

Abstract
Background:
Elevated advanced glycation end products (AGE) in diabetes mellitus (DM) are implicated in the progression of DM-associated tissue injury, including diabetic nephropathy. The intrarenal renin-angiotensin system, in particular augmentation of angiotensinogen (AGT) in proximal tubular cells (PTC), plays a crucial role in the development of diabetic nephropathy. This study investigated hypothesis that AGE stimulates AGT production in PTC.
Materials and Methods:
Urinary AGT and AGE levels in streptozotocin-induced DM mice were measured by enzyme-linked immunosorbent assays. AGT expression and secretion were evaluated in cultured rat PTC receiving 0-200 μg/ml AGE-BSA treatments for 24 hours. Furthermore, intracellular signaling pathways activated by AGE were elucidated.
Results:
DM mice exhibited greater urinary AGT and AGE levels compared to control mice (AGT: 21.6 ± 5.5 ng/day vs. 190.1 ± 57.8 ng/day, AGE: 139.1 ± 21.6 μg/day vs. 332.8 ± 102.7 μg/day). In cultured PTC, treatment with AGE-BSA enhanced AGT mRNA expression (3.43 ± 0.11-fold compared to control), intracellular AGT protein levels (3.60 ± 0.38-fold), and secreted AGT levels (2.11 ± 0.18-fold). On the other hand, AGT levels were not altered in PTC receiving nonglycated BSA. Recombinant soluble AGE receptor, which competes with endogenous AGE receptor, diminished the AGE-induced AGT upregulation, suggesting that AGE-BSA stimulates AGT expression via activation of the AGE receptor. Enhanced phosphorylation of ERK½ and c-Jun, but not p38 MAP kinase, were observed in AGE-BSA-treated PTC. AGE-induced AGT augmentation was attenuated by an ERK inhibitor.
Conclusions:
The findings indicate that AGE enhances proximal tubular AGT expression via ERK1/2, which can exacerbate the development of diabetic related kidney injury.
Keywords: Advanced glycation end products, Angiotensinogen, Kidney, Renal proximal tubules, Diabetes mellitus
INTRODUCTION
Both type-1 and -2 diabetes mellitus (DM) are common diseases with increasing prevalence that often lead to the development of kidney injury and diabetic nephropathy with consequent end-stage renal disease. About one-third of all patients with DM develop nephropathy, with type-1 DM patients at greater risk for its development.1,2 Excessive generation of reactive oxygen species (ROS) and activation of inflammatory responses under hyperglycemic conditions are major contributors to DM related kidney injury and other complications.3,4 In addition to these pathologic factors, elevated advanced glycation end products (AGE) contribute to tissue injury in DM.5 AGEs are produced when sugars react with proteins via the formation of Schiff bases and Amadori products. This reaction is strongly enhanced in DM.6 The binding of AGE to their receptors (RAGE) activates various signal transducers including intracellular ROS,7 Janus kinase 2,8 protein kinase C,9 p38 mitogen-activated protein kinase (p38 MAPK),10 and extracellular signal-regulated kinases 1/2 (ERK 1/2),11 thus, mediating activation of signal transducer and activator of transcription 1 and 3 (STAT3),8,12 NF-κB,13 and activator protein 1 (AP-1) formed by c-Jun/c-Fos complex.14 Activated RAGE signaling pathways augment transcripts of pathophysiological factors and promote the development of tissue injury in DM. Indeed, diabetic nephropathy is exacerbated in RAGE-overex-pressed DM mice and ameliorated by RAGE inhibition,15 indicating involvement of the AGE-RAGE axis in the development of kidney damage in DM.
In DM, inappropriate activation of the intrarenal renin-angiotensin system (RAS), which augments intrarenal angiotensin II levels, leads to elevated sodium retention and increased arterial pressure and tissue injury. A key factor in the activation of the intrarenal RAS is stimulation of renal proximal tubular angiotensinogen (AGT), which is the precursor of angiotensin peptides.16 In animal models of both type-1 and type-2 DM, hyperglycemia is accompanied by elevated intrarenal AGT and urinary AGT levels.17–19 Immunohistologic analyses demonstrated that augmentation of AGT was observed mainly in proximal tubules in the kidneys of DM models. Furthermore, direct treatment of rat renal proximal tubular cells (PTC) with high glucose results in elevated ROS generation and consequent stimulation of AGT production,20 indicating that high glucose-induced oxidative stress stimulates intrarenal AGT production during the development of diabetic nephropathy.
The 5′ flanking region of AGT and molecular mechanisms underlying regulation of AGT expression via its promoter have been well delineated. Many transcription factors, which are activated by AGE-RAGE pathway, including STAT1, STAT3, NF-κB and AP-1 have been shown to bind to AGT promoter and accelerate AGT expression.21–24 These molecular biological characteristics suggest that not only high glucose, but also elevated AGE, can stimulate AGT expression via RAGE signaling pathways in PTC under diabetic conditions, augmenting intrarenal angiotensin II formation and leading to the progression of kidney injury. However, the effects of AGE/RAGE pathway on AGT regulation in PTC have not been demonstrated. Therefore, the present study tested the hypothesis that AGE increases AGT production in PTC and elucidated the signaling pathways.
MATERIALS AND METHODS
Animals and Tissue Samples
All protocols were evaluated and approved by the Tulane University Institutional Animal Care and Use Committee and conformed to the guidelines of the National Institutes of Health on the care and use of laboratory animals. Male C57bl/6 mice, 10-12 weeks of age (Jackson Laboratories), were cage housed and maintained in a temperature-controlled room on a 12-hour light to dark cycle, with free access to tap water and mouse chow during acclimation. Induction of diabetes in mice was performed as previously described.19 Mice received a single intraperitoneal injection of 200 mg/kg streptozotocin (STZ, Fisher Health Care) dissolved in 0.05 M citrate buffer, pH 4.5 (n = 5, day 0). Citrate buffer alone was used in control mice (n = 6). Twenty-four hour urine samples were collected in metabolic cages. On day 14, mice were euthanized by conscious decapitation, and trunk blood, liver and kidney tissue were collected.
