

Abstract
We have shown that Na/K-ATPase interacts with Src. Here, we test the role of this interaction in H2O2-induced activation of Src and ERK. We found that exposure of LLC-PK1 cells to H2O2 generated by the addition of glucose oxidase into the culture medium activated Src and ERK1/2. It also caused a modest reduction in the number of surface Na/K-ATPases and in ouabain-sensitive Rb+ uptake. These effects of H2O2 seem similar to those induced by ouabain, a specific ligand of Na/K-ATPase, in LLC-PK1 cells. In accordance, we found that the effects of H2O2 on Src and ERK1/2 were inhibited in α1 Na/K-ATPase-knockdown PY-17 cells. Whereas expression of wild-type α1 or the A420P mutant α1 defective in Src regulation rescued the pumping activity in PY-17 cells, only α1, and not the A420P mutant, was able to restore the H2O2-induced activation of protein kinases. Consistent with this, disrupting the formation of the Na/K-ATPase/Src complex with pNaKtide attenuated the effects of H2O2 on the kinases. Moreover, a direct effect of H2O2 on Na/K-ATPase-mediated regulation of Src was demonstrated. Finally, H2O2 reduced the expression of E-cadherin through the Na/K-ATPase/Src-mediated signaling pathway. Taken together, the data suggest that the Na/K-ATPase/Src complex may serve as one of the receptor mechanisms for H2O2 to regulate Src/ERK protein kinases and consequently the phenotype of renal epithelial cells.
Keywords: Na/K-ATPase, Mutant, Hydrogen peroxide, Src, ERK, Free radicals
The Na/K-ATPase is a transmembrane enzyme responsible for pumping Na+ and K+ ions across the plasma membrane by hydrolyzing ATP [1]. Recent studies have revealed many non-pumping functions of the Na/K-ATPase, especially the α1 isoform [2]. For example, the α1 Na/K-ATPase is involved in the formation of membrane structures such as caveolae and tight junctions [3,4]. Moreover, the α1 Na/K-ATPase tethers and regulates multiple protein and lipid kinases as well as membrane receptors (e.g., Src, epidermal growth factor receptor (EGFR), and PI3K) [2,5]. Specifically, we have reported that the α1 subunit of Na/K-ATPase interacts with Src via two pairs of domain interactions [6]. Functionally, these interactions allow a cell to regulate Src and Src effectors through at least two potential pathways. First, changes in the expression of either α1 or Src would alter the interaction equilibrium between these two proteins, resulting in changes in Src activity because these interactions keep Src in an inactive state [7]. In accordance, a decrease in the α1 expression correlates with a large increase in Src activity in cultured cells as well as in vivo [7–9]. Moreover, we have recently demonstrated that the expression of α1 Na/K-ATPase in the plasma membrane is significantly reduced in highly proliferative cells in which an increase in active Src is detected [10]. Second, the Na/K-ATPase/ Src complex functions as a receptor for cardiotonic steroids and other pump ligands [11]. Based on early studies, the α1 Na/K-ATPase adopts two major distinct conformations, namely E1 and E2 [12]. The equilibrium between E1 and E2 could be regulated by many factors including pump substrates and cardiotonic steroids such as ouabain [12]. Whereas E1 Na/K-ATPase keeps Src in an inactive state, conversion of E1 to E2 by ouabain activates the Na/ K-ATPase-associated Src [11]. It is important to note that normal epithelial cells express millions of α1 Na/K-ATPases (roughly five times the amount of Src) [13]. Therefore, the Na/K-ATPase/Src interaction could represent a critical mechanism of Src regulation [13,14], working in concert with the other well-established pathways, including tyrosine phosphorylation of Src [15,6].
Src is the prototypic member of a family of nonreceptor tyrosine kinases that associate with the plasma membrane. It contributes to the regulation of several important cellular activities such as cell proliferation, survival, motility, and migration through activation of multiple protein and lipid kinase cascades via phosphorylation-mediated pathways [16]. Src contains a positive-regulatory phosphorylation site at Tyr418 (Y418) and a negative phosphorylation site at Tyr529 (Y529). Phosphorylation of Y418 induces a full activation of Src. Many intracellular and extracellular stimuli affect Src activities [15]. One class of these regulators is reactive oxygen species (ROS) [17].
“Reactive oxygen species” refers to a group of oxygen derivatives, including superoxide, H2O2, and hydroxyl radical. Although excessive production of ROS could induce DNA damage, oxidation of protein and lipids, and cell death, recent studies have demonstrated ROS to be important second messengers [18]. For example, we have shown that increases in ROS are required for ouabain-induced growth stimulation of cardiac myocytes [19,20] and marinobufugenin-induced collagen production of cardiac fibroblasts [21]. ROS have also been shown to regulate renal proximal tubule sodium handling and contribute to renal fibrosis, diabetic nephropathy, and hypertension [22,23]. Furthermore, as second messengers, ROS exerts many regulatory effects on key signaling proteins [17]. For example, increases in cellular ROS could activate protein kinase cascades, including the transactivation of EGFR and consequently the activation of extracellular signal-regulated kinase 1/2 (ERK1/2). Interestingly, inhibition of Src by chemical Src inhibitors (such as PP2) or the expression of dominant-negative mutant of Src or Src RNAi attenuates ROS-induced activation of these kinases [17,24]. These findings point to Src as a key upstream target of ROS-initiated signal transduction [25,26]. Although direct oxidation/reduction of Src by ROS appears to be involved in regulation of Src activity, there is also evidence that ROS could regulate Src by affecting the interaction between Src and its regulatory proteins [17]. To this end, it is known that ROS affects the Na/K-ATPase directly [20,27–30]. The fact that the Na/K-ATPase is a major regulator of Src in epithelial cells led us to propose that the Na/K-ATPase could be involved in ROS-mediated regulation of Src and consequently other protein kinases such as ERK1/2. To test this hypothesis, we have measured the effects of H2O2 on cellular Na/K-ATPase, Src, and ERK1/2 in control LLC-PK1 cells, Na/K-ATPase-knockdown cells, and both wild-type and Src regulation-defective α1 mutant-rescued cells. These studies suggest that the Na/K-ATPase/Src complex plays an important role in ROS-induced activation of Src and ERK1/2.
Material and methods
Materials
The antibodies used in this work and their resources were as follows: the polyclonal anti-ERK1/2 antibody, the monoclonal anti-phospho-ERK1/2 antibody, the monoclonal anti-c-Src antibody (B12), the monoclonal anti-insulin receptor β subunit antibody, and the anti-rabbit- and the anti-mouse-conjugated horseradish peroxidase second antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA); the polyclonal anti-phospho-Src pY418 and the polyclonal anti-phospho-Src pY529 antibodies were obtained from Invitrogen (Carlsbad, CA. USA); the monoclonal anti-α1 antibody α6 f was obtained from the Developmental Studies Hybridoma Bank at the University of Iowa and used for Western blot; the recombinant human Src was from Upstate (Lake Placid, NY, USA). Chemiluminescence ECL kit was purchased from PerkinElmer (Rockford, IL, USA). [3H]Ouabain was purchased from DuPont NEN (Boston, MA, USA). Radioactive 86Rb+ was from PerkinElmer Life Science Products (Boston, MA, USA). Other chemicals of the highest purity were all obtained from Sigma–Aldrich (St. Louis, MO, USA).
Cell culture and treatment
The pig renal proximal tubule cell line, LLC-PK1, was obtained from the American Type Culture Collection and cultured as described before [31]. After reaching 80–90% confluence, cells were serum-starved overnight before they were used for experiments. PY-17 cells were generated from LLC-PK1 cells that were stably transfected with α1-specific small interfering RNA (siRNA) [7]. AAC-19 cells were derived from PY-17 cells rescued by wild-type rat α1 carrying silent mutations of siRNA-targeting sequence [7]. LL-A416P-4 cells and LL-A420P-20 cells were derived from PY-17 cells rescued by A416P and A420P rat α1 mutants, respectively [14].
