, Himisha Beltran
Collection and assembly of data: Verena Sailer, Kenneth Wa Eng, David J. Pisapia, David Wilkes, Joanna Cyrta, Sheena Sahota, Chantal Pauli, Shaham Beg, Samaneh Motanagh, Myriam Kossai, Jacqueline Fontugne, Jenny Zhaoying Xiang, Noah Greco, Rob Kim, Theresa Y. MacDonald, Terra McNary, Mirjam Blattner-Johnson, Marc H. Schiffman, Bishoy M. Faltas, Jeffrey P. Greenfield, David Rickman, Eleni Andreopoulou, Kevin Holcomb, Linda T. Vahdat, Douglas S. Scherr, Koen van Besien, Christopher E. Barbieri, Brian D. Robinson, Howard Alan Fine, Allyson J. Ocean, Ana Molina, David M. Nanus, Scott T. Tagawa, Juan Miguel Mosquera, Mark A. Rubin, Olivier Elemento, Himisha Beltran
Data analysis and interpretation: Verena Sailer, Kenneth Wa Eng, Tuo Zhang, Rohan Bareja, David J. Pisapia, Alexandros Sigaras, Bhavneet Bhinder, Alessandro Romanel, David Wilkes, Evan Sticca, Rema Rao, Shaham Beg, Loredana Puca, Rob Kim, Mirjam Blattner-Johnson, Eleni Andreopoulou, Linda T. Vahdat, Douglas S. Scherr, Howard Alan Fine, Manish A. Shah, Qiulu Pan, Francesca Demichelis, Scott T. Tagawa, Wei Song, Juan Miguel Mosquera, Andrea Sboner, Mark A. Rubin, Olivier Elemento, Himisha Beltran
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Alexandros Sigaras
Employment: Weill Cornell Medical College
Loredana Puca
Employment: Petra Pharma Corporation
Bishoy M. Faltas
Honoraria: Digital Science Press publications
Research Funding: Eli Lilly
David Rickman
Research Funding: Janssen Oncology
Eleni Andreopoulou
Honoraria: AstraZeneca, Sirtex, SVB Leerink, AbbVie, Eisai, NanoString Technologies
Consulting or Advisory Role: AstraZeneca, Sirtex, SVB Leerink, AbbVie, Eisai, NanoString Technologies
Speakers' Bureau: R-Pharm US
Travel, Accommodations, Expenses: AstraZeneca, Sirtex, SVB Leerink, AbbVie, Eisai, NanoString Technologies
Kevin Holcomb
Research Funding: Fujirebio Diagnostics
Expert Testimony: Johnson & Johnson
Linda T. Vahdat
Honoraria: Polyphor, R-Pharm, Athenex, Seattle Genetics, Osmol Therapeutics
Consulting or Advisory Role: Berg Pharma
Speakers' Bureau: Eisai
Research Funding: Polyphor (Inst), Immunomedics (Inst), Seattle Genetics (Inst)
Patents, Royalties, Other Intellectual Property: Patent pending on the application for BMD progenitor cells (Inst)
Douglas S. Scherr
Research Funding: Urogen Pharma (Inst), Cepheid (Inst), Anchiano (Inst), CryoLife (Inst)
Koen van Besien
Stock and Other Ownership Interests: Hemogenyx
Consulting or Advisory Role: Hemogenyx, Actinium Pharmaceuticals, Cellectis, Tessa Therapeutics
Research Funding: Miltenyi Biotec, Actinium Pharmaceuticals, Juno Therapeutics, Fate Therapeutics
Christopher E. Barbieri
Patents, Royalties, Other Intellectual Property: Coinventor on a patent application filed regarding SPOP mutations in prostate cancer by Weill Cornell Medicine
Brian D. Robinson
Consulting or Advisory Role: Bristol-Myers Squibb
Patents, Royalties, Other Intellectual Property: Methods for diagnosing and treating prostate cancer
Allyson J. Ocean
Consulting or Advisory Role: Celgene, Tyme
Speakers' Bureau: Daiichi Sankyo
Travel, Accommodations, Expenses: Daiichi Sankyo
Ana Molina
Honoraria: American Society of Clinical Oncology
Consulting or Advisory Role: Eisai, Exelixis, Novartis, Janssen Oncology
Manish A. Shah
Consulting or Advisory Role: Astellas Pharma, Eli Lilly
Research Funding: Gilead Sciences (Inst), Merck (Inst), Boston Biomedical (Inst), Oncolys BioPharma (Inst), Bristol-Myers Squibb (Inst)
