

Abstract
As antibiotic resistance rises, there is a need for strategies such as antibiotic adjuvants to conserve already-established antibiotics. A family of bacterial kinases known as the penicillin-binding-protein and serine/threonine kinase-associated (PASTA) kinases has attracted attention as targets for antibiotic adjuvants for β-lactams. Here, we report that the pyrazolopyridazine GW779439X sensitizes methicillin-resistant Staphylococcus aureus (MRSA) to various β-lactams through inhibition of the PASTA kinase Stk1. GW779439X potentiates β-lactam activity against multiple MRSA and MSSA isolates, including the sensitization of a ceftaroline-resistant isolate to ceftaroline. In silico modeling was used to guide the synthesis of GW779439X derivatives. The presence and orientation of GW779439X’s methylpiperazine moiety was crucial for robust biochemical and microbiologic activity. Taken together, our data provide a proof of concept for developing the pyrazolopyridazines as selective Stk1 inhibitors which act across S. aureus isolates.
Keywords: MRSA, PASTA kinase, β-lactam, drug discovery, pyrazolopyridazine, antibiotic
Graphical Abstract
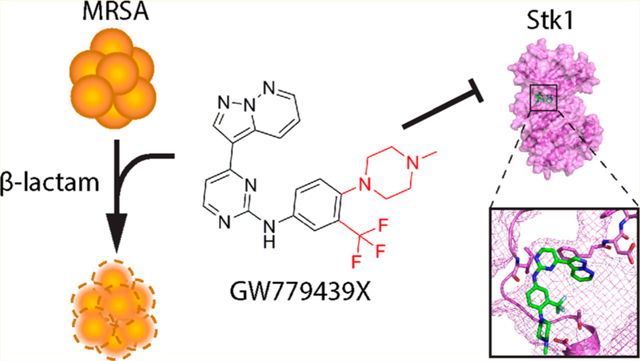
The β-lactams are one of the oldest and most successful classes of antibiotics; however, they are also arguably the most affected by antibiotic resistance. Methicillin-resistant Staphylococcus aureus (MRSA) quickly evolved as a form of β-lactam resistance, and it continues to be a relevant threat in the clinic and the community.1 Unfortunately, resistance to front-line MRSA treatments such as vancomycin, daptomycin, ceftaroline, and linezolid has begun to appear in the clinic,2–5 highlighting the need for novel antibiotic strategies. One such strategy is the utilization of antibiotic adjuvants to preserve the efficacy of current antibiotics.6 This strategy has already proven successful in the case of β-lactam/β-lactamase inhibitor combination therapy to treat various β-lactamase-expressing Gram-negative infections and penicillin-resistant S. aureus.7 However, because MRSA strains utilize the intrinsically β-lactam resistant penicillin-binding protein PBP2A as their primary resistance mechanism, alternative adjuvants are needed to augment β-lactams in these strains.
Over the past decade, there has been an increased interest in exploring bacterial signal transduction mechanisms, in particular those regulated by protein kinases, as potential antibiotic and antivirulence targets.8,9 While prokaryotic protein kinases were originally thought to only phosphorylate proteins on histidine and aspartate residues,10,11 it is now well-established that eukaryotic-like serine/threonine protein kinases (eSTKs) are near ubiquitous in bacteria.12 Of note, a subset of eSTK known as the penicillin-binding protein and Ser/Thr kinase-associated (PASTA) kinases has been demonstrated to be crucial for cell wall homeostasis,13,14,23,15–22 virulence,9,23,24 biofilm formation,25 germination,26,27 and metabolism28,29 in a variety of Firmicutes and Actinobacteria. Intriguingly, while the PASTA kinase PknB is essential for the viability of Mycobacterium tuberculosis,30,31 deletion of PknB homologues in the Firmicutes decreases resistance to the β-lactam antibiotics (reviewed in ref 9). This characteristic makes the PASTA kinases attractive targets for potential β-lactam adjuvants.
Eukaryotic protein kinases have been extensively studied and successfully exploited in the clinic as therapeutic targets for anticancer treatments.32 As such, there is established knowledge and resources such as small-molecule protein kinase inhibitor libraries that can be repurposed for the identification of eSTK-targeting scaffolds. We and others have published multiple PASTA kinase inhibitors.20,22,33–37 Here, we report that GW779439X, a pyrazolopyridazine identified in a small-molecule kinase inhibitor library screen, increases the sensitivity of the MRSA strain LAC to β-lactam antibiotics via inhibition of the PASTA kinase Stk1. In addition, GW779439X also sensitizes a variety of other MRSA and further enhances β-lactam activity against methicillin-sensitive S. aureus (MSSA) isolates in a similar fashion. Finally, using a series of GW779439X congeners, we propose a preliminary structure–activity relationship (SAR) for the pyrazolopyridazine pharmacophore’s activity against the Stk1 kinase domain. Taken together, our data demonstrate that GW779439X and the pyrazolopyridazines are promising lead compounds for further optimization as Stk1 inhibitors as adjuvants to β-lactams for certain MRSA strains.
RESULTS AND DISCUSSION
GW779439X Sensitizes MRSA to β-Lactams.
In a variety of Gram-positive pathogens, including S. aureus, genetic deletion of the PASTA kinases results in increased sensitivity to β-lactam antibiotics.13,18,20,23 Through a kinase inhibitor library screen, we previously identified the compound GSK690693 as a potent inhibitor of the Listeria monocytogenes PASTA kinase PrkA which sensitizes the organism to β-lactams.22 However, GSK690693 showed only minor biochemical activity against the S. aureus PASTA kinase Stk1 and no appreciable activity microbiologically as a β-lactam adjuvant against S. aureus. Therefore, to identify inhibitors of Stk1 that could function as β-lactam adjuvants, we screened 1147 small molecule kinase inhibitors from the GlaxoSmithKline Published Kinase Inhibitor Sets 1 and 2 (PKIS1 and PKIS2)38,39 and Selleck kinase inhibitor libraries against the wild-type MRSA strain LAC in the presence of a sublethal dose of ceftriaxone. Thirty-one compounds potentiated ceftriaxone activity three standard deviations or greater than the library average (Figure 1A). Of these, 25 compounds failed to show a dose response in secondary screens, and an additional 3 demonstrated β-lactam independence. The three remaining compounds were NVP-ADW742, BI-2536, and GW779439X (Supplemental Figure S1). Of these, we selected GW779439X (Figure 1A, cyan dot, and B) for further analysis both because it was the most potent compound in our secondary screens and due to the availability of molecular derivatives (Figure 1A, red dots).
Figure 1.
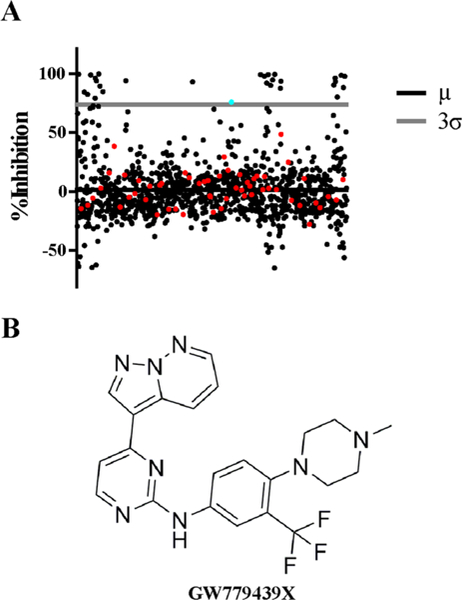
Library screen identifies GW779439X as a compound that potentiates ceftriaxone activity against MRSA. (A) Scatter plot representing percent growth inhibition of WT LAC in the presence of a combination of a sublethal concentration (l0 μg/mL) of ceftriaxone and compounds in the screen. The solid black line represents the library average (μ), and the gray line represents the three standard deviations (3σ) above the library average. The cyan and red dots represent GW779439X and other compounds from the pyrazolopyridazine family, respectively. (B) Skeletal structure of GW779439X.
Genetic deletion of pknB (which expresses Stk1 and will be referred to as stk1 throughout this manuscript) has been shown to decrease resistance of MRSA to a variety of β-lactams.13,23,35 We therefore hypothesized that if GW779439X acts through an Stk1-dependent mechanism, it should be able to potentiate other β-lactams beyond ceftriaxone. To test this hypothesis, we determined the minimum inhibitory concentration (MIC) values of a variety of antibiotics against LAC in the presence and absence of 5 μM GW779439X. Importantly, GW779439X has no intrinsic effects on bacterial growth below 20 μM (Supplemental Figure S2). As can be seen in Table 1, GW779439X was able to potentiate the activity of all β-lactams tested, particularly the penicillinase-resistant penicillins oxacillin and nafcillin to an MIC considered susceptible to these agents. Intriguingly, GW779439X also increased sensitivity twofold to ceftaroline, one of the few clinically available β-lactams with anti-MRSA activity. Importantly, GW779439X was unable to further sensitize a Δstk1 mutant to any of the β-lactams tested. Consistent with the PASTA kinase literature, the MICs of non-β-lactams such as vancomycin or chloramphenicol remained unaffected in any condition tested. Taken together, these data support the hypothesis that GW779439X potentiates a variety of β-lactam antibiotics in an Stk1-dependent manner and shifts the β-lactam class collectively toward sensitivity.
Table 1.
MIC (μg/mL) of Various Antibiotics against WT LAC and Δstk1 Strains ±5 μM GW779439Xa
| LAC (WT) |
Δstk1 |
|||
|---|---|---|---|---|
| GW779439X | − | + | − | + |
| ceftriaxone | 32 [32, 32] | 16 [16, 16] | 16 [16, 16] | 16 [16, 32] |
| oxacillin | 16 [16, 32] | 1 [1, 4] | 1 [1, 4] | 1 [1, 4] |
| nafcillin | 16 [16, 16] | 2 [1, 2] | 2 [1, 2] | 2 [1, 2] |
| meropenem | 0.25 [0.25, 0.5] | 0.125 [0.125, 0.125] | 0.125 [0.0625, 0.125] | 0.125 [0.0625, 0.125] |
| ceftaroline | 1 [0.5, 1] | 0.5 [0.5, 0.5] | 0.5 [0.5, 0.5] | 0.5 [0.5, 0.5] |
| vancomycin | 1 [1, 2] | 1 [1, 2] | 1 [1, 2] | 1 [1, 2] |
| chloramphenicol | 4 [8, 4] | 4 [8, 4] | 4 [8, 4] | 4 [8, 4] |
Data represented as median of at least three independent trials with the range in brackets.