Assay of cell surface expression of Na/K-ATPase by [3H]ouabain binding and pumping activity by ouabain-sensitive 86Rb+ uptake
Cell surface Na/K-ATPase content was determined using the [3H]ouabain binding assay as we previously reported [8]. Briefly, cells were cultured in 12-well plates, serum-starved, and treated with glucose oxidase. Afterward, the cells were incubated in K+-free Krebs solution (NaCl 137 mM, CaCl2 2.8 mM, NaH2PO4 0.6 mM, MgSO4 1.2 mM, dextrose 10 mM, Tris 15 mM, pH 7.4) for 15 min and then exposed to 200 nM [3H]ouabain for 30 min at 37 °C in the presence of 20 μM monensin. At the end of incubation, the cells were washed three times with ice-cold K+-free Krebs solution, solubilized in 0.1 M NaOH–0.2% (w/v) SDS, and counted in a scintillation counter for [3H]ouabain. Nonspecific binding was measured in the presence of 1 mM unlabeled ouabain and subtracted from total binding. [3H]Ouabain binding data were calculated as binding sites per milligram of protein. To assess the pumping activity, cells were cultured in 12-well plates and 86Rb+ uptake was performed as previously described [8]. All counts were normalized to protein amount.
Western blot and immunoprecipitation
Immunoblotting and Src immunoprecipitation were performed as described previously [32,33]. Activation of Src and ERK1/2 was determined by using anti-phospho-Src and anti-phospho-ERK1/2 antibodies as we previously described [8]. The same blots were stripped and probed with antibodies that recognize the total amount of Src and ERK1/2 to account for equal loading. Detection was performed using the chemiluminescence ECL kit. Films were scanned and quantified using ImageJ software.
Cell surface biotinylation and streptavidin precipitation
Biotinylation assay was performed according to Liang et al. [8]. Cells were cultured on 60-mm petri dishes until they reached 90% confluence. The cells were then rinsed twice with ice-cold PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) and incubated with 2 ml of NHS-SS-biotin (1.5 mg/ml) freshly dissolved into biotinylation buffer (10 mM triethanolamine, 150 mM NaCl, pH 9.0) for 25 min at 4 °C on a gently shaking rocker. After being rinsed twice with PBS containing 100 mM glycine, the cells were incubated in the same buffer for 20 min at 4 °C to quench the unreacted biotin and solubilized in 400 μl of lysis buffer (1% (v/v) Triton X-100, 150 mM NaCl, 5 mM EDTA, 50 mM Tris, pH 7.5) for 60 min on a rocker with gentle motion. Cell lysates were collected by centrifugation at 16,000g for 10 min. The cell lysates (250 μg) were incubated with 150 μl of streptavidin– agarose beads in lysis buffer in a total volume of 800 μl at 4 °C with end-over-end rotation overnight. The pellet was collected, rinsed, resuspended in 50 μl of Laemmli loading buffer, and subjected to Western blot analysis.
Preparation of purified Na/K-ATPases
Purified Na/K-ATPase preparations from pig kidney outer medulla, with specific activities between 1000 and 1400 μmol Pi/mg/h, were obtained using a modified Jorgensen method as previously described [34].
Assays of Src autophosphorylation
Purified Na/K-ATPase was exposed to 10 mM H2O2 in 50 mM Tris–HCl (pH 7.4) buffer for 30 min at 37 °C. One unit of catalase was added into the system to stop the reaction. Treated Na/K-ATPase was then incubated with 1 μl of purified Src for 15 min at 37 °C in a solution containing 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 5 mM KCl, and 100 μM vanadate. Reaction was started by the addition of 2 mM Mg2+/ATP. The reaction continued for 10 min and was stopped by adding SDS sample buffer. The Src activity was determined by phosphorylation of Src Tyr418 using immunoblot analysis.
Statistical analysis
Data are expressed as the mean±SE. Statistical analysis was performed using the Student t test and significance was accepted at p<0.05. When more than two groups were compared, one-way analysis of variance was performed before comparison of individual groups with the unpaired Student t test. Each presented immunoblot is a representative of similar results of at least three separate experiments. SPSS software (Chicago, IL, USA) was used for all analyses.
Results
Effects of H2O2 on the α1 Na/K-ATPase in LLC-PK1 cells
To study the effect of an increase in cellular H2O2 on LLC-PK1 cells, we adopted a previously established protocol by treating cells with various concentrations of glucose oxidase [20,35]. Glucose oxidase produces H2O2 in the presence of glucose in culture medium, which results in a modest but sustained rise in intracellular H2O2. Control experiments confirmed that glucose oxidase produced sustained increases in intracellular H2O2 in a dose- and time-dependent manner in LLC-PK1 cells (Supplementary Figs. 1A and B). The maximal H2O2 concentration in cell culture produced by 3 mU/ml of glucose oxidase was about 4 μM, much less than that of the addition of a bolus 25 μM H2O2 as we reported in the culture of cardiac myocytes [20].
To select a nontoxic dose of glucose oxidase for the experiments in LLC-PK1 cells, we measured lactate dehydrogenase (LDH) release after cells were exposed to various doses of glucose oxidase as a function of time. As depicted in Supplementary Fig. 1C, no increase in LDH release was detected when cells were exposed to 3 mU/ml glucose oxidase for up to 24 h. However, when cells were treated with 5 or 10 mU/ml glucose oxidase, a significant increase in LDH release was detected in a dose- and time-dependent manner. Based on the above findings, we used 3 mU/ml glucose oxidase as the highest dose for the following experiments.
In our previous studies we have shown that the amount of α1 Na/ K-ATPase on the plasma membrane regulates cellular Src activity [7]. Knockdown of α1 Na/K-ATPase by siRNA led to increased Src activity, and knock-in of α1 Na/K-ATPase later brought the Src activity to the normal level [7]. To test the role of α1 Na/K-ATPase in H2O2-induced activation of protein kinases, we determined the effect of H2O2 on α1 Na/K-ATPase in LLC-PK1 cells. We previously showed that H2O2 produced by glucose oxidase caused a time-dependent inhibition of Na/K-ATPase in the culture of cardiac myocytes where at least two different isoforms of Na/K-ATPase are expressed [20]. In contrast to cardiac myocytes, LLC-PK1 cells express only the α1 isoform of Na/K-ATPase. First, we measured ouabain-sensitive 86Rb+ uptake to assess the effect of H2O2 on the pumping function of Na/K-ATPase. As depicted in Fig. 1A, 3 mU/ml glucose oxidase produced a modest inhibition of ouabain-sensitive 86Rb+ uptake in a time-dependent manner. The maximal inhibition was seen at 60 min of exposure and the inhibition lasted for 24 h. To assess whether H2O2 is capable of reducing the surface expression of Na/K-ATPase in LLC-PK1 cells, we measured [3H]ouabain binding. Like its effect on 86Rb+ uptake, a small but statistically significant reduction was detected after 60 min of glucose oxidase exposure (Fig. 1B). Again, this decrease persisted for at least 24 h. To verify the effect of H2O2 on the surface expression of Na/K-ATPase, we treated cells with glucose oxidase for 24 h and performed biotinylation analyses. As shown in Fig. 1C, surface expression of Na/K-ATPase was reduced about 30%, which is comparable to that as measured by [3H]ouabain binding (Fig. 1B). Moreover, this H2O2-induced membrane receptor downregulation was specific to the Na/K-ATPase because the amount of insulin receptor β subunit remained unaltered as evidenced by the same biotinylation analysis (Fig. 1C). Interestingly, H2O2 exposure up to 24 h had no effect on total cellular content of α1 subunit as measured by Western blot analyses of cell lysates (Fig. 1D). Decreases in total cellular amount of Na/K-ATPase did occur after the cells were exposed to glucose oxidase for 48 h (Fig. 1D).
Fig. 1.
