

Abstract
Purpose
Fuchs' endothelial corneal dystrophy (FECD) is a major cause of vision loss and the most common nucleotide repeat disorder, affecting 4% of United States population greater than 40 years of age. Seventy percent of FECD cases are due to an intronic CTG expansion within the TCF4 gene, resulting in accumulation of CUG repeat RNA nuclear foci in corneal endothelium. Each endothelial cell has approximately two sense foci, and each focus is a single RNA molecule. This study aimed to obtain a better understanding of how rare repeat RNA species lead to disease.
Methods
We quantitatively examined muscleblind-like (MBNL) proteins and their interaction with foci in both patient-derived corneal endothelial cell lines and human corneal endothelial tissue.
Results
Using fluorescent in situ hybridization and immunofluorescence, we found that depletion of both MBNL1 and MBNL2 reduces nuclear RNA foci formed by the repeat, suggesting that both are necessary for foci. Quantitative studies of RNA and protein copy number revealed MBNLs to be abundant in the total cellular pool in endothelial cell lines but are much lower in human corneal endothelial tissue. Studies using human tissue nuclear and cytoplasmic fractions indicate that most MBNL proteins are localized to the cytoplasm.
Conclusions
The low levels of MBNL1/2 in corneal tissue, in combination with the small fraction of protein in the nucleus, may make corneal endothelial cells especially susceptible to sequestration of MBNL1/2 by CUG repeat RNA. These observations may explain how a limited number of RNA molecules can cause widespread alteration of splicing and late-onset degenerative FECD.
Keywords: DNA, trinucleotide repeat disease, RNA, intronic RNA, RNA degradation, RNA binding protein, molecular genetics, eye, cornea, Fuchs' dystrophy
More than 20 diseases are caused by mutations that expand regions of repetitive DNA.1 Although these diseases have similar degenerative cellular phenotypes, they affect different cell types, are characterized by the expansion of different repeats, and can code for different RNA species. For example, Huntington's disease is caused by a CAG expansion within the coding region of the huntingtin gene. By contrast, a major cause of familial amyotrophic lateral sclerosis (ALS) is a hexanucleotide CCGGGG expansion within an intron on C9orf72 mRNA.
Explanations for how these expanded repeats trigger disease also vary. In cases where expression of the parent gene is affected, a deficiency of protein or an excess of a mutant protein might cause disease. Mutant RNAs can be translated into peptides by repeat-associated non-AUG (RAN) translation,2 and these peptides may contribute to disease phenotypes. It is also possible that the mutant RNA molecules themselves may form associations that disrupt normal cellular processes.3 A better understanding of the molecular causes of trinucleotide repeat disease and their similarities and differences will improve opportunities for diagnosis and treatment.
Fuchs' endothelial corneal dystrophy (FECD) is the most common trinucleotide repeat expansion disorder. FECD affects 4% of the population in the United States over the age of 40.4 FECD is an age-related degenerative disease of the postmitotic corneal endothelium. Patients are often diagnosed between 50 and 60 years of age, and initial blurred vision and glare can progress to profound loss of vision.
Currently, the only effective therapy for FECD is corneal transplantation. Although surgical outcomes have improved markedly over the last decade, current endothelial keratoplasty techniques have complications including graft detachment and primary graft failure in the immediate postoperative period, as well as increased risk for glaucoma and graft rejection or decompensation years after the initial successful surgery.5,6 To benefit from surgery, patients need to have access to advanced surgical centers and eye banks that can supply donor corneas and be willing to accept the risks and discomforts of the surgery.
Furthermore, before surgery, patients may endure many years of declining quality of vision before surgery becomes an acceptable option. There is a significant unmet need for less invasive treatment strategies that can benefit patients at an earlier age before significant vision is lost and that will be accessible to a wider population who may not now have access to advanced treatment centers. A detailed understanding of molecular mechanism will facilitate discovery of new treatments.
A CTG trinucleotide repeat expansion within intron 2 of the TCF4 gene (CTG18.1 triplet repeat polymorphism) accounts for up to 70% of FECD cases.7–10 Mutant CUG repeat transcripts accumulate as nuclear foci in corneal endothelial tissue of affected subjects11,12 without reducing mRNA levels expressed by the parent TCF4 gene.11,13 These data implicate mutant noncoding regions of RNA as the cause of FECD.
The TCF4 gene encodes the E2-2 protein, a ubiquitously expressed class 1 basic-helix-loop-helix transcription factor.14 Unlike other trinucleotide repeat diseases, mutant TCF4 does not cause apparent neurodegenerative disease. However, neurons and corneal endothelial cells share important similarities that impact our understanding of disease pathology and treatment.15 During embryonic development, corneal endothelial cells are derived from neural crest cells, and adult corneal cells retain peripheral neuronal markers.16 Like neurons, corneal endothelial cells are postmitotic and terminally differentiated. Both neurons and corneal endothelial cells are not replaced, and degeneration slowly degrades function over a patient's lifetime. There is currently no explanation for the restriction of disease phenotype to corneal tissue in FECD.
Myotonic dystrophy type 1 (DM1) is a multisystem disorder caused by a CUG repeat expansion within the 3′ UTR of DMPK mRNA.17,18 Importantly, this mutation has also been associated with FECD.19,20 This remarkable finding that FECD can be caused by the same expanded repeat within noncoding regions of RNAs associated with two different genes reinforces the conclusion that the mutant expanded CUG repeat RNA is the cause of FECD. A key issue for therapeutic intervention is understanding how mutant RNA molecules can cause a severe degenerative disease.
