

ABSTRACT
Prion diseases are fatal transmissible neurodegenerative disorders that affect animals and humans. Prions are proteinaceous infectious particles consisting of a misfolded isoform of the cellular prion protein PrPC, termed PrPSc. PrPSc accumulates in infected neurons due to partial resistance to proteolytic digestion. Using compounds that interfere with the production of PrPSc or enhance its degradation cure prion infection in vitro, but most drugs failed when used to treat prion-infected rodents. In order to synergize the effect of anti-prion drugs, we combined drugs interfering with the generation of PrPSc with compounds inducing PrPSc degradation. Here, we tested autophagy stimulators (rapamycin or AR12) and cellulose ether compounds (TC-5RW or 60SH-50) either as single or combination treatment of mice infected with RML prions. Single drug treatments significantly extended the survival compared to the untreated group. As anticipated, also all the combination therapy groups showed extended survival compared to the untreated group, but no combination treatment showed superior effects to 60SH-50 or TC-5RW treatment alone. Unexpectedly, we later found that combining autophagy stimulator and cellulose ether treatment in cultured neuronal cells mitigated the pro-autophagic activity of AR12 and rapamycin, which can in part explain the in vivo results. Overall, we show that it is critical to exclude antagonizing drug effects when attempting combination therapy. In addition, we identified AR-12 as a pro-autophagic drug that significantly extends survival of prion-infected mice, has no adverse side effects on the animals used in this study, and can be useful in future studies.
KEYWORDS: Prion, prion disease, combination therapy, autophagy, AR12, rapamycin, cellulose ethers, TC-5RW, 60SH-50
Introduction
Prion diseases are neurodegenerative disorders in humans and animals that are invariably fatal. Human prion diseases occur in sporadic, genetic, and acquired forms. The autocatalytic conversion of the non-infectious and endogenously expressed isoform PrPC to the infectious isoform PrPSc is attributed to be the cause of the disease [1,2]. The human forms of prion disease include familial, sporadic, iatrogenic and variant Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), and fatal familial insomnia (FFI). Animal forms are scrapie in sheep and goat, bovine spongiform encephalopathy (BSE) in cattle and other species, and chronic wasting disease (CWD) in deer, reindeer, elk and moose [3–7]. Loss of neurons, vacuolation, astrogliosis and microglial activation is the major histopathological findings in prion disease. This results in spongiform degeneration of the central nervous system (CNS), leading to ataxia, behavioural changes and highly progressive loss of intellectual propensity [8,9].
Prions use living cells for propagation by conformational conversion of endogenous PrPC, and newly generated PrPSc undergoes subcellular trafficking events and is exposed to cellular degradation and recycling machineries. Mechanistically, there are many steps in the life cycle of prions which can be used as therapeutic targets, ranging from PrPC substrate, PrPC/PrPSc interaction to lysosomal PrPSc clearance [10,11]. A huge variety of chemical compounds was tested in vitro and in vivo for anti-prion activities. Historically, PrPSc was aimed as therapeutic target [12,13], with an emphasis early on lysosomal degradation [14–16]. Another strategy was targeting PrPC, by binding to chemical compounds, other ligands or antibodies [11]. Interestingly, PrPSc itself is a difficult pharmacological target because of its poorly defined structure and the ability to generate different structural conformers that can confound treatment. As a consequence of the quasi-species nature of prions [17], prions are sensitive to the development of drug resistance [18–20]. Although combination therapy could be used to create synergies by targeting different steps in the life cycle of prions, this was hardly done for mammalian prions [21].
Autophagy is the natural, regulated mechanism that allows the orderly degradation and recycling of cellular components [22,23]. We and others have established pharmacological induction of autophagic degradation as a potent anti-prion strategy [24–28]. Our recent work demonstrated that AR12 (OSU-03012, PDK1 inhibitor), a novel autophagy-inducing drug, enhanced the degradation of PrPSc and reduced prion seeding activity in cell lines infected with various prion strains in vitro [29]. Here we test the in vivo effect of AR12. Additionally, we used the autophagy stimulator and mTOR inhibitor rapamycin which we have shown previously to extend the survival time of prion-infected mice [26].
Recently, cellulose ethers (CEs) have emerged as promising anti-prion compounds. The CE compounds are non-ionic, non-digestible, water soluble and widely used in the pharmaceutical industry as inert additives. They showed remarkable anti-prion effects with only a single subcutaneous injection [30]. Mechanistically, they are thought to inhibit the conversion of PrPC into PrPSc.
Here, we studied the anti-prion effect of the CE compounds TC-5RW and 60SH-50 when combined with the autophagy stimulators AR12 or rapamycin, under the premise that these drugs target different steps and might therefore induce additive anti-prion effects. We tested four different combination groups (AR12, TC-5RW), (AR12, 60SH-50), (rapamycin, TC-5RW) and (rapamycin, 60SH-50) in prion-infected mice. Our results demonstrate that all combination therapy groups significantly extended survival compared to the untreated group. However, no combination treatment showed superior effects to 60SH-50 or TC-5RW treatment alone. To exclude antagonizing drug effects, we revisited these drug combinations in prion-infected cultured cells. These studies showed that combining autophagy stimulators and cellulose ethers significantly alleviated the autophagic activity of AR12 and rapamycin. This could explain the results obtained in vivo and shows that it is critical to exclude antagonizing drug effects when attempting combination therapy.
Results
Treatment with autophagy stimulators AR12 or rapamycin significantly prolonged the survival of RML infected mice
The role of autophagy in modulating prion disease has been the focus of our research for more than a decade [24–26,31–33]. Here, we examined the effect of treatment with the autophagy inducer AR12 on the survival times of FVB mice infected intra-cerebrally with RML prions. AR12 treatment started 30 days after prion inoculation and was administered through i.p. injection (5 mg/kg body weight) [34] for 4 weeks, twice weekly. After 4 weeks of i.p. treatment, AR12 was administered in drinking water (50 µg/ml) until the experimental endpoint (Figure 1). Our data showed markedly improved survival of the animals treated with AR12 compared with untreated animals (p = 0.0008) (Figure 2(a)). In addition, we used rapamycin to compare its effect to AR12. Rapamycin was administered to the animals via i.p. route (20 mg/kg body weight) [35], twice weekly for 4 weeks (Figure 1(a)). Due to the poor solubility of rapamycin in water, we did not continue rapamycin treatment in drinking water. Our results show significant extended survival of the animals treated with rapamycin compared to untreated ones (p = 0.0002) (Figure 2(b)). There was no significant difference between the effect of AR12 and rapamycin on the survival time. Mean survival times are displayed in Table 1. Notably, this is the first study to demonstrate the anti-prion effect of AR12 in vivo.