David M. Nanus
Consulting or Advisory Role: Genentech
Research Funding: Novartis (Inst), Boehringer Ingelheim (Inst), Zenith Epigenetics (Inst)
Qiulu Pan
Employment: Caris Life Sciences
Stock and Other Ownership Interests: Caris Life Sciences
Francesca Demichelis
Patents, Royalties, Other Intellectual Property: Co-inventor on a patent filed by the University of Michigan and Brigham and Women’s Hospital covering the diagnostic and therapeutic fields for ETS fusions in prostate cancer, diagnostic field licensed to Gen-Probe
Scott T. Tagawa
Consulting or Advisory Role: Medivation, Astellas Pharma, Dendreon, Janssen Oncology, Bayer, Genentech, Endocyte, Immunomedics, Karyopharm Therapeutics, AbbVie, Tolmar, QED, Amgen, Sanofi, Pfizer
Research Funding: Eli Lilly (Inst), Sanofi (Inst), Janssen Oncology (Inst), Astellas Pharma (Inst), Progenics (Inst), Millennium Pharmaceuticals (Inst), Amgen (Inst), Bristol-Myers Squibb (Inst), Dendreon (Inst), Rexahn Pharmaceuticals (Inst), Bayer (Inst), Genentech (Inst), Newlink Genetics (Inst), Inovio Pharmaceuticals (Inst), AstraZeneca (Inst), Immunomedics (Inst), Novartis (Inst), AVEO (Inst), Boehringer Ingelheim (Inst), Merck (Inst), Stem CentRx (Inst), Karyopharm Therapeutics (Inst), AbbVie (Inst), Medivation (Inst), Endocyte (Inst), Exelixis (Inst), Clovis Oncology (Inst)
Travel, Accommodations, Expenses: Sanofi, Immunomedics, Amgen
Wei Song
Employment: Genentech (I), Cytokinetics (I)
Honoraria: Foundation Medicine, Loxo
Consulting or Advisory Role: Foundation Medicine, Loxo
Juan Miguel Mosquera
Research Funding: Personal Genome Diagnostics
Travel, Accommodations, Expenses: Personal Genome Diagnostics
Mark A. Rubin
Honoraria: F Hoffmann-La Roche, Novartis, Astellas Pharma
Research Funding: Eli Lilly, Janssen Oncology, Millennium Pharmaceuticals, Sanofi
Patents, Royalties, Other Intellectual Property: US Patent (7,767,393 and 7,229,774), Expression Profile of Prostate Cancer, 2007; US Patent (7,332,290), Detection of AMACR Cancer Markers in Urine, 2008; US Patent (7,718,369), Recurrent Gene Fusions in Prostate Cancer, 2010; US Patent (7,803,552 and 7,666,595), Biomarkers for Predicting Prostate Cancer Progression, 2010; US Patent (7,981,609 B2), Methods for Identifying and Using SNP Panels, 2011; US Patent (8,106,037 B2), Identification and Treatment of Estrogen Responsive PCa, 2012; US Patent (9,090,899 B2), Methods of Diagnosing and Treating Prostate Cancer Characterized by NDRG1-ERG Fusion, 2015; US Patent (9,458,213 B2), Compositions and Methods for Diagnosing Prostate Cancer Based on Detection of SLC45A3-ELK4 Fusion Transcript, 2016; US Patent (9,568,483 B2), Molecular Subtyping, Prognosis and Treatment of Prostate Cancer, 2017; US Patent (9,678,077 B2), ERG/TFF3/HMWCK Triple Immunostain for Detection of Prostate Cancer, 2017; US Patent (61,408,341), Exploration of Novel Gene Fusion in Prostate Cancer by RNA-Seq
Travel, Accommodations, Expenses: F Hoffmann-La Roche, Novartis, Astellas Pharma
Olivier Elemento
Stock and Other Ownership Interests: Volastra, Owkin, One Three Biotech
Himisha Beltran
Consulting or Advisory Role: Janssen Oncology, Genzyme, GlaxoSmithKline, AbbVie, Astellas Pharma, AstraZeneca
Research Funding: Janssen Oncology (Inst), AbbVie (Inst), Stemcentrx (Inst), Eli Lilly (Inst)
Travel, Accommodations, Expenses: Janssen Oncology
No other potential conflicts of interest were reported.