GW779439X Biochemically Inhibits Stk1 in Vitro
Because GW779439X was originally designed to be a eukaryotic kinase inhibitor40 and its effects on β-lactam MICs are Stk1-dependent, we hypothesized that GW779439X acts through direct inhibition of the Stk1 kinase domain. Using autoradio-graphy, we monitored the in vitro kinase activity of the purified Stk1 kinase domain in the presence of increasing concentrations of GW779439X. Strikingly, we observed a robust inhibition of Stk1 autophosphorylation and phosphorylation of the non-specific phosphosubstrate myelin basic protein (MBP) at concentrations of GW779439X as low as 2 μM (Figure 2). This is in contrast to the compound SB-747651A, a compound which failed to sensitize LAC to ceftriaxone in our library screen and which we’ve previously demonstrated has no intrinsic activity against the L. monocytogenes PASTA kinase PrkA (Figure 2).22 When taken with our broth culture experiments, these data support the hypothesis that GW779439X potentiates β-lactam activity via direct inhibition of Stk1.
Figure 2.
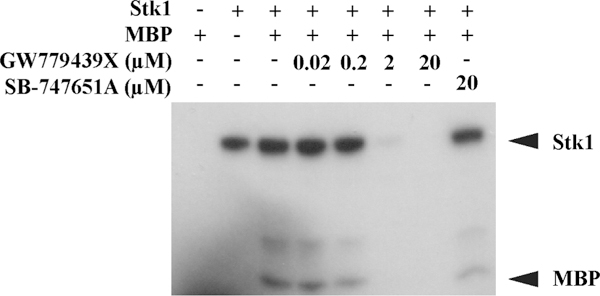
GW779439X directly inhibits Stk1 kinase activity in vitro. Autoradiography blot of purified Stk1 kinase domain and the nonspecific phosphosubstrate MBP untreated or in increasing concentrations of GW779439X. SB-747651A is an imidazopyridine aminofurazan kinase inhibitor present in the initial library screen which shows no Stk1 activity. Blot is representative of three independent trials.
GW779439X Potentiates Oxacillin Activity against Various S. aureus Strains.
LAC was chosen as the MRSA strain for our primary screen for multiple reasons. It is a representative of the USA300 clonal lineage, and it is a particularly virulent and prevalent pulsed-field gel electrophoresis (PFGE) type among community-acquired MRSA infections in the United States.41 To determine if GW779439X would be effective against additional clonal lineages, we determined the MIC of oxacillin against a variety of MRSA and MSSA isolates in the presence and absence of GW779439X (Table 2). At 5 μM GW779439X, we observed potentiation of oxacillin against representatives of PFGE types USA400, USA600, and USA800 as well as the hospital-acquired MRSA strain COL. Strikingly, GW779439X lowered the MIC at least 512-fold against ATCC BAA-2686, a MRSA isolate isolated from the blood of a patient with cystic fibrosis which evolved high-level resistance to ceftaroline; this phenotype was also observed when tested with ceftaroline (Figure 3A) in a ceftaroline-dependent manner (Figure 3B). Finally, GW779493X was also able to modestly potentiate oxacillin activity against the MSSA isolates Newman and NCTC8325 (Table 2). These data demonstrate that abroad range of S. aureus isolates are susceptible to β-lactam potentiation by GW779439X, and combination therapy with kinase inhibitors may represent a particularly powerful therapeutic option in the context of ceftaroline-resistant S. aureus.
Table 2.
MIC (μg/mL) of Oxacillin for Various S. aureus Isolates ±5 μM GW779439Xa
| GW779439X |
− |
+ |
|---|---|---|
| USA400 (MW2) | 16 [16, 32] | 2 [1, 4] |
| USA600 | 16 [16, 16] | 8 [8, 8] |
| USA800 | 4 [4, 8] | 1 [0.5, 2] |
| COL | 256 [256, 256] | 64 [64, 64] |
| ATCC BAA-2686 | 128 [64,128] | 0.25 [0.25, 0.5] |
| Newman | 0.25 [0.125, 0.5] | 0.125 [0.0625, 0.125] |
| NCTC8325 | 0.125 [0.125, 0.25] | 0.0625 [0.0625, 0.125] |
Data represented as median of at least three independent trials with the range in brackets.
Figure 3.
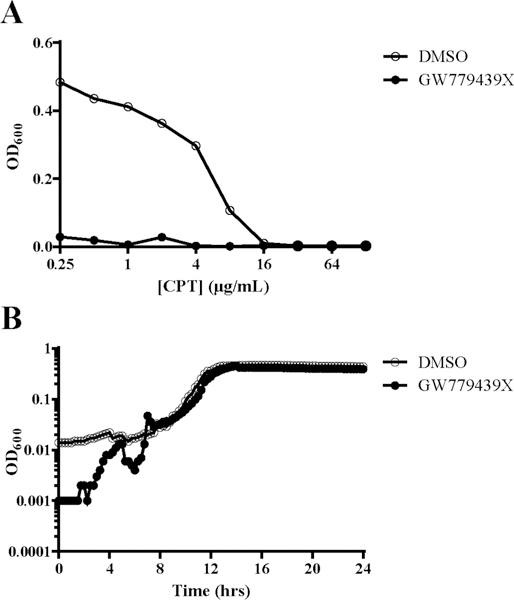
GW779439X potentiates ceftaroline activity against a ceftaroline-resistant MRSA strain. (A) MRSA strain BAA-2686 was back-diluted into increasing concentrations of ceftaroline in the presence or absence of 5 μM GW779439X. (B) BAA-2686 was back-diluted into 5 μM GW779439X and growth was monitored for 24 h. Dose–response curves and growth curves are representative of three independent trials.
Subset of Pyrazolopyridazines Retain Biochemical and Microbiologic Activity.
Because GW779439X was originally designed to target human CDK4, its direct utility as an antibiotic adjuvant is limited. To rationally design modifications which would minimize off-target effects, a better understanding of the GW779439X-Stk1 SAR is needed. To focus our efforts, we utilized the program Autodock4.2 to dock GW779439X into the active site of the Stk1 kinase domain (Figure 4A). GW779439X’s core scaffold consistently docks into the Stk1 active site in a fashion similar to that of other pyrazolopyridazines in CDK240 with the aminopyrimidine moiety participating in a hydrogen bonding pattern with the backbone atoms of Ile90. The pyrazolopyridazine head pivots almost perpendicular to the active site floor, allowing for a potential parallel-displaced π-stacking interaction with Phe150. The p-N-methyl piperazine tail reaches out of the active site and appears to anchor to Glu97 via salt bridge. Finally, the m-trifluoromethyl group comes to rest against Thr94. This model gave us an initial reference for selecting chemical modifications of GW779439X.
Figure 4.
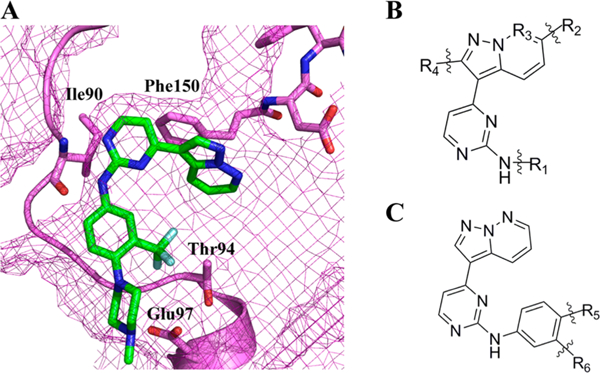
The pyrazolopyridazine scaffold docks in the Stk1 active site. (A) GW779439X docked in silico into the Stk1 active site. GW779439X is displayed as green sticks; Stk1 residues of note are displayed as violet sticks. Stk1 backbone is displayed as violet cartoon, and Stk1 van der Waals radii are displayed as violet mesh. (B) Skeletal structure of the pyrazolopyridazine scaffold that was present in the kinase inhibitor library screen. (C) Skeletal structure of our restricted pyrazolopyridazine scaffold used for medicinal chemistry.
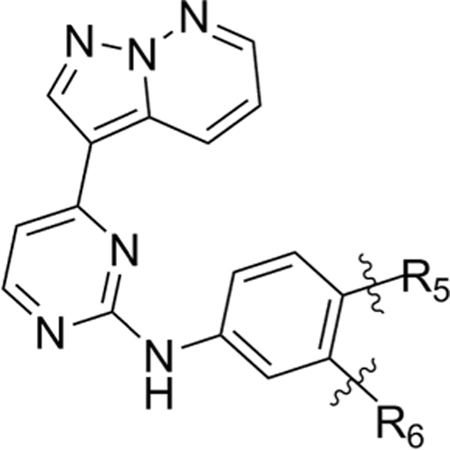
In our primary kinase inhibitor library screen (Figure 1), the pyrazolopyridazine family members as a group possessed modifications at four locations (Figure 4B). Based on our in silico model, we speculated that addition of side chains at positions R2 and R4 would not be tolerated, so we chose to leave these positions unmodified. We retained the pyridazine nitrogen at the R3 position to remain consistent with GW779439X. Because we hypothesized the p-N-methyl piperazine is important for a stabilizing salt bridge with Glu97, we restricted the R1 position to the original aniline and allowed for modifications on this moiety at an R5 and R6 position which would come within proximity of Glu97 (Figure 4C). Utilizing Scheme 1, we ultimately synthesized seven compounds with R5 and R6 modifications to test both biochemically and micro-biologically (Table 3).
Scheme 1. Synthesis of Pyrazolopyridazine Analoguesa.
