

Abstract
Background
Botulinum neurotoxin type A, an FDA-approved prophylactic drug for chronic migraine, is thought to achieve its therapeutic effect through blocking activation of unmyelinated meningeal nociceptors and their downstream communications with myelinated nociceptors and potentially the vasculature and immune cells. Prior investigations to determine botulinum neurotoxin type A effects on meningeal nociceptors were carried out in male rats and tested with stimuli that act outside the blood brain barrier. Here, we sought to explore the effects of extracranial injections of botulinum neurotoxin type A on activation of meningeal nociceptors by cortical spreading depression, an event which occurs inside the blood brain barrier, in female rats.
Material and methods
Using single-unit recording, we studied myelinated C- and unmyelinated Aδ-meningeal nociceptors' responses to cortical spreading depression 7–14 days after injection of botulinum neurotoxin type A or saline along calvarial sutures.
Results
In female rats, responses to cortical spreading depression were typically more prolonged and, in some cases, began at relatively longer latencies post-cortical spreading depression, than had been observed in previous studies in male rats. Extracranial administration of botulinum neurotoxin type A reduced significantly the prolonged firing of the meningeal nociceptors, in the combined sample of Aδ- and C-fiber, but not their response probability.
Discussion
The findings suggest that the mechanism of action by which botulinum neurotoxin type A prevents migraine differ from the one by which calcitonin gene-related peptide monoclonal antibodies prevent migraine and that even when the origin of migraine is central (i.e. in the cortex), a peripherally acting drug can intercept/prevent the headache.
Keywords: Migraine, headache, botox, CGRP, trigeminal, pain
Introduction
Migraine is a complex brain disorder, characterized by intense and long-lasting throbbing headaches, that in most cases is thought to originate in abnormal functioning of multiple brain regions and neuronal pathways that regulate homeosthesis, sensory, autonomic, affective and cognitive functions (1,2). While the mechanisms by which abnormal functioning of the brain gives rise to the unique perception of migraine headache is largely unknown, it is widely accepted that the headache begins with activation of nociceptors that supply the meninges and that drugs that prevent the activation of these nociceptors may be effective in reducing migraine. OnabotulinumtoxinA (BoNT-A) is an FDA-approved prophylactic drug for chronic migraine (3,4). When used for migraine prevention, BoNT-A is injected into discrete pericranial, neck and shoulder muscles (5). Until recently, it was not clear how administration of BoNT-A to peripheral sites outside the head reduces the sensitivity, responsiveness or activity of nociceptors supplying the meninges, inside the head. A significant advance in answering this question was made by the discovery of two populations of sensory fibers that cross the bones of the calvaria, one originating in the trigeminal ganglion and crossing from inside to outside through sutures of the calvaria (i.e. belonging to the trigeminovascular pathway) (6–8), and a second originating in C2-C3 dorsal root ganglia and crossing from outside to inside through small foramen and emissary canals (i.e. belonging to the cervicovascular pathway) (9).
Our overall understanding of the mechanism of action of BoNT-A derives from its ability to disrupt sensory transmission by preventing the adhesion of synaptic vesicles to the synaptic membrane and consequently the release of neurotransmitters and/or insertion of membrane-bound ion channels and receptors (10). For reasons that are not yet understood, BoNT-A appears to inhibit mainly unmyelinated C- and to a far lesser extent myelinated Aδ-nociceptors. This conclusion was first reached in a series of elegant studies of cutaneous nociceptors (11–14), and later observed in studies of meningeal nociceptors (16). When administered topically to the dura, BoNT-A decreased significantly the mechanosensitivity of C- but not Aδ-meningeal nociceptors and reversed their sensitization by inflammatory mediators (15). When injected extracranially 7 days earlier, BoNT-A lowered the incidence and magnitude of activation of C- but not Aδ-meningeal nociceptors by TRPV1 and TRPA1 agonists (16). Common to these studies was that the employed stimuli were all administered locally/topically to the outside, rather than inside layer of the dura (i.e. outside the blood brain barrier (bbb)) and that their administration modeled intracranial nociception (i.e. prolonged activation of meningeal nociceptors) rather than migraine. One of the most commonly used and best characterized animal models of migraine is the induction of cortical spreading depression (CSD) (17). CSD is a slowly propagating wave of neuronal depolarization thought to underlie the aura phase of migraine (18). Because CSD is an event that occurs inside the bbb, our previous studies could not predict the effects of extracranial administration of BoNT-A on activation of meningeal nociceptors by CSD. To better understand BoNT-A mechanisms of action in migraine prevention, in the current study we injected rats with BoNT-A and 7 days later determined the extent of activation of C- and Aδ meningeal nociceptors by CSD.