REFERENCES
- 1.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Prasad V. Perspective: The precision-oncology illusion. Nature. 2016;537:S63. doi: 10.1038/537S63a. [DOI] [PubMed] [Google Scholar]
- 3.Beltran H, Eng K, Mosquera JM, et al. Whole-exome sequencing of metastatic cancer and biomarkers of treatment response. JAMA Oncol. 2015;1:466–474. doi: 10.1001/jamaoncol.2015.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pauli C, Hopkins BD, Prandi D, et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017;7:462–477. doi: 10.1158/2159-8290.CD-16-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [Erratum: Nat Med 23:1004, 2017] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Massard C, Michiels S, Ferté C, et al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: Results of the MOSCATO 01 trial. Cancer Discov. 2017;7:586–595. doi: 10.1158/2159-8290.CD-16-1396. [DOI] [PubMed] [Google Scholar]
- 7.Mody RJ, Wu YM, Lonigro RJ, et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA. 2015;314:913–925. doi: 10.1001/jama.2015.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sholl LM, Do K, Shivdasani P, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight. 2016;1:e87062. doi: 10.1172/jci.insight.87062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–453. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schrader KA, Cheng DT, Joseph V, et al. Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol. 2016;2:104–111. doi: 10.1001/jamaoncol.2015.5208. [Erratum: JAMA Oncol 2:279, 2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holter S, Borgida A, Dodd A, et al. Germline BRCA mutations in a large clinic-based cohort of patients with pancreatic adenocarcinoma. J Clin Oncol. 2015;33:3124–3129. doi: 10.1200/JCO.2014.59.7401. [DOI] [PubMed] [Google Scholar]
- 12.Robinson DR, Wu YM, Lonigro RJ, et al. Integrative clinical genomics of metastatic cancer. Nature. 2017;548:297–303. doi: 10.1038/nature23306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pisapia DJ, Salvatore S, Pauli C, et al. Next-generation rapid autopsies enable tumor evolution tracking and generation of preclinical models. JCO Precis Oncol. 2017 doi: 10.1200/PO.16.00038. 10.1200/PO.16.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sailer V, Schiffman MH, Kossai M, et al. Bone biopsy protocol for advanced prostate cancer in the era of precision medicine. Cancer. 2018;124:1008–1015. doi: 10.1002/cncr.31173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faltas BM, Prandi D, Tagawa ST, et al. Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nat Genet. 2016;48:1490–1499. doi: 10.1038/ng.3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rennert H, Eng K, Zhang T, et al. Development and validation of a whole-exome sequencing test for simultaneous detection of point mutations, indels and copy-number alterations for precision cancer care. NPJ Genom Med. 2016;1:16019. doi: 10.1038/npjgenmed.2016.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sailer V, Pauli C, Merzier EC, et al. On-site cytology for development of patient-derived three-dimensional organoid cultures: A pilot study. Anticancer Res. 2017;37:1569–1573. doi: 10.21873/anticanres.11486. [DOI] [PubMed] [Google Scholar]
- 18.Huang L, Fernandes H, Zia H, et al. The cancer precision medicine knowledge base for structured clinical-grade mutations and interpretations. J Am Med Inform Assoc. 2017;24:513–519. doi: 10.1093/jamia/ocw148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romanel A, Zhang T, Elemento O, et al. EthSEQ: Ethnicity annotation from whole exome sequencing data. Bioinformatics. 2017;33:2402–2404. doi: 10.1093/bioinformatics/btx165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hashim D, Manczuk M, Holcombe R, et al. Cancer mortality disparities among New York City’s Upper Manhattan neighborhoods. Eur J Cancer Prev. 2017;26:453–460. doi: 10.1097/CEJ.0000000000000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demichelis F, Greulich H, Macoska JA, et al. SNP panel identification assay (SPIA): A genetic-based assay for the identification of cell lines. Nucleic Acids Res. 2008;36:2446–2456. doi: 10.1093/nar/gkn089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landrum MJ, Lee JM, Riley GR, et al. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lavoine N, Colas C, Muleris M, et al. Constitutional mismatch repair deficiency syndrome: Clinical description in a French cohort. J Med Genet. 2015;52:770–778. doi: 10.1136/jmedgenet-2015-103299. [DOI] [PubMed] [Google Scholar]
- 26.Brown JS, O’Carrigan B, Jackson SP, et al. Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discov. 2017;7:20–37. doi: 10.1158/2159-8290.CD-16-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilkes DC, Sailer V, Xue H, et al. A germline FANCA alteration that is associated with increased sensitivity to DNA damaging agents. Cold Spring Harb Mol Case Stud. 2017;3:a001487. doi: 10.1101/mcs.a001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romei C, Ciampi R, Elisei R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat Rev Endocrinol. 2016;12:192–202. doi: 10.1038/nrendo.2016.11. [DOI] [PubMed] [Google Scholar]
- 29.Daly MB, Pilarski R, Berry M, et al. NCCN guidelines insights: Genetic/familial high-risk assessment—Breast and ovarian, version 2.2017. J Natl Compr Canc Netw. 2017;15:9–20. doi: 10.6004/jnccn.2017.0003. [DOI] [PubMed] [Google Scholar]