The molecular mechanisms for DM1 have been extensively studied and may offer lessons for understanding FECD. In DM1 cells derived from affected tissues, expanded DMPK transcripts accumulate as nuclear foci,21 and the expanded CUG repeat region is thought to sequester muscleblind-like (MBNL) proteins.22–24 MBNL normally acts to regulate splicing, and perturbing the concentration of available MBNL may account for the widespread splicing changes observed in DM1 cells and tissue.25–27
MBNL1 proteins colocalize with the expanded CUG repeat RNA in FECD patient-derived corneal endothelial cells with either TCF4 or DMPK expansions.12,20 Additionally, MBNL2 has been shown to colocalize in cultured endothelial cells of FECD subjects with the TCF4 expansion.28 In parallel with the suggested mechanism explaining altered splicing in DM1, one hypothesis to explain how RNA might cause FECD suggests that the expanded repeat within the TCF4 gene binds MBNL proteins and reduces the pool of free cellular MBNL proteins, thereby inducing global splicing changes that ultimately lead to cellular malfunction and degeneration. This hypothesis has been supported by observations that FECD cells or tissue with TCF4 expansions exhibit changes in the alternative splicing of critical MBNL-sensitive genes relative to normal cells.12,29
Complicating this hypothesis, we previously observed that, in cultured corneal endothelial cells or in tissue, each cell has only a limited number of foci and each focus is a single RNA molecule.30 This observation raised a critical question underlying the mechanism of disease action: how can a small number of mutant RNA molecules affect splicing to cause a late-onset disease?
In this report, we characterize MBNL1/2 expression and examine the association of MBNL proteins with mutant RNA in both patient-derived corneal endothelial cell lines and human corneal endothelial tissue. We found that both MBNL1/2 colocalize with the nuclear foci in FECD endothelial tissue. Depletion of MBNL1 or MBNL2 individually did not impact the number of foci in cultured cells. Knockdown of both MBNL proteins resulted in a reduction of foci suggesting that they play a redundant role in stabilizing the mutant RNA. We found that MBNL1/2 is less expressed in corneal endothelial tissue from human donors than in cultured corneal endothelial cells. In addition, only a small minority of the cellular MBNL1/2 is present in the nuclei of cells in donor tissue. Taken together, these observations make it more plausible that sequestration of MBNL1/2 by expanded CUG repeat may contribute to the observed abnormal splicing that is a hallmark of advanced FECD.
Materials and Methods
Corneal Tissue Samples
The study was approved by the institutional review board at the University of Texas Southwestern Medical Center and conducted in adherence with the tenets of the Declaration of Helsinki. Written informed consent was obtained from subjects prior to inclusion in study.
Surgically explanted endothelium–Descemet's membrane monolayers were immediately freeze-snapped in liquid nitrogen or alternatively fixed in a 4% phosphate-buffered formaldehyde, equilibrated in a 30% sucrose solution for cytoprotection, and frozen in Tissue-Tek Optimal Cutting Tissue compound (Sakura, Torrance, CA, USA). Genomic DNA from subjects' peripheral blood leukocytes was extracted using Autogen Flexigene (Qiagen, Germantown, MD, USA).
Corneal endothelial samples from donors preserved in Optisol GS corneal storage media (Bausch & Lomb, Rochester, NY, USA) were obtained from the eye bank of Transplant Services at UT Southwestern. Endothelium––Descemet's membrane monolayers from donor corneas were dissected and stored as previously described.11 DNA from the remaining corneal tissue was extracted with TRIzol reagent (Thermo Scientific, Waltham, MA, USA).
Genomic DNA from subjects' peripheral leukocytes or corneal tissue was used for genotyping the CTG18.1 and DMPK trinucleotide repeat polymorphism using a combination of short tandem repeat, triplet repeat primed PCR, and Southern blot assays as we have previously described (Supplementary Table S1).10
Cell Culture and siRNA Transfection
F35T corneal endothelial cell line (a generous gift of Dr. Albert Jun, Johns Hopkins, Baltimore, MD, USA), F45SV corneal endothelial cell line (SV 40-mediated immortalization by ALSTEM, Richmond, CA, USA), and a control corneal endothelial cell line HCN19 (W4056-16-000418 primary culture) were cultured as described.30 siRNAs were transfected into cells with Lipofectamine RNAiMax (Invitrogen, Carlsbad, CA, USA) per the manufacturer's protocols. After 4 days of transfection, cells were collected and used for further analysis. siRNAs were obtained from Integrated DNA Technologies (IDT, Coralville, IA, USA), and the siRNA sequences were described previously.31–33
Antibodies and Immunoblotting
The commercial antibodies used for protein detection were: MBNL1 (Santa Cruz sc-47740 or EMD Millipore ABE241), MBNL2 (Santa Cruz sc-136167), CUGBP1 (Abcam ab9549), TCF4 (Abnova H00006925-M03), tubulin (Cell Signaling 3873P), GAPDH (G9545 Sigma), and Histone H3 (Cell Signaling 2650S). Cells were lysed with the lysis buffer containing 25 mM Tris-HCl at pH 7.5, 100 mM NaCl, 5 mM MgCl2, 5 mM NaF, 0.1% TritonX-100, 10 mM β-glycerophosphate, 1 mM dithiothreitol and 1X protease inhibitor cocktail (Promega). After incubation on ice and sonication, SDS sample buffer was added. Lysates were boiled for 5 min, separated by SDS-PAGE, and blotted with the appropriate antibodies.
Fluorescence In Situ Hybridization and Colocalization With MBNL Proteins
Fluorescence in situ hybridization (FISH) of cultured cells and tissue monolayers was performed as previously described.20 Briefly, cells were fixed and immersed in 70% ethanol at 4°C overnight. After treatment with wash buffer and prehybridization buffer, slides were incubated with (CAG)6CA-5′ Texas red-labeled 2-O-methyl RNA 20-mers probe (IDT). The next day, slides were washed twice and then stained with mounting media with DAPI (H-1500; Vector Labs, Burlingame, CA, USA). For detection of MBNL protein, prior to ethanol treatment, permeabilization with 0.2% Triton 100 in 2× SCC was performed. After probe incubation, cells were incubated with anti-MBNL1 or anti-MBNL2 antibody.
The slides were sealed and imaged at 60× magnification using a Widefield Deltavision microscope. Images were processed by deconvolution with AutoQuant X3 (Media Cybernetics, Rockville, MD, USA) software. Visualization of RNA foci and co-localization of MBNLs proteins were made using ImageJ (National Institutes of Health, Bethesda, MD, USA). For quantification, data were analyzed from at least 10 pictures containing greater than 50 cells each. For cells, data were analyzed from two batches of F35T cells. For tissues, data were analyzed from five FECD tissues.