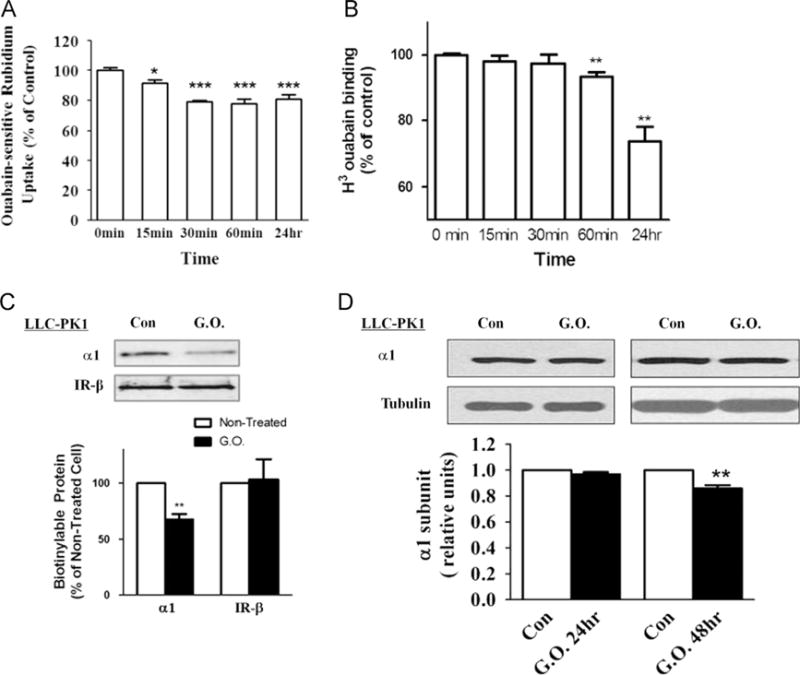
Effects of glucose oxidase (G.O.) treatment on Na/K-ATPase. (A) 86Rb+ uptake assay. Cells were treated with G.O. (3 mU/ml) for indicated times and ouabain-sensitive 86Rb+ uptake was measured as described under Material and methods. (B) [3H]Ouabain binding assay. Cells were treated with G.O. (3 mU/ml) for indicated times and surface expression of Na/K-ATPase was measured by ouabain binding as described under Material and methods. (C) Surface biotinylation assay. Cells were treated with G.O. (3 mU/ ml) for 24 h and biotinylated as described under Material and methods. The same volume of the bound fraction was analyzed by Western blot. (D) Representative immunoblots and quantification of Na/K-ATPase α1 subunit amount in total cell lysates. n=3–5. Values are expressed as the mean±SE. *p<0.05; **p<0.01; ***p<0.001 compared with control.
In short, the above control experiments showed that H2O2 produced by glucose oxidase was capable of affecting α1 Na/K-ATPase in LLC-PK1 cells, resulting in a decrease in surface expression of Na/K-ATPase.
H2O2 activates Src and ERK1/2 in LLC-PK1 cells
To monitor the changes in Src activity after the cells were exposed to glucose oxidase, we measured Src Y418 phosphorylation, an indicator of Src activation [6,7], by Western blot. As depicted in Fig. 2A and B, there appeared to be a two-phase regulation of Src activity by glucose oxidase. The initial detectable increase occurred after the cells were exposed to glucose oxidase for 15 min and peaked after 30–60 min, following the similar time course of H2O2-induced inhibition of Na/K-ATPase (Fig. 1A). Afterward, the activation of Src gradually decreased after 6 h of exposure. The second phase of further activation occurred in 24 h (Fig. 2B). The similar time course pattern of H2O2 effect on both Src and Na/K-ATPase could be due to the increased endocytosis of α1 Na/K-ATPase by Src stimulation [36]. Consequently, reduced membrane α1 Na/K-ATPase would lead to a persistent Src activation as shown in Fig. 2B.
Fig. 2.
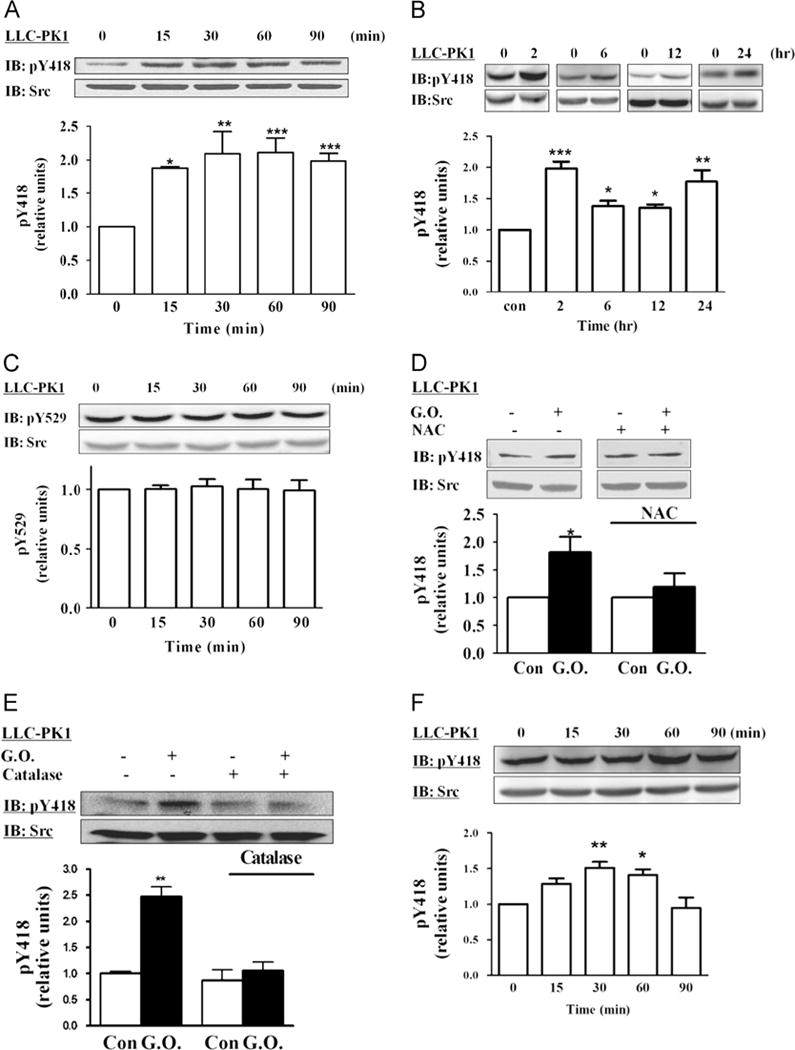
Effects of glucose oxidase (G.O.) on Src. (A) Time course of Src activation. LLC-PK1 cells were exposed to G.O. (3 mU/ml) for up to 90 min. Cell lysates were assayed for Src (pY418) activation by Western blot as described under Material and methods. Membranes were first probed for pY418 Src, then stripped, and reprobed by an anti-Src antibody that recognizes total Src. The active Src values were normalized against the total Src signal. The ratio was then expressed relative to control value of 1. (B) Long-term effects of G.O. on Src. Cells were exposed to G.O. (3 mU/ml) for up to 24 h, and cell lysates were assayed as for (A). (C) Cells were exposed to G.O. (3 mU/ml) for up to 90 min. Cell lysates were assayed for Src (pY529). (D) Cells were pretreated with 5 mM N-acetylcysteine (NAC) for 30 min and exposed to G.O. for 60 min, and cell lysates were assayed as for (A). (E) Cells were pretreated with catalase (50 U/ml) for 15 min and exposed to 3 mU/ml G.O. for 2 h, and cell lysates were assayed as for (A). (F) Cells were exposed to 50 μM H2O2 for up to 90 min, and cell lysates were assayed as for (A). Representative blots and quantitative data are shown. n=3–5. Values are expressed as the mean±SE. *p<0.05; **p<0.01; ***p<0.001 compared with control.
It has been suggested that H2O2 could inactivate protein phosphatase and therefore indirectly upregulate kinase activity [37]. However, there was no detectable change in the phosphorylation level of Src Y529 by the same treatment (Fig. 2C). To further verify the effect of H2O2 on Src, we probed for pY418 Src after we immunoprecipitated Src from the cell lysates by using a Src-specific monoclonal antibody. As depicted in Supplementary Fig. 2, the addition of glucose oxidase to culture medium produced a significant increase in the Src activation.
To be sure that the effect of glucose oxidase on Src is mediated by an increase in cellular ROS stress, we measured whether glucose oxidase-induced activation of Src is sensitive to N-acetylcysteine (NAC) and catalase. Cells were pretreated with either 5 mM NAC or 50 U/ml catalase and then exposed to glucose oxidase. As depicted in Figs. 2D and E, both NAC and catalase were effective in abolishing the effect of glucose oxidase on cellular Src. Moreover, pretreatment with α-tocopherol also blocked H2O2-induced Src activation (Supplementary Fig. 3). This specific effect of intracellular ROS was further confirmed when glucose was omitted from the culture medium (Supplementary Fig. 4) and by the addition of a bolus H2O2 to the medium (Fig. 2F). Taken together, these findings indicate that H2O2 produced by glucose oxidase could activate Src in LLC-PK1 cells.
It has been shown that the activation of Src by ouabain could consequently increase the recruitment of additional Src to the signaling complex [32,33,38]. To begin assessing the role of Na/K-ATPase in H2O2-induced Src activation, we treated the cells with glucose oxidase, immunoprecipitated Src from cell lysates, and probed for α1. As depicted in s, addition of glucose oxidase significantly increased the formation of Na/K-ATPase/Src signaling complex, like ouabain [21,32,36].