Cell Culture
Immortalized rat PTC were kindly provided by Dr. Ingelfinger (Harvard Medical School) and used in this study.25 The cells were cultured in DMEM medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen) and plated in 6-well plates. Prior to stimulation, the cells were serum-starved for 24 hours. PTC were subsequently treated with 0-200 μg/ml AGE-BSA (BioVision) or nonglycated BSA (control, BioVision) for 24 hours in serum-free medium. Recombinant soluble RAGE (sRAGE, Aviscera), which has been identified as an antagonist of membrane RAGE,26 was used to inhibit RAGE-specific action on AGT regulation.
Antibodies
A rabbit anti-RAGE antibody from Novus and a rabbit anti-AGT antibody from IBL were used in this study. A mouse anti-phospho p47phox (ser359) antibody and a rabbit anti-total p47phox antibody were purchased from Thermo Fisher Scientific. To elucidate RAGE signaling pathways involved in AGT regulation in PTC, mouse antiphospho-p42/44 MAPK (ERK 1/2, Thr202/Tyr204), a rabbit anti-p42/44 MAPK, mouse anti-phospho-STAT3 (Tyr705), rabbit anti-STAT3, rabbit anti-phospho-STAT1 (Tyr701), rabbit anti-STAT1, rabbit anti-phospho-p38 MAPK (Thr180/Tyr182), rabbit anti-p38 MAPK, rabbit anti-p65, rabbit anti-phospho-c-Jun (Ser73), rabbit anti-c-Jun, rabbit antiphospho-c-Fos (Ser32), and rabbit anti-c-Fos antibodies were purchased from Cell Signaling Technology and used in this study. In addition, rabbit anti-phospho-p65 (Ser536) antibody from Signalway Antibody was also used. Mouse anti-GAPDH antibody from Abcam was used as an internal control. IRDye-labeled anti-mouse IgG and anti-rabbit IgG antibodies were obtained from Li-Cor as secondary antibodies in Western blot analyses. Alexa Fluor 488 goat anti-rabbit IgG (H+L) antibody from Life Technologies was used as a secondary antibody in immunostaining.
Enzyme-Linked Immunosorbent Assays
AGT levels in urine, plasma and cell culture medium were measured as previously described27 using the Mouse and Rat Total Angiotensinogen Assay Kits (IBL). Urinary AGE levels in diabetic mice were determined using AGE Competitive enzyme-linked immunosorbent assays (ELISA) (Cell Biolab). Each urinary AGT and AGE levels were normalized based on 24-hour urine volume. To analyze AGT levels in medium of cultured PTC, 0.5 × 106 cells were seeded in each well of a 6-well plate containing 1.5 ml of medium. After treatment of cells with AGE, the medium was collected at 24 hours. AGT protein levels in unconcentrated medium were measured by ELISA (IBL). The total volume of medium was measured; total AGT protein levels were calculated based on the medium volume.
Quantitative Real-Time Polymerase Chain Reaction
Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) was performed to evaluate AGT mRNA expression using the TaqMan PCR system as previously described.28 For total RNA isolation, tissues and cells were washed with 3 ml of phosphate-buffered saline (PBS). PBS was aspirated and total RNA was isolated from the cells using the RNeasy Mini Kit (Qiagen). Subsequently, qRT-PCR was performed. The data were normalized based on GAPDH mRNA expression.
Western Blot Analysis
AGT levels, phosphorylation levels of p47phox (a subunit of NADPH oxidase), and activities of signal transducers and transcription factors were determined using Western blot analysis. The Western blots were performed as previously described.21,28 Tissues and cells were homogenized with 60 μl lysis buffer containing 1% Triton X-100, 150 mmol/l NaCl, 1 mmol/l EDTA, 1% Nonidet P-40, 1 mmol/l Na3VO4 and 0.25% Protease Inhibitor Cocktail (Sigma). The lysates were sonicated 3 times for 10 seconds each on ice. Total protein concentration of the supernatant was quantified using Micro BCA Protein Assay Kit (Pierce). Then, 20 μg of total protein was applied to a precast NuPAGE 4%-12% gel (Invitrogen). The separated proteins were transferred to a nitrocellulose membrane (Bio-Rad). After incubation of the membrane with primary and secondary antibodies, detection and analysis were performed using the Odyssey Infrared Imaging System (Li-Cor). Data were normalized based on GAPDH protein expression. Phosphorylation levels of signal transducers and transcription factors were normalized based on total protein levels of each target.
Immunohistochemical Studies
In addition to immunoblotting, the expression of RAGE protein in PTC was confirmed by immunostaining. PTC were cultured in 4-well chambers (Lab-Tek). The cells were rinsed with PBS and then fixed for 20 minutes by 4% paraformaldehyde. After 4 minutes incubation with 0.2% Triton X-100, the blocking agent Image-iT FX signal enhancer (Invitrogen) was added to the chambers. The cells were incubated with RAGE antibody overnight at 4°C. After washing with PBS, the cells were incubated with an Alexa Fluor 594-labeled secondary antibody. Vectashield HardSet mounting medium with DAPI from Vector Laboratories was used as a nuclear stain and mounting reagent. The RAGE staining was observed and photographed using a fluorescence Nikon Eclipse 50i microscope. A similar staining protocol was used in the kidney sections.
Statistical Analysis
Data are expressed as means ± standard error (SE). The data were analyzed using Student t-test or 1-way analysis of variance (ANOVA) followed by post hoc Bonferroni/Dunn multiple comparison test. A value of P < 0.05 was considered statistically significant.