Real-Time Quantitative PCR Analysis of TCF4 Transcripts
Quantitative PCR (qPCR) was performed to analyze TCF4 transcripts level with iTaq SYBR Green Supermix. Sequences of primers were described previously.30 Cells were harvested using Trizol agent (Sigma, St. Louis, MO, USA), and the TCF4 mRNA levels were analyzed by qPCR.
Transcript Copy Number Measured by qPCR
Transcript copy number measurement was performed as previously described.30 Primers and probes used in this experiment are shown in Supplementary Table S2. RNA standards of MBNL1 and MBNL2 were synthesized by in vitro transcription and purity, or RNAs were measured with Bioanalyzer (Agilent, Santa Clara, CA, USA). A serial dilution of purified standard RNA, ranging from 10 to 105 copies, was used to construct standard curve for qPCR efficiency by qPCR following reverse transcription. For each transcript, a serial dilution of cDNA was used. Ct values in each dilution were measured in triplicate. Copy number per cell was calculated according to the standard curve for qPCR efficiency of each transcript. For cells, data were analyzed from two batches of F35T cells. For tissues, data were analyzed from three control tissues and three FECD tissues. The number of F35T cells and endothelial cells from collected tissues were calculated based on central endothelial cell density measurements as previously reported.30 Results are showed as the mean ± SD.
Estimation of Protein Copy Number by Immunoblotting
Recombinant human MBNL1 (ab114825; Abcam, Cambridge, MA, USA) and MBNL2 (ab160843; Abcam) proteins were used to make standard curves of protein copy number. To determine the protein copy number in F35T cells and control endothelial tissue, two different amounts of cell lysate were loaded, and a serial dilution of each recombinant protein was used to construct standard curve. BSA (50 ng/μL) was also added in the standard recombinant proteins to avoid adherence of proteins to the wall of tube. The intensity of the band was determined and analyzed by ImageJ. The number of F35T cells and endothelial cells from collected tissues were calculated as described above. Data were analyzed from two batches of F35T cells and three different control tissues. Results are showed as the mean ± SD.
Nuclear and Cytoplasmic Extraction
Briefly, a cell pellet was lysed in hypotonic lysis buffer (10 mM Tris, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 10% glycerol, 0.3% NP-40) with 1 mM dithiothreitol and 1× protease inhibitor cocktail (Promega, Madison, WI, USA). After centrifugation, supernatant was kept as cytoplasmic extract after addition of NaCl to 0.15 M. Pelleted nuclei were washed three times with hypotonic lysis buffer. To make nuclear extract, nuclei were resuspended in nuclear lysis buffer (same as hypotonic lysis buffer but containing 0.15 M NaCl) with 1 mM dithiothreitol and 1× protease inhibitor cocktail (Promega). After sonication, the supernatant was kept as nuclear extract.
Results
Association of MBNL1/2 With Expanded CUG RNA in FECD Cultured Cells and Patient Tissue
Previous reports have shown that MBNL1 and MBNL2 proteins associate with mutant TCF4 intronic RNA at foci within FECD patient-derived cells in tissue and cell culture, respectively.12,28 To confirm this observation and extend it to patient tissue, we used both the patient-derived immortalized F35T corneal endothelial cell line and corneal endothelial tissue from FECD patients that was obtained after removal of the tissue during transplant surgery.
It is important to emphasize that patient endothelial tissue removed at surgery or the analogous control tissue dissected from donor cornea from the eye bank is a homogeneous single-cell monolayer composed of ∼25,000 to 100,000 individual endothelial cells, respectively. These disease-relevant samples, not the entire cornea, were used for all experiments with human tissue.
RNA foci were visualized with anti-CUG RNA probe by FISH assay. Binding of MBNL1 or MBNL2 was detected using anti-MBNL1 or MBNL2 antibodies followed by immunofluorescence staining (Figs. 1A, 1B). We analyzed at least 50 cells per sample for colocalization of foci and MBNL.
Figure 1.

Colocalization of MBNL proteins with CUG repeat RNA foci in FECD corneal endothelial cells detected by RNA FISH and immunofluorescence staining. MBNL1 and MBNL2 colocalize with CUG repeat RNA nuclear foci in (A) F35T patient-derived corneal endothelial cell line (22/1500 CTG18.1 alleles) and (B) corneal endothelial tissue from an FECD patient with the TCF4 expansion (27/51 CTG18.1 alleles). Percentage of RNA foci that colocalize with MBNL proteins is analyzed in (C) F35T patient-derived corneal endothelial cells (n = 2 independent experiments) and (D) endothelial tissues from FECD patients with TCF4 expansion (n = 5 FECD tissues). Results are shown as the mean ± SD. At least 50 cells were analyzed for each sample. Scale bars: 10 μm.
Microscopy revealed that many CUG repeat foci associated with MBNL1 or MBNL2 (Figs. 1A, 1B). We found that in the F35T cell line, 82% of the foci colocalized with MBNL1 and 39% of the foci colocalized with MBNL2 (Fig. 1C). Examination of five corneal endothelial tissue samples from FECD patients revealed that approximately 30% and 46% of the foci colocalized with MBNL1 and MBNL2, respectively (Fig. 1D). Failure to observe colocalization of all foci with MBNL may reflect inefficient FISH analysis of MBNL binding or may suggest that MBNL does not associate with all RNA foci.
Of note, a majority of the MBNL1 and MBNL2 proteins detected in the nuclei of FECD patient tissues showed localization of MBNL proteins to the foci (Fig. 1B). We also detected these MBNL proteins in the cytoplasm of patient endothelial tissues in a diffuse pattern.
Effect of MBNL Proteins on TCF4 Foci
We hypothesized that binding of MBNL protein might affect the persistence of CUG RNA foci. To test this hypothesis, we used MBNL siRNAs to examine the effect of reducing the amount of MBNL1 and/or MBNL2 protein on CUG RNA foci in two FECD cell lines (Fig. 2A and Supplementary Figs. S1, S2). We analyzed at least 100 cells per sample to quantify foci reduction after knockdown of MBNL proteins.