We have shown that activation of Na/K-ATPase/Src complex by ouabain stimulates ERK1/2 in LLC-PK1 cells. To further assess the role of Na/K-ATPase in H2O2-induced activation of protein kinases, we measured the effect of glucose oxidase on ERK1/2 activation by Western blot. As shown in Fig. 3A, H2O2 produced by glucose oxidase stimulated ERK1/2 in a time-dependent manner in LLC-PK1 cells. A significant increase was observed after cells were exposed to 3 mU/ml glucose oxidase for 30 min and this activation lasted for at least 2 h. Long-term exposure to glucose oxidase induced a modest but sustained ERK activation from 6 h up to 24 h in cells (Fig. 3B). Again, pretreatment of cells with NAC (Fig. 3C) or removal of glucose from medium (Supplementary Fig. 6) abolished H2O2-induced ERK1/2 activation, whereas the addition of a bolus H2O2 into the culture medium (Fig. 3D) was sufficient to activate ERK1/2. To further determine whether the Src activation was an upstream step of ERK1/2 signaling, we pretreated cells with PP2, a specific Src inhibitor. Western blot analyses indicate that PP2 completely blocked the activation of ERK1/2 induced by H2O2 in LLC-PK1 cells (Fig. 3E).
Fig. 3.
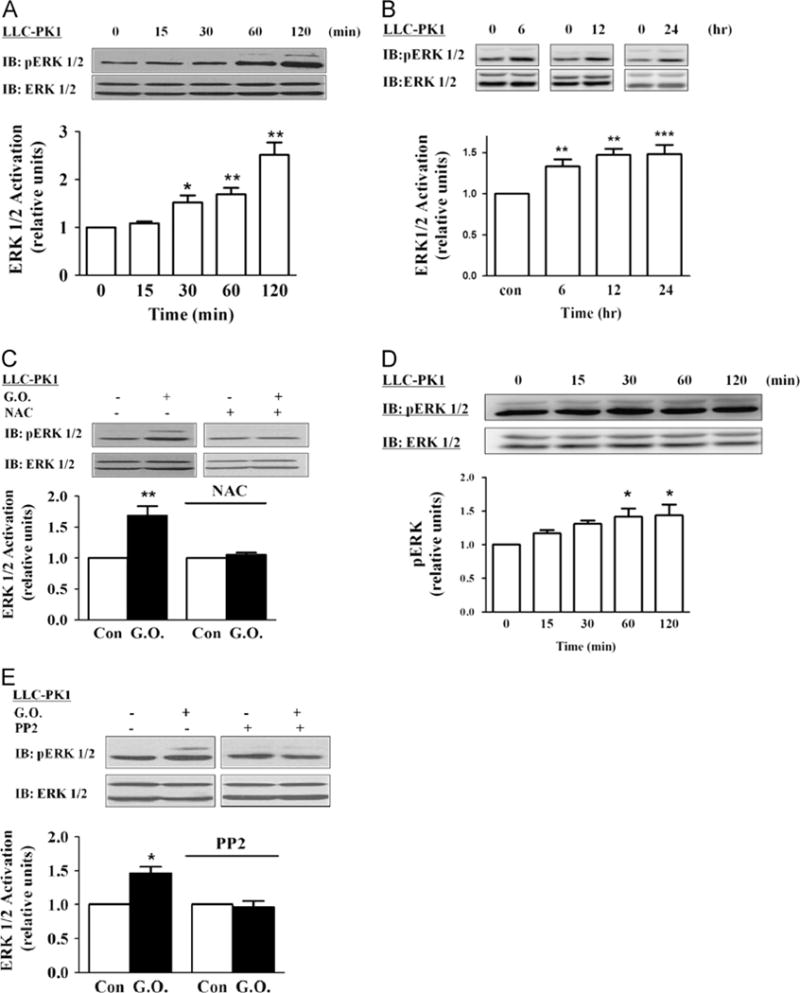
Effects of glucose oxidase (G.O.) on ERK1/2 activation. (A) Time course of ERK activation. Cells were exposed to G.O. (3 mU/ml) for the indicated times and assayed for ERK1/2 activation as described under Material and methods. Membranes were first probed with anti-phosphorylated (active) ERK antibody, then stripped, and reprobed by an anti-ERK antibody that recognizes total ERK. The active ERK values were normalized against the total ERK signal. The ratio was then expressed relative to the control value as 1. (B) Long-term effects of G.O. on ERK. Cells were exposed to G.O. (3 mU/ml) for up to 24 h, and cell lysates were assayed as for (A). (C) Cells were pretreated with 5 mM N-acetylcysteine (NAC) for 30 min and exposed to G.O. for 60 min, and cell lysates were assayed as for (A). (D) Cells were exposed to 50 μM H2O2 for up to 120 min. Cell lysates were assayed as for (A). (E) The effects of PP2 on G.O.-induced activation of ERK1/2. Cells were pretreated PP2 (2 μM) and exposed to 3 mU/ml G.O. for 60 min, and cell lysates were assayed as for (A). Representative immunoblots and quantitative data are shown. n=3–5. Values are expressed as the mean±SE. *p<0.05; **p<0.01; ***p<0.001 compared with control.
Antagonist of receptor Na/K-ATPase/Src complex is effective in inhibition of H2O2-induced activation of ERK1/2
To explore the potential involvement of Na/K-ATPase/Src receptor complex in H2O2-induced activation of Src and ERK1/2, we loaded cells with pNaKtide, a peptide derived from the N domain of Na/K-ATPase α1 subunit [39]. Our previous studies have demonstrated that the N domain of α1 subunit binds Src and inhibits Src by preventing Y418 phosphorylation. Mapping of the N domain led to the development of a cell-permeative pNaKtide that competes with the α1 Na/K-ATPase for Src binding, thus preventing the formation of a functional Na/K-ATPase/Src receptor complex [39]. As depicted in Fig. 4A and B, pNaKtide caused a large decrease in H2O2-induced activation of Src and ERK1/2 in LLC-PK1 cells.
Fig. 4.
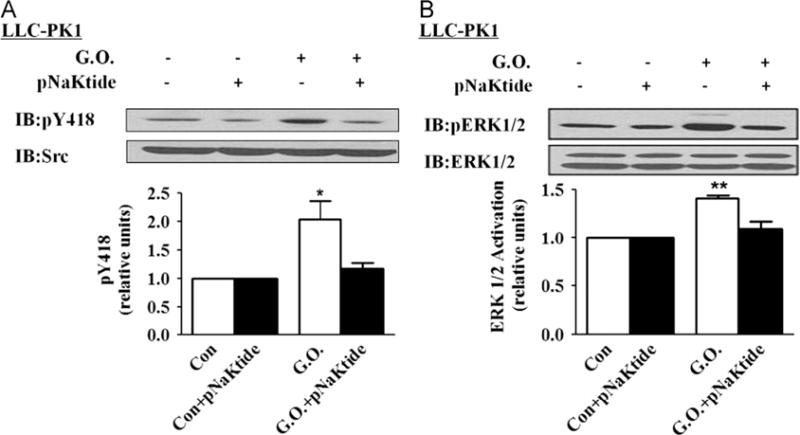
Effects of pNaKtide on glucose oxidase (G.O.)-induced Src and ERK1/2 activation in LLC-PK1 cells. Cells were exposed to pNaKtide (1 μM) for 1 h and then exposed to G.O. for 60 min. (A) A representative immunoblot and quantitative data of Src activation. (B) A representative immunoblot and quantitative data of ERK1/2 activation. n=5. Values are expressed as the mean±SE. *p<0.05; **p<0.01 compared with control.
Activation of the Src/ERK1/2 cascade by H2O2 is dependent on the expression of Na/K-ATPase
The above findings indicate that H2O2 may stimulate the Src/ERK1/2 cascade through the α1 Na/K-ATPase. If this is true, knockdown of α1 Na/K-ATPase should attenuate H2O2-induced activation of Src and ERK pathways. PY-17 cells were derived from LLC-PK1 cells stably transfected with α1-specific siRNA and the expression of endogenous Na/K-ATPase was reduced more than 90% (Fig. 5A and [8]). As depicted in Fig. 5B, knockdown of Na/K-ATPase completely abolished H2O2-induced activation of Src. Moreover, whereas EGF induced a robust activation of ERK1/2 in PY-17 cells, H2O2 failed to stimulate these kinases (Figs. 5C and D). Thus, the failure of H2O2 to activate ERK is unlikely to be due to a general defect in cellular signal transduction. It is also important to note that PY-17 cells expressed the same amount of total Src and ERK1/2 as in the control LLC-PK1 cells [7].
Fig. 5.