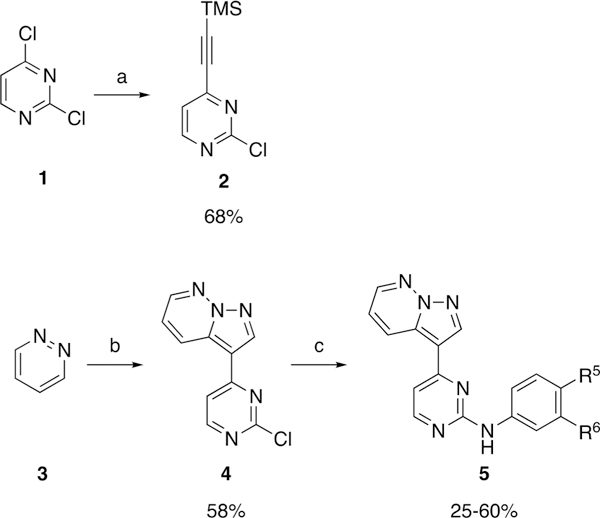
aReagents and conditions. (a) pd(dppf)Cl2 (5 mol %), PPh3 (10 mol %), Et3N (0.2 M), Cul (10 mol %), trimethylsilylacetylene (1.1 equiv), THF (0.3 M), 70 °C, 10 min; (b) HOSA (4.5 equiv), KHCO3 pH 5, 70 °C, 2 h, then KHCO3 pH 9, 2 (1.0 equiv), CH2Cl2 (0.6 M), KOH (aq) (6.0 equiv), rt, 18 h; (c) ArNH2 (1.3 equiv), TFA (3.7 M), sec-BuOH (0.05 M), 100 °C, 18 h.
Table 3.
Structures of Various Pyrazolopyridazine Compounds
 | ||
|---|---|---|
| Compound | R5 | R6 |
| GW779439X |  |
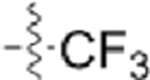 |
| CAF045 | 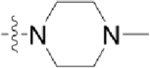 |
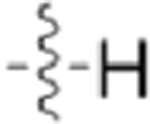 |
| CAF052 | 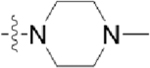 |
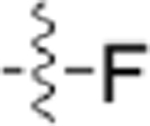 |
| CAF070 | 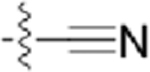 |
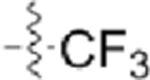 |
| CAF075 | 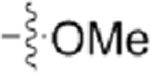 |
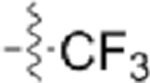 |
| CAF077 | 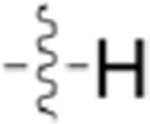 |
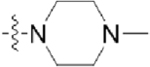 |
| CAF078 | 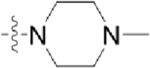 |
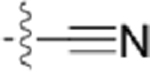 |
| CAF089 | 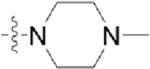 |
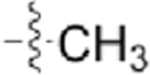 |
Using autoradiography, we first tested the ability of our compound series to inhibit Stk1 activity in vitro. At an inhibitor concentration of 2 μM, the compounds CAF052 and CAF078 robustly inhibited Stk1 kinase activity like the GW779439X control (Figure 5A). In contrast, CAF070, CAF075, and CAF089 possessed very little activity at this concentration. The final two compounds, CAF045 and CAF077, displayed an intermediate phenotype with partial Stk1 inhibition. From these data, we hypothesized that the inhibition profile of our compound series would directly translate to the potentiation of oxacillin in broth culture. When tested against LAC with a sublethal concentration of oxacillin and an inhibitor concentration of 20 μM, the compounds CAF045, CAF052, CAF077, and CAF078 mimicked the GW779439X control with a near 100% inhibition of growth relative to bacteria treated with oxacillin alone (Figure 5B). In agreement with our autora-diography data, the compounds CAF070, CAF075, and CAF089 were significantly less effective at potentiating oxacillin in broth culture. To determine the potencies of the four most-efficacious compounds relative to GW779439X, we treated LAC with a sublethal dose of oxacillin and decreasing concentrations of the kinase inhibitors. We discovered that CAF078 had a potency similar to GW779439X, achieving complete inhibition of bacterial growth at submicromolar concentrations (Figure 5C). On the other hand, CAF052 was 4-fold less potent than GW779439X in broth culture. CAF045 and CAF077 were both 32-fold less potent than GW779439X with near-complete inhibition of growth occurring only as low as l0 μM. Taken together, our data demonstrate that multiple family members of the pyrazolopyridazine class of compounds retain both biochemical and microbiologic activity, though with a wide variation in potency. Importantly, our data suggest that the presence of the positive charge on the p-N-methyl piperazine tail is important for biochemical activity.
Figure 5.
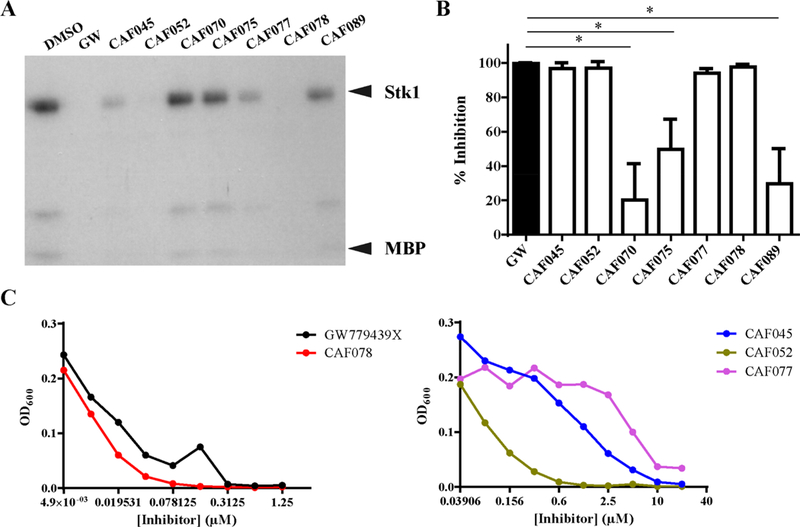
GW779439X’s p-N-methyl piperazine side chain is important for biochemical and microbiologic activity. (A) Autoradiography blot of Stk1 kinase domain and MBP untreated or in the presence of 2 μM GW779439X (GW) or other kinase inhibitors. Blot is representative of three independent trials. (B) WT LAC was back-diluted into cation-adjusted Mueller–Hinton media with a sublethal concentration (8 μg/mL) of oxacillin in the presence of 20 μM GW or otherkinase inhibitors. Percent inhibition for each compound was calculated relative to bacteria grown in oxacillin and DMSO. Error bars represent the standard deviation of 3 independent trials. * = statistically significant by Student’s t test. (C) WT LAC was back- diluted into a sublethal concentration (8 μg/mL) of oxacillin in increasing concentrations of (left) GW779439X or CAF078 or (right) CAF045, CAF052, or CAF077. Dose–response curves are representative of three independent trials.
With the current rise of antibiotic resistance, it has become imperative that novel antibiotic strategies are developed for use in the clinic. The concept of antibiotic adjuvants (i.e., compounds which enhance the activity of established antibiotics) has gained considerable attention within the past decades,6 particularly after the introduction and clinical success of the β-lactamase inhibitors. Due to their roles in virulence and β-lactam resistance, a highly conserved family of bacterial kinases known as the PASTA kinases are attractive targets for the development of β-lactam adjuvants.9 Here, we present our findings on GW779439X, a pyrazolopyridazine protein kinase inhibitor which potentiates β-lactam activity against MRSA via inhibition of the PASTA kinase Stk1.
The intracellular kinase domain of the PASTA kinases has high structural homology to eukaryotic protein kinases, allowing for the knowledge and resources used in the eukaryotic kinase field to be easily repurposed for identification and development of bacterial PASTA kinase inhibitors. However, this also means that extra care must be taken to avoid nonspecific activity against eukaryotic kinases. Nonetheless, it is feasible to generate kinase inhibitors with relative selectivity among eukaryotic kinase targets,42,43 and we have previously identified an inhibitor that can discriminate between S. aureus Stk1 and L. monocytogenes PrkA, two PASTA kinases with greater similarity than that between eukaryotic kinases and PASTA kinases.22 This strongly suggests that PASTA kinase inhibitors with minimized off-target effects can be generated. Furthermore, by coadministrating a relatively selective antibiotic adjuvant, there is at least the theoretical potential of focusing the β-lactam activity against the target pathogenic species and blunting the effect of the β-lactam on microbiota that do not have a PASTA kinase whose catalytic active site is highly similar to the target PASTA kinase.
To identify inhibitors of Stk1, we screened three small-molecule ATP-competitive kinase inhibitor libraries (1147 compounds total) against wild-type LAC in the presence of sublethal ceftriaxone. We chose a microbiologic approach rather than a biochemical approach as our primary screen to focus our efforts on lead compounds which could access the bacterial cytosol, a hurdle that has proven challenging in biochemical screens for inhibitors of the M. tuberculosis PASTA kinase PknB.36 Of the 3l hits in our primary screen, we selected GW779439X as our lead compound due to its dependence on the presence of a β-lactam and Stk1, its dose responsiveness, and the accessibility of synthesized derivatives. GW779439X possesses a pyrazolo-pyridazine scaffold that is structurally unique compared to other previously identified Stk1 inhibitors.33–35 The sulfonamide and quinazoline scaffolds identified by Vornhagen et al. and Kant et al., respectively, were absent from the inhibitor libraries we screened. On the other hand, SB-202190, an inhibitor identified by Boudreau et al., was present in the Selleck library but failed to potentiate ceftriaxone activity against LAC, potentially due to differences in screening conditions (ceftriaxone in tryptic soy broth [TSB] reported here and oxacillin in cation-adjusted Mueller–Hinton media reported in Boudreau et al.). We validated this hypothesis by testing the compound in both conditions and found that these conditions were indeed the cause of the discrepancy (Supplemental Figure S3). These data suggest that more potential Stk1 inhibitors could be identified by screening small molecule libraries under various growth conditions.
GW779439X potentiates the activity of a variety of β-lactam antibiotics in an Stk1-dependent manner. Interestingly, GW779439X can sensitize LAC to β-lactams to the same extent as a Δstk1 mutant. This is in contrast to our previous findings with the compound GSK690693 against L. monocytogenes which could not reach the same levels of sensitivity as a ΔprkA mutant.22 This suggests that either GW779439X has a greater efficacy against Stk1 than GSIK690693 against PrkA or that the β-lactam phenotypes of Stk1 are solely dependent on its kinase activity while PrkA regulates additional effects which are independent of its enzymatic activity. The latter case would suggest that the functions of the Stk1 and PrkA signaling circuits do not completely overlap. Introduction of kinase-dead complements into their respective kinase mutants would shed light on this hypothesis.