Figure 2.

Depletion of both MBNL1 and MBNL2 reduces RNA foci in F35T patient-derived corneal endothelial cells. (A) FISH images of CUG repeat RNA foci in F35T patient-derived corneal endothelial cells transfected with the indicated siRNA oligonucleotides. (B) Immunoblotting images of lysates from F35T cells showing the efficiency of siRNA-mediated depletion of MBNL1 or MNBL2 protein by siRNAs complementary to MNBL1 or MBNL2 mRNA. GAPDH was used as a loading control (C). Percentage of cells containing foci and number of foci per 100 cells are shown. Results are shown as the mean ± SD, n = 2 independent experiments. P-value was obtained by t-tests analysis of CM compared with siMBNL1+ siMBNL2. At least 100 cells were analyzed for each sample. CM, scrambled control siRNA; MBNL1mm and MBNL2mm, mismatched control siRNAs based on the sequence of the active siRNAs. Scale bars: 10 μm.
Silencing MBNL1 or MBNL2 individually had no effect on the percentage of cells containing foci or on the number of foci per cell (Figs. 2B, 2C). Minor levels of MBNL1 proteins remaining despite siRNA knockdown may contribute to incomplete disappearance of RNA foci. When both MBNL1 and MBNL2 were silenced, the percentage of cells containing foci and number of foci per cell were significantly decreased in F35T corneal endothelial cells (Fig. 2C). We observed a ∼38% reduction in the percentage of cells with foci and a ∼54 % reduction in the number of foci per 100 cells. Reduction of foci was also observed in the patient-derived F45SV corneal endothelial cell line (Supplementary Fig. S2). We observed no change in foci number when cells were treated with mismatched or scrambled control siRNAs. These data are consistent with the conclusion that MBNL1 and MBNL2 have redundant roles in foci stabilization.
Effect of MBNL Expression on TCF4 RNA
After observing a potential linkage between MNBL levels and CUG RNA foci, we used qPCR to analyze the impact of MBNL levels on the expression of TCF4 mRNA (Supplementary Fig. S3A). Duplex RNAs were used to knockdown MBNL1, MBNL2 or TCF4 mRNA.
Reducing the expression level of MBNL1 or MBNL2 individually or together had no effect on levels of mature TCF4 mRNA (Supplementary Fig. S3A). It suggests that MBNL1 and MBNL2 do not affect mature TCF4 mRNA expression. We also noted that knockdown of TCF4 mRNA has no impact on RNA foci number (Supplementary Figs. S3B–S3D).
Measurement of MBNL RNA and Protein Copy Number
We previously showed that the average copy number of mutant TCF4 intronic RNA in patient-derived corneal endothelial cells or tissue is one to two copies per cell.30 The copy number of mutant RNA is equivalent to the average number of foci, suggesting that each focus is composed of one RNA molecule.
The number of MBNL proteins that might bind to the repeat RNA is limited by the small number of mutant RNA molecules per cell. To determine whether this number might significantly affect the cellular pool of MBNL protein, we measured the approximate number of MBNL mRNA and protein molecules per endothelial cell in both FECD endothelial cell line and FECD patient endothelial tissues (Figs. 3, 4).
Figure 3.

Measurement of the copy number of MBNL1 and MBNL2 mRNA. Standard curves for qPCR efficiency of (A) MBNL1 and (B) MBNL2 were constructed with a serial dilution of purified standard RNAs. Ct values in each dilution were measured in triplicate. The copy numbers of MBNL1 and MBNL2 transcripts per cell in (C) F35T patient-derived corneal endothelial cells (n = 2 independent experiments), (D) control corneal endothelial tissues (n = 3), and (E) patient-derived expanded TCF4 repeat FECD corneal endothelial tissues (n = 3). Results are shown as the mean ± SD.
Figure 4.

Experimental estimates for the protein copy number per cell for MBNL1 and MBNL2. (A) Representative images for quantification of MBNL1 and MBNL2 proteins in F35T patient-derived corneal endothelial cells. Recombinant tagged MBNL1 (N-terminal GST tag) and MBNL2 (N-terminal proprietary tag) were used for standardization and moved slower in the gel because of the tags. (B) Protein copy number per cell in F35T patient-derived corneal endothelial cells (n = 2 independent experiments). Results are shown as the mean ± SD. (C) Representative images for quantification of MBNL1 and MBNL2 proteins in control corneal endothelial tissue. (D) Protein copy number per cell in control corneal endothelial tissues (n = 3 control tissues). Results are shown as the mean ± SD.
Using qPCR to determine the number of MBNL RNA molecules per cell requires determination of standard curves for qPCR efficiency using purified RNAs (Figs. 3A, 3B; Supplementary Fig. S4). Using these standard curves, we estimated the number of MBNL1 and MBNL2 transcripts in F35T corneal endothelial cells at 54 and 120 per cell, respectively (Fig. 3C).
In human corneal endothelial tissue from eye bank donors (Fig. 3D) and mutant endothelial tissue from FECD patients (Fig. 3E), however, the number of transcripts per endothelial cell was more than 10-fold lower. We speculate that the lower RNA copy number in human endothelial tissue versus immortalized corneal endothelial cell lines may be due to differences in proliferative capacity because endothelial cells of patient tissue are considered postmitotic.34
The lower levels of MBNL RNA in human tissue suggested that protein levels might also be lower. To test this hypothesis, we established standard curves for protein concentration using known amounts of commercial purified tagged-MBNL1 or MBNL2 (Supplementary Figs. S5A, S5B; Fig. 4A) and used these curves to evaluate protein levels from known numbers of F35T cells. We estimated levels of MBNL1 and MBNL2 in F35T cells at 1,390,000 and 660,000 copies per cell, respectively (Fig. 4B).