RESULTS
Regulation of Intrarenal AGT and AGE in Diabetic Mice
Fourteen days after STZ injection, blood glucose levels in STZ-induced DM mice were greater than in control mice. As shown in Figure 1A, blood glucose levels were 130.6 ± 4.9 mg/dl in the control group and 553.0 ± 21.8 mg/dl in the STZ group. Water intake (2.2 ± 0.5 ml/day vs. 20.3 ± 3.4 ml/day) and urine (2.6 ± 0.5 ml/day vs. 15.9 ± 2.8 ml/day) volumes were much greater in the STZ group. There were no significant differences in body weights between the control and the STZ groups. Urinary protein excretion levels in the STZ group were slightly, but not significantly, higher than control group on day 14. Since intrarenal AGT is expression mainly in proximal tubules, as aforementioned, AGT mRNA levels in RNA isolated from dissected renal cortex were evaluated. The STZ mice had augmented renal cortical AGT mRNA levels (Figure 1B, 1.93 ± 0.4-fold, ratio to control group) as previously reported in CD-1 mice.19 Levels of both urinary AGT (Figure 1C) and AGE (Figure 1D) were also greater in the STZ group compared to the control group (urinary AGT: 21.6 ± 5.5 ng/day vs. 190.1 ± 57.8 ng/day, urinary AGE: 136.7 ± 20.7 μg/day vs. 246.6 ± 40.9 mg/day). Furthermore, renal oxidative stress levels were evaluated by measuring urinary 8-isoprostane levels (Figure 1E) and phosphorylation levels of renal p47phox (Figure 1F), which is a subunit of NADPH oxidase. Both levels of urinary 8-isoprostane and phosphorylation of renal p47phox were greater in the STZ group compared to control group.
FIGURE 1. Regulation of intrarenal AGT and AGE in diabetic mice.
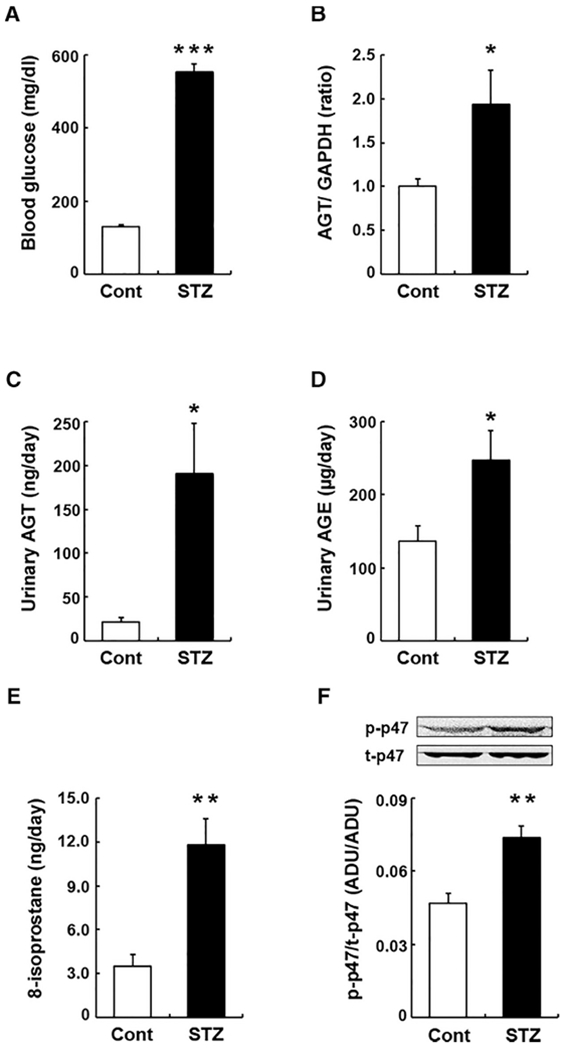
Levels of blood glucose (A), renal cortical AGT mRNA (B), urinary AGT protein (C), and urinary AGE (D) in control group (Cont, n = 6) and STZ-induced DM mice (STZ, n = 5). Graphs E and F show levels of urinary 8-isoprostane and phosphorylation of intrarenal p47phox protein. Data are expressed as mean ± SE. Asterisks (*P < 0.05, ** P < 0.01 and *** P < 0.001) indicate significant difference compared to control group. Abbreviations: AGT, angiotensinogen; AGE, advanced glycation end products; DM, diabetes mellitus; SE, standard error; STZ, streptozotocin.
Expression of RAGE in PTC
Since several studies have reported that RAGE expression levels in renal proximal tubules are varied by disease and/or experimental conditions and the levels are relatively low under normal conditions,29,30 we first confirmed the expression of RAGE in PTC used in the present study. Western blot analysis using PTC lysates and an anti-RAGE antibody showed immunoreactive bands between 40 and 50 kDa (Figure 2A) matching the expected molecular weight of rat RAGE (48 kDa). Almost 100% of PTC showed immunoreactivity against the antiRAGE antibody (Figure 2B), and the signal was observed in cytoplasm and membrane, supporting previous findings in other types of cells.31,32
FIGURE 2. Expression of RAGE in PTC.
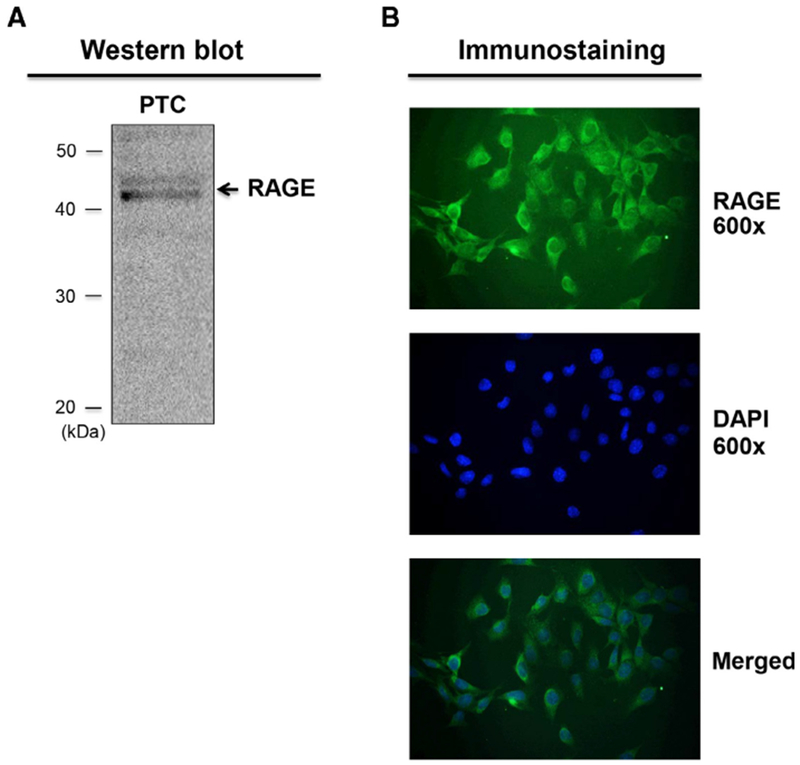
Western blot analysis of RAGE in PTC lysate (A) and immunostaining of RAGE in PTC (B). Arrow in the Western blot indicates immunoreactive bands. Abbreviation: PTC, proximal tubular cells.