It is intriguing that pharmacologic inhibition or genetic deletion of Stk1 potentiates different β-lactams to varying degrees. Ceftriaxone is only augmented 2-fold, while nafcillin and oxacillin (the mainstay therapies for MSSA infections) are more strikingly potentiated to below their respective clinical break points. This data suggests that while Stk1 inhibition may not be able to clinically sensitize MRSA to all β-lactams, it could allow for treatment of MRSA with a traditional MSSA therapy.
GW779439X is able to potentiate the activity of oxacillin against various S. aureus isolates, including both MRSA and MSSA isolates, but the potentiation is clearly strongest in PBP2A-containing strains. With the exception of USA800, the hospital-acquired MRSA isolates (COL, USA600) appear relatively recalcitrant toward GW779439X treatment, while the community-acquired MRSA isolates (USA300, USA400) are sensitized by GW779439X to below the oxacillin clinical breakpoint. USA800 is also sensitized to oxacillin though it is a hospital-acquired MRSA isolate. Further investigation is needed to elucidate the mechanism responsible for the variations in β-lactam sensitization among MRSA isolates.
GW779439X’s ability to sensitize the ceftaroline-resistant MRSA strain BAA-2686 to both oxacillin and ceftaroline is quite striking and brings their MICs well below the clinical susceptibility breakpoints of ≤2 and ≤l μg/mL, respectively. Ceftaroline’s unique mechanism of action involves binding of a ceftaroline molecule into an allosteric binding site on PBP2A, leading to a conformational change which exposes the transpeptidase active site to acylation by a second ceftaroline molecule.44 Low-level resistance to ceftaroline (MIC > l—8 μg/mL) can be achieved by select mutations in the allosteric binding site.45,46 High-level resistance (MIC > 32 μg/mL) can be achieved directly by mutations in the PBP2A active site5 or indirectly by PBP2A-independent mechanisms such as mutations in PBP2, PBP4, LytD, and GdpP.47,48 Although the exact resistance mechanism is unknown in BAA-2686, the exquisite augmentation of ceftaroline by GW779439X might hint at underlying players in the Stk1 signaling cascade.
To begin developing a GW779439X-Stk1 SAR, we synthesized seven pyrazolopyridazine derivatives with a focus on modifications to the p-N-methyl piperazine tail and m-trifluoromethyl side chain. In general, we discovered that modifications to the p-N-methyl piperazine were detrimental to biochemical activity as demonstrated by the greatly reduced activity of CAF070 and CAF075 in both our in vitro and microbiology assays. This could be due to the loss of a stabilizing salt bridge with Glu97 by swapping the positively charged piperazine for an electronegative side chain (CAF070) and/or by the inability of a side chain that can reach Glu97 (CAF075) (Supplemental Figure S4). This could be further validated by reversing or removing the charge at position 97. CAF077 is unique in the fact that it simply shifts the positive charge into the meta-position on the aniline moiety rather than completely eliminate it. While this is ultimately a detriment to biochemical activity, some intermediate inhibition is salvaged possibly by the formation of a hydrogen bond with Thr94 (Supplemental Figure S4). However, this moderate decrease in biochemical activity is enough to potently decrease the compound’s microbiologic potency. These findings with CAF077 support the hypothesis that it is not only the presence of a positive charge, but also its optimal positioning on the side chain that is important to stabilize the compound in the active site. On the other hand, modifications to the m-trifluoromethyl side chain appear to be generally more forgiving; replacement with other electronegative side chains such as fluorine (CAF052) or nitrile (CAF078) retain both biochemical and microbiologic activity on par with GW779439X. Interestingly, replacement with a nonpolar methyl side chain (CAF089) greatly diminishes biochemical activity and abolishes microbiologic activity, while a complete lack of a side chain (CAF045) creates an intermediate phenotype. We speculate that an electronegative side chain at this position might add an additional stabilizing hydrogen bond with Thr94 (Supplemental Figure S4). A mutation ofThr94 to a nonpolar residue such as alanine might shed light on this hypothesis. Future work will be needed to elucidate the effects of modifications to the pyrazolopyridazine head.
Besides β-lactam sensitivity, Stk1 also regulates virulence in S. aureus (reviewed in ref 9). Overall, there is conflicting evidence on the effects of Stk1 interference, with certain strains and infection models showing Stk1 as a positive regulator of virulence while others reveal Stk1 to be a negative regulator of virulence. While an increase in virulence due to Stk1 inhibition may be problematic, the trade-off with increased sensitivity to antibiotics still needs to be considered. In any case, there is yet to be conclusive evidence about the effects of dysregulation of Stk1 on β-lactam therapy in an animal model of infection, a crucial experiment needed for further development of PASTA kinase inhibitors as β-lactam adjuvants.
Although it is well established that interference with PASTA kinase signaling augments β-lactam activity, our understanding of the exact signaling circuits required to maintain β-lactam resistance remains incomplete. Bacterial eSTKs have been found to phosphorylate a wide variety of candidate proteins across species such as DivIVA/Wag31,49 Yvck/CuvA,50,51 MurC,52 VraR,53 IreB,19 and GpsB.14 Of note, Stk1 has been found to play a role in regulating the activity of the class-A β-lactamase BlaZ in select S. aureus strains.33 However, bacteria which lack a β-lactamase (such as L. monocytogenes and the S. aureus strains COL and Newman) are still sensitized to β-lactams when PASTA kinase signaling is disrupted,20,23 notably against β-lactams that are resistant to hydrolysis by BlaZ like oxacillin and nafcillin. Overall, the current literature suggests that Stk1 and other PASTA kinases most likely act as master regulators of various components of the cell wall synthesis and division machinery, further emphasizing their attractiveness as antibiotic adjuvant targets.
CONCLUSIONS
In summary, we identified the compound GW779439X as a novel inhibitor of the S. aureus PASTA kinase Stk1. We have demonstrated that GW779439X potentiates the activity of β-lactam antibiotics against various MRSA and MSSA isolates, some even crossing the breakpoint from resistant to sensitive. Finally, we have determined the importance of the p-N-methyl piperazine side chain for biochemical and microbiologic activity. Future work with the pyrazolopyridazine scaffold may lead to the design of compounds with better selectivity for staphylococcal PASTA kinases as antibiotic adjuvants to restore β-lactam activity and prolong the clinical utility of newer agents such as ceftaroline.
METHODS
Bacterial Strains and Growth Conditions.
All bacterial strains used in this study are listed in Table S1. Generation ofthe Δstk1 mutant strain was achieved as previously described.22 All S. aureus strains were grown in TSB medium at 37 °C shaking overnight until stationary phase. Cultures were then back-diluted to an OD600 of 0.06 for broth growth experiments. Escherichia coli strains XL-1Blue and Rosetta BL21 were used for subcloning and protein expression, respectively. When needed, chloramphenicol (Sigma-Aldrich) was used at 10 μg/mL and carbenicillin (Sigma-Aldrich) was used at 100 μg/mL.
Library Screen.
The PKIS1, PKIS2, and Selleck libraries were obtained via the University of Wisconsin Carbone Cancer Center’s Small Molecule Screening Facility. Overnight cultures were back-diluted 1:100 into fresh TSB medium containing 10 μg/mL ceftriaxone and either library compounds (final concentration: 20 μM in 2% DMSO) or DMSO (final concentration: 2%). Growth was measured as an optical density at 600 nm (OD600) on 15 min intervals for 12 h in a 96-well format using an Eon microplate spectrophotometer or Synergy HT microplate spectrophotometer (BioTek Instruments, Inc., Winooski, VT) (growth conditions: 37 °C, linear shaking). Each compound was screened twice. Percent inhibition was calculated as (1 – (ODX/ODCRO)) × 100, where ODX is the end point OD600 for a culture treated with both ceftriaxone and compound X, and ODCRO is the end point OD600 for a culture treated with ceftriaxone alone. Compounds that inhibited growth three standard deviations greater than the library mean were further verified for dose responsiveness and β-lactam dependence.
MIC Determination.
S. aureus overnight cultures were back-diluted to an OD600 of 0.06 into cation-adjusted Mueller–Hinton (CA-MH) medium containing 2-fold dilutions of the tested antibiotics in the presence or absence of 5 μM GW779439X. The CA-MH was supplemented with 2% NaCl when determining the MICs of nafcillin, oxacillin, or ceftaroline as per recommendation by the Clinical and Laboratory Standards Institute.54 OD600 was measured to monitor growth of the microdilutions for 12 h (with the exception of the S. aureus strain BAA-2686, which was grown for 24 h due to its relatively long lag phase). MICs were defined as the lowest concentration of antibiotic required to prevent turbidity in broth visible by eye. Each MIC experiment was performed at least three times.
Kinase Domain Protein Expression and Purification.
The stk1 kinase domain (residues 1–348) was subcloned into the expression vector pGEX-2T as previously described.20 Expression and purification of the GST-tagged Stk1 kinase domain was performed as previously described.22 Briefly, protein expression was induced by addition of 1 mM Isopropyl β-D-l-thiogalactopyranoside (IPTG) overnight at 23 °C. Cells were lysed by sonication, and the lysates were processed through GS4FF affinity resin columns. The GST tag was then cleaved by thrombin digestion, and liberated Stk1 kinase domain was further processed through a HiPrep Q16 10FF anionic exchange column and a Sephadex75 size-exclusion column to purity.
In Vitro Protein Phosphorylation.
Two micromolar kinase domain, 10 μM MBP (Novatein Biosciences, Woburn, MA), and various concentrations of kinase inhibitors were incubated at 37 °C for 10 min, then added to a mixture of10 mM Tris pH 7.4, 150 mM NaCl, 50 μM MnCl2, 50 μM ATP, and 1 μCi of [γ−32P] ATP. Reactions were incubated at 37 °C for 30 min and terminated by the addition of 6×SDS loading buffer. Samples were run on an SDS-PAGE gel then fixed for 2 h in fixation solution (40% methanol, 5% glycerol, 10% glacial acetic acid). Fixed gels were dried for 1 h and blots visualized by autoradiography.
In Silico Modeling.