Materials and methods
BoNT-A suture injections and surgical preparation for neural recording
Experiments were approved by the Beth Israel Deaconess Medical Center and Harvard Medical School standing committees on animal care and were in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. Female Sprague-Dawley rats weighing 210–250 g were briefly anesthetized (2% isoflurane) and injected with BoNT-A (final dose = 5 units) or vehicle (normal saline). Four injections of BoNT-A (each containing 1.25 units diluted in 5 μl saline) or saline (5 μl) were made along the lambdoid (two injection sites) and sagittal (two injection sites) sutures (16).
Seven to 14 days following the injection of BoNT-A or saline, the rats were surgically prepared for neural recording in the trigeminal ganglion. Rats were anesthetized with urethane (1.5 g/kg i.p.) and treated with atropine (0.4 ml i.p.) to reduce intraoral secretions. Core temperature was maintained at 37℃ using a feedback-controlled heating pad, and pO2 was maintained above 98% by O2 inhalation. The rat was mounted in a stereotaxic apparatus. A craniotomy was made to expose the left transverse sinus, to allow electrical and mechanical stimulation of the receptive fields of dural primary afferent nociceptors. A small burr hole was drilled in the calvarium on the left side, at approximately 2 mm rostral to bregma and approximately 2mm lateral, to allow insertion of a glass micropipette for recording of electrocorticogram activity. A second burr hole was drilled approximately 5 mm caudal to the first one (approximately 5 mm rostral to the transverse sinus), for induction of cortical spreading depression by pinprick (brief insertion of a metal microelectrode into the cortex). The dura exposed by these burr holes was not removed. All of the exposed dura was kept moist using a modified synthetic interstitial fluid (135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 5 mM CaCl2, 10 mM glucose and 10 mM Hepes, pH 7.2). For neural recording from the left trigeminal ganglion, a craniotomy was made on the right (contralateral) side to allow the microelectrodes to be advanced through the contralateral cortex to reach the ganglion, using an angled approach. This angled approach was used to avoid damage to the ipsilateral cortex. The craniotomy was made to allow electrode insertions into the right cortex covering an area of 1.5–3 mm caudal to bregma, and 2–3 mm lateral. The dura covering this area of cortex was removed to allow microelectrode insertion. The experimental setup is shown in Figure 1.
Figure 1.
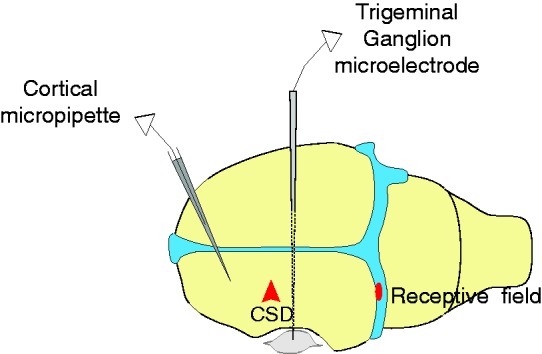
Experimental setup. Diagram showing position of: Cortical micropipette for electrocorticogram recording; trigeminal ganglion microelectrode, which was advanced through the contralateral (right) cortex with a medial angle to reach the left trigeminal ganglion, for unit recording; and a representative mechanical receptive field on the transverse sinus.