Figure 1.
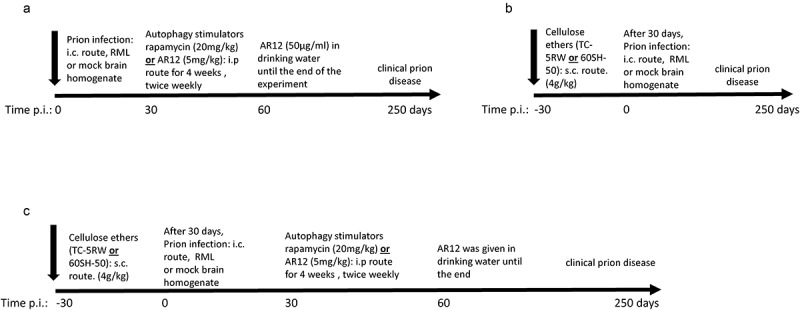
Schematic representation of animal experiments. (a) Scheme of the animal experiments using autophagy inducers AR12 and rapamycin. (b) Schematic representation of animal experiments using cellulose ethers TC-5RW or 60SH-50. (c) Animal experiments using drug combinations of cellulose ethers and autophagy stimulators. FVB mice were inoculated with either RML prions from terminally sick mice or mock brain homogenate.
Figure 2.
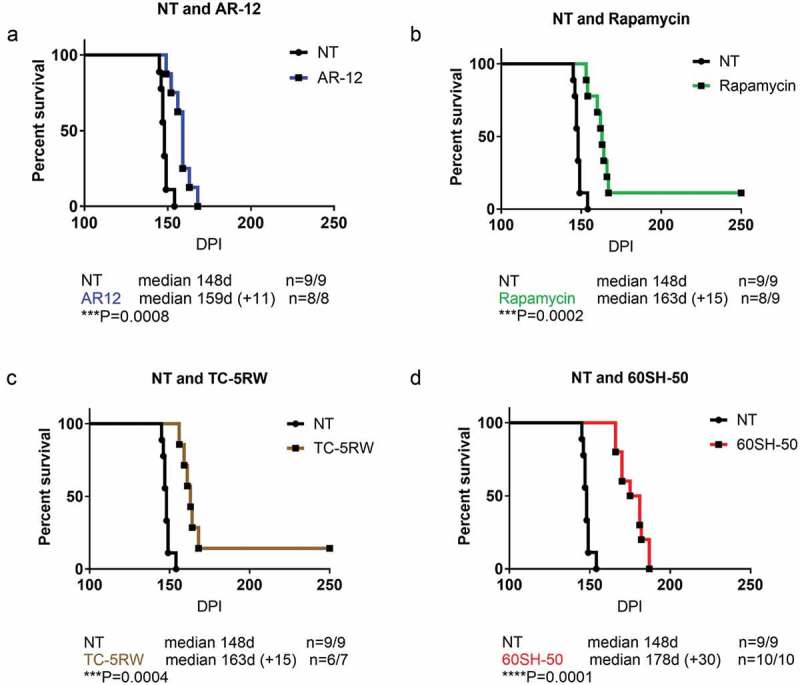
Effect of autophagy stimulators or CE compounds in RML infected FVB mice. Survival analysis of FVB mice intra-cerebrally infected with RML prion was performed. (a) Thirty days after i.c. inoculation mice were treated with AR-12 (5 mg/kg, i.p.) twice per week for 4 weeks, then AR-12 was added to the drinking water (50 µg/ml) until the end of the experiment. (b) Rapamycin treatment started also at 30 dpi. The mice were injected i.p. twice per week (20 mg/kg) for 4 weeks. (c and d) Mice were treated with single subcutaneous injection of either CE-5RW or 60SH-50 at 4g/kg body weight 30 days before i.c. inoculation. Experiments were stopped at 250 dpi.
Table 1.
Mean survival time of mice groups.
| Animal group | #of mice/group | Treatment | Survival time (DPI) (Mean ± SD) |
|---|---|---|---|
| Mock | 9 | - | 250 |
| Mock-AR12 | 4 | AR12 | 250 |
| Mock-Rapamycin | 4 | Rapamycin | 250 |
| RML | 9 | - | 148.11 ± 2.57 |
| RML-AR12 | 8 | AR12 | 158.12 ± 5.96 |
| RML-Rapamycin | 9* | Rapamycin | 161.83 ± 5.19 |
| RML-5RW | 7* | 5RW | 161.83 ± 4.16 |
| RML-60SH | 10 | 60SH | 173.87 ± 6.79 |
| RML-(AR12 + 5RW) | 10* | AR12 + 5RW | 162.11 ± 3.68 |
| RML-(AR12 + 5RW) | 8 | AR12 + 60HS | 165.75 ± 4.65 |
| RML-(Rapamycin+5RW) | 10 | Rapamycin+5RW | 167 ± 2.74 |
| RML-(Rapamycin+60SH) | 8 | Rapamycin+60SH | 173 ± 9.82 |
*One animal in the group did not show any prion signs until the end of the experiment and sacrificed at 250 DPI. This one animal is not considered in the calculation of mean since the calculations are only for prion sick mice.
FVB mice were either mock or RML infected. Mock groups were sacrificed at 250 days post mock i.c. inoculation.
AR-12 treatment was started 4 weeks after i.c. inoculation. The drug was I.P. injected for 4 weeks (5mg/ml) then shifted to dissolving the drug in water (50ug/ml, 15,000ug) until the end of the experiment.
Rapamycin treatment started 4 weeks after i.c. inoculation. The drug was I.P. injected for 4 weeks (20mg/ml) only.
Cellulose ethers (5RW and 60SH) were given as a single subcutaneous dose (4g/kg) 4 weeks before i.c. inoculation.