In silico modeling was performed as previously described.22 Briefly, the primary sequence of the Stk1 kinase domain (residues 1–270) was threaded onto the crystal structure of the kinase domain of PknB from M. tuberculosis (PDB ID 1O6Y) using the Phyre2 server’s one-to-one threading.55 Gatsieger–Huckel charges were added to Stk1 and compounds using SYBYL-X1.2.56 Compounds were docked into a 66 × 66 × 66 unit grid encompassing the kinase’s active site cleft using the docking program Autodock’s Lamarckian genetic algorithm.57 Models were visualized using PyMOL.58
COMPOUND SYNTHESIS
Synthesis of 2-Chloro-4-((trimethylsilyl)ethynyl)-pyrimidine (Compound 2).
To a solution of Pd(dppf)2Cl2 (200 mg, 0.3 mmol), PPh3 (200 mg, 0.7 mmol), and 2,4-dichloropyrimidine (1.0 g, 7.0 mmol) (Compound 1) in anhydrous THF (25 mL) was added Et3N (33 mL) under a N2 atmosphere. Nitrogen was bubbled into the solution for 20 min before the addition of (trimethylsilyl)acetylene (1.0 mL, 7.0 mmol) followed by Cul (100 mg, 0.7 mmol). The mixture was then heated at 70 °C for 10 min. Upon completion, the mixture was cooled to room temperature and filtered, and the solvent was removed under rotary evaporation. Purification by silica gel flash column chromatography (hexane:EtOAc 8:2) afforded 2-chloro-4-((trimethylsilyl)ethynyl)pyrimidine (Compound 2) (920 me. 4.4 mmol, 68%) as a brown solid.
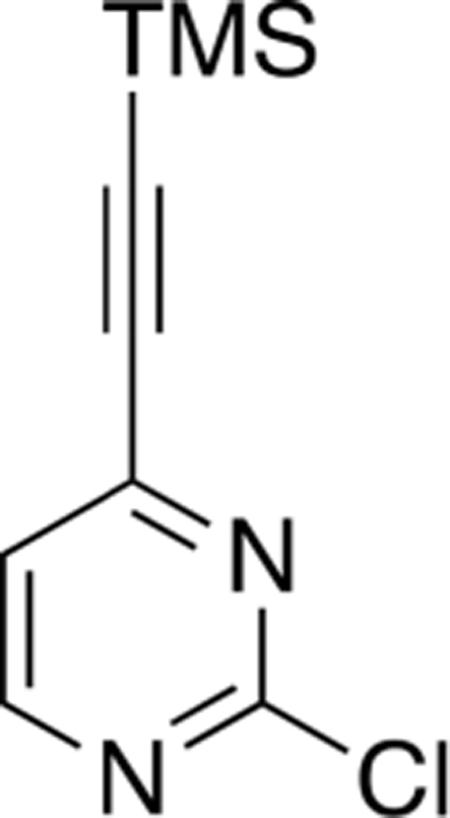
Mp 49–50 °C; 1H NMR (400 MHz, CDCl3) δ 8.58 (d, J = 5.0 Hz, 1H), 7.31 (d, J = 5.0 Hz, 1H), 0.28 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 161.6, 159.6, 152.8, 122.0, 103.7, 100.2, 0.5; LRMS m/z 211 [M + H]+.
Synthesis of 3-(2-Chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (Compound 4).
Hydroxylamine-O-sulfonic acid (1.2 g, 10.7 mmol) was dissolved in H2O (3 mL), and the pH was adjusted to 5–6 by the addition of a 2.5 M solution of NaHCO3. Pyridazine (Compound 3) (514 μL, 7.1 mmol) was added dropwise, and the solution was stirred at 70 °C for 2 h. After cooling at room temperature, the solution was neutralized to pH 8–9 by the addition of a 2.5 M solution of NaHCO3. Then, a solution of 2-chloro-4-[2-(trimethylsilyl)ethynyl]-pyrimidine (Compound 2) (0.5 g, 2.4 mmol) in CH2Cl2 (4 mL) was added, followed by a solution of KOH (0.8 g, 14.2 mmol) in H2O (7 mL). The resulting solution turned dark red, and the stirring was continued for 18 h at room temperature. Upon completion, the mixture was diluted with CH2Cl2 (15 mL), and layers were separated. The organics were further extracted from the aqueous with CH2Cl2 (3 × 20 mL), and the combined organics were washed with brine (50 mL), dried over Na2SO4, and filtered, and the solvent was removed by rotary evaporation. Purification by silica gel flash column chromatography (hexane:EtOAc 7:3) afforded 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (Compound 4) (0.3 g, 1.4 mmol, 58%) as a brown solid.
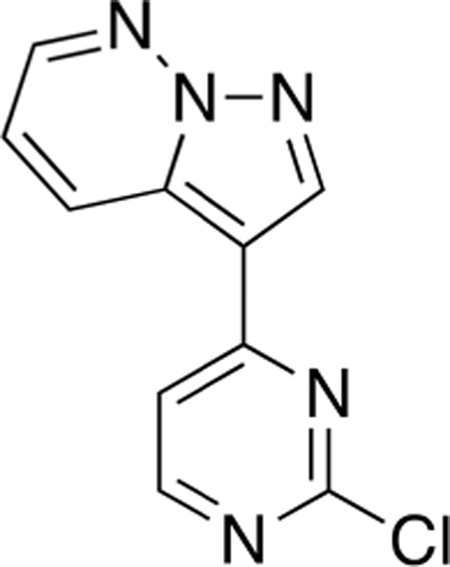
Mp >250 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.03 (s, 1H), 8.90 (dd, J =9.0,1.9 Hz, 1H), 8.73 (d, J = 5.3 Hz, 1H), 8.69 (dd, J = 4.5, 1.9 Hz, 1H), 8.06 (d, J = 5.4 Hz, 1H), 7.58 (dd, J = 9.1, 4.5 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 161.8, 160.3, 144.6, 140.7, 132.9, 128.9, 120.2, 115.4, 108.3; LRMS m/z 232.0 [M + H]+.
General Procedure 1.
To a solution of 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (Compound 4) (1.0 mmol) in sec-BuOH (0.05 M) was added the aniline of interest (1.3 mmol) followed by TFA (3.7 M). The resulting mixture was stirred at 100 °C for 18 h. Upon completion, the mixture was diluted with EtOAc, and a saturated solution of NaHCO3 was added. The layers were separated, and the organics were further extracted from the aqueous with EtOAc (2×). The combined organics were washed with brine, dried over Na2SO4, and filtered, and the solvent was removed by rotary evaporation. Purification by silica gel flash column chromatography (hexane:EtOAc or CH2Cl2:MeOH mixtures) afforded the desired Compound 5.
N-(4-(4-Methylpiperazin-1-yl)phenyl)-4-(pyrazolo-[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (CAF045).
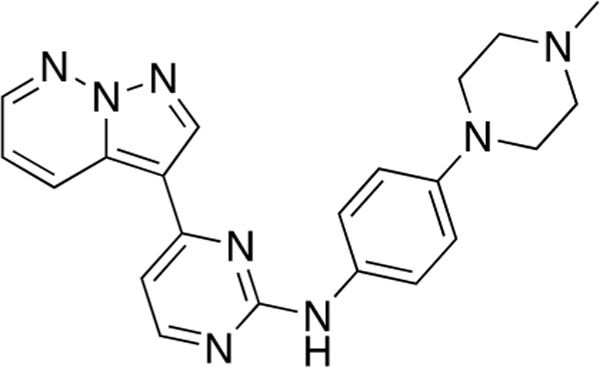
Reaction of 3-(2-chloropyrimidin-4-yl)pyrazolo[l,5-b]-pyridazine (Compound 4) (50 mg, 0.22 mmol), 4-(4-methylpiperazin-l-yl)aniline (45 mg, 0.24 mmol) and TFA (58 μL) in sec-BuOH (4 mL) according to General Procedure 1 afforded the title compound after purification by silica gel flash column chromatography (CH2Cl2:MeOH 9:1) (48 mg, 0.12 mmol, 70%) as a yellow solid.
Mp 150 °C decomp; 1H NMR (400 MHz, Methanol-d4) δ 9.03 (dd, J = 9.1, 1.9 Hz, 1H), 8.67 (s, 1H), 8.50 (dd, J = 4.5, 2.0 Hz, 1H), 8.36 (d, J = 5.4 Hz, 1H), 7.56–7.54 (m, 2H), 7.30 (dd, J = 9.1, 4.5 Hz, 1H), 7.20 (d, J = 5.3 Hz, 1H), 7.09–7.06 (m, 2H), 3.29–3.26 (m, 4H), 2.77–2.74 (m, 4H), 2.46 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 161.0, 159.9, 158.4, 148.1, 143.1, 139.4, 133.2, 131.9, 130.1, 123.2, 117.8, 116.9, 111.0, 107.4, 55.3, 49.9,46.3; LRMS m/z 387 [M + H]+; HRMS (ESI) calculated for [M + H]+: 387.1967, found 387.20447
N-(3-Fluoro-4-(4-methylpiperazin-1-yl)phenyl)-4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (CAF052).
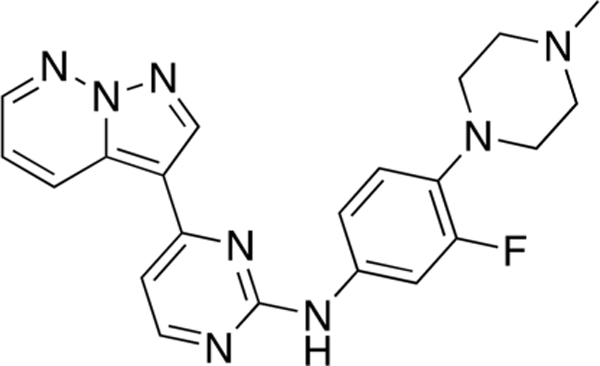
Reaction of 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]-pyridazine (Compound 4) (35 mg, 0.15 mmol), 3-fluoro-4-(4-methylpiperazin-1-yl)aniline (35 mg, 0.17 mmol), and TFA (41 μL) in sec-BuOH (3 mL) according to General Procedure 1 afforded the title compound after purification by silica gel flash column chromatography (CH2Cl2:MeOH 9:1) (30 mg, 0.15 mmol, 49%) as a brown solid.