Neural recording
A glass micropipette filled with 0.9% saline (∼1 megohm, 7 µm tip) was inserted through the rostral burr hole 500 µm below the cortical surface for electrocorticogram recording. Platinum-coated tungsten microelectrodes (impedance 150–300 kΩ) were used for single-unit recording in the trigeminal ganglion. To reach the ganglion via a contralateral approach, the electrode was angled medially 21° and was advanced through the contralateral cortex (see above for coordinates). Dural afferent neurons in the ganglion were identified by their constant latency response to single shock stimulation delivered to the dura overlying the ipsilateral transverse sinus with a bipolar stimulating electrode (0.5 ms pulse, 5 mA, 0.75 Hz) (Figure 2(a),(b)). The response latency was used to calculate conduction velocity (c.v.), based on a conduction distance to the trigeminal ganglion of 12 mm. Neurons were classified as either C- (c.v. ≤1.5 ms) or Aδ-units (c.v. 1.5–5 m/s). Neurons with conduction velocity greater than 5 m/s were not studied. Spike 2 software (CED, Cambridge, UK) was used for acquisition and waveform discrimination of the electrically evoked spikes and for offline analysis.
Figure 2.

Electrophysiological characterization of meningeal nociceptors and CSD. (a), (b). Electrophysiological recordings of action potentials in C-fiber (a) and Aδ-fiber (b) meningeal nociceptors evoked by electrical and mechanical stimulation of their receptive fields on the transverse sinus. (c) Electrocorticogram showing CSD wave induced by pinprick in the cortex.
Mechanical receptive fields of dural afferents were identified by probing the dura overlying the transverse sinus with blunt forceps and von Frey hairs. After mapping of the receptive field, responses were recorded to threshold and suprathreshold intensities of mechanical stimulation delivered with a servo force-controlled mechanical stimulator (Series 300B Dual Mode Servo System, Aurora Scientific) (Figure 2(a),(b)). Only neurons for which a mechanical receptive field could be identified were selected for study.
Ongoing discharge activity was recorded continuously throughout the experiment. After mapping the neuron's dural receptive field, ongoing activity was recorded for a period of 40–60 mins for determination of baseline firing rate. A single wave of CSD was then induced by pinprick (briefly inserting a metal microelectrode manually into the cortex) through the caudal burr hole. Induction of CSD was confirmed by recording of the propagating CSD wave with the recording micropipette in the frontal cortex, approximately 5 mm rostral to the site of the pinprick (Figure 2(c)). Recording of ongoing discharge continued for 2–3 hours following induction of CSD.
Data analysis
Firing rate was analyzed in 1-min bins. A neuron was considered to have a response to CSD if its firing rate increased above baseline by at least two standard deviations for a period of at least 10 min (Figures 3, 4, 5(a)). The total firing that exceeded this level was calculated as a measure of response amplitude. Response latency was also measured. Neurons from BoNT-A-treated versus saline-treated animals were compared for percentage of neurons with a CSD response (Chi-square) and response amplitude (Mann-Whitney).
Figure 3.

Responses of C-fibers to CSD. Plots of firing rate before and after induction of CSD, for two C-fibers with CSD responses in a saline-treated (upper) and a BoNT-A-treated (middle) animal, and for a C-fiber that showed no CSD response (lower). The horizontal dotted line marks the firing level two standard deviations above the baseline rate.
Figure 4.

Responses of Aδ-fibers to CSD. Same format as Figure 3.
Figure 5.

Long-latency responses to CSD. (a) Example of a C-fiber with a relatively long latency response to CSD. The horizontal dotted line marks the firing level two standard deviations above the baseline rate. (b) Percentage of neurons that had latencies shorter or longer than 30 mins. (c) Scattergram plotting the latencies for all of the neurons with CSD responses.