Treatment with cellulose ethers TC-5RW or 60SH-50 significantly increased survival of RML infected mice
Next, we tested the prophylactic effect of two cellulose ethers, TC-5RW and 60SH-50. Both CE compounds were used in a previous study; however, C57Bl/6 mice were used for these experiments [30]. Given the proposed dependence of CE effects on the genetic background of mice [30], we first wanted to confirm the anti-prion effects of CEs in FVB mice. CEs were administered as a single subcutaneous dose (4g/kg), 30 days before i.c. RML prion inoculation (Figure 1(b)). In line with previous results, we show that treatment with either TC-5RW or 60SH-50 results in a significantly increased survival time (p = 0.0004) and p = 0.0001, respectively (Figure 2(c,d); Table 1).
Combination therapy using autophagy stimulators and cellulose ethers
The promising outcome using the aforementioned compounds motivated us to test a combination therapy of autophagy stimulators and CEs. We had four groups of combinations, AR12 with TC-5RW, AR12 with 60SH-50, rapamycin with TC-5RW, and rapamycin with 60SH-50. Of note, the same regimen of treatment that has been used with the single drug treatments was applied for the combinations. CEs (TC-5RW or 60SH-50) were given as a single subcutaneous dose, 30 days before i.c. inoculation with RML prions. Thirty days after inoculation, animals received either AR12 or rapamycin twice per week (i.p.) for 4 weeks. After 4 weeks, AR12 treatment was continued until the experimental endpoint by applying the drug with the drinking water (Figure 1(c)). This was not possible for rapamycin due to its poor solubility in water. All mice treated with the drug combinations showed significantly extended survival times compared to the untreated group (Figure 3(a–d)). However, when we compared the survival times of mice co-treated with AR12 and TC-5RW to those treated with either of the drug alone, we did not find a significant difference (Figure 4(a,b)). Surprisingly, combining AR12 and 60SH-50 resulted in a significant decrease in the survival compared to the group treated only with 60SH-50 (p = 0.0032) (Figure 4(c)). Yet, there was a significant increase in the survival of the AR12 and 60SH-50 co-treated group compared to the AR12 only treated group (p = 0.011) (Figure 4(d)). Similar results were obtained with the combination of rapamycin and TC-5RW. While treatment with the drug combination did not result in a significantly different survival time compared to the TC-5RW treated group, a significant increase of the survival compared to the rapamycin only treated group was found (p = 0.03) (Figure 5(b)). Furthermore, comparing the combination of rapamycin with 60SH-50 to treatment with rapamycin only or 60SH-50 only, similar survival times were observed (Figure 5(c,d)).
Figure 3.
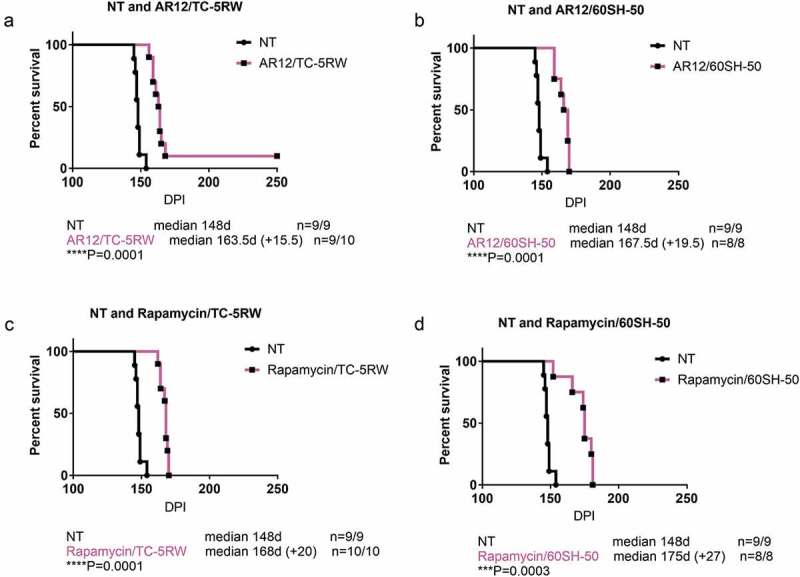
Effect of combined treatment with autophagy stimulators and CEs compared to the mock-treated group. Survival analysis of FVB mice i.c. infected with RML prions. (a) Combination of AR-12 (5 mg/kg i.p twice per week for 4 weeks, then AR-12 was added to the drinking water (50 µg/ml) until the end of the experiment) and single subcutaneous injection of TC-5RW one month before prion inoculation. (b) Combination of AR-12 (treatment as in a) and single subcutaneous injection of 60SH-50 one month before prion inoculation. (c) Combination of rapamycin (starting at 30 dpi, treatment (i.p.) twice per week (20 mg/kg) for 4 weeks only) with TC-5RW given as a single subcutaneous injection 30 days before prion inoculation. (d) Combination of rapamycin (treatment as in c) with 60SH-50 given as a single subcutaneous injection 30 days before prion inoculation. CEs were given at a dose of 4 g/kg body weight. Experiments were stopped at 250 dpi.
Figure 4.
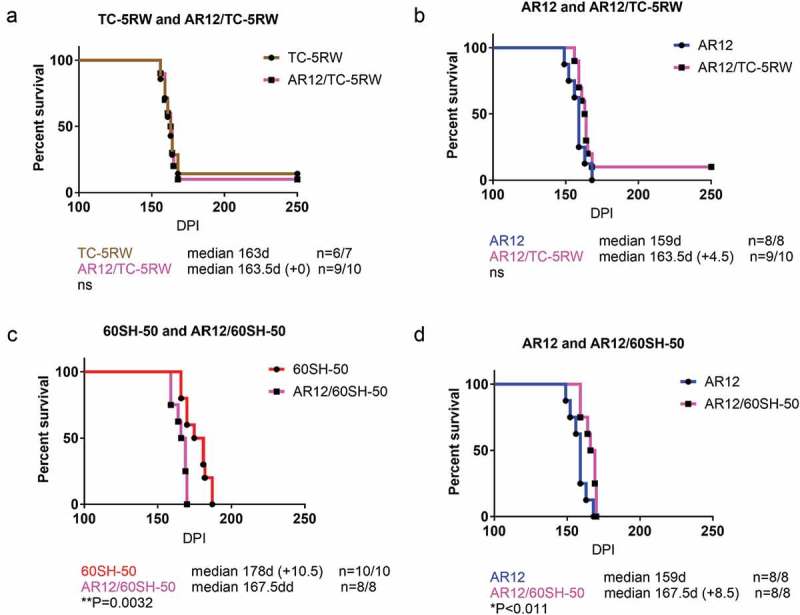
Combination of autophagy stimulator AR-12 and cellulose ethers. Survival analysis of FVB mice infected i.c. with RML prions. For combination therapy, the mice were treated with a single subcutaneous injection of either CE-5RW (a, b) or 60SH-50 (c, d) 30 days before prion inoculation. Thirty days after prion inoculation, the mice were treated with AR-12 as above, until the end of the experiment (250 dpi).