Mp 170 °C decomp; 1H NMR (400 MHz, Methanol-d4) δ 9.15 (dd, J = 9.2, 1.9 Hz, 1H), 8.70 (s, 1H), 8.53 (dd, J = 4.4, 2.0 Hz, 1H), 8.43 (d, J =5.3 Hz, 1H), 7.85 (bs, 1H), 7.68 (dd, J =14.8, 2.5 Hz, 1H), 7.38–7.32 (m, 2H), 7.26 (d, J =5.6 Hz, 1H), 7.08 (d, J = 9.2 Hz, 1H), 3.18 (bs, 4H), 2.74 (bs, 4H), 2.44 (s, 3H); 13C NMR (100 MHz, CDC13) δ 160.3, 160.0, 158.3, 155.8 (d, J =245.2 Hz), 143.2, 139.5, 135.8 (d, J =9.2 Hz), 134.7 (d, J = 10.7Hz), 133.1, 129.7, 119.2 (d, J = 4.8Hz), 118.1, 116.4 (d, J = 3.1 Hz), 110.8, 109.6 (d, J = 25.3 Hz), 108.1, 55.4, 51.0 (d, J = 2.8 Hz), 46.3; LRMS m/z 405 [M + H]+; HRMS (ESI) calculated for [M + H]+: 405.1873, found 405.19572.
4-((4-(Pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)-amino)-2-(trifluoromethyl)benzonitrile (CAF070).
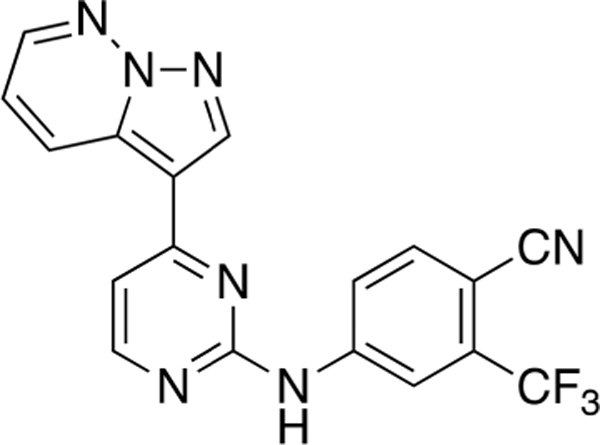
Reaction of 3-(2-chloropyrimidin-4-yl)pyrazolo[l,5-b]-pyridazine (Compound 4) (36 mg, 0.16 mmol), 4-amino-2-(trifluoromethyl)benzonitrile (32 mg, 0.17 mmol), and TFA (42 μL) in sec-BuOH (3 mL) according to General Procedure 1 afforded the title compound after purification by silica gel flash column chromatography (hexane:EtOAc 3:7) (15 mg, 0.16mmol, 25%) as a yellow solid.
Mp >250 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1H), 9.13 (dd, J = 9.1,2.0 Hz, 1H), 8.95 (s, 1H),8.65(dd, J = 4.5, 2.0 Hz, 1H), 8.63 (d, J = 5.3 Hz, 1H), 8.49 (d, J = 2.1 Hz, 1H), 8.23 (dd, J = 8.7,2.2 Hz, 1H), 8.08 (d, J = 8.6 Hz, 1H), 7.59 (d, J = 5.4 Hz, 1H), 7.51 (dd, J = 9.1, 4.5 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 159.7, 159.0, 158.2, 145.3, 144.2, 140.1, 136.3, 132.6, 132.0 (d, J = 3.1 Hz), 131.5 (d, J = 9.8 Hz), 129.3, 128.7 (d, J = 11.7 Hz), 122.7 (d, J = 273.6 Hz), 120.8, 119.2, 116.3, 115.5 (q, J = 5.1 Hz), 109.7 (d, J = 30.7 Hz);LRMS m/z 382 [M + H]+; HRMS (ESI) calculated for [M + H]+: 382.0950, found 382.10433.
N-(4-Methoxy-3-(trifluoromethyl)phenyl)-4-(pyrazolo-[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (CAF075).
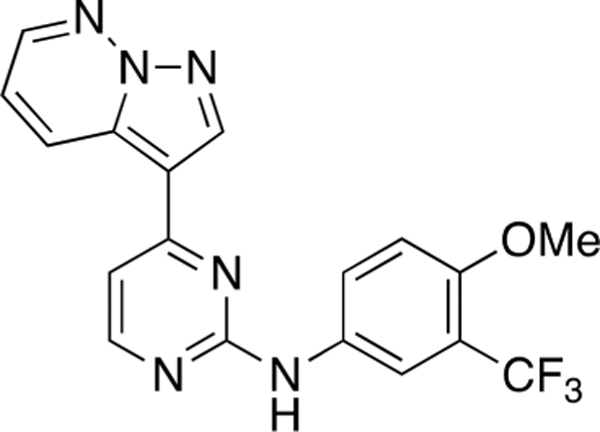
Reaction of 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]-pyridazine (Compound 4) (45 mg, 0.19 mmol), 4-methoxy-3-(trifluoromethyl)aniline (41 mg, 0.21 mmol), and TFA (53 μL) in sec-BuOH (4 mL) according to General Procedure 1 afforded the title compound after purification by silica gel flash column chromatography (hexane:EtOAc 3:7) (32 mg, 0.08 mmol, 43%) as a yellow solid.
Mp 220 °C decomp; 1H NMR (400 MHz, DMSO-d6) δ 9.65 (s, 1H), 9.06 (bd, J = 8.8 Hz, 1H), 8.90 (s, 1H), 8.62 (dd, J = 4.5, 1.9 Hz, 1H), 8.48 (d, J = 5.3 Hz, 1H), 8.09 (d, J = 2.7 Hz, 1H), (dd, J =9.2,2.6 Hz, 1H), 7.42 (dd, J =9.1,4.5 Hz, 1H), 7.38 (d, J = 5.3 Hz, 1H), 7.28 (d, J = 9.0 Hz, 1H), 3.88 (s, 3H); 13C NMR (100 MHz, Methanol-d4) δ 160.9, 160.1, 157.1, 154.0, 143.8, 139.8, 133.6, 132.2, 130.0, 126.8,123.9 (d, J = 272.5 Hz), 121.4 (q, J = 5.7 Hz), 119.0, 113.1, 110.9, 107.9, 56.5; LRMS m/z 387 [M + H]+; HRMS (ESI) calculated for [M + H]+: 387.1103, found 387.11939.
N-(3-(4-Methylpiperazin-1-yl)phenyl)-4-(pyrazolo-[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (CAF077).
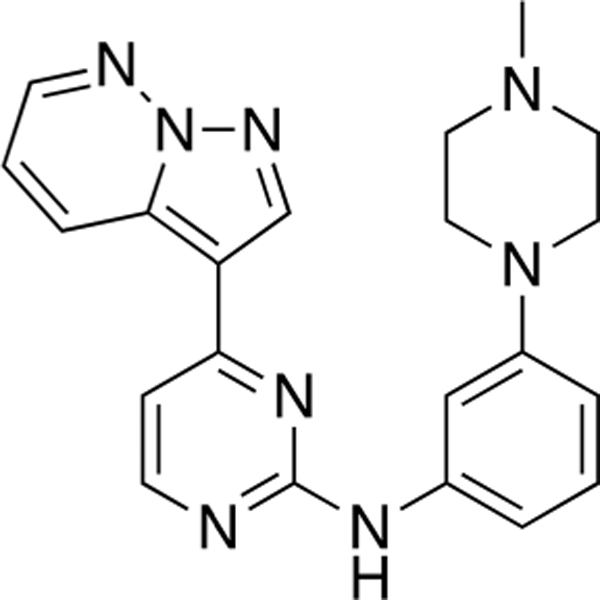
Reaction of 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]-pyridazine (Compound 4) (40 mg, 0.17 mmol), 3-(4-methylpiperazin-1-yl)aniline (36 mg, 0.19 mmol), and TFA (47 μL) in sec-BuOH (3 mL) according to General Procedure 1 afforded the title compound after purification by silica gel flash column chromatography (CH2Cl2:MeOH 9:1) (31 mg, 0.08 mmol, 46%) as a yellow solid.
Mp 139–140 °C; 1H NMR (400 MHz, CDCl3) δ 8.90 (dd, J = 9.0, 2.0 Hz, 1H), 8.49 (s, 1H), 8.40 (d, J = 5.3 Hz, 1H), 8.37 (dd, J = 4.4,2.0 Hz, 1H), 7.33 (bs, 1H), 7.27–7.23 (m, 2H), 7.11 (dd, J =9.1, 4.4 Hz, 1H), 7.07 (dd, J = 7.6, 1.5 Hz, 1H), 7.03 (d, J = 5.2 Hz, 1H), 6.69 (ddd, J = 8.2, 2.5, 0.9 Hz, 1H), 3.24–3.22 (m, 4H), 2.57–2.54 (m, 4H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 160.6, 159.9, 158.4, 152.0, 143.2, 140.4, 139.5, 133.2, 130.0, 129.6,118.0,112.4,111.0,110.9,108.5, 108.0, 55.2,48.9, 46.1; LRMS m/z 387 [M + H]+; HRMS (ESI) calculated for [M + H]+: 387.1967, found 387.20611.
2-(4-Methylpiperazin-1-yl)-5-((4-(pyrazolo[1,5-b]-pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (CAF078).
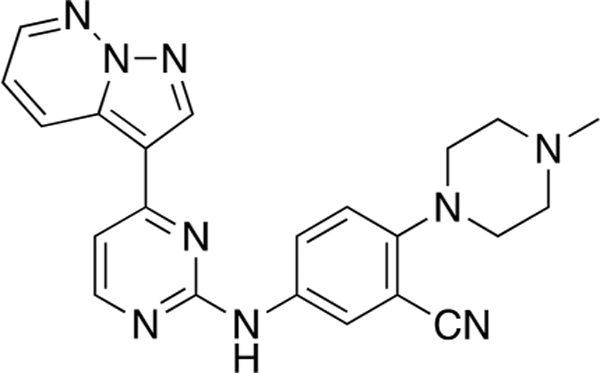
Reaction of 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]-pyridazine (Compound 4) (35 mg, 0.15 mmol), 5-amino-2-(4-methylpiperazin-1-yl)benzonitrile (36 mg, 0.17 mmol), and TFA (41 μL) in sec-BuOH (3 mL) according to General Procedure 1 afforded the title compound after purification by silica gel flash column chromatography (CH2Cl2:MeOH 9:1) (38 mg, 0.09 mmol, 61%) as a yellow solid.