Effects of AGE on AGT Expression in PTC
In PTC receiving 0 (control group), 2, 20 and 200 μg/ml AGE-BSA for 24 hours (n = 4 in each group), intracellular AGT protein levels were augmented in a dose dependent manner (Figure 3A, 1.40 ± 0.31-fold by 2 mg/ml AGE-BSA, 2.24 ± 0.24-fold by 20 μg/ml AGE-BSA, 5.63 ± 0.67-fold by 200 μg/ml AGE-BSA, ratio to control group). As a control, nonglycated BSA (200 μg/ml) did not induce elevation of intracellular AGT protein levels (Figure 3B). Accumulated AGT protein levels in culture medium of AGE-treated PTC were also greater compared with the control group and nonglycated BSA-treated group (Figure 3C, 0.83 ± 0.07 ng/ml in the control group, 1.76 ± 0.15 ng/ml in the AGE-treated group, and 0.96 ± 0.01 ng/ml in the nonglycated BSA-treated group). Moreover, treatment of PTC with AGE-BSA resulted in enhanced AGT mRNA transcription (Figure 3D).
FIGURE 3. Effects of AGE on AGT expression in PTC.
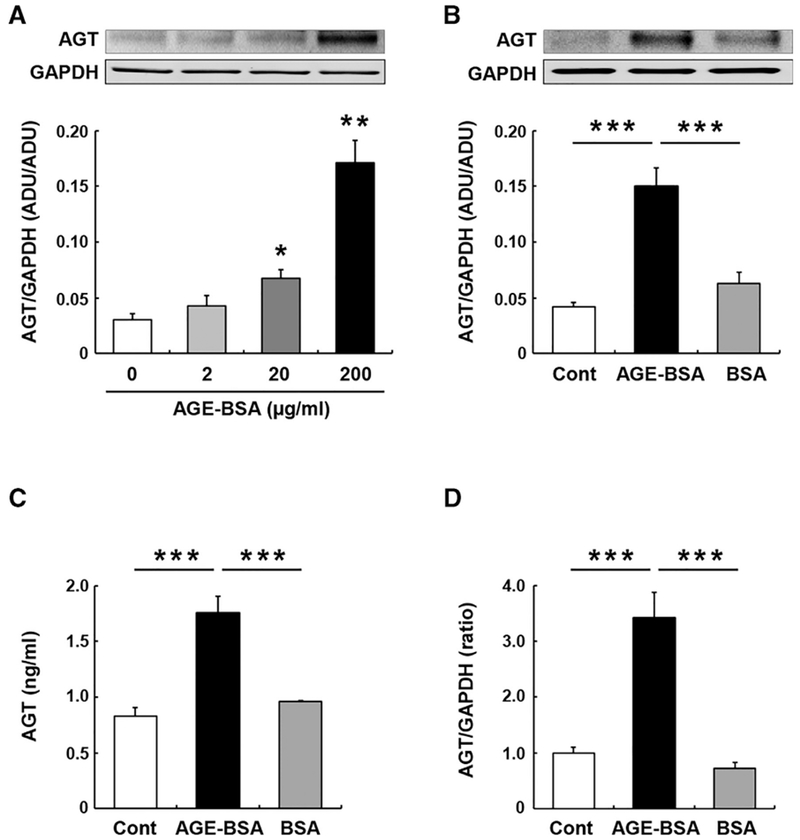
The figures summarize effects of different concentrations of AGE (0-200 μg/ml) on intracellular AGT protein expression at 24 hour in PCT (A, n = 4 in each group), effects of AGE (200 μg/ml) and non-glycated BSA (BSA) on intracellular AGT protein expression at 24 hour (B, n = 4 in each group), effects of AGE (200 μg/ml) and non-glycated BSA on secreted AGT protein in the medium during 24-hour treatments (C, n = 4 in each group), and effects of AGE (200 μg/ml) and non-glycated BSA on AGT mRNA expression at 24 hours (D, n = 4 in each group). Data are expressed as mean ± SE. Asterisks (*P < 0.05, ** P < 0.01 and *** P < 0.001) indicate significant difference compared to control group (Cont) or among groups. Abbreviations: ADU, arbitrary densitometry units; AGE, advanced glycation end products; AGT, angiotensinogen; PTC, proximal tubular cells.
Effects of sRAGE on AGE-Induced AGT Upregulation in PTC
To test if AGE-induced AGT augmentation in PTC is caused by RAGE activation, recombinant sRAGE was used as a competitor of membrane RAGE.26 AGE-BSA (200 μg/ml) increased AGT protein levels at 24 hours (Figure 4) as shown in Figure 3. The augmentation of AGT production was normalized by applying 20 μg/ml sRAGE (Figure 4, 2.42 ± 0.32-fold by AGE-BSA and 1.09 ± 0.24-fold by AGE-BSA+sRAGE, ratio to control group). Treatment of PTC with sRAGE alone did not alter AGT levels.
FIGURE 4. Effects of sRAGE on AGE-induced AGT upregulation in PTC.