Results
The effect of extracranial injection of BoNT-A along the suture lines on responses of meningeal nociceptors to CSD was examined in female rats. Single unit recordings were obtained from meningeal nociceptors (29 C-fiber and 26 Aδ) in the trigeminal ganglion 7 to 14 days after treatment with BoNT-A (n = 16 C-fibers, 15 Aδ) or saline (n = 13 C-fibers, 11 Aδ).
Baseline firing rates prior to CSD did not significantly differ between neurons recorded in BoNT-A versus saline-treated animals (BoNT-A, 0.7 Hz, [IQR: 0.3–1.2]; saline, 0.8 Hz, [IQR: 0.3–1.7]; p = 0.58). There was no significant difference in the baseline firing rates between BoNT-A-treated Aδ neurons (0.7 spikes/sec [IQR: 0.5–1.2]) and saline-treated Aδ neurons (1.0 zspikes/sec [IQR: 0.7–1.7]) (p = 0.16). Similarly, the BoNT-A treated C-fibers (0.7 spikes/sec [IQR: 0.2–1.2]) were comparable to the saline-treated group (0.7 spikes/sec [IQR: 0.06–1.8]) (p = 0.64).
Incidence of CSD responses
Pretreatment with BoNT-A had no significant effect on the percentage of neurons that were activated by CSD, in either the Aδ or the C-fiber population. Following CSD, an increase in firing of at least 2 standard deviations above baseline (see Methods) was observed in 12/31 (38%) neurons (combining C and Aδ) in BoNT-A-treated animals and 11/24 (45%) neurons in saline-treated animals (χ2, p = 0.60). Responses to CSD were observed in 5/15 (33%) Aδ neurons in the BoNT A-treated group and 4/11 (36%) Aδ neurons in the saline-treated group [χ2 (1) = 0.0257, p = 0.8725]. Time to onset of activation was shorter than 10 min in three (33%) of the nine activated Aδ neurons, and longer than 10 min in the remining six (66%) neurons. CSD activated 7/16 (43%) C-fibers in the BoNT A-treated group and 7/13 (53%) C-fibers in the saline-treated group [χ2 (1) = 0.292, p = 0.588]. Time to onset of activation was shorter than 10 min in nine (64%) of the 14 activated C neurons, and longer than 10 min in the remining five (36%) neurons.
Amplitude of CSD responses
Although there was no effect of BoNT-A on the percentage of neurons that met our criterion for a CSD response, it was apparent that there was a large variation in the amplitude of these responses, in that some CSD responses seemed to be larger than had been observed in previous studies of meningeal nociceptors (which were all done in male rats, rather than females as in the present study). We therefore investigated whether there might be an effect of the drug on CSD response amplitude, which was not done in previous studies. It was also noticed during the experiments that some responses were exceptionally prolonged, continuing and even increasing beyond the typical 1hr post-CSD recording period that had been used in prior studies, and so after seeing this we increased our standard recording period to 3 hours post-CSD. Inspection of activity profiles obtained with these longer recording periods revealed that the CSD responses could be complex, showing multiple phases or “waves” of activation separated by intervals at baseline levels, rather than a single episode of increased firing (Figures 3, 4). Some neurons had longer response latencies than had been found in previous studies ( > 40 min post-CSD) (e.g. Figure 5(a)), and the greatest firing typically did not occur until later than 1 hour.
Since all neurons that were eventually classified as activated had started their increase in firing frequency by around 1 hour post-CSD, we decided to examine the interval of 60–180 mins for the analysis of response amplitude. This was done to avoid comparing data from a time period in which some activated neurons are in their pre-activation stage while others are already activated. Responses are reported as the total firing (total number of spikes) during the 2-hour recording period that was more than two standard deviations above the baseline level (as analyzed in 1-min bins, see Methods). In this time period, there was a reduced level of activation in the BoNT-A-treated animals (median 597, [IQR: 413–1188], n = 11) as compared to the saline-treated animals (median 2452, [IQR 989–3667], n = 7) (p < 0.01, Mann-Whitney), in the combined sample of Aδ and C-fibers (Figure 6). When separated by class, a significant effect of treatment was found in the C-fibers where there was a reduced level of activation in the BoNT-A treated animals (520 spikes [IQR: 261–1326], n = 6) as compared to the saline-treated animals (1990 spikes [IQR: 1168–3299], n = 4) (p = 0.036). The sample size for the Aδ−fibers was not powered sufficiently to allow statistical analysis.