Figure 5.
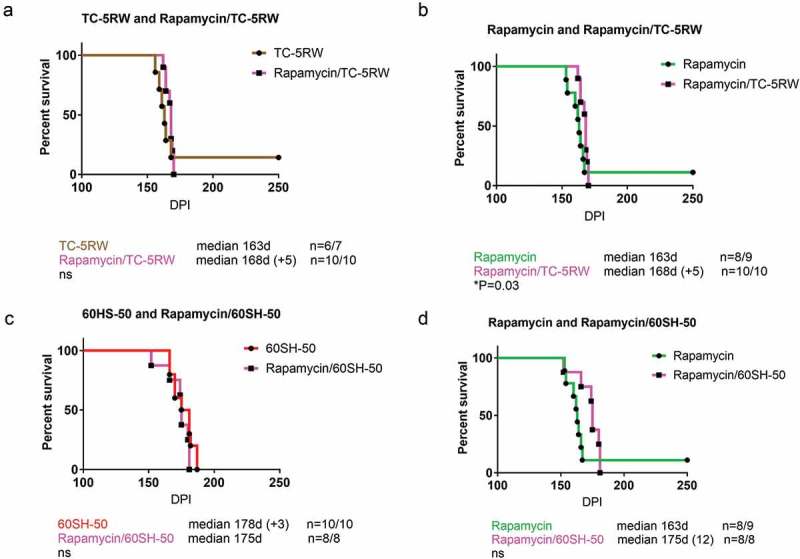
Combination of autophagy stimulator rapamycin and cellulose ethers. For combination therapy, the mice were treated with a single subcutaneous injection of either CE-5RW (a, b) or 60SH-50 (c, d) 30 days before prion inoculation. Thirty days after prion inoculation, the mice were treated with rapamycin as above (4 weeks). Experiments were stopped 250 dpi.
Taken together, combination therapy resulted in prolonged survival compared to the untreated group. However, none of the combination groups showed better effects than 60SH-50 or TC-5RW treatment alone.
Combining autophagy stimulators and cellulose ethers alleviates the autophagy-inducing activity of AR12 and rapamycin
Our previous experiments have shown that treatment with either autophagy stimulators or CEs significantly increased the survival; however, the combination of those drugs did not result in an improvement of survival times. Rather, the survival times of mice treated with the drug combinations were similar to those treated only with CEs. Therefore, we hypothesized that CE treatment might negatively interfere with autophagy stimulation. To verify this, we treated cultured neuronal N2a cells with either AR12, rapamycin, TC-5RW, and 60SH-50 alone, or a combination of one autophagy stimulator with one of the CEs, and analysed autophagy stimulation using LC3-II levels as a readout. Treatment with the lysosomal inhibitor Bafilomycin A1 was used as positive control, resulting in the accumulation of LC3-II levels inside cells (Figure 6(a,b)). As expected, treatment with AR12 or rapamycin showed a significant increase in LC3-II levels (Figure 6(a,c,d)). Interestingly, we found that TC-5RW treatment significantly decreased the LC3-II levels (Figure 6(a,e)), however 60SH-50 did not (Figure 6(a,f)). Our results indicate that combining autophagy stimulators and cellulose ethers significantly decreased the LC3-II levels when compared to LC3-II levels in cells treated with AR12 or rapamycin alone (Figure 6(a,g,h)).
Figure 6.
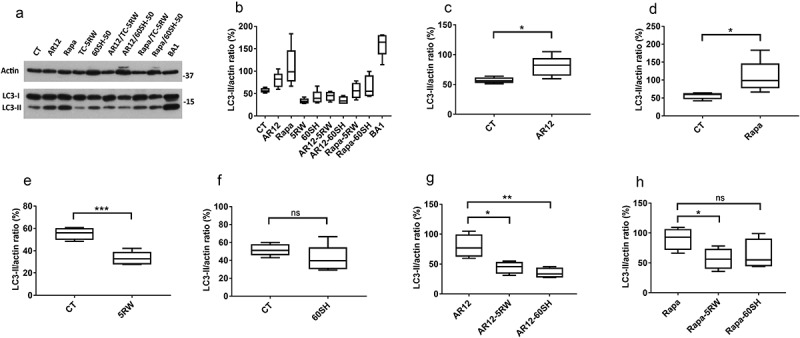
Impairment of the autophagy stimulatory effect of AR12 and rapamycin upon combining with cellulose ethers in N2a cells. (a) Neuronal cells (N2a) were treated with either AR12 (3 µM), rapamycin (500 nM), TC-5RW (3 mg/ml), 60SH-50 (3 mg/ml), bafilomycin A1 (100 nM), or DMSO (vehicle control) for 6 h. Cells were then lysed and LC3-I/II levels assessed in immunoblot analysis. Actin was used as a loading control. (b-h). Densitometry for panel A. LC3-II levels were normalized to actin and compared. *p = 0.05, **p = 0.01, ***p = 0.001, ns = non-significant. n = 5.
Taken together, our in vitro studies in cultured neuronal cells show a mitigation of the autophagy-inducing activity of AR12 and rapamycin upon combining them with cellulose ether compounds. This might explain the lack of additive or synergistic effects of the combinations in vivo.