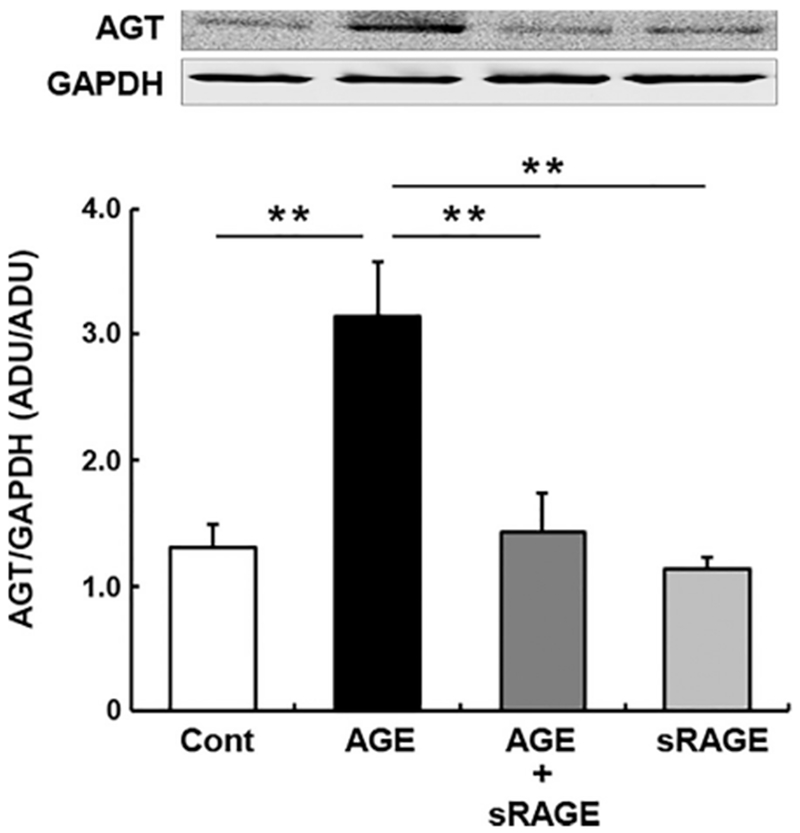
Intracellular AGT protein levels at 24 hour in PTC receiving AGE with/without sRAGE (n = 4 in each group). Data are expressed as mean ± SE. Asterisks (*P < 0.05, ** P < 0.01 and *** P < 0.001) indicate significant difference among groups. Abbreviations: ADU, arbitrary densitometry units; AGE, advanced glycation end products; AGT, angiotensinogen; PTC, proximal tubular cells.
Signaling Pathways Involved in AGE-Induced AGT Augmentation in PTC
Activation of p38 MAPK and ERK 1/2, which has been shown in other cell types treated with AGE, was investigated in PTC following AGE treatment (n = 4 in each group). AGE-BSA did not alter phosphorylation levels of p38 MAPK in PTC at 10 and 30 minutes (Figure 5A). Phosphorylation levels of ERK 1/2 in control group were decreased at 30 minutes compared to at 10 minutes. Importantly, AGE-BSA augmented phosphorylation levels of ERK 1/2, especially ERK 2, at 30 minutes (Figure 5A, 1.99 ± 0.18-fold by AGE-BSA). Activities of AGE/RAGE-associated transcription factors were also determined in these samples. Phosphorylation levels of STAT3, NF-κB (p65 subunit) and c-Fos were not changed by AGE-BSA treatment in PTC at 10 and 30 minutes (Figure 5A). In contrast, c-Jun was activated by AGE-BSA treatment at 30 minutes (Figure 5A, 1.70 ± 0.05-fold by AGE-BSA). U0126, an ERK 1/2 inhibitor, was employed to investigate a role of AGE-activated ERK 1/2 in AGT upregulation in PTC. The control group received DMSO, a solvent of the inhibitor. Pretreatment of PTC with 1.5 μM U0126 partially, but significantly, attenuated AGE-induced AGT augmentation (Figure 5B, 2.05 ± 0.04-fold by AGE-BSA, 1.26 ± 0.04-fold by AGE-BSA+U0126 and 1.08 ± 0.05-fold by U0126, at 24 hours). Treatment with U0126 alone (group U in Figure 5B) did not change AGT expression levels.
FIGURE 5. Signaling pathways involved in AGE-induced AGT augmentation in PTC.
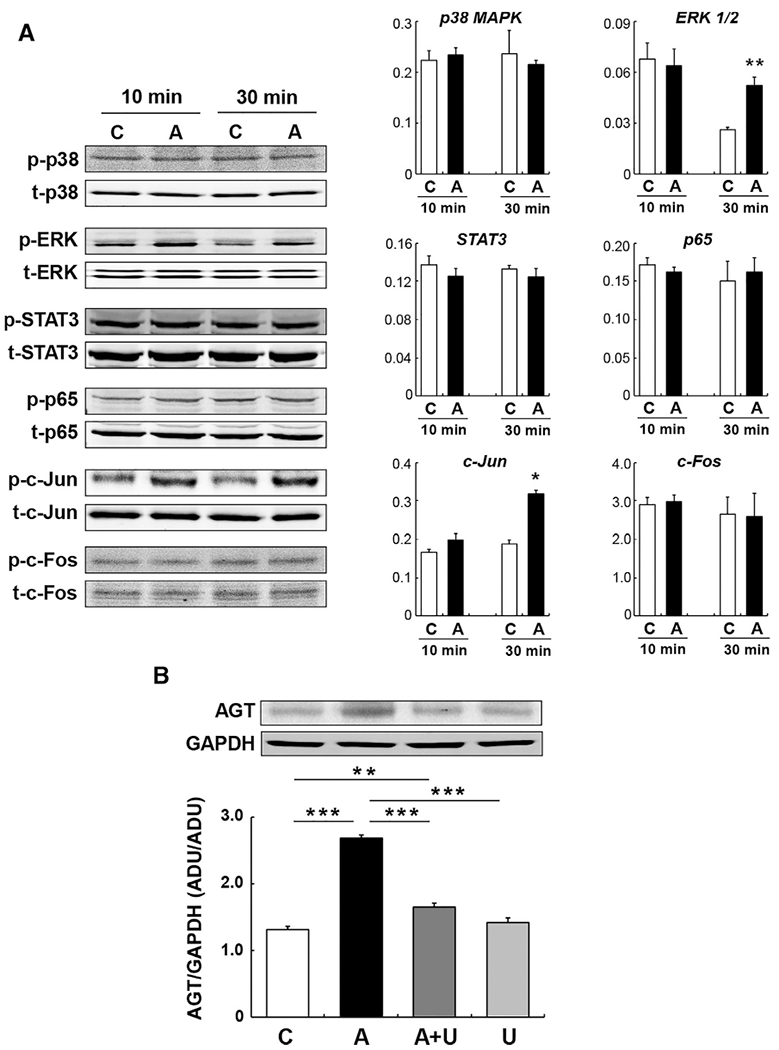
Phosphorylation levels of signal transducers and transcription factors 10 minutes and 30 minutes after AGE treatment in PTC (A, n = 4 in each group). C: control and A: AGE-treated PTC. Y-axes of bar graphs are ratio of phosphorylated target protein to total target protein. Panel B indicates effects of U0126 (U), an ERK inhibitor, on AGE-induced AGT upregulation at 24 hours in PTC (n = 4 in each group). Data are expressed as mean ± SE. Asterisks (*P < 0.05, ** P < 0.01 and *** P < 0.001) indicate significant difference compared to control group or among groups. Abbreviations: ADU, arbitrary densitometry units; AGE, advanced glycation end products; AGT, angiotensinogen; PTC, proximal tubular cells; SE, standard error.