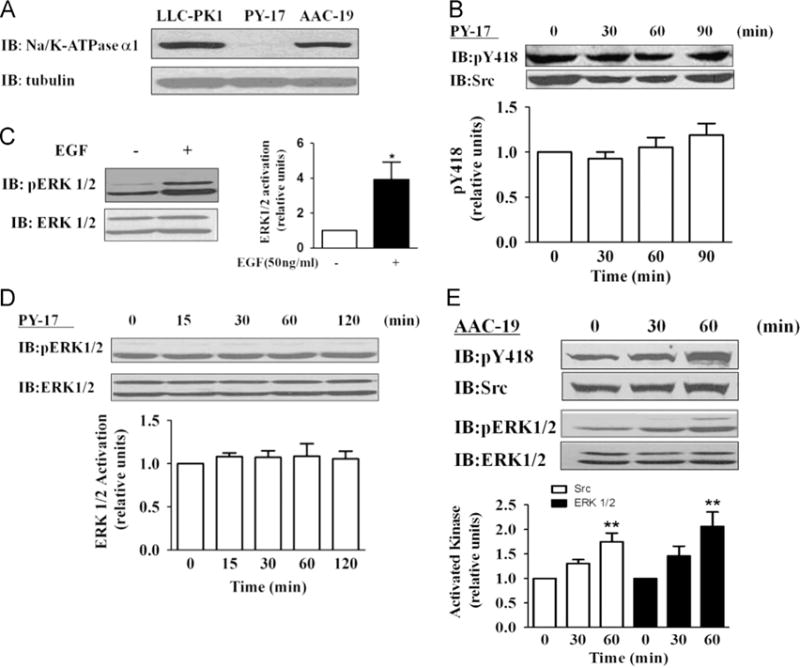
Effects of glucose oxidase (G.O.) on Src and ERK1/2 activation in α1-knockdown and -rescued cells. (A) A representative immunoblot showing α1 Na/K-ATPase expression in LLC-PK1, PY-17, and AAC-19 cells. (B) Effect of 3 mU/ml G.O. on Src in PY-17 cells. Cells were treated and assayed as for Fig. 2. EGF (10 ng/ml and 2 min exposure) was used as a control and its effect on ERK1/2 is presented in (C). (D) Effect of 3 mU/ml G.O. on ERK in PY-17 cells. Cells were treated and assayed as for Fig. 3. Similar studies were conducted in AAC-19 cells and the results are shown in (E). Representative immunoblots and quantitative data from at least three independent experiments are shown. Values are expressed as the mean±SE. n=3–5. *p<0.05; **p<0.01 compared with control.
To be sure that the inability of PY-17 cells to respond to H2O2 is because of the downregulation of α1 Na/K-ATPase, we repeated this set of experiments in AAC-19 cells. AAC-19 cells were derived from PY-17 cells and the expression of Na/K-ATPase was restored to a comparable level of LLC-PK1 by knocking in rat α1 cDNA (Fig. 5A). As depicted in Fig. 5E, expression of rat α1 restored H2O2-induced activation of both Src and ERK1/2 in AAC-19 cells.
Disruption of Na/K-ATPase/Src interaction abolishes H2O2-induced Src/ERK activation
We showed that knockdown of α1 Na/K-ATPase resulted in an increase in intracellular Na+ concentration and an overall inhibition of transmembrane ion transport activity [8]. Thus, it is plausible that these changes could affect Src regulatory mechanisms other than the Na/K-ATPase. To seek further support that the α1 Na/K-ATPase is directly involved in H2O2-induced activation of Src and ERK pathways, we determined the effect of glucose oxidase on these kinases in LL-A416P-4 and LL-A420P-20 cells. LL-A416P-4 and LL-A420P-20 cells were generated from PY-17 cells stably transfected with A416P and A420P mutant α1, respectively. The amount of mutant α1 Na/K-ATPase expressed in these cells was comparable to that of wild-type α1 in AAC-19 cells [14]. Moreover, they had the same pumping activity as measured by 86Rb+ uptake and showed the same ouabain sensitivity [14]. Thus, both A416P and A420P mutants exhibit normal pumping capacity. However, unlike the A416P mutant, the A420P mutant failed to interact with Src because A420P mutation alters the helical structure that is required for the Src interaction [14]. As such, the mutant α1 Na/K-ATPase could not form a functional receptor with Src as evidenced by the fact that ouabain failed to stimulate Src in LL-A420P-20 cells [14]. As depicted in Fig. 6A and B, when these cells were exposed to glucose oxidase, Src and ERK were activated in LL-A416P-4, but not LL-A420P-20 cells.
Fig. 6.
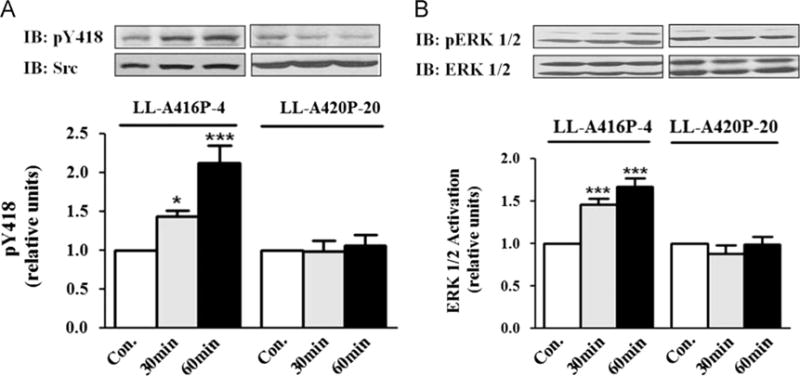
Effects of glucose oxidase (G.O.) on Src and ERK1/2 in mutant-rescued cells. (A) Cells were treated with 3 mU/ml G.O. and assayed for Src as for Fig. 2. Similar studies were conducted to detect ERK1/2 activation and the results are shown in (B). Representative immunoblots and quantitative data from at least three independent experiments are shown. Values are expressed as the mean±SE. n=3–5. *p<0.05; ***p<0.001 compared with control.
H2O2 activates Src in a cell-free system
To complement the above studies in cultured cells and further explore whether H2O2-modified Na/K-ATPase has altered properties of Src regulation, we adapted a previously established cell-free assay [6] and measured the effect of purified Na/K-ATPase on Src after H2O2 treatment. Prior experiments showed that 30 min exposure of purified kidney Na/K-ATPase to 10 mM H2O2 produced about 55% inhibition of ATPase activity [40], similar to 10 μM ouabain. Therefore, the same protocol was followed and afterward, 1 U of catalase was added to the reaction mixture to remove excess H2O2. The control and H2O2-pretreated enzyme were then incubated with purified Src to assess the effect of Na/K-ATPase on Src activity. To exclude the possibility that Src activity is affected because of reduced ATP hydrolysis capacity of H2O2-treated Na/K-ATPase, we performed the analyses in the presence of 100 μM vanadate, which completely inhibited the ATPase activity of Na/K-ATPase [6]. Prior kinetic studies have suggested that H2O2 induces E2-Na/K-ATPase accumulation, a conformation that is less effective at inhibiting Src [11,40,41]. Indeed, as depicted in Fig. 7, increases in Src activity were detected when Src was incubated with H2O2-pretreated Na/K-ATPase, which is similar to the result from ouabain-treated Na/K-ATPase.
Fig. 7.
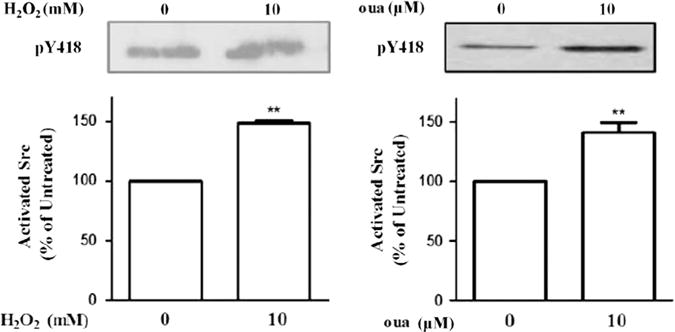
Effects of H2O2 on the α1 Na/K-ATPase-associated Src. Purified Na/K-ATPase was first exposed to 10 mM H2O2 for 30 min, then to 1 U catalase to decompose the excess H2O2. Pretreated Na/K-ATPase was incubated with purified Src in the presence of 150 mM NaCl, 5 mM KCl, 100 μM vanadate, 10 μM ouabain, as indicated, and assayed for Src Tyr418 phosphorylation as described under Material and methods. Representative immunoblots and quantitative data from at least three independent experiments are shown. n=3–5. Values are expressed as the mean±SE. **p<0.01 compared with control.