Discussion
Prion diseases are prototypic neurodegenerative disorders which manifest in humans as sporadic, genetic and acquired-by-infection forms, all being strictly fatal [2]. They come with an incidence of about one in a million worldwide. Although rare, about 6,000 to 7,000 individuals die every year from such diseases. The vast majority of human prion diseases are sporadic and have a clinical onset peak around 60 years of age, and rapidly progress when symptomatic [36]. Forms acquired by infection come with a distinct exposition risk to exogenous factors and can have epidemic character [37]. Classical examples are kuru infection, iatrogenic CJD and vCJD. Since prion diseases are characterized by a long incubation time followed by rapid clinical phase, therapy should be initiated before clinical symptoms manifest and before major damage is already present in the central nervous system. The human form with probably the longest incubation time is genetic prion disease. Here, a defined and disease-associated mutation is present from birth in the gene encoding the human prion protein [38]. Being a rare and fatal disease with a fast progression, and the absence of reliable preclinical markers dampened the search for therapeutic options. Only a few clinical trials were done, and mostly had outcomes which were not encouraging [39,40].
On the other hand, a huge variety of compounds with anti-prion properties or of other approaches targeting prion infection has been described [41,42]. Most of these compounds were established in cultured cells persistently infected with prions, using effects on prion propagation as main read-out. Cell death features usually are absent in such cell models. A significant proportion of such compounds were further validated in animal models of prion disease. Main read-out here is extension of incubation time to clinical disease, with describing an extension of 10–20% as already very successful and promising [25,42].
What are the reasons that the results obtained in vitro and in animal models are poor predictors for therapy in humans? First of all, these are strictly diseases of the central nervous system, and compounds have to pass the blood-brain barrier at concentrations which are effective, but not yet toxic. Of note, most anti-prion compounds described so far contain positive or negative charges and are less likely to effectively cross the blood-brain barrier [42,43]. Another hindrance is that in vitro and in vivo studies are usually done with mouse-adapted scrapie strains, which can react differently to therapeutic approaches than would do human prions [44]. Furthermore, long-term treatment could induce the development of resistant prions, as described previously [45]. Some of these obstacles could be addressed by combination therapy. As done for chemotherapy in infectious diseases and in cancer, a sophisticated combination of compounds which target different steps can achieve additive effects and reduce required concentrations of individual drugs and side effects.
Using this premise, we combined in this study compounds which seemed to address different molecular targets in prion pathogenesis. One group of substances covered inducers of autophagic activity to increase prion clearance (rapamycin and AR12 [25]), and was administered starting at day 30 after prion infection. These drugs were combined with cellulose ethers (TC-5RW and 60SH-50 [30]), which were applied as a single dose 30 days before infection. This combination seemed therefore less likely to be prone to drug interactions. As animal model, we used wild-type mice infected with RML prions, at a high dose and intracerebrally. We feel this is a model with aggressive disease development which well addresses a pre-clinical therapy scenario.
AR12 (OSU-03012) is a derivative of the anti-inflammatory compound celecoxib; however, it lacks the anti-inflammatory effect. AR12 was reported to have anti-cancer, antifungal and anti-microbial activity [46–49]. AR12 can induce autophagy activity [29,50,51]. We showed that treatment with AR12 enhanced the degradation of PrPSc and cleared prion seeding activity in cell line models infected with different prion strains [29]. Of note, AR12 has been reported to cross the blood-brain barrier effectively [52]. In the present study, we show an improved survival time of infected mice treated with AR12 compared with mock-treated controls. This is the first study which shows that AR12 has anti-prion effects in vivo.
The second autophagy stimulator was rapamycin. Rapamycin is a macrolide compound with immunosuppressant functions, which is used in humans for preventing the rejection of organ transplants [53,54]. It is a classical mTOR inhibitor and established stimulator of autophagy. Rapamycin was studied in various neurological and neurodegenerative disorders [55–59]. For prion diseases, we have shown that rapamycin treatment extended the survival of prion-infected mice when administered orally starting at day 100 post-infection [26]. Another study showed that rapamycin-treatment of Tg(PrP-A116V) mice, a model of Gerstmann-Sträussler-Scheinker syndrome, delayed disease onset and prolonged survival [35]. In the present study, we demonstrate that treatment with rapamycin for 4 weeks only extended the survival compared to the mock-treated group by 9%, corroborating the previous findings that rapamycin is a candidate for anti-prion therapy.
Over the last decades polymers showed up as drugs with anti-prion potential. Sulfated glycans [60,61], cationic dendrimers [62,63] and polyamines [64–66] have been used as anti-prion compounds. Recently, work from one of us identified cellulose ethers as a promising candidate. The most remarkable feature of CEs is that it is sufficient to administer a single dose, even long before prion infection occurs [30]. CEs do not have similarity in structure or chemical properties with previously described anti-prion polymers. Although the addition of TC-5RW inhibited 263K prion amplification in vitro in protein misfolding cyclic amplification reaction (PMCA), it is not clear yet how such polymers suppress prion disease in vivo and prolong the animal survival. Effects on autophagy were excluded [30]. Of note, the effectiveness of CEs was affected by the type of prion strain and animal model used [30].
In the current work, we speculated that combining CEs with stimulators of autophagy would result in additive effects. Unfortunately, this combination therapy did not result in a significant prolongation of incubation time when compared to mice treated with CEs alone. There are several reasons for this. One could be that the administration of autophagy inducers was sub-optimal, as being rather early in disease pathogenesis (day 30–60 p.i. applied i.p.). Only AR12 treatment was continued via drinking water, which might be less effective than i.p. application. For follow-up studies, drug application should be started later and should be prolonged. In addition, infecting the mice with a lower dose of prions might show protective effects better. Since we also had one combination treatment group which succumbed to disease earlier, we investigated the possibility that one drug group could antagonize the effect exerted by the other group. We focused on the well-defined autophagy-inducing effects of rapamycin and AR12, and performed a detailed in vitro analysis in cultured neuronal N2a cells. To our surprise, this showed that the autophagy-inducing activity of AR12 and rapamycin was neutralized upon combining them with CE compounds. Interestingly, TC-5RW application even resulted in a significant decrease of the levels of the autophagy marker LC3-II in N2a cells. This might provide an explanation of why our in vivo experiments did not work out as expected.