DISCUSSION
Kidney disease leading to end-stage renal disease is a major complication in DM occurring in approximately 40% of patients.33 Since RAS blockers have shown protective effects in the development of diabetic nephropathy, intrarenal RAS has been a target to treat diabetic nephropathy.34 Thus, elucidating the regulatory mechanisms of the intrarenal RAS augmentation is essential for the development of novel strategies to prevent and treat DM associated kidney injury. In such investigations, stimulation of AGT expression by high glucose/ROS axis in PTC has been well established.20,35,36 On the other hand, the role of the AGE/RAGE pathway on intrarenal AGT regulation is poorly understood despite AGE’s role as a crucial risk factor for DM complications. Direct treatment with AGE increased AGT expression approximately 1.6-fold via activated phosphoinositide 3-kinase in cultured podocytes.37 This is important evidence for the upregulation of AGT expression by AGE. However, it has been shown that AGT expression levels in podocytes are very low and that elevation of glomerular AGT production was induced mainly in mesangial cells of DM mice.38 Therefore, although elevated AGT expression in podocytes has the potential to partially contribute to glomerular RAS activation and the progression of glomerular damage, it is unlikely that podocyte AGT is a major source of intrarenal angiotensin. Many studies using different approaches revealed that intrarenal AGT is primarily produced in PTC.39–42 The pathogenic significance of proximal tubular AGT has been established by showing that renal proximal tubule-specific overexpression of AGT by genetic manipulations amplified intrarenal angiotensin II formation leading to hypertension and kidney injury.43,44 The present study demonstrated elevated urinary AGE levels, which are accompanied with augmented renal oxidative stress, in STZ-induced DM mice and that direct treatments of cultured PTC resulted in augmentation of AGT mRNA, intracellular AGT protein and secreted AGT protein levels. Thus, AGT augmentation in renal proximal tubules by elevated AGE as well as by hyperglycemic conditions can be important, contributing to the progression of kidney injury in DM. The contribution of the intrarenal AGE-AGT axis to the development of kidney injury including renal inflammation and fibrosis in diabetes will be investigated in further studies.
Numerous forms of sRAGE, the truncated versions of membrane RAGE, are endogenously produced as spliced variants.26 Antagonistic actions of sRAGE against AGE/RAGE pathway and protective effects of sRAGE on DM complications including diabetic nephropathy have been shown in many studies.26,45–50 Applying sRAGE attenuated AGE-induced AGT upregulation in PTC shown in the present study, so administration of sRAGE may be a therapy to suppress intrarenal RAS activation and associated kidney injury in DM. Further studies are required to investigate suppressing effects of sRAGE on intrarenal RAS. It has been reported that gene deletion of RAGE did not significantly change intrarenal AGT mRNA levels in DM mice,51 which does not support our conclusion in the present study. A RAGE gene knockout that diminishes both pathogenicity of RAGE and protective actions by sRAGE may account for this different findings.
Various signal transducers and transcription factors have been identified as mediators of the pathophysiological actions caused by the AGE/RAGE pathway. In the present study, phosphorylation levels of ERK 1/2 in the control group exhibited temporal downregulation. This may by the result of temporal oscillations of ERK activity which have been well characterized.52,53 Importantly, AGE enhanced activation of ERK 1/2, and ERK inhibition attenuated AGT upregulation in PTC, indicating that AGE-induced AGT augmentation in PTC is mediated by ERK 1/2 activation. In tested transcription factors, only c-Jun was activated by AGE treatment in PTC. c-Jun is one of the downstream factors of ERK 1/254,55 and forms AP-1 complex, which promotes AGT transcription.24STAT3 and NF-κB, which can be activated by AGE8,12,13 and have been identified as AGT regulating transcription factors in PTC,21–23 failed to demonstrate activation during AGE treatment under our experimental conditions.
CONCLUSIONS
In conclusion, the findings in the present study suggest activation of the AGE/RAGE pathway augments renal proximal tubular AGT production via ERK 1/2, which can contribute to the development of diabetic nephropathy. The findings provide a mechanistic rationale for targeting AGE/RAGE pathway axis to suppress intrarenal RAS activity and prevent or treat kidney injury in DM.
ACKNOWLEDGMENTS
The authors thank Dr. Julie R. Ingelfinger (Massachusetts General Hospital and Harvard Medical School) for providing established PTC. The authors are grateful to Drs. Warren R. Bourgeois III and Usha Ramadhyani Bourgeois for supporting a student involved in this study.