Mp 231–232 °C, 1H NMR (400 MHz, CDCl3) δ 8.88 (dd, J = 9.1, 2.0 Hz, 1H), 8.49 (s, 1H), 8.40–8.38 (m, 2H), 8.17 (d, J = 2.6 Hz, 1H), 7.50 (dd, J = 8.8, 2.7 Hz, 1H), 7.42 (bs, 1H), 7.24 (dd, J = 9.1, 4.4 Hz, 1H), 7.1 (d, J = 5.3 Hz, 1H), 7.04 (d, J = 8.8 Hz, 1H), 3.23–3.20 (m, 4H), 2.67–2.65 (m, 4h), 2.38 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 160.2, 160.1, 158.3, 151.3, 143.3, 139.7,134.2,133.1,129.4,126.0,125.7,119.7,118.5,118.3,110.6, 108.7, 106.8, 55.2, 51.9,46.1; LRMS m/z 412 [M + H]+; HRMS (ESI) calculated for [M + H]+: 412.1920, found 412.20171.
N-(3-Methyl-4-(4-methylpiperazin-1-yl)phenyl)-4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (CAF089).
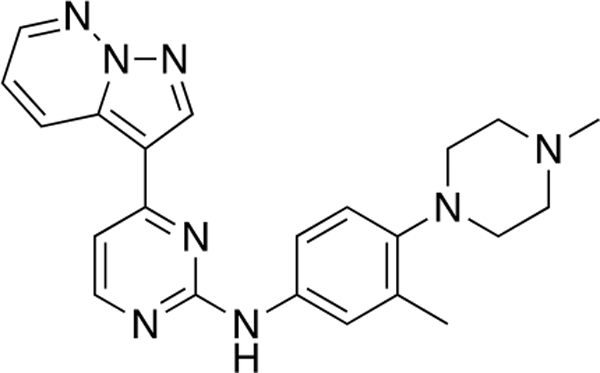
Reaction of 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]-pyridazine (Compound 4) (33 mg, 0.14 mmol), 3-methyl-4-(4-methylpiperazin-1-yl)aniline (38 mg, 0.16 mmol), and TFA (39 μL) in sec-BuOH (3 mL) according to General Procedure 1 afforded the title compound after purification by silica gel flash column chromatography (CH2Cl2:MeOH 9:1) (27 mg, 0.14 mmol, 47%) as a brown solid.
Mp > 250 °C; 1H NMR (400 MHz, CDCl3) δ 8.90 (dd, J = 9.1, 2.0 Hz, 1H), 8.49 (s, 1H), 8.39–8.37 (m, 2H), 7.45 (d, J 2.6 = Hz, 1H), 7.35 (dd, J =8.5, 2.7 Hz, 1H), 7.10–7.06 (m, 3H), 7.02 (d, J =5.3 Hz, 1H), 3.00–2.97 (m, 4H), 2.66 (bs, 4h), 2.42 (s, 3H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 160.7, 159.9, 158.4, 143.1, 139.5, 135.1, 133.6, 133.2, 129.9, 123.9, 119.9, 119.6, 117.9, 111.0, 107.8, 55.6, 51.5, 45.8, 18.0; LRMS m/z 401 [M + H]+; HRMS (ESI) calculated for [M + H]+: 401.2124, found 401.22196.
Supplementary Material
ACKNOWLEDGMENTS
The PKIS was supplied by GlaxoSmithKline, LLC and the Structural Genomics Consortium under an open access Material Transfer and Trust Agreement: http://www.sgc-unc.org. The SGC is a registered charity (Number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) (ULTRA-DD Grant 115766), Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, Sao Paulo Research Foundation-FAPESP, Takeda, and Wellcome Trust (106169/ZZ14/Z). This work was supported (or supported in part) by Merit Review Award Number I01 BX004221 from the United States Department of Veterans Affairs Biomedical Laboratory R&D (BLRD) Service via a VA Merit Award I0BX004089 to R.S. It was also supported by Grant UL1TR000427 from the Clinical and Translational Science Award (CTSA) program of the National Center for Advancing Translational Sciences, NIH and Grant AI121704 from the National Institute of Allergy and Infectious Diseases, NIH. R.S. and N.W. were supported by the Hartwell Foundation and the above Veteran’s Affairs Merit Award. A.S. was supported by the PhRMA Foundation. J.D.S. wassupported by The Wisconsin Partnership Program.
ABBREVIATIONSUSED
- PASTA
penicillin-binding-protein and serine/threonine kinase-associated
- PKIS
published kinase inhibitor set
- MIC
minimum inhibitory concentration
- TSB
tryptic soy broth
- MRSA
methicillin-resistant S. aureus
- MSSA
methicillin-sensitive S. aureus
- IPTG
Isopropyl β-D-1-thiogalactopyrano-side
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.8b00136.
Complete list of kinase inhibitor library percent inhibition values (XLSX)
Structures of validated kinase inhibitor hits from kinase inhibitor library screen (Figure S1); GW779439X bacterial toxicity data (Figure S2); dose–response curves of SB-202190 (Figure S3); in silico docking models of pyrazolopyridazine analogs (Figure S4); bacterial strains used in this study (Table S1) (PDF)
1H and 13C NMR validation of synthesized compounds (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Frieden T (2013) Antibiotic resistance threats. CDC, 22–50. [Google Scholar]
- (2).Appelbaum PC (2006) The emergence of vancomycin-intermediate and vancomycin-resistant Staphylococcus aureus. Clin. Microbiol. Infect. 12, 16–23. [DOI] [PubMed] [Google Scholar]
- (3).Marty FM, Yeh W, Wennersten CB, Venkataraman L, Albano E, Alyea EP, Gold HS, Baden LR, and Pillai SK (2006) Emergence of a Clinical Daptomycin Resistant Staphylococcus aureus Isolate during Treatment of Methicillin Resistant Staphylococcus aureus Bacteremia and Osteomyelitis. J. Clin. Microbiol. 44, 595–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gu B, Kelesidis T, Tsiodras S, Hindler J, and Humphries RM (2013) The emerging problem of linezolid-resistant Staphylococcus. J. Antimicrob. Chemother. 68,4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Long SW, Olsen RJ, Mehta SC, Palzkill T, Cernoch PL, Perez KK, Musick WL, Rosato AE, and Musser JM (2014) PBP2a mutations causing high-level ceftaroline resistance in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 58, 6668–6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wright GD (2016) Antibiotic Adjuvants: Rescuing Antibiotics from Resistance. Trends Microbiol. 24, 862–871. [DOI] [PubMed] [Google Scholar]
- (7).Lee N, Yuen K-Y, and Kumana CR (2003) Clinical role of beta-lactam/beta-lactamase inhibitor combinations. Drugs 63, 1511–1524. [DOI] [PubMed] [Google Scholar]
- (8).Kurosu M, and Begari E (2010) Bacterial protein kinase inhibitors. DrugDev. Res. 71, 168–187. [Google Scholar]
- (9).Pensinger DA, Schaenzer AJ, and Sauer J (2018) Do Shoot the Messenger: PASTA Kinases as Virulence Determinants and Antibiotic Targets. Trends Microbiol. 26, 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Stock JB, Stock a M., and Mottonen JM (1990) Signal transduction in bacteria. Nature 344, 395–400. [DOI] [PubMed] [Google Scholar]
- (11).Hoch J. a. (2000) Two-component and phosphorelay signal transduction. Curr. Opin. Microbiol. 3, 165–170. [DOI] [PubMed] [Google Scholar]
- (12).Shi L, Potts M, and Kennelly PJ (1998) The serine, threonine, and/or tyrosine-specific protein kinases and protein phosphatases of prokaryotic organisms: a family portrait. FEMS Microbiol. Rev. 22, 229–253. [DOI] [PubMed] [Google Scholar]
- (13).Beltramini AM, Mukhopadhyay CD, and Pancholi V (2009) Modulation of cell wall structure and antimicrobial susceptibility by a Staphylococcus aureus eukaryote-like serine/threonine kinase and phosphatase. Infect. Immun. 77, 1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Foulquier E, Pompeo F, Freton C, Cordier B, Grangeasse C, and Galinier A (2014) PrkC-mediated phosphorylation of overexpressed YvcK regulates PBP1 localization in Bacillus subtilis mreB mutant cells. J. Biol. Chem. 289, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Fleurie A, Manuse S, Zhao C, Campo N, Cluzel C, Lavergne JP, Freton C, Combet C, Guiral S, Soufi B, Macek B, Kuru E, VanNieuwenhze MS, Brun YV, Di Guilmi AM, Claverys JP, Galinier A, and Grangeasse C (2014) Interplay of the Serine/Threonine-Kinase StkP and the Paralogs DivIVA and GpsB in Pneumococcal Cell Elongation and Division. PLoS Genet. 10, e1004275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Liebeke M, Meyer H, Donat S, Ohlsen K, and Lalk M (2010) A metabolomic view of staphylococcus aureus and its ser/thr kinase and phosphatase deletion mutants: Involvement in cell wall biosynthesis. Chem. Biol. 17, 820–830. [DOI] [PubMed] [Google Scholar]
- (17).Fridman M, Williams GD, Muzamal U, Hunter H, Siu KWM, and Golemi-Kotra D (2013) Two unique phosphorylation-driven signaling pathways crosstalk in staphylococcus aureus to modulate the cell-wall charge: Stk1/Stp1 meets GraSR. Biochemistry 52, 7975–7986. [DOI] [PubMed] [Google Scholar]
- (18).Kristich CJ, Wells CL, and Dunny GM (2007) A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence. Proc. Natl. Acad. Sci. U. S.A. 104, 3508–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hall CL, Tschannen M, Worthey EA, and Kristich CJ (2013) IreB, a Ser/Thr kinase substrate, influences antimicrobial resistance in Enterococcus faecalis. Antimicrob. Agents Chemother. 57, 6179–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pensinger D. a., Aliota MT, Schaenzer AJ, Boldon KM, Ansari IUH, Vincent WJB, Knight B, Reniere ML, Striker R, and Sauer JD (2014) Selective pharmacologic inhibition of a PASTA kinase increases Listeria monocytogenes susceptibility to β-lactam antibiotics. Antimicrob. Agents Chemother. 58, 4486–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Pensinger DA, Boldon KM, Chen GY, Vincent WJB, Sherman K, Xiong M, Schaenzer AJ, Forster ER, Coers J, Striker R, and Sauer JD (2016) The Listeria monocytogenes PASTA Kinase PrkA and Its Substrate YvcK Are Required for Cell Wall Homeostasis, Metabolism, and Virulence. PLoS Pathog. 12, 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Schaenzer AJ, Wlodarchak N, Drewry DH, Zuercher WJ, Rose WE, Striker R, and Sauer JD (2017) A screen for kinase inhibitors identifies antimicrobial imidazopyridine aminofurazans as specific inhibitors ofthe Listeria monocytogenes PASTA kinase PrkA. J. Biol. Chem. 292, 17037–17045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tamber S, Schwartzman J, and Cheung AL (2010) Role of PknB kinase in antibiotic resistance and virulence in community-acquired methicillin-resistant Staphylococcus aureus strain USA300. Infect. Immun. 