We then evaluated the amount of MBNL1 or MBNL2 protein in control endothelial tissue samples from eye bank donor corneas (Supplementary Figs. S5C, S5D; Fig. 4C). Quantitation in FECD tissue is severely restricted by the fact that each FECD sample has ∼75% attrition of endothelial cell density compared with control tissue, and the supply of FECD tissue is limited. Therefore, we estimated the protein copy number of MBNL 1 and 2 at ∼30,000 per cell in control endothelial tissue (Fig. 4D), which is ∼10- to 30-fold lower than in cultured cells.
Cellular Distribution of MBNL
The total number of MBNL1 and MBNL2 proteins measured in human tissue likely exceeds the sequestration capacity of the handful of mutant expanded CUG repeat RNA molecules present in diseased tissue. MBNL proteins, however, act both in the cytoplasm and nucleus. It is possible that only nuclear MBNL might be relevant when considering the importance of protein sequestration as a disease mechanism.
To investigate this possibility, we examined the distribution of MBNL1 and MBNL2 between cytoplasm and nuclei of both culture cells and human corneal endothelial tissue. Using Western blot analysis of the cytoplasmic and nuclear fractions, we determined that the majority of MBNL1 and MBNL 2 are found in the cytoplasm in both the FECD endothelial cell line F35T and control endothelial cell line HCN19 (Fig. 5A).
Figure 5.

Distribution of MBNL1 and MBNL2 in FECD and control corneal endothelial cells and in corneal tissue. Immunoblotting images of cytoplasmic (C) and nuclear (N) fractions in (A) Lysates from FECD patient-derived corneal endothelial cell line F35T and control donor-derived corneal endothelial cell line HCN19. (B) Lysates from control corneal endothelial tissues. Two independent replicate experiments are shown. Cytoplasmic and nuclear lysates from equal amounts of cells were loaded in each lane for each protein. GAPDH was used as a cytoplasmic marker. Histone was used as a nuclear marker. Estimation by densitometry measurements showed 3% or less MBNLs proteins to be present in nuclei in control endothelial tissues. (C) Immunofluorescence images of MBNL1 and MBNL2 in control corneal endothelial tissue. Nuclear DAPI staining (blue) defines cell nuclei. Scale bars: 10 μm.
We then examined MBNL1 and MBNL2 levels in human control corneal endothelial tissues. Western blot analysis revealed that a substantial majority of MBNL protein resides in the cytoplasm, with 3% MBNL1 protein and no MBNL2 protein detected in cell nuclei (Fig. 5B). These data are consistent with the observation by immunofluorescence (Fig. 5C). Given the measured value of 65,000 MBNL proteins per cell, we estimate that approximately 1950 MBNL molecules localize to cell nuclei in endothelial tissue.
Estimating the Number of MBNL1 and MBNL2 Proteins Bound to Mutant TCF4 Foci
The mean repeat CUG repeat length of FECD patients' peripheral leukocytes is approximately 100, with some individuals harboring over a thousand triplet repeats.10 We examined the reported crystal structure of MBNL1 to estimate the capacity of the mutant expanded repeat to bind MBNL protein.
MBNL1 has four conserved ZnF RNA binding domains arranged in tandem pairs (ZnF1/2 and ZnF3/4).35 These ZnF tandem domains target two separated GC(U) sites within RNA with an optimal spacer length of 10 to 15 nucleotides. Using single-molecule binding assays to assess the MBNL1–(CUG)n interaction, (CUG)4 is the shortest validated repeat model that can be used in analyses.36 Therefore, a mutant RNA with 100 CUG repeats can bind approximately 25 MBNL proteins.
We previously showed that each focus detected in human corneal endothelial tissue corresponds to one mutant TCF4 intronic RNA transcript.30 To determine the number of foci/mutant RNA transcripts within a population of cells in FECD corneal endothelial tissue, we used FISH with an anti-CUG repeat probe to visualize the distribution of foci with cells (Figs. 6A–6D). We observe that most cells have one or two foci. A few cells had as many as 10 foci. This observation, combined with our estimate that each mutant RNA molecule with 100 CUG repeats can bind ∼25 MBNL proteins, suggests a potential to bind from 25 to 250 MBNL proteins within different cells.
Figure 6.

The number of CUG repeat RNA foci varies in FECD corneal endothelial cells in patient tissue detected by RNA FISH. (A) Image of CUG repeat RNA nuclear foci in FECD patient corneal tissue (CA054). Right: enlarged pictures of two cell nuclei with 7 or 10 foci. (B) Analysis of foci distribution in FECD corneal tissue CA054 showing that the number of foci per cell varies greatly among cells. (C) Image of CUG repeat RNA nuclear foci in FECD corneal tissue (CA056). Right: enlarged pictures of two cell nuclei with nine or five foci. (D) Analysis of foci distribution in FECD corneal tissue CA056. Two tissue samples are shown to demonstrate reproducibility. Scale bars: 10 μm.
Discussion
FECD is a common age-related degenerative disorder,and improved treatments are necessary to complement existing surgical interventions and help prevent loss of vision. Developing such interventions will be facilitated by insights into the molecular mechanism of disease pathogenesis.
Previous reports on DM1 have hypothesized that the expanded CUG repeats in the 3′ UTR transcript of DMPK gene bind MBNL proteins, sequester them from the cellular protein pool, and cause global splicing changes at MBNL-target genes. Similar splicing abnormalities are observed in FECD where the mutant CUG repeat is located in an intron within the TCF4 gene.12,29 The central question for understanding the molecular basis for disease causation is how expression of a mutant expanded repeat RNA can lead to global changes in splicing. The goal of this study was to develop a quantitative framework for analyzing the strength of the sequestration hypothesis.
MBNL Protein Interaction With Mutant TCF4 RNA Foci
Our results confirm that MBNL1 and MBNL2 associate with expanded CUG foci in patient-derived cell lines and patient tissue (Fig. 1). Association of MBNL1 and MBNL2 may stabilize the RNA containing the CUG repeat (Fig. 2; Supplementary Figs. S1, S2).
In a previous report of DM1 cultured myoblasts, siRNA knockdown of MBNL1 resulted in ∼70% of reduction of foci compared with ∼25% reduction with MBNL2 knockdown.31 Simultaneous depletion of both MBNLs resulted in only an incremental greater reduction of foci (∼80%), resulting the authors to conclude that MBNL1 is the primary determinant of DM1 foci.