Although our trial failed to create additive effects, it illustrates both the need to empirically test promising drug combinations in vivo, and the requirement to exclude antagonistic mechanistic effects in vitro, if possible. The concept of establishing and validating appropriate combination therapies for prion diseases in vivo is valid, and there clearly will be a place for combination therapy in the future.
Materials and methods
Reagents
AR12 (also known as OSU-03012) was purchased from Medkoo Bioscience (200272). Rapamycin was purchased from LC Laboratories (R-5000), and Bafilomycin A1 from Sigma Aldrich (B1793). CE compounds TC-5RW and 60SH-50 were dissolved at a concentration of 5% in water. Compound properties and details are as previously described [30]. Anti-β-actin mAb was obtained from Sigma Aldrich (A5441) and anti-LC3 mAb (Clone 2G6) from NanoTools (0260–100). Peroxidase-conjugated immunoglobulins (goat anti-mouse HRP) was from Jackson Immuno-research Lab (115-035-003).
Ethics statement
All animal experiments were performed strictly following the Canadian Council for Animal Care guidelines and were approved by the University of Calgary Health Sciences Animal Care Committee (protocol number AC18-0030 for CE treatment). The experiments involving the propagation of RML prions in FVB mice obtained from Charles River Laboratories were approved under protocol number AC14-0165.
Drug treatment
Rapamycin was dissolved in 950 µl pure ethanol/50 µl DMSO as a 20 mg/ml stock solution and diluted in injection buffer (4% ethanol, 5% Tween 80, 5% PEG400 in dH2O) on the day of injection to a dose of 20 mg/kg in a final volume of 100 µl. Mice were injected twice per week. Control mice received a similar volume of injection buffer without active drug [67]. Treatment continued for 4 weeks. AR12 was dissolved in 950 µl pure ethanol/50 µl DMSO as a 5 mg/ml stock solution and diluted in injection buffer on the day of injection to a dose of 5 mg/kg in a final volume of 100 µl. Mice were injected twice per week. Control mice received a similar volume of injection buffer without active drug. Treatment continued for 4 weeks. Then, AR12 was given in drinking water at a concentration of 50 µg/ml until the end of the experiment. Cellulose ethers TC-5RW and 60SH-50 were applied as a single subcutaneous dose of 4 g/kg 30 days before prion inoculation.
Mouse bioassay
Six-week-old female FVB mice obtained from Charles River Laboratories were treated with a single subcutaneous dose of cellulose ethers or left untreated. After 30 days, mice were inoculated under anaesthesia in the parietal lobe with 20 µl of 1% brain homogenate from terminally sick mice inoculated with prion strain RML, or with non-infected brain. For the intracerebral inoculation, 25-gauge disposable needles were used. After inoculation, mice were observed daily for any adverse conditions. Thirty days after prion inoculation, the animals were injected intraperitoneally with either rapamycin or AR12 for 4 weeks. Treatment with rapamycin was stopped and AR12 treatment was administered in drinking water until the end of the experiment. The animals were monitored for progression of clinical prion disease. At the terminal stage of disease, animals were sacrificed under anaesthesia, and the survival time of each animal was recorded.
Maintenance of cell culture
The mouse neuroblastoma cell line N2a was obtained from ATCC (CCL-131) and cultured in Gibco OptiMEM Glutamax medium obtained from GIBCO, 51985–34 containing 10% fetal bovine serum obtained from Sigma Aldrich (F1051), and penicillin/streptomycin in a 5% CO2 atmosphere. N2a cells were plated overnight. The next day, cells were treated with either AR12 (3 µM), rapamycin (500 nM), TC-5RW (3 mg/ml), and 60SH-50 (3 mg/ml) alone, or a combination of one autophagy stimulator with one of the CEs. Bafilomycin A1 (100 nM) was used to block the lysosomal fusion resulting in pronounced accumulation of LC3-II. Drug treatment was done for 6 h followed by cell lysis.
Cell lysis
N2a cells were lysed in cold lysis buffer (10 mM Tris-HCl, pH 7.5; 100 mM NaCl; 10 mM EDTA; 0.5% Triton X-100; 0.5% sodium deoxycholate (DOC)) for 10 min. Then, cell lysates were centrifuged to remove the cell debris followed by methanol precipitation. Lysates were frozen at −20°C until used for Western blotting.
Western blotting
Methanol precipitated cell lysates were centrifuged, and pellets resuspended in TNE buffer (50 mM Tris-HCl pH 7.5; 150 mM NaCl; 5 mM EDTA). 12.5% SDS-PAGE was used. Gels were electro-blotted on Amersham Hybond P 0.45 PVDF membranes (Amersham, 10600023) and incubated with the desired antibodies. Luminata Western Chemiluminescent HRP Substrates from Millipore (WBLUF0100) was used for development. Densitometry was done using ImageJ program (NIH, USA). LC3-II signals were normalized to actin for quantification.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad Software, version 7.03). For survival, the percent survival was plotted in a Kaplan–Meier plot, and a log-rank (Mantel–Cox) test was performed. Statistical significance for immunoblots was expressed as mean ± S.D. LC3-II levels were normalized to actin and compared using either the unpaired two-tailed t-test for pair-wise comparisons or the One-way ANOVA analysis with Tukey post test for multiple comparisons. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Funding Statement
This work was supported by the Alberta Innovates/Alberta Prion Research Institute [201600010, 201700012].
Acknowledgments
This work was supported by grants from Alberta Innovates/Alberta Prion Research Institute (APRI) (201700012), and was performed within the framework of the Calgary Prion Research Unit (APRI grant 201600010). S.G. is supported by the Canada Research Chairs program. B.A.A. was supported by a postdoctoral fellowship from Alberta Innovates. We thank Dr. Lilian Oribhabor and Bukola Alli for taking excellent care of the mice used in this study.