Source of Funding: This study was supported by grants from the National Institute of General Medical Sciences IDeA Program (COBRE, P30GM103337), the National Institute of Diabetes and Digestive and Kidney Diseases (R01DK107694-01), and Tulane Bourgeois Ramadhyani Summer Fellowship Award.
Footnotes
Conflict of Interest: The authors have nothing to disclose.
REFERENCES
- 1.Ozieh MN, Dismuke CE, Lynch CP, et al. Medical care expenditures associated with chronic kidney disease in adults with diabetes: United States 2011. Diabetes Res Clin Pract. 2015;109:185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schena FP, Gesualdo L. Pathogenetic mechanisms of diabetic nephropathy. J Am Soc Nephrol. 2005;16(Suppl 1):S30–S33. [DOI] [PubMed] [Google Scholar]
- 3.Elmarakby AA, Abdelsayed R, Yao Liu J, et al. Inflammatory cytokines as predictive markers for early detection and progression of diabetic nephropathy. EPMA J. 2010;1:117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varga ZV, Giricz Z, Liaudet L, et al. Interplay of oxidative, nitrosative/nitrative stress, inflammation, cell death and autophagy in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1852:232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goutos I, Nicholas RS, Pandya AA, et al. Diabetes mellitus and burns. Part I-basic science and implicationsfor management. Int J Burns Trauma. 2015;5:1–12. [PMC free article] [PubMed] [Google Scholar]
- 6.Singh R, Barden A, Mori T, et al. Advanced glycation end-products: a review. Diabetologia. 2001;44:129–146. [DOI] [PubMed] [Google Scholar]
- 7.Vincent AM, Perrone L, Sullivan KA, et al. Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology. 2007;148:548–558. [DOI] [PubMed] [Google Scholar]
- 8.Grimm S, Ott C, Horlacher M, et al. Advanced-glycation-end-productinduced formation of immunoproteasomes: involvement of RAGE and Jak2/STAT1. Biochem J. 2012;448:127–139. [DOI] [PubMed] [Google Scholar]
- 9.Yu L, Zhao Y, Xu S, et al. Advanced Glycation End Product (AGE)-AGE Receptor (RAGE) System Upregulated Connexin43 Expression in Rat Car-diomyocytes via PKC and Erk MAPK Pathways. Int J Mol Sci. 2013; 14:2242–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu P, Lai D, Lu P, et al. ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int J Mol Med. 2012;29:613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu C, He JC, Cai W, et al. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc Natl Acad Sci USA. 2004;101:11767–11772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen H, Liu W, Wu X, et al. Advanced glycation end products induced IL-6 and VEGF-A production and apoptosis in osteocyte-like MLO-Y4 cells by activating RAGE and ERK1/2, P38 and STAT3 signalling pathways. Int Immunopharmacol. 2017;52:143–149. [DOI] [PubMed] [Google Scholar]
- 13.Cheng A, Dong Y, Zhu F, et al. AGE-LDL activates Toll like receptor 4 pathway and promotes inflammatory cytokines production in renal tubular epithelial cells. Int J Biol Sci. 2013;9:94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mercado-Pimentel ME, Onyeagucha BC, Li Q, et al. The S100P/RAGE signaling pathway regulates expression of microRNA-21 in colon cancer cells. FEBS lett. 2015;589:2388–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto Y, Kato I, Doi T, et al. Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J Clin Invest. 2001;108:261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Navar LG, Prieto MC, Satou R, et al. Intrarenal angiotensin II and its contribution to the genesis of chronic hypertension. Curr Opin Pharmacol. 2011;11:180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyata K, Ohashi N, Suzaki Y, et al. Sequential activation of the reactive oxygen species/angiotensinogen/renin-angiotensin system axis in renal injury of type 2 diabetic rats. Clin Exp Pharmacol Physiol. 2008;35:922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park S, Bivona BJ, Kobori H, et al. Major role for ACE-independent intrarenal ANG II formation in type II diabetes. Am J Physiol Renal Physiol. 2010;298:F37–F48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamiyama M, Zsombok A, Kobori H. Urinary angiotensinogen as a novel early biomarker of intrarenal renin-angiotensin system activation in experimental type 1 diabetes. J Pharmacol Sci. 2012;119: 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsieh TJ, Fustier P, Wei CC, et al. Reactive oxygen species blockade and action of insulin on expression of angiotensinogen gene in proximal tubular cells. J Endocrinol. 2004;183:535–550. [DOI] [PubMed] [Google Scholar]
- 21.Satou R, Miyata K, Gonzalez-Villalobos RA, et al. Interferon-gamma biphasically regulates angiotensinogen expression via a JAK-STAT pathway and suppressor of cytokine signaling 1 (SOCS1) in renal proximal tubularcells.FASEB J.2012;26:1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Satou R, Gonzalez-Villalobos RA, Miyata K, et al. Costimulation with angiotensin II and interleukin 6 augments angiotensinogen expression in cultured human renal proximal tubular cells. Am J Physiol Renal Physiol. 2008;295:F283–F289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Acres OW, Satou R, Navar LG, et al. Contribution of a nuclear factor-kappaB binding site to human angiotensinogen promoter activity in renal proximal tubular cells. Hypertension. 2011;57:608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uhal BD, Zhang H, Abdul-Hafez A, et al. Amiodarone induces angiotensinogen gene expression in lung alveolar epithelial cells through activation protein-1. Basic Clin Pharmacol Toxicol. 2007;100:59–66. [DOI] [PubMed] [Google Scholar]
- 25.Ingelfinger JR, Jung F, Diamant D, et al. Rat proximal tubule cell line transformed with origin-defective SV40 DNA: autocrine ANG II feedback. Am J Physiol. 1999;276:F218–F227. [DOI] [PubMed] [Google Scholar]