- 30.Antoniou AC, Casadei S, Heikkinen T, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371:497–506. doi: 10.1056/NEJMoa1400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tung N, Lin NU, Kidd J, et al. Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J Clin Oncol. 2016;34:1460–1468. doi: 10.1200/JCO.2015.65.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cherniack AD, Shen H, Walter V, et al. Integrated molecular characterization of uterine carcinosarcoma. Cancer Cell. 2017;31:411–423. doi: 10.1016/j.ccell.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khan I, Ansari IA, Singh P, et al. Prediction of functionally significant single nucleotide polymorphisms in PTEN tumor suppressor gene: An in silico approach. Biotechnol Appl Biochem. 2017;64:657–666. doi: 10.1002/bab.1483. [DOI] [PubMed] [Google Scholar]
- 34.McConechy MK, Hoang LN, Chui MH, et al. In-depth molecular profiling of the biphasic components of uterine carcinosarcomas. J Pathol Clin Res. 2015;1:173–185. doi: 10.1002/cjp2.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tschui J, Vassella E, Bandi N, et al. Morphological and molecular characteristics of HER2 amplified urothelial bladder cancer. Virchows Arch. 2015;466:703–710. doi: 10.1007/s00428-015-1729-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Wang M, MacLennan GT, et al. Evidence for common clonal origin of multifocal lung cancers. J Natl Cancer Inst. 2009;101:560–570. doi: 10.1093/jnci/djp054. [DOI] [PubMed] [Google Scholar]
- 37.Murphy SJ, Aubry MC, Harris FR, et al. Identification of independent primary tumors and intrapulmonary metastases using DNA rearrangements in non–small-cell lung cancer. J Clin Oncol. 2014;32:4050–4058. doi: 10.1200/JCO.2014.56.7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kris MG, Gaspar LE, Chaft JE, et al. Adjuvant systemic therapy and adjuvant radiation therapy for stage I to IIIA completely resected non–small-cell lung cancers: American Society of Clinical Oncology/Cancer Care Ontario clinical practice guideline update. J Clin Oncol. 2017;35:2960–2974. doi: 10.1200/JCO.2017.72.4401. [DOI] [PubMed] [Google Scholar]
- 39.Klempner SJ, Ou SH, Costa DB, et al. The clinical use of genomic profiling to distinguish intrapulmonary metastases from synchronous primaries in non-small-cell lung cancer: A mini-review. Clin Lung Cancer. 2015;16:334–339.e1. doi: 10.1016/j.cllc.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 40.Ueda T, Nakata Y, Yamasaki N, et al. ALK(R1275Q) perturbs extracellular matrix, enhances cell invasion and leads to the development of neuroblastoma in cooperation with MYCN. Oncogene. 2016;35:4447–4458. doi: 10.1038/onc.2015.519. [DOI] [PubMed] [Google Scholar]
- 41.Garofalo A, Sholl L, Reardon B, et al. The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med. 2016;8:79. doi: 10.1186/s13073-016-0333-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hyman DM, Solit DB, Arcila ME, et al. Precision medicine at Memorial Sloan Kettering Cancer Center: Clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discov Today. 2015;20:1422–1428. doi: 10.1016/j.drudis.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cooper LA, Demicco EG, Saltz JH, et al. PanCancer insights from The Cancer Genome Atlas: The pathologist’s perspective. J Pathol. 2018;244:512–524. doi: 10.1002/path.5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.AACR Project GENIE Consortium AACR Project GENIE: Powering precision medicine through an international consortium. Cancer Discov. 2017;7:818–831. doi: 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cancer Genome Atlas Research Network The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Isaka M, Serizawa M, Kenmotsu H, et al. Comparison of clinically relevant mutation profiles between preoperative biopsy and corresponding surgically resected specimens in Japanese patients with non-small-cell lung cancer by amplicon-based massively parallel sequencing. Clin Lung Cancer. 2017;18:519–526.e1. doi: 10.1016/j.cllc.2016.11.022. [DOI] [PubMed] [Google Scholar]
- 47.Meric-Bernstam F, Frampton GM, Ferrer-Lozano J, et al. Concordance of genomic alterations between primary and recurrent breast cancer. Mol Cancer Ther. 2014;13:1382–1389. doi: 10.1158/1535-7163.MCT-13-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wyatt AW, Annala M, Aggarwal R, et al. Concordance of circulating tumor DNA and matched metastatic tissue biopsy in prostate cancer. J Natl Cancer Inst. 2017;109:109. doi: 10.1093/jnci/djx118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kang HJ, Hwangbo B, Lee JS, et al. Comparison of epidermal growth factor receptor mutations between metastatic lymph node diagnosed by EBUS-TBNA and primary tumor in non-small cell lung cancer. PLoS One. 2016;11:e0163652. doi: 10.1371/journal.pone.0163652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robson M, Im SA, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377:523–533. doi: 10.1056/NEJMoa1706450. [DOI] [PubMed] [Google Scholar]
- 51.Wu YM, Cieslik M, Lonigro RJ, et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell. 2018;173:1770–1782.e14. doi: 10.1016/j.cell.2018.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]