Author contributions
B.A.A. designed, conducted the experiments and wrote the manuscript. W.T. and B.A.A. conducted the animal experiments. K. D. provided the cellulose ether compounds and revised the manuscript. S.G. and H.M.S designed the study and revised the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science [Internet] 1982. [cited 2016 November3];216:136–144. Available from: http://www.ncbi.nlm.nih.gov/pubmed/6801762 [DOI] [PubMed] [Google Scholar]
- [2].Prusiner SB. Prions. Proc Natl Acad Sci U S A [Internet] 1998. [cited 2016 November3];95:13363–13383. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9811807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gilch S, Chitoor N, Taguchi Y, et al. Chronic wasting disease. Top Curr Chem [Internet] 2011. [cited 2016 November3]; 51–77. Available from: http://link.springer.com/10.1007/128_2011_159 [DOI] [PubMed] [Google Scholar]
- [4].Hannaoui S, Schatzl HM, Gilch S, et al. Chronic wasting disease: emerging prions and their potential risk. True HL, editor PLoS Pathog. [Internet] 2017. [cited 2019 June18];13:e1006619 Available from: https://dx.plos.org/10.1371/journal.ppat.1006619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Knight R. Infectious and sporadic prion diseases. 2017. [cited 2019 June12];293–318. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1877117317300911 [DOI] [PubMed] [Google Scholar]
- [6].Telling GC. The importance of prions. True-Krob H, editor PLoS Pathog [Internet] 2013. [cited 2016 November3];9:e1003090 Available from: http://dx.plos.org/10.1371/journal.ppat.1003090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Watts JC, Balachandran A, Westaway D. The expanding universe of prion diseases. PLoS Pathog [Internet] 2006. [cited 2016 November3];2:e26 Available from: http://dx.plos.org/10.1371/journal.ppat.0020026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Collinge J. Molecular neurology of prion disease. J Neurol Neurosurg Psychiatry [Internet] 2005. [cited 2016 November3];76:906–919. Available from: http://jnnp.bmj.com/cgi/doi/10.1136/jnnp.2004.048660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Weissmann C. The state of the prion. Nat Rev Microbiol [Internet] 2004. [cited 2016 November3];2:861–871. Available from: http://www.nature.com/doifinder/10.1038/nrmicro1025 [DOI] [PubMed] [Google Scholar]
- [10].Nunziante M, Gilch S, Schätzl HM. Prion diseases: from molecular biology to intervention strategies. Chem Bio Chem [Internet] 2003. [cited 2017 October2];4:1268–1284. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14661267 [DOI] [PubMed] [Google Scholar]
- [11].Krammer C, Vorberg I, Schätzl HM, et al. Therapy in prion diseases: from molecular and cellular biology to therapeutic targets. Infect Disord Drug Targets [Internet] 2009. [cited 2017 April26];9:3–14. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19200010 [DOI] [PubMed] [Google Scholar]
- [12].Caughey B. Scrapie associated PrP accumulation and its prevention: insights from cell culture. Br Med Bull [Internet] 1993. [cited 2019 July10];49:860–872. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8137133 [DOI] [PubMed] [Google Scholar]
- [13].Diringer H, Ehlers B. Chemoprophylaxis of scrapie in mice. J Gen Virol [Internet] 1991. [cited 2019 July10];72:457–460. Available from: http://jgv.microbiologyresearch.org/content/journal/jgv/10.1099/0022-1317-72-2-457 [DOI] [PubMed] [Google Scholar]
- [14].Doh-Ura K, Iwaki T, Caughey B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol [Internet] 2000. [cited 2019 June19];74:4894–4897. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10775631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Goold R, McKinnon C, Tabrizi SJ. Prion degradation pathways: potential for therapeutic intervention. Mol Cell Neurosci [Internet] 2015. [cited 2017 May30];66:12–20. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25584786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Korth C, May BCH, Cohen FE, et al. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci [Internet] 2001. [cited 2017 May30];98:9836–9841. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11504948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Weissmann C. Mutation and selection of prions. True-Krob H, editor PLoS Pathog [Internet] 2012. [cited 2019 July10];8:e1002582 Available from: http://www.ncbi.nlm.nih.gov/pubmed/22479179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bian J, Kang H-E, Telling GC. Quinacrine promotes replication and conformational mutation of chronic wasting disease prions. Proc Natl Acad Sci U S A [Internet] 2014. [cited 2017 May30];111:6028–6033. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.1322377111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ghaemmaghami S, Ahn M, Lessard P, et al. Continuous quinacrine treatment results in the formation of drug-resistant prions. Mabbott N, editor PLoS Pathog [Internet] 2009. [cited 2019 June18];5:e1000673 Available from: https://dx.plos.org/10.1371/journal.ppat.1000673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li J, Browning S, Mahal SP, et al. Darwinian evolution of prions in cell culture. Science [Internet] 2010. [cited 2019 June18];327:869–872. Available from: http://www.sciencemag.org/cgi/doi/10.1126/science.1183218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Spilman P, Lessard P, Sattavat M, et al. A gamma-secretase inhibitor and quinacrine reduce prions and prevent dendritic degeneration in murine brains. Proc Natl Acad Sci U S A [Internet] 2008. [cited 2017 May30];105:10595–10600. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.0803671105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Klionsky DJ. Why do we need autophagy? A cartoon depiction. Autophagy [Internet] 2018. [cited 2019 June11];14:739–742. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29782213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Puri D, Subramanyam D. Stress – (self) eating: epigenetic regulation of autophagy in response to psychological stress. FEBS J [Internet] 2019. [cited 2019 June11];febs.14826 Available from: http://www.ncbi.nlm.nih.gov/pubmed/30927484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Aguib Y, Heiseke A, Gilch S, et al. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy [Internet] 2009. [cited 2016 November3];5:361–369. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19182537 [DOI] [PubMed] [Google Scholar]
- [25].Abdelaziz DH, Abdulrahman BA, Gilch S, et al. Autophagy pathways in the treatment of prion diseases. Curr Opin Pharmacol [Internet] 2019. [cited 2019 June13];44:46–52. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31096117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Heiseke A, Aguib Y, Riemer C, et al. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J Neurochem [Internet] 2009. [cited 2016 November3];109:25–34. Available from: http://doi.wiley.com/10.1111/j.1471-4159.2009.05906.x [DOI] [PubMed] [Google Scholar]