In contrast, our data show that siRNA-mediated knockdown of MBNL1 or MBNL2 individually results in only nominal reduction of expanded CUG foci in FECD. Simultaneous depletion of both MBNL 1 and 2 reduce foci formation in FECD patient-derived cultured cells, suggesting that MBNL1 and MBNL2 play a redundant role in stabilizing mutant TCF4 RNA in corneal endothelium.
Additionally, we found that knockdown of mature TCF4 mRNA has no impact on RNA foci number. The splicing of introns occurs with retention of CUG intronic RNA in the nucleus followed by export of the mature mRNA into the cytoplasm. Our results are consistent with the conclusion that siRNA-mediated knockdown of mature TCF4 mRNA primarily occurs in the cytoplasm. This conclusion is consistent with a thorough study of long noncoding RNAs that reported much higher activity of siRNAs in the cytoplasm relative to the nucleus.37
Quantitative Assessment of Mutant RNA and MBNL Protein
Alteration of splicing is a hallmark of FECD. The sequestration of MBNL proteins through binding the expanded CUG repeat offers a logical explanation for the observed splicing abnormalities in both DM1 and FECD.
However, in both DM121,38,39 and FECD,30 the number of disease-causing RNA molecules per cell has been measured to be less than five per cell. Similar low numbers of RNA molecules have also been implicated in amyotrophic lateral sclerosis/frontotemporal dementia, another disease associated with an expanded repeat within mutant intronic RNA.40
It is a challenge to understand how a limited number of RNA molecules might bind a sufficient number of MBNL proteins to affect global splicing because a low number of mutant RNA molecules necessarily restricts the capacity to bind MBNL proteins. For example, an average of ∼100 CUG repeats are detected in peripheral leukocytes of FECD subjects,10 and we estimate that each mutant RNA has the capacity to directly bind approximately 25 MBNL proteins.
Understanding whether sequestration of MBNL proteins is a viable hypothesis to explain the connection between expanded repeat mutations and altered splicing requires quantitative determination of the expression number of MBNL1 and MBNL2 proteins per cell (Fig. 7). Two million MNBL proteins are found in each cultured endothelial cell, but only 65,000 copies were identified per cell in patient tissue. We acknowledge that these estimates of MBNL protein levels per cell are subject to errors in the measurement of standard recombinant protein concentrations and endothelial cell number. We measured MBNL protein number in tissue using triplicated determination and present our results as mean ± SD. Our assessments of both mRNA and protein copy number combine to suggest that the MBNL family of splicing factors are less abundant in postmitotic endothelial tissue compared with cultured human corneal endothelial cells.
Figure 7.
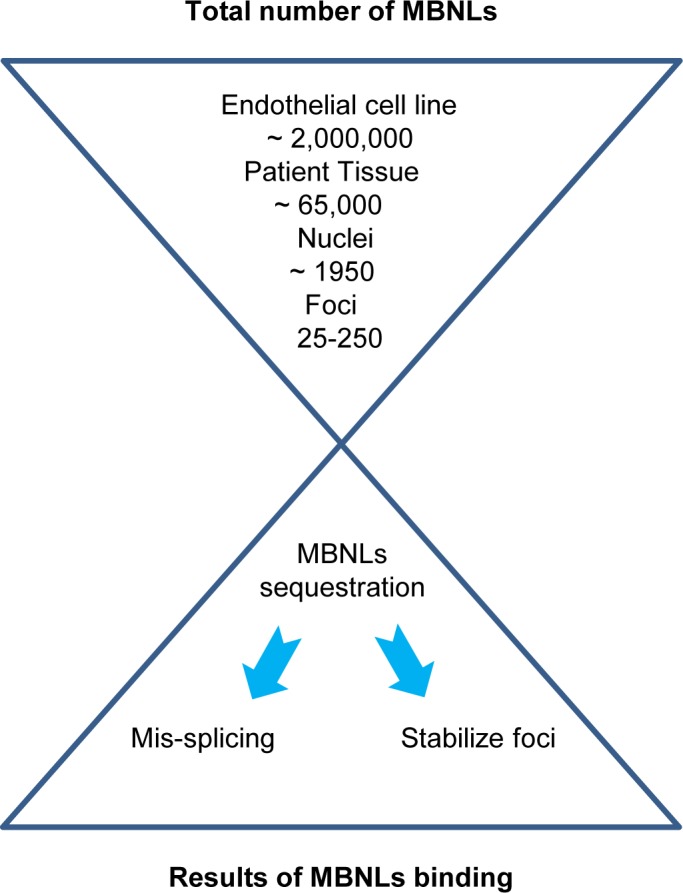
Estimating the quantitative relationship between cellular MBNL protein and the capacity for protein recognition by expanded RNA within intronic TCF4 foci. There are approximately 2 million copies of MBNL1 and MBNL2 proteins in patient-derived cultured endothelial cells but only 65,000 copies per cells in corneal endothelium taken from human donor corneal tissue. In human endothelial tissue, most MNBL1 and MBNL2 protein are in the cytoplasm, with only a fraction (estimated at 1950 copies per cell) remaining in cell nuclei. Depending on the number of repeats and the number of foci per cell, expanded CAG repeat foci have the capacity to bind 25-250 MBNL1 and MBNL2 proteins.
Although the muscleblind-like family of RNA-binding proteins are splicing factors in the nuclear compartment, they also play an important role in mRNA stability and localization in the cytoplasmic pool.41 Our data suggest that just 3% of MBNL protein is in the nucleus in human corneal endothelial tissue, leading us to estimate that there are ∼1950 copies of MBNL protein per cell nuclei (Fig. 7).
Mutant RNA, MBNL, and Molecular Disease Mechanism
Not only are MBNL proteins expressed at a relatively low level in corneal tissue, the relatively little MBNL that is present is overwhelmingly localized to the cytoplasm. Cells have between 1 and 10 mutant RNA foci per cell, giving mutant RNA the capacity to bind as many as 250 MBNL proteins (Fig. 7). Based on these calculations, mutant RNA may have the ability to bind approximately 10% of nuclear MBNL protein.