- 26.Koyama H, Yamamoto H, Nishizawa Y. RAGE and soluble RAGE: potential therapeutic targets for cardiovascular diseases. Mol Med. 2007;13:625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobori H, Katsurada A, Miyata K, et al. Determination of plasma and urinary angiotensinogen levels in rodents by newly developed ELISA. Am J Physiol Renal Physiol. 2008;294:F1257–F1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.OĽeary R, Penrose H, Miyata K, et al. Macrophage-derived IL-6 contributes to ANG II-mediated angiotensinogen stimulation in renal proximal tubular cells. Am J Physiol Renal Physiol. 2016;310:F1000–F1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukami K, Yamagishi S, Coughlan MT, et al. Ramipril inhibits AGE-RAGE-induced matrix metalloproteinase-2 activation in experimental diabetic nephropathy. Diabetol Metab Syndr. 2014;6:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Huang K, Cai GY, et al. Receptor for advanced glycation end products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cell signal. 2014;26:110–121. [DOI] [PubMed] [Google Scholar]
- 31.Hermani A, De Servi B, Medunjanin S, et al. S100A8 and S100A9 activate MAP kinase and NF-kappaB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp Cell Res. 2006; 312:184–197. [DOI] [PubMed] [Google Scholar]
- 32.Wei W, Lampe L, Park S, et al. Disulfide bonds within the C2 domain of RAGE play key roles in its dimerization and biogenesis. PloS One. 2012;7: e50736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tuttle KR, Bakris GL, Bilous RW, et al. Diabetic kidney disease: a report from an ADA Consensus Conference. Am J Kidney Dis. 2014;64:510–533. [DOI] [PubMed] [Google Scholar]
- 34.Yacoub R, Campbell KN. Inhibition of RAS in diabetic nephropathy. Int J Nephrol Renovasc Dis. 2015;8:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abdo S, Lo CS, Chenier I, et al. Heterogeneous nuclear ribonucleoproteins F and K mediate insulin inhibition of renal angiotensinogen gene expression and prevention of hypertension and kidney injury in diabetic mice. Diabetologia. 2013;56:1649–1660. [DOI] [PubMed] [Google Scholar]
- 36.Wang J, Shibayama Y, Kobori H, et al. High glucose augments angiotensinogen in human renal proximal tubular cells through hepatocyte nuclear factor-5. PLoS One. 2017;12 e0185600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng CL, Tang Y, Zheng Z, et al. Advanced glycation end-products activate the renin-angiotensin system through the RAGE/PI3-K signaling pathway in podocytes. Clin Invest Med. 2012;35:E282. [DOI] [PubMed] [Google Scholar]
- 38.Ohashi N, Urushihara M, Satou R, et al. Glomerular angiotensinogen is induced in mesangial cells in diabetic rats via reactive oxygen species–ERK/JNK pathways. Hypertens Res. 2010;33:1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ingelfinger JR, Zuo WM, Fon EA, et al. In situ hybridization evidence for angiotensinogen messenger RNA in the rat proximal tubule. An hypothesis for the intrarenal renin angiotensin system. J Clin Invest. 1990; 85:417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobori H, Harrison-Bernard LM, Navar LG. Expression of angiotensinogen mRNAand protein in angiotensin II-dependent hypertension. J Am Soc Nephrol. 2001;12:431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Terada Y, Tomita K, Nonoguchi H, et al. PCR localization of angiotensin II receptor and angiotensinogen mRNAs in rat kidney. Kidney Int. 1993;43:1251–1259. [DOI] [PubMed] [Google Scholar]
- 42.Pohl M, Kaminski H, Castrop H, et al. Intrarenal renin angiotensin system revisited: role of megalin-dependent endocytosis along the proximal nephron. J Biol Chem. 2010;285:41935–41946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sachetelli S, Liu Q, Zhang SL, et al. RAS blockade decreases blood pressure and proteinuria in transgenic mice overexpressing rat angiotensinogen gene in the kidney. Kidney Int. 2006;69:1016–1023. [DOI] [PubMed] [Google Scholar]
- 44.Ying J, Stuart D, Hillas E, et al. Overexpression of mouse angiotensinogen in renal proximal tubule causes salt-sensitive hypertension in mice. Am J Hypertens. 2012;25:684–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park L, Raman KG, Lee KJ, et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–1031. [DOI] [PubMed] [Google Scholar]
- 46.Kislinger T, Tanji N, Wendt T, et al. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2001;21:905–910. [DOI] [PubMed] [Google Scholar]
- 47.Wendt TM, Tanji N, Guo J, et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol. 2003;162:1123–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y, Yan SS, Colgan J, et al. Blockade of late stages of autoimmune diabetes by inhibition ofthe receptorfor advanced glycation end products. J Immunol. 2004;173:1399–1405. [DOI] [PubMed] [Google Scholar]
- 49.Bucciarelli LG, Wendt T, Qu W, et al. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation. 2002;106:2827–2835. [DOI] [PubMed] [Google Scholar]
- 50.Goh SY, Cooper ME. Clinical review: The role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab. 2008;93:1143–1152. [DOI] [PubMed] [Google Scholar]
- 51.Sourris KC, Morley AL, Koitka A, et al. Receptor for AGEs (RAGE) blockade may exert its renoprotective effects in patients with diabetic nephropathy via induction of the angiotensin II type 2 (AT2) receptor. Dia-betologia. 2010;53:2442–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shankaran H, Ippolito DL, Chrisler WB, et al. Rapid and sustained nuclear-cytoplasmic ERK oscillations induced by epidermal growth factor. MolSyst Biol. 2009;5:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shin SY, Rath O, Choo SM, et al. Positive- and negative-feedback regulations coordinate the dynamic behavior of the Ras-Raf-MEK-ERK signal transduction pathway. J Cell Sci. 2009;122:425–435. [DOI] [PubMed] [Google Scholar]
- 54.Kim SH, Chang JW, Kim SB, et al. Myoglobin induces vascular cell adhesion molecule-1 expression through c-Src kinase-activator protein-1/nuclear factor-kappaB pathways. Nephron Exp Nephrol. 2010;114 :e48–e60. [DOI] [PubMed] [Google Scholar]
- 55.Gough DJ, Sabapathy K, Ko EY, et al. A novel c-Jun-dependent signal transduction pathway necessary for the transcriptional activation of interferon gamma response genes. J Biol Chem. 2007;282:938–946. [DOI] [PubMed] [Google Scholar]