- [27].Heiseke A, Aguib Y, Schatzl HM. Autophagy, prion infection and their mutual interactions. Curr Issues Mol Biol [Internet] 2010. [cited 2017 October2];12:87–97. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19767652 [PubMed] [Google Scholar]
- [28].Phadwal K, Kurian D, Salamat MKF, et al. Spermine increases acetylation of tubulins and facilitates autophagic degradation of prion aggregates. Sci Rep [Internet] 2018. [cited 2019 June11];8:10004 Available from: http://www.ncbi.nlm.nih.gov/pubmed/29968775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Abdulrahman BA, Abdelaziz D, Thapa S, et al. The celecoxib derivatives AR-12 and AR-14 induce autophagy and clear prion-infected cells from prions. Sci Rep [Internet] 2017. cited 2017 December22;7:17565 Available from: http://www.ncbi.nlm.nih.gov/pubmed/29242534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Teruya K, Oguma A, Nishizawa K, et al. A single subcutaneous injection of cellulose ethers administered long before infection confers sustained protection against prion diseases in rodents. PLoS Pathog [Internet] 2016. [cited 2019 June11];12:e1006045 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27973536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Abdulrahman BA, Abdelaziz DH, Schatzl HM. Autophagy regulates exosomal release of prions in neuronal cells. J Biol Chem [Internet] 2018. [cited 2019 June11];293:8956–8968. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29700113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ertmer A, Gilch S, S-W Y, et al. The tyrosine kinase inhibitor STI571 induces cellular clearance of PrPSc in prion-infected cells. J Biol Chem [Internet] 2004. [cited 2017 October3];279:41918–41927. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15247213 [DOI] [PubMed] [Google Scholar]
- [33].Ertmer A, Huber V, Gilch S, et al. The anticancer drug imatinib induces cellular autophagy. Leukemia [Internet] 2007. [cited 2017 October3];21:936–942. Available from: http://www.nature.com/doifinder/10.1038/sj.leu.2404606 [DOI] [PubMed] [Google Scholar]
- [34].Collier MA, Peine KJ, Gautam S, et al. Host-mediated Leishmania donovani treatment using AR-12 encapsulated in acetalated dextran microparticles. Int J Pharm [Internet] 2016. [cited 2017 May 30];499:186–194. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0378517316300084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cortes CJ, Qin K, Cook J, et al. Rapamycin delays disease onset and prevents PrP plaque deposition in a mouse model of Gerstmann-Straussler-Scheinker disease. J Neurosci [Internet] 2012. [cited 2016 November3];32:12396–12405. Available from: http://www.jneurosci.org/cgi/doi/10.1523/JNEUROSCI.6189-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Parchi P, Strammiello R, Giese A, et al. Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future. Acta Neuropathol [Internet] 2011. [cited 2019 July11];121:91–112. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21107851 [DOI] [PubMed] [Google Scholar]
- [37].Haïk S, Brandel J-P. Infectious prion diseases in humans: cannibalism, iatrogenicity and zoonoses. Infect Genet Evol [Internet] 2014. [cited 2019 July11];26:303–312. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1567134814002135 [DOI] [PubMed] [Google Scholar]
- [38].Lloyd SE, Mead S, Collinge J. Genetics of prion diseases. Curr Opin Genet Dev [Internet] 2013. [cited 2019 July11];23:345–351. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0959437X13000300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Forloni G, Artuso V, Roiter I, et al. Therapy in prion diseases. Curr Top Med Chem [Internet] 2013. [cited 2017 April26];13:2465–2476. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24059336 [DOI] [PubMed] [Google Scholar]
- [40].Zerr I. Therapeutic trials in human transmissible spongiform encephalo-pathies: recent advances and problems to address. Infect Disord Drug Targets [Internet] 2009. [cited 2017 April26];9:92–99. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19200019 [DOI] [PubMed] [Google Scholar]
- [41].Aguzzi A, Lakkaraju AKK, Frontzek K. Toward therapy of human prion diseases. Annu Rev Pharmacol Toxicol [Internet] 2018. [cited 2019 July11];58:331–351. Available from: http://www.annualreviews.org/doi/10.1146/annurev-pharmtox-010617-052745 [DOI] [PubMed] [Google Scholar]
- [42].Vorberg I, Chiesa R. Experimental models to study prion disease pathogenesis and identify potential therapeutic compounds. Curr Opin Pharmacol [Internet] 2019. [cited 2019 July11];44:28–38. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1471489218301735 [DOI] [PubMed] [Google Scholar]
- [43].Gilch S, Krammer C, Schätzl HM. Targeting prion proteins in neurodegenerative disease. Expert Opin Biol Ther [Internet] 2008. [cited 2017 April26];8:923–940. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18549323 [DOI] [PubMed] [Google Scholar]
- [44].Giles K, Olson SH, Prusiner SB. Developing therapeutics for PrP prion diseases. Cold Spring Harb Perspect Med [Internet] 2017. [cited 2019 July11];7:a023747 Available from: http://perspectivesinmedicine.cshlp.org/lookup/doi/10.1101/cshperspect.a023747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Berry DB, Lu D, Geva M, et al. Drug resistance confounding prion therapeutics. Proc Natl Acad Sci U S A [Internet] 2013. [cited 2019 July11];110:E4160–E4169. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.1317164110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Booth L, Cruickshanks N, Ridder T, et al. OSU-03012 interacts with lapatinib to kill brain cancer cells. Cancer Biol Ther [Internet] 2012. [cited 2017 May30];13:1501–1511. Available from http://www.tandfonline.com/doi/abs/10.4161/cbt.22275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chen -H-H, Chen -C-C, Lin Y-S, et al. AR-12 suppresses dengue virus replication by down-regulation of PI3K/AKT and GRP78. Antiviral Res [Internet] 2017. [cited 2017 May4];142:158–168. Available from http://linkinghub.elsevier.com/retrieve/pii/S0166354216305800 [DOI] [PubMed] [Google Scholar]
- [48].Chiu H-C, Kulp SK, Soni S, et al. Eradication of intracellular Salmonella enterica serovar Typhimurium with a small-molecule, host cell-directed agent. Antimicrob Agents Chemother [Internet] 2009. [cited 2017 May30];53:5236–5244. Available from http://aac.asm.org/cgi/doi/10.1128/AAC.00555-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yacoub A, Park MA, Hanna D, et al. OSU-03012 promotes caspase-independent but PERK-, Cathepsin B-, BID-, and AIF-dependent killing of transformed cells. Mol Pharmacol. [Internet] 2006. [