Ten percent may seem like a minor fraction of MBNL protein, but FECD is a late-onset disease that requires five to six decades before symptoms begin to manifest themselves. It may not be necessary to dramatically reduce MBNL1/2 protein levels to cause a gradual deterioration of cellular function over time. It is also possible that the disease is triggered in the subset of cells with the highest expression of mutant TCF4 transcript and the highest capacity to sequester MBNL protein. Additionally, the nucleation of multiple adjacent proteins on the repeat may create the opportunity for deleterious interactions.
Somatic instability of the repeat locus in affected tissues has been well documented in other triplet repeat disorders such as DM1.42 Tibialis anterior muscle biopsies of DM1 subjects have shown repeat expansions of approximately 4000 to 6000 compared with several hundred repeats in the peripheral leukocytes of these subjects.43 Somatic mutations in corneal endothelial tissue resulting in larger triplet repeat lengths may further enhance the sequestration of MBNLs by a small number of mutant repeat RNA molecules in FECD patient corneal cells.
Conclusions
FECD is an age-related degenerative caused by a limited number of mutant RNA molecules per cell. Previous studies have demonstrated that FECD is characterized by global splicing defects. DM1, another disease caused by an expanded CUG repeat, is caused by similar splicing defects, and reduced MBNL1 and MBNL2 function has been implicated as a cause for both diseases. Our data suggest that MBNL1/2 may play redundant roles in stabilizing the mutant TCF4 RNA.
Any explanation for disease causation must confront the observation that a small number of mutant RNA molecules disrupt normal gene splicing. We find that the numbers of MBNL1 and MBNL2 protein in the nuclei of corneal endothelial tissue are a fraction of the total pool of cellular MBNL1 and MBNL2. It is plausible that, over time, sequestration of MBNL1 and MBNL2 by the mutant repeat RNA could perturb splicing and eventually lead to age-related corneal disease pathology.
Supplementary Material
Acknowledgments
The authors thank the patients for their participation in this study; Albert Jun for generously sharing the F35T cell line; and collaborating corneal specialists Wayne R. Bowman, Brad Bowman, and Walter Beebe.
Supported by Grants R01EY022161 (VVM), P30EY020799 (VVM), and R35GM118103 (DRC) from the National Institutes of Health, Bethesda, MD, USA; an unrestricted grant from Research to Prevent Blindness, New York, NY, USA (VVM); Harrington Scholar-Innovator Award from Harrington Discovery Institute (VVM); the Alfred and Kathy Gilman Special Opportunities in Pharmacology Fund (DRC); and the Robert A. Welch Foundation I-1244 (DRC). VVM is the Paul T. Stoffel/Centex Professor in Clinical Care. DRC is the Rusty Kelley Professor of Biomedical Science.
Disclosure: Z. Rong, None; J. Hu, None; D.R. Corey, None; V.V. Mootha, None
References
- 1.Rohilla KJ, Gagnon KT. RNA biology of disease-associated microsatellite repeat expansions. Acta Neuropathol Commun. 2017;5:63. doi: 10.1186/s40478-017-0468-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao FB, Richter JD, Cleveland DW. Rethinking unconventional translation in neurodegeneration. Cell. 2017;171:994–1000. doi: 10.1016/j.cell.2017.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jain A, Vale RD. RNA phase transitions in repeat expansion disorders. Nature. 2017;546:243–247. doi: 10.1038/nature22386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorenzetti DW, Uotila MH, Parikh N, Kaufman HE. Central cornea guttata. Incidence in the general population. Am J Ophthalmol. 1967;64:1155–1158. [PubMed] [Google Scholar]
- 5.Deng SX, Lee WB, Hammersmith KM, et al. Descemet membrane endothelial keratoplasty: safety and outcomes: a report by the American Academy of Ophthalmology. Ophthalmology. 2018;125:295–310. doi: 10.1016/j.ophtha.2017.08.015. [DOI] [PubMed] [Google Scholar]
- 6.Ang M, Soh Y, Htoon HM, Mehta JS, Tan D. Five-year graft survival comparing Descemet stripping automated endothelial keratoplasty and penetrating keratoplasty. Ophthalmology. 2016;123:1646–1652. doi: 10.1016/j.ophtha.2016.04.049. [DOI] [PubMed] [Google Scholar]
- 7.Wieben ED, Aleff RA, Tosakulwong N, et al. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS One. 2012;7:e49083. doi: 10.1371/journal.pone.0049083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mootha VV, Gong X, Ku HC, Xing C. Association and familial segregation of CTG18.1 trinucleotide repeat expansion of TCF4 gene in Fuchs' endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2014;55:33–42. doi: 10.1167/iovs.13-12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xing C, Gong X, Hussain I, et al. Transethnic replication of association of CTG18.1 repeat expansion of TCF4 gene with Fuchs' corneal dystrophy in Chinese implies common causal variant. Invest Ophthalmol Vis Sci. 2014;55:7073–7078. doi: 10.1167/iovs.14-15390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soliman AZ, Xing C, Radwan SH, Gong X, Mootha VV. Correlation of severity of Fuchs endothelial corneal dystrophy with triplet repeat expansion in TCF4. JAMA Ophthalmol. 2015;133:1386–1391. doi: 10.1001/jamaophthalmol.2015.3430. [DOI] [PubMed] [Google Scholar]
- 11.Mootha VV, Hussain I, Cunnusamy K, et al. TCF4 triplet repeat expansion and nuclear RNA Foci in Fuchs' endothelial corneal dystrophy. Invest Ophthmol Vis Sci. 2015;56:2003–2011. doi: 10.1167/iovs.14-16222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du J, Aleff RA, Soragni E, et al. RNA toxicity and missplicing in the common eye disease fuchs endothelial corneal dystrophy. J Biol Chem. 2015;290:5979–5990. doi: 10.1074/jbc.M114.621607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okumura N, Hayashi R, Nakano M, et al. Effect of trinucleotide repeat expansion on the expression of TCF4 mRNA in Fuchs' endothelial corneal dystrophy. Invest Ophthmol Vis Sci. 2019;60:779–786. doi: 10.1167/iovs.18-25760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cano A, Portillo F. An emerging role for class I bHLH E2-2 proteins in EMT regulation and tumor progression. Cell Adhesion Migration. 2010;4:56–60. doi: 10.4161/cam.4.1.9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu AY, Eberhart CG, Jun AS. Fuchs endothelial corneal dystrophy: a neurodegenerative disorder? JAMA Ophthalmol. 2014;132:377–378. doi: 10.1001/jamaophthalmol.2013.7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatou S, Yoshida S, Higa K, et al. Functional corneal endothelium derived from corneal stroma stem cells of neural crest origin by retinoic acid and Wnt/beta-catenin signaling. Stem Cells Dev. 2013;22:828–839. doi: 10.1089/scd.2012.0286. [DOI] [PubMed] [Google Scholar]