78, 3637–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Burnside K, Lembo A, de los Reyes M, Iliuk A, BinhTran NT, Connelly JE, Lin WJ, Schmidt BZ, Richardson AR, Fang FC, Tao WA, and Rajagopal L (2010) Regulation of hemolysin expression and virulence of staphylococcus aureus by a serine/threonine kinase and phosphatase. PLoS One 5, e11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Liu Q, Fan J, Niu C, Wang D, Wang J, Wang X, Villaruz AE, Li M, Otto M, and Gao Q (2011) The eukaryotic-type serine/threonine protein kinase stk is required for biofilm formation and virulence in Staphylococcus epidermidis. PLoS One 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ortega C, Liao R, Anderson LN, Rustad T, Ollodart AR, Wright AT, Sherman DR, and Grundner C (2014) Mycobacterium tuberculosis Ser/Thr Protein Kinase B Mediates an Oxygen-Dependent Replication Switch. PLoS Biol. 12, e1001746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shah IM, Laaberki M-H, Popham DL, and Dworkin J (2008) A Eukaryotic-like Ser/Thr Kinase Signals Bacteria to Exit Dormancy in Response to Peptidoglycan Fragments. Cell 29, 997–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lomas-Lopez R, Paracuellos P, Riberty M, Cozzone AJ, and Duclos B (2007) Several enzymes of the central metabolism are phosphorylated in Staphylococcus aureus. FEMS Microbiol. Lett. 272, 35–42. [DOI] [PubMed] [Google Scholar]
- (29).Leiba J, Hartmann T, Cluzel ME, Cohen-Gonsaud M, Delolme F, Bischoff M, and Molle V (2012) A novel mode of regulation of the Staphylococcus aureus catabolite control protein A (CcpA) mediated by Stk1 protein phosphorylation. J. Biol. Chem. 287, 43607–43619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chawla Y, Upadhyay S, Khan S, Nagarajan SN, Forti F, and Nandicoori VK (2014) Protein kinase B (PknB) of Mycobacterium tuberculosis is essential for growth of the pathogen in vitro as well as for survival within the host. J. Biol. Chem. 289, 13858–13875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Fernandez P, Saint-Joanis B, Barilone N, Jackson M, Gicquel B, Cole ST, and Alzari PM (2006) The Ser/Thr protein kinase PknB is essential for sustaining mycobacterial growth. J. Bacteriol 188, 7778–7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Liao JJ-L (2007) Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem. 50, 409–424. [DOI] [PubMed] [Google Scholar]
- (33).Boudreau MA, Fishovitz J, Llarrull LI, Xiao Q, and Mobashery S (2015) Phosphorylation of BlaR1 in Manifestation of Antibiotic Resistance in Methicillin-Resistant Staphylococcus aureus and Its Abrogation by Small Molecules. ACS Infect. Dis. 1, 454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kant S, Asthana S, Missiakas D, and Pancholi V (2017) A novel STK1-targeted small- molecule as an “ antibiotic resistance breaker ” against aureus. Sci. Rep. 7, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Vornhagen J, Burnside K, Whidbey C, Berry J, Qin X, and Rajagopal L (2015) Kinase Inhibitors that Increase the Sensitivity of Methicillin Resistant Staphylococcus aureus to β-Lactam Antibiotics. Pathog. Pathogens 4, 708–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lougheed K. E. a., Osborne S. a., Saxty B, Whalley D, Chapman T, Bouloc N, Chugh J, Nott TJ, Patel D, Spivey VL, Kettleborough C. a., Bryans JS, Taylor DL, Smerdon SJ, and Buxton RS (2011) Effective inhibitors of the essential kinase PknB and their potential as anti-mycobacterial agents. Tuberculosis 91, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chapman TM, Bouloc N, Buxton RS, Chugh J, Lougheed KEA, Osborne SA, Saxty B, Smerdon SJ, Taylor DL, and Whalley D (2012) Substituted aminopyrimidine protein kinase B (PknB) inhibitors show activity against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 22, 3349–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Drewry DH, Willson TM, and Zuercher WJ (2014) Seeding collaborations to advance kinase science with the GSK Published Kinase Inhibitor Set (PKIS). Curr. Top. Med. Chem. 14, 340–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Elkins JM, et al. (2016) Comprehensive characterization of the Published Kinase Inhibitor Set. Nat. Biotechnol 34, 95–103. [DOI] [PubMed] [Google Scholar]
- (40).Stevens KL, Reno MJ, Alberti JB, Price DJ, Kane-Carson LS, Knick VB, Shewchuk LM, Hassell AM, Veal JM, Davis ST, Griffin RJ, and Peel MR (2008) Synthesis and evaluation of pyrazolo[1,5-b]pyridazines as selective cyclin dependent kinase inhibitors. Bioorg. Med. Chem. Lett. 18, 5758–5762. [DOI] [PubMed] [Google Scholar]
- (41).Diekema DJ, Richter SS, Heilmann KP, Dohrn CL, Riahi F, Tendolkar S, McDanel JS, and Doern GV (2014) Continued Emergence of USA300 Methicillin-Resistant Staphylococcus aureus in the United States: Results from a Nationwide Surveillance Study. Infect. Control Hosp. Epidemiol. 35, 285–292. [DOI] [PubMed] [Google Scholar]
- (42).Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, and Zarrinkar PP (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- (43).Knight ZA, and Shokat KM (2005) Features of selective kinase inhibitors. Chem. Biol. 12, 621–637. [DOI] [PubMed] [Google Scholar]
- (44).Otero LH, Rojas-Altuve a., Llarrull LI, Carrasco-Lopez C, Kumarasiri M, Lastochkin E, Fishovitz J, Dawley M, Hesek D, Lee M, Johnson JW, Fisher JF, Chang M, Mobashery S, and Hermoso J. a. (2013) How allosteric control ofStaphylococcus aureus penicillin binding protein 2a enables methicillin resistance and physiological function. Proc. Natl.Acad. Sci. U. S.A. 110,16808–16813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Alm RA, McLaughlin RE, Kos VN, Sader HS, Iaconis JP, and Lahiri SD (2014) Analysis of Staphylococcus aureus clinical isolates with reduced susceptibility to ceftaroline: An epidemiological and structural perspective. J. Antimicrob. Chemother. 69, 2065–2075. [DOI] [PubMed] [Google Scholar]
- (46).Mendes RE, Tsakris A, Sader HS, Jones RN, Biek D, McGhee P, Appelbaum PC, and Kosowska-Shick K (2012) Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J. Antimicrob. Chemother. 67, 1321–1324. [DOI] [PubMed] [Google Scholar]
- (47).Lahiri SD, and Alm R A. (2016) Identification of non-PBP2a resistance mechanisms in Staphylococcus aureus after serial passage with ceftaroline: Involvement of other PBPs. J. Antimicrob. Chemother. 71, 3050–3057. [DOI] [PubMed] [Google Scholar]
- (48).Chan LC, Basuino L, Diep B, Hamilton S, Chatterjee SS, and Chambers HF (2015) Ceftobiprole- and ceftaroline-resistant methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 59, 2960–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Jani C, Eoh H, Lee JJ, Hamasha K, Sahana MB, Han JS, Nyayapathy S, Lee JY, Suh JW, Lee SH, Rehse SJ, Crick DC, and Kang CM (2010) Regulation ofpolar peptidoglycan biosynthesis by Wag31 phosphorylation in mycobacteria. BMC Microbiol. 10, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Mir M, Prisic S, Kang CM, Lun S, Guo H, Murry JP, Rubin EJ, and Husson RN (2014) Mycobacterial gene cuvA is required for optimal nutrient utilization and virulence. Infect. Immun. 82,4104–4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Kang C, Abbott DW, Park ST, Dascher CC, Cantley LC, and Husson RN (2005) The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: substrate identification and regulation of cell shape. Genes Dev. 19, 1692–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Fiuza M, Canova MJ, Patin D, Letek M, Zanella-Cléon I, Becchi M, Mateos LM, Mengin-Lecreulx D, Molle V, and Gil JA (2008) The MurC ligase essential for peptidoglycan biosynthesis is regulated by the serine/threonine protein kinase PknA in Coryne-bacterium glutamicum. J. Biol. Chem. 283, 36553–36563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Canova MJ, Baronian G, Brelle S, Cohen-Gonsaud M, Bischoff M, and Molle V (2014) A novel mode of regulation of the Staphylococcus aureus Vancomycin-resistance-associated response regulator VraR mediated by Stk1 protein phosphorylation. Biochem. Biophys. Res. Commun. 447, 165–171. [DOI] [PubMed] [Google Scholar]
- (54).Weinstein MP Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard, 9th ed; Clinical and Laboratory Standards Institute: 2012. [Google Scholar]
- (55).Kelley LA, Mezulis S, Yates C, Wass M, and Sternberg M (2015) The Phyre2 web portal for protein modelling, prediction, and analysis. Nat. Protoc. 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).G6G Tech. SYBYL-X; Tripos International: St. Louis, MO. [Google Scholar]
- (57).Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, and Olson AJ (1998) Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 19, 1639–1662. [Google Scholar]
- (58).Schrödinger L The PyMOL Molecular Graphics System, Version 2.0; Schrödinger LLC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.