Figure 6.
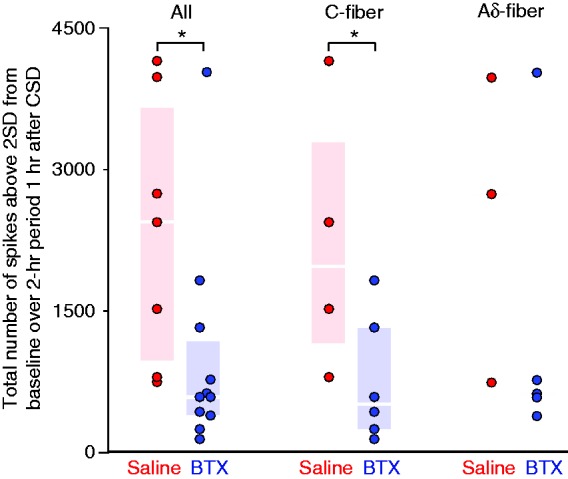
CSD response amplitude. Plots of the values for the individual neurons are shown for the C-fibers and Aδ-fibers combined (a), the C-fibers (b) and the Aδ-fibers (c). Box-and-whisker plots (median and inter-quartile range) are omitted from (c) because the Aδ sample was too small for statistical analysis. Asterisks indicate significant differences between the saline and BoNT-A-treated neurons. Only neurons that were recorded for at least 3 hours post-CSD are included in the analysis of response amplitude. Response amplitude was quantified as the activity that was more than two standard deviations above the baseline rate, during the interval 60–180 min post-CSD (see Methods), and is expressed here as total spikes (in other words, the number of spikes that equals two standard deviations above the mean, calculated during the baseline period in 1-min bins, was subtracted from each 1-min bin. The values from all bins that showed a positive number after this subtraction were summed for the interval 60–180 mins post-CSD).
Discussion
This study demonstrates that extracranial injections of BoNT-A inhibit the activation of meningeal nociceptors by CSD, a cortically mediated event occurring inside, rather than outside, the bbb. It also documents CSD effects on meningeal nociceptors in female rather than male rats. Although based on a small number of neurons in female rats, responses to CSD were typically more prolonged and, in some cases, began at relatively longer latencies post-CSD than had been observed in previous studies, which were all done in male rats (19–21). Relevant to our understanding of the BoNT-A mechanism of action in migraine prevention are the findings that extracranial administration of BoNT-A reduces significantly the prolonged firing of the unmyelinated C-fibers, while our findings on A-δ fibers are inconclusive. This finding of an effect on C-fibers is in agreement with a number of clinical (12–14) and pre-clinical (11, 14–16) studies that point to an inhibitory effect of BoNT-A on unmyelinated C-fiber nociceptors. Given our recently-published studies showing that an anti-CGRP monoclonal antibody prevents CSD-induced activation of Aδ- but not C-fibers (20), BoNT-A direct effects on the C-fibers may justify attempts to invent multi-therapy approaches that combine drugs that directly inhibit all, rather than one class of meningeal nociceptors.