- 17.Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;69:385. doi: 10.1016/0092-8674(92)90418-c. [DOI] [PubMed] [Google Scholar]
- 18.Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 19.Gattey D, Zhu AY, Stagner A, Terry MA, Jun AS. Fuchs endothelial corneal dystrophy in patients with myotonic dystrophy: a case series. Cornea. 2014;33:96–98. doi: 10.1097/ICO.0000000000000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mootha VV, Hansen B, Rong Z, et al. Fuchs' endothelial corneal dystrophy and RNA foci in patients with myotonic dystrophy. Invest Ophthalmol Vis Sci. 2017;58:4579–4585. doi: 10.1167/iovs.17-22350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taneja KL, Mccurrach M, Schalling M, Housman D, Singer RH. Foci of trinucleotide repeat transcripts in nuclei of myotonic-dystrophy cells and tissues. J Cell Biol. 1995;128:995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller JW, Urbinati CR, Teng-Umnuay P, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Timchenko LT, Miller JW, Timchenko NA, et al. Identification of a (CUG)(n) triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 1996;24:4407–4414. doi: 10.1093/nar/24.22.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fardaei M, Rogers MT, Thorpe HM, et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet. 2002;11:805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 25.Lin X, Miller JW, Mankodi A, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Human Molec Genet. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- 26.Koshelev M, Sarma S, Price RE, Wehrens XH, Cooper TA. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Human Molec Genet. 2010;19:1066–1075. doi: 10.1093/hmg/ddp570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ward AJ, Rimer M, Killian JM, Dowling JJ, Cooper TA. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Human Molec Genet. 2010;19:3614–3622. doi: 10.1093/hmg/ddq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zarouchlioti C, Sanchez-Pintado B, Hafford Tear NJ, et al. Antisense therapy for a common corneal dystrophy ameliorates TCF4 repeat expansion-mediated toxicity. Am J Human Genet. 2018;102:528–539. doi: 10.1016/j.ajhg.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wieben ED, Aleff RA, Tang X, et al. Trinucleotide repeat expansion in the transcription factor 4 (TCF4) gene leads to widespread mRNA splicing changes in Fuchs' endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2017;58:343–352. doi: 10.1167/iovs.16-20900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu J, Rong Z, Gong X, et al. Oligonucleotides targeting TCF4 triplet repeat expansion inhibit RNA foci and mis-splicing in Fuchs' dystrophy. Human Molec Genet. 2018;27:1015–1026. doi: 10.1093/hmg/ddy018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dansithong W, Paul S, Comai L, Reddy S. MBNL1 is the primary determinant of focus formation and aberrant insulin receptor splicing in DM1. J Biol Chem. 2005;280:5773–5780. doi: 10.1074/jbc.M410781200. [DOI] [PubMed] [Google Scholar]
- 32.Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA. Muscleblind proteins regulate alternative splicing. EMBO J. 2004;23:3103–3112. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forrest MP, Waite AJ, Martin-Rendon E, Blake DJ. Knockdown of human TCF4 affects multiple signaling pathways involved in cell survival, epithelial to mesenchymal transition and neuronal differentiation. PLoS One. 2013;8:e73169. doi: 10.1371/journal.pone.0073169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joyce NC. Proliferative capacity of corneal endothelial cells. Exp Eye Res. 2012;95:16–23. doi: 10.1016/j.exer.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teplova M, Patel DJ. Structural insights into RNA recognition by the alternative-splicing regulator muscleblind-like MBNL1. Nat Struct Mol Biol. 2008;15:1343–1351. doi: 10.1038/nsmb.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haghighat Jahromi A, Honda M, Zimmerman SC, Spies M. Single-molecule study of the CUG repeat-MBNL1 interaction and its inhibition by small molecules. Nucl Acids Res. 2013;41:6687–6697. doi: 10.1093/nar/gkt330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lennox KA, Behlke MA. Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucl Acids Res. 2016;44:863–877. doi: 10.1093/nar/gkv1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ho TH, Savkur RS, Poulos MG, Mancini MA, Swanson MS, Cooper TA. Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J Cell Sci. 2005;118:2923–2933. doi: 10.1242/jcs.02404. [DOI] [PubMed] [Google Scholar]
- 39.Gudde AEEG, Gonzalez-Barriga A, van den Broek WJAA, Wieringa B, Wansink DG. A low absolute number of expanded transcripts is involved in myotonic dystrophy type 1 manifestation in muscle. Hum Mol Genet. 2016;25:1648–1662. doi: 10.1093/hmg/ddw042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu J, Hu J, Ludlow AT, et al. c9orf72 Disease-related foci are each composed of one mutant expanded repeat RNA. Cell Chem Biol. 2017;24:141–148. doi: 10.1016/j.chembiol.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang ET, Cody NAL, Jog S, et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–724. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ashizawa T, Dubel JR, Harati Y. Somatic instability of CTG repeat in myotonic dystrophy. Neurology. 1993;43:2674–2678. doi: 10.1212/wnl.43.12.2674. [DOI] [PubMed] [Google Scholar]
- 43.Nakamori M, Sobczak K, Puwanant A, et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol. 2013;74:862–872. doi: 10.1002/ana.23992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.