H2O2 represses E-cadherin expression
Finally, we assessed the effects of H2O2 on E-cadherin expression in LLC-PK1 cells. E-cadherin is a cell–cell adhesion molecule, and loss of E-cadherin is a key feature of the epithelial–mesenchymal transition and renal fibrosis. Long-term treatment with glucose oxidase for up to 24–48 h reduces the E-cadherin amount in LLC-PK1 cells (Fig. 8A). Similar to the activation of Src/ERK, ROS were the major mediators of glucose oxidase treatment, as the loss of E-cadherin was abolished by NAC (Fig. 8B). Both Src (PP2) and ERK (PD98059) inhibitors could prevent glucose oxidase-induced downregulation of E-cadherin as depicted in Figs. 8C and D, suggesting that the ROS-activated protein kinase cascades regulate E-cadherin expression. The ROS effect was further tested in AAC-19 cells and PY-17 cells. As expected, the reduction in E-cadherin was observed only in α1 Na/K-ATPase-rescued AAC-19 cells and not in the PY-17 cells (Fig. 8E and F).
Fig. 8.
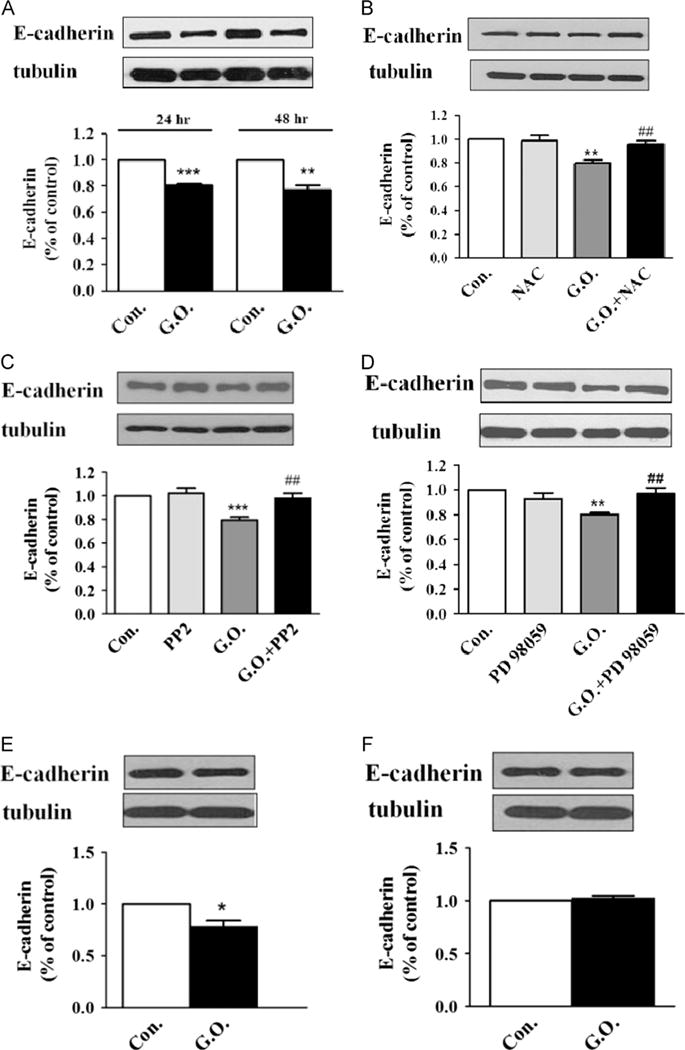
Effects of H2O2 on E-cadherin expression. (A) LLC-PK1 cells were exposed to glucose oxidase (G.O.; 3 mU/ml) for up to 48 h. Cell lysates were assayed for E-cadherin by Western blot. The E-cadherin amount was normalized against tubulin signal. The ratio was then expressed relative to a control value of 1. (B) Cells were pretreated with 5 mM N-acetylcysteine (NAC) for 30 min and exposed to G.O. for 24 h, and cell lysates were assayed as for (A). (C) and (D) Cells were pretreated with (C) PP2 (2 μM) or (D) PD 98059 and then exposed to 3 mU/ml G.O. for 24 h. Cell lysates were assayed as for (A). (E) and (F) Effects of 3 mU/ml G.O. and 24-h exposure on E-cadherin in (E) AAC-19 cells and (F) PY-17 cells. Cells lysates were assayed as for (A). Representative blots and quantitative data are shown. n=3–5. Values are expressed as the mean±SE. *p<0.05; **p<0.01; ***p<0.001 compared with control. ##p<0.05 compared with G.O.-treated group.
Discussion
In this work, we show that H2O2 activates Src/ERK1/2 in LLC-PK1 cells. Like ouabain, the effects of H2O2 on Src and ERK1/2 depend on the expression of Na/K-ATPase. Inhibition of the formation of Na/K-ATPase/Src complex by pNaKtide or by expression of A420P mutant α1 attenuates H2O2-induced activation of protein kinases. Moreover, the direct effect of H2O2 on Na/K-ATPase–Src interaction is detected in a cell-free system. Finally, H2O2 also induced functional changes in the cells as indicated by the loss of epithelial marker E-cadherin after glucose oxidase treatment. Taking these results together, we suggest that H2O2 may, at least partially, activate the Na/K-ATPase-associated Src and consequently protein kinase cascades through a mechanism similar to that of ouabain, namely the stabilization of E2-like conformation as schematically illustrated in Fig. 9. Unlike ouabain, however, the stabilization of the E2-like conformation is most likely through the oxidation of key residues in the α1 subunit as demonstrated by prior studies [22,29]. This and other important issues are further discussed below.
Fig. 9.
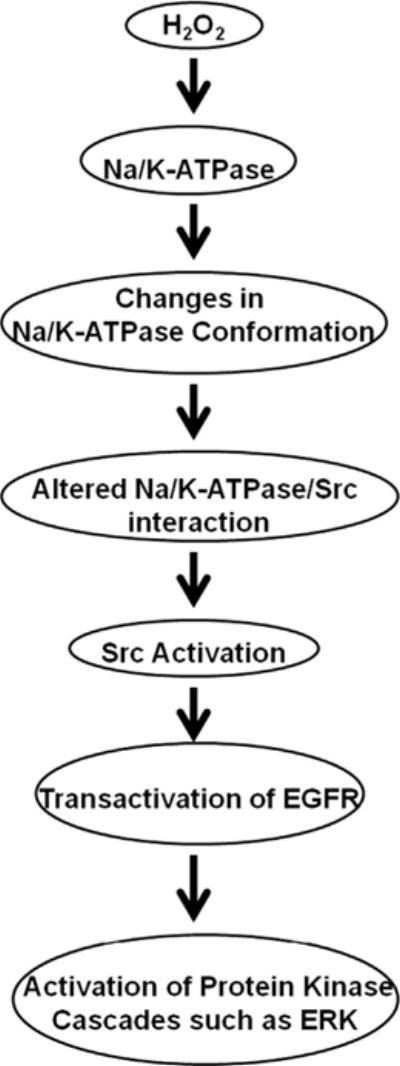
Schematic presentation of H2O2-induced signal transduction through the Na/K-ATPase/Src receptor complex.
Effects of ROS on Na/K-ATPase and Src
Like ouabain, ROS inhibits Na/K-ATPase. Early evidence comes from the studies of Na/K-ATPase in cardiac and renal tissues after ischemia [42,43]. This inhibition was further demonstrated in cell cultures and by using the purified Na/K-ATPase [27,44,45]. Whereas hydroxyl radicals fragment the α1 subunit of Na/K-ATPase, H2O2 appears to induce both inter- and intramolecular oxidation [44]. Interestingly, H2O2 seems to prefer the E2-like conformation of the Na/K-ATPase and inhibits E2 to E1 transition [40,41]. Moreover, several recent studies have demonstrated reversible modifications of residues of Na/K-ATPase by ROS [29]. Thus, H2O2 could have a ligand-like effect on the Na/K-ATPase. Needless to say, this remains to be tested experimentally.
Reports from several laboratories during the past 10 years have supported the notion that Src may be regulated by ROS. A reversible oxidation of Cys residues by H2O2 has been demonstrated in cells in which ROS production is increased, which seems to affect Src activity either directly or indirectly via its interacting proteins [17]. Interestingly, some of the oxidized residues are located in either the SH2 or the kinase domains of Src kinase, where the interaction between Na/K-ATPase and Src is also known to occur. Thus, it is conceivable that H2O2-induced changes in either Na/K-ATPase or Src or both might affect the interaction between Na/K-ATPase and Src, resulting in the activation of the receptor Na/K-ATPase/Src complex and consequently the downstream kinase cascade (e.g., ERK1/2) by transactivation of EGFR, like the actions of ouabain [32]. The findings depicted in Fig. 7 seem to support the notion that the direct effect of H2O2 on Na/K-ATPase is sufficient to stimulate the Na/K-ATPase-associated Src.