This is the first scientific attempt to determine whether activation of meningeal nociceptors by a stimulus that originates inside (i.e. CSD), rather than outside (e.g. inflammatory soup) the bbb can be attenuated by extracranial injections of BoNT-A. Unlike BoNT-A's robust attenuation of responses to dural stimulation with inflammatory mediators (15), capsaicin and mustard oil (16), the attenuation of neuronal responses to CSD was far more moderate. Rather than reducing the percentage of activated nociceptors, BoNT-A reduced their total firing over a 2-hour period that started 1 hour after induction of CSD. A logical way to explain this moderate BoNT-A effect on the studied nociceptors is that, unlike common analgesics and CGRP monoclonal antibodies or CGRP antagonists, BoNT-A is not a systemically-acting drug. As such, it is expected to attenuate only those meningeal nociceptors whose axons bifurcate from their parent axons in the dura and issue collateral branches that exit the skull through the sutures (6–8) or axons of C2-3 dorsal root ganglion that issue branches that enter the skull and innervate the posterior dura (9). Such a scenario will dilute the overall impact of BoNT-A on the studied population of meningeal nociceptors.
In spite of the mild effect of BoNT-A on the unmyelinated C-fibers in this model, the reduction in the total number of action potentials that reached > 2 SD (of the baseline mean) during the analyzed period (60–180 min post CSD) was 1416 (1980 spikes in the saline group vs. 564 spikes in the BoNT-A group) – a 72% reduction. If meningeal C-fibers maintain this low level of activity throughout the headache phase of a migraine attack (which, although lacking direct evidence, is reasonable to suggest as peripherally acting drugs and procedures commonly terminate acute migraine attacks), these numbers translate to a reduction of nearly 17,000 action potentials (23,760 spikes in the saline group vs. 6768 spikes in the BoNT-A group) for attacks that last 24 hours. Given that C-fiber activity of more than 0.4 spikes per second is sufficient to induce the perception of pain (22), it is reasonable to propose that, although seemingly mild, BoNT-A's ability to significantly reduce the total number of action potentials that are generated in the C-fibers and eventually transmitted to the spinal trigeminal nucleus is likely to be therapeutically relevant.
Finally, it is worth noting that BoNT-A did not prevent the occurrence of CSD. Translating these findings to possible clinical implications, one would predict that if such a scenario were to hold true in patients, it is likely that some migraine aura patients may continue to experience aura but not the headache that follows. The current study justifies conducting a parallel clinical study as it may strengthen the translational power of the current (preclinical) findings.
In summary, this study raises the possibility that the BoNT-A mechanism of action in preventing the headache involves mild but prolonged reduction in the overall firing of unmyelinated C-, while the question of a possible involvement of the thinly-myelinated Aδ-fibers is not yet resolved and requires further study.
Article highlights
The present study demonstrates that activation of meningeal nociceptors by CSD, an event that occurs inside the bbb, is attenuated by extracranial injections of BoNT-A.
BoNT-A's ability to reduce the prolonged firing of the nociceptors but not their response probability defines fundamental differences between the mechanism of action of systemically administered migraine prophylactics such as CGRP monocloncal antibodies and the locally administered BoNT-A.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Allergan funded parts of the study. MFB is an employee of Allergan and RB and SA are consultants to Allergan.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by a grant from Allergan, and NIH grants R37-NS079678, RO1 NS069847, RO1 NS094198 (RB).
References
- 1.Burstein R, Noseda R, Borsook D. Migraine: Multiple processes, complex pathophysiology. J Neurosci 2015; 35: 6619–6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dodick DW. A phase-by-phase review of migraine pathophysiology. Headache 2018; 58: S4–S16. [DOI] [PubMed] [Google Scholar]
- 3.Aurora SK, Dodick DW, Turkel CC, et al. OnabotulinumtoxinA for treatment of chronic migraine: Results from the double-blind, randomized, placebo-controlled phase of the PREEMPT 1 trial. Cephalalgia 2010; 30: 793–803. [DOI] [PubMed] [Google Scholar]
- 4.Diener HC, Dodick DW, Aurora SK, et al. OnabotulinumtoxinA for treatment of chronic migraine: Results from the double-blind, randomized, placebo-controlled phase of the PREEMPT 2 trial. Cephalalgia 2010; 30: 804–814. [DOI] [PubMed] [Google Scholar]
- 5.Blumenfeld AM, Silberstein SD, Dodick DW, et al. Insights into the functional anatomy behind the PREEMPT injection paradigm: Guidance on achieving optimal outcomes. Headache 2017; 57: 766–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kosaras B, Jakubowski M, Kainz V, et al. Sensory innervation of the calvarial bones of the mouse. J Comp Neurol 2009; 515: 331–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schueler M, Messlinger K, Dux M, et al. Extracranial projections of meningeal afferents and their impact on meningeal nociception and headache. Pain 2013; 154: 1622–1631. [DOI] [PubMed] [Google Scholar]
- 8.Schueler M, Neuhuber WL, De Col R, et al. Innervation of rat and human dura mater and pericranial tissues in the parieto-temporal region by meningeal afferents. Headache 2014; 54: 996–1009. [DOI] [PubMed] [Google Scholar]
- 9.Noseda R, Melo-Carrillo A, Nir RR, et al. Non-trigeminal nociceptive innervation of the posterior dura: Implications to occipital headache. J Neurosci 2019; 39: 1867–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dolly JO, Aoki KR. The structure and mode of action of different botulinum toxins. Eur J Neurol 2006; 13: S1–S9. [DOI] [PubMed] [Google Scholar]
- 11.Gazerani P, Au S, Dong X, et al. Botulinum neurotoxin type A (BoNTA) decreases the mechanical sensitivity of nociceptors and inhibits neurogenic vasodilation in a craniofacial muscle targeted for migraine prophylaxis. Pain 2010; 151: 606–616. [DOI] [PubMed] [Google Scholar]
- 12.Gazerani P, Pedersen NS, Staahl C, et al. Subcutaneous botulinum toxin type A reduces capsaicin-induced trigeminal pain and vasomotor reactions in human skin. Pain 2009; 141: 60–69. [DOI] [PubMed] [Google Scholar]
- 13.Gazerani P, Staahl C, Drewes AM, et al. The effects of botulinum toxin type A on capsaicin-evoked pain, flare, and secondary hyperalgesia in an experimental human model of trigeminal sensitization. Pain 2006; 122: 315–325. [DOI] [PubMed] [Google Scholar]
- 14.Paterson K, Lolignier S, Wood JN, et al. Botulinum toxin-A treatment reduces human mechanical pain sensitivity and mechanotransduction. Ann Neurol 2014; 75: 591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burstein R, Zhang X, Levy D, et al. Selective inhibition of meningeal nociceptors by botulinum neurotoxin type A: Therapeutic implications for migraine and other pains. Cephalalgia 2014; 34: 853–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Strassman AM, Novack V, et al. Extracranial injections of botulinum neurotoxin type A inhibit intracranial meningeal nociceptors' responses to stimulation of TRPV1 and TRPA1 channels: Are we getting closer to solving this puzzle?. Cephalalgia 2016; 36: 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pietrobon D, Moskowitz MA. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci 2014; 15: 379–393. [DOI] [PubMed] [Google Scholar]
- 18.Lauritzen M, Strong AJ. ‘Spreading depression of Leao’ and its emerging relevance to acute brain injury in humans. J Cereb Blood Flow Metab 2017; 37: 1553–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao J, Levy D. The CGRP receptor antagonist BIBN4096 inhibits prolonged meningeal afferent activation evoked by brief local K(+) stimulation but not cortical spreading depression-induced afferent sensitization. Pain Rep 2018; 3: e632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melo-Carrillo A, Strassman AM, Nir RR, et al. Fremanezumab-A humanized monoclonal anti-CGRP antibody-inhibits thinly myelinated (Adelta) but not unmyelinated (C) meningeal nociceptors. J Neurosci 2017; 37: 10587–10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X, Levy D, Noseda R, et al. Activation of meningeal nociceptors by cortical spreading depression: Implications for migraine with aura. J Neurosci 2010; 30: 8807–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Hees J, Gybels J. C nociceptor activity in human nerve during painful and non painful skin stimulation. J Neurol Neurosurg Psychiatry 1981; 44: 600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]