Na/K-ATPase signalosome and H2O2-induced activation of protein kinases
We have shown that there is a large nonpumping pool (about 67%) of Na/K-ATPase in LLC-PK1 cells [8]. This pool of Na/K-ATPase interacts with many signaling proteins and forms a signalosome that receives and propagates extra- and intracellular stimuli [6]. So far we have identified several extracellular and intracellular ligands of Na/K-ATPase that could use the identified Na/K-ATPase/Src receptor complex to initiate signaling events and then amplify/branch out the initial signal through other participants of this signalosome. These ligands include cardiotonic steroids, intracellular Na+, and extracellular K+ [11]. Our new findings presented in this report suggest that ROS, specifically H2O2, may also use this signalosome to stimulate the protein kinase cascades. First, as discussed above, H2O2 is known to affect Na/K-ATPase, resulting in the inhibition of E2/E1 transition. Second, we demonstrate here that the knockdown of Na/K-ATPase abolished H2O2-induced activation of Src and ERK1/2. Because ligands of receptor tyrosine kinases (e.g., EGF, Fig. 5D) and G-protein-coupled receptors are still able to activate ERK1/2 in PY-17 cells, we suggest that Na/K-ATPase is a specific target for H2O2-induced activation of protein kinases. Third, addition of pNaKtide, which blocks the formation of Na/K-ATPase/Src receptor complex, was effective at attenuating effects of H2O2 on Src and ERK1/2, further confirming the involvement of the Na/K-ATPase/Src receptor complex in H2O2 signaling. Moreover, the mutant A416P, which is normal in both pumping and signaling functions, retained full response to ouabain and H2O2 stimuli; on the other hand, the mutant A420P Na/K-ATPase, which has normal pumping function but is defective in Src association, fails to respond to ouabain and H2O2. The opposite findings from these two mutants further indicate the role of an intact and functional Na/K-ATPase/Src complex in cellular H2O2 signaling. Finally, H2O2 appears to work similar to ouabain or other Na/K-ATPase ligands, but not EGF, in the stimulation of ERK1/2 in LLC-PK1 cells (Fig. 5D and [32]).
Several uncertainties, however, should be noted. First, it is unclear whether modification of Src (e.g., Cys oxidation) by H2O2 contributes to the altered interaction between Na/K-ATPase and Src and consequently the activation of Src. Similarly, the molecular basis of regulation by H2O2 on Na/K-ATPase remains to be determined. Both the sulfhydryl (SH)-dependent antioxidant NAC and SH-independent antioxidant α-tocopherol significantly attenuated H2O2-induced signaling, suggesting multiplexed modifications of Na/K-ATPase/Src are involved. Indeed, one recent report showed that both NAC and α-tocopherol blocked H2O2-induced carbonylation in the Na/K-ATPase α1 subunit, indicating that carbonylation may be important in H2O2 signaling [22]. It is important to mention that the 10 mM H2O2 we used in the cell-free system is much higher than the H2O2 amount produced by 3 mU/ml glucose oxidase in culture [35]. This difference between a cell-free system and live cells may be due to the limitation of our in vitro kinase assay, which also was present when we measured the effect of ouabain on Na/K-ATPase/Src receptor complex [6,7]. For example, the effect of ouabain on Src was detected in the cell-free system at the concentration of 10 μM, whereas it stimulated Src at 10–100 nM in LLC-PK1 cells. Second, the role of H2O2-induced reduction of surface expression of Na/K-ATPase in this signaling event requires further investigation. Because decreases in the expression of Na/K-ATPase are sufficient to stimulate Src, this event, at least, could establish a feed-forward regulatory system to amplify the signal. It is also of interest to note the two-phase regulation of Src by H2O2. The downregulation of Na/K-ATPase expression is probably a major contributor of Src activation after 24 h of H2O2 exposure (Fig. 1B). Finally, because the signaling function of Na/K-ATPase is cell specific, the applicability of our new findings in LLC-PK1 cells has to be tested in other cells. Nevertheless, our new findings suggest a novel role of Na/ K-ATPase in ROS-induced activation of protein kinase cascades.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants HL-109015 and HL-105649 from the NHLBI and by Chinese National Science Foundation Grant 81041001. We thank Tillekeratne Manoranjani for her technical support.
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freeradbiomed.2014.03.036.
References
- 1.Lingrel JB, Kuntzweiler T. Na+, K+-ATPase. J Biol Chem. 1994;269:19659–19662. [PubMed] [Google Scholar]
- 2.Li Z, Xie Z. The Na/K-ATPase/Src complex and cardiotonic steroid-activated protein kinase cascades. Pflugers Arch. 2009;457:635–644. doi: 10.1007/s00424-008-0470-0. [DOI] [PubMed] [Google Scholar]
- 3.Cai T, Wang H, Chen Y, Liu L, Gunning WT, Quintas LE, Xie ZJ. Regulation of caveolin-1 membrane trafficking by the Na/K-ATPase. J Cell Biol. 2008;182:1153–1169. doi: 10.1083/jcb.200712022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rajasekaran AK, Rajasekaran SA. Role of Na-K-ATPase in the assembly of tight junctions. Am J Physiol Renal Physiol. 2003;285:F388–396. doi: 10.1152/ajprenal.00439.2002. [DOI] [PubMed] [Google Scholar]
- 5.Tian J, Xie ZJ. The Na-K-ATPase and calcium-signaling microdomains. Physiology (Bethesda) 2008;23:205–211. doi: 10.1152/physiol.00008.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tian J, Cai T, Yuan Z, Wang H, Liu L, Haas M, Maksimova E, Huang XY, Xie ZJ. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol Biol Cell. 2006;17:317–326. doi: 10.1091/mbc.E05-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang M, Cai T, Tian J, Qu W, Xie ZJ. Functional characterization of Src-interacting Na/K-ATPase using RNA interference assay. J Biol Chem. 2006;281:19709–19719. doi: 10.1074/jbc.M512240200. [DOI] [PubMed] [Google Scholar]
- 8.Liang M, Tian J, Liu L, Pierre S, Liu J, Shapiro J, Xie ZJ. Identification of a pool of non-pumping Na/K-ATPase. J Biol Chem. 2007;282:10585–10593. doi: 10.1074/jbc.M609181200. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Cai T, Wang H, Li Z, Loreaux E, Lingrel JB, Xie Z. Regulation of intracellular cholesterol distribution by Na/K-ATPase. J Biol Chem. 2009;284:14881–14890. doi: 10.1074/jbc.M109.003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Z, Zhang Z, Xie JX, Li X, Tian J, Cai T, Cui H, Ding H, Shapiro JI, Xie Z. Na/K-ATPase mimetic pNaKtide inhibits the growth of human cancer cells. J Biol Chem. 2011;286:32394–32403. doi: 10.1074/jbc.M110.207597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ye Q, Li Z, Tian J, Xie JX, Liu L, Xie Z. Identification of a potential receptor that couples ion transport to protein kinase activity. J Biol Chem. 2011;286:6225–6232. doi: 10.1074/jbc.M110.202051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jorgensen PL, Hakansson KO, Karlish SJ. Structure and mechanism of Na, K-ATPase: functional sites and their interactions. Annu Rev Physiol. 2003;65:817–849. doi: 10.1146/annurev.physiol.65.092101.142558. [DOI] [PubMed] [Google Scholar]
- 13.Ye Q, Lai F, Banerjee M, Duan Q, Li Z, Si S, Xie Z. Expression of mutant alpha1 Na/K-ATPase defective in conformational transition attenuates Src-mediated signal transduction. J Biol Chem. 2013;288:5803–5814. doi: 10.1074/jbc.M112.442608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai F, Madan N, Ye Q, Duan Q, Li Z, Wang S, Si S, Xie Z. Identification of a mutant alpha1 Na/K-ATPase that pumps but is defective in signal transduction. J Biol Chem. 2013;288:13295–13304. doi: 10.1074/jbc.M113.467381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown MT, Cooper JA. Regulation, substrates and functions of src. Biochim Biophys Acta. 1996;1287:121–149. doi: 10.1016/0304-419x(96)00003-0. [DOI] [PubMed] [Google Scholar]
- 16.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 17.Sun G, Kemble DJ. To C or not to C: direct and indirect redox regulation of Src protein tyrosine kinase. Cell Cycle. 2009;8:2353–2355. doi: 10.4161/cc.8.15.9225. [DOI] [PubMed] [Google Scholar]
- 18.Rhee SG. Cell signaling: H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 19.Xie Z, Kometiani P, Liu J, Li J, Shapiro JI, Askari A. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J Biol Chem. 1999;274:19323–19328. doi: 10.1074/jbc.274.27.19323. [DOI] [PubMed] [Google Scholar]
- 20.Liu L, Li J, Liu J, Yuan Z, Pierre SV, Qu W, Zhao X, Xie Z. Involvement of Na+/K+-ATPase in hydrogen peroxide-induced hypertrophy in cardiac myocytes. Free Radic Biol Med. 2006;41:1548–1556. doi: 10.1016/j.freeradbiomed.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 21.Elkareh J, Kennedy DJ, Yashaswi B, Vetteth S, Shidyak A, Kim EG, Smaili S, Periyasamy SM, Hariri IM, Fedorova L, Liu J, Wu L, Kahaleh MB, Xie Z, Malhotra D, Fedorova OV, Kashkin VA, Bagrov AY, Shapiro JI. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension. 2007;49:215–224. doi: 10.1161/01.HYP.0000252409.36927.05. [DOI] [PubMed] [Google Scholar]
- 22.Yan Y, Shapiro AP, Haller S, Katragadda V, Liu L, Tian J, Basrur V, Malhotra D, Xie ZJ, Abraham NG, Shapiro JI, Liu J. Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J Biol Chem. 2013;288:34249–34258. doi: 10.1074/jbc.M113.461020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J, Kennedy DJ, Yan Y, Shapiro JI. Reactive oxygen species modulation of Na/K-ATPase regulates fibrosis and renal proximal tubular sodium handling. Int J Nephrol. 2012;2012:381320. doi: 10.1155/2012/381320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiarugi P, Buricchi F. Protein tyrosine phosphorylation and reversible oxidation: two cross-talking posttranslation modifications. Antioxid Redox Signaling. 2007;9:1–24. doi: 10.1089/ars.2007.9.1. [DOI] [PubMed] [Google Scholar]
- 25.Yoshizumi M, Abe J, Haendeler J, Huang Q, Berk BC. Src and Cas mediate JNK activation but not ERK1/2 and 38 kinases by reactive oxygen species. J Biol Chem. 2000;275:11706–11712. doi: 10.1074/jbc.275.16.11706. [DOI] [PubMed] [Google Scholar]
- 26.Zhuang S, Schnellmann RG. H2O2-induced transactivation of EGF receptor requires Src and mediates ERK1/2, but not Akt, activation in renal cells. Am J Physiol Renal Physiol. 2004;286:F858–865. doi: 10.1152/ajprenal.00282.2003. [DOI] [PubMed] [Google Scholar]
- 27.Huang WH, Wang Y, Askari A, Zolotarjova N, Ganjeizadeh M. Different sensitivities of the Na+/K+-ATPase isoforms to oxidants. Biochim Biophys Acta. 1994;1190:108–114. doi: 10.1016/0005-2736(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 28.Xie Z, Jack-Hays M, Wang Y, Periyasamy SM, Blanco G, Huang WH, Askari A. Different oxidant sensitivities of the α1 and α2 isoforms of Na+/K+-ATPase expressed in baculovirus-infected insect cells. Biochem Biophys Res Commun. 1995;207:155–159. doi: 10.1006/bbrc.1995.1166. [DOI] [PubMed] [Google Scholar]
- 29.Figtree GA, Liu CC, Bibert S, Hamilton EJ, Garcia A, White CN, Chia KK, Cornelius F, Geering K, Rasmussen HH. Reversible oxidative modification: a key mechanism of Na+-K+ pump regulation. Circ Res. 2009;105:185–193. doi: 10.1161/CIRCRESAHA.109.199547. [DOI] [PubMed] [Google Scholar]
- 30.Dada LA, Chandel NS, Ridge KM, Pedemonte C, Bertorello AM, Sznajder JI. Hypoxia-induced endocytosis of Na, K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-zeta. J Clin Invest. 2003;111:1057–1064. doi: 10.1172/JCI16826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu J, Periyasamy SM, Gunning W, Fedorova OV, Bagrov AY, Malhotra D, Xie Z, Shapiro JI. Effects of cardiac glycosides on sodium pump expression and function in LLC-K1 and MDCK cells. Kidney Int. 2002;62:2118–2125. doi: 10.1046/j.1523-1755.2002.00672.x. [DOI] [PubMed] [Google Scholar]
- 32.Haas M, Wang H, Tian J, Xie Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J Biol Chem. 2002;277:18694–18702. doi: 10.1074/jbc.M111357200. [DOI] [PubMed] [Google Scholar]
- 33.Wang H, Haas M, Liang M, Cai T, Tian J, Li S, Xie Z. Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J Biol Chem. 2004;279:17250–17259. doi: 10.1074/jbc.M313239200. [DOI] [PubMed] [Google Scholar]
- 34.Liu L, Gable ME, Garlid KD, Askari A. Interactions of K+ATP channel blockers with Na+/K+-ATPase. Mol Cell Biochem. 2007;306:231–237. doi: 10.1007/s11010-007-9574-7. [DOI] [PubMed] [Google Scholar]
- 35.Lai C, Peng M, Huang L, Huang WH, Chiu T. Chronic exposure of neonatal cardiac myocytes to hydrogen peroxide enhances the expression of catalase. J Mol Cell Cardiol. 1996;28:1157–1163. doi: 10.1006/jmcc.1996.0106. [DOI] [PubMed] [Google Scholar]
- 36.Liu J, Kesiry R, Periyasamy SM, Malhotra D, Xie Z, Shapiro JI. Ouabain induces endocytosis of plasmalemmal Na/K-ATPase in LLC-K1 cells by a clathrin-dependent mechanism. Kidney Int. 2004;66:227–241. doi: 10.1111/j.1523-1755.2004.00723.x. [DOI] [PubMed] [Google Scholar]
- 37.Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 38.Kotova O, Al-Khalili L, Talia S, Hooke C, Fedorova OV, Bagrov AY, Chibalin AV. Cardiotonic steroids stimulate glycogen synthesis in human skeletal muscle cells via a Src- and ERK1/2-dependent mechanism. J Biol Chem. 2006;281:20085–20094. doi: 10.1074/jbc.M601577200. [DOI] [PubMed] [Google Scholar]
- 39.Li Z, Cai T, Tian J, Xie JX, Zhao X, Liu L, Shapiro JI, Xie Z. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J Biol Chem. 2009;284:21066–21076. doi: 10.1074/jbc.M109.013821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Askari A, Huang WH. Properties of the oxidized Na+/K+-ATPase. Proceedings of the 7th International Conference on the Sodium Pump; Todtmoose, Germany. September 1993; pp. 880–883. [Google Scholar]
- 41.Goldshleger R, Karlish SJ. Fe-catalyzed cleavage of the alpha subunit of Na/ K-ATPase: evidence for conformation-sensitive interactions between cytoplasmic domains. Proc Natl Acad Sci USA. 1997;94:9596–9601. doi: 10.1073/pnas.94.18.9596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bersohn MM, Philipson KD, Fukushima JY. Sodium–calcium exchange and sarcolemmal enzymes in ischemic rabbit hearts. Am J Physiol. 1982;242:C288–295. doi: 10.1152/ajpcell.1982.242.5.C288. [DOI] [PubMed] [Google Scholar]
- 43.Kako K, Kato M, Matsuoka T, Mustapha A. Depression of membrane-bound Na+-K+-ATPase activity induced by free radicals and by ischemia of kidney. Am J Physiol. 1988;254:C330–337. doi: 10.1152/ajpcell.1988.254.2.C330. [DOI] [PubMed] [Google Scholar]
- 44.Huang WH, Wang Y, Askari A. Na+ +K+)-ATPase: inactivation and degradation induced by oxygen radicals. Int J Biochem. 1992;24:621–626. doi: 10.1016/0020-711x(92)90337-z. [DOI] [PubMed] [Google Scholar]
- 45.Xie ZJ, Wang YH, Askari A, Huang WH, Klaunig JE. Studies on the specificity of the effects of oxygen metabolites on cardiac sodium pump. J Mol Cell Cardiol. 1990;22:911–920. doi: 10.1016/0022-2828(90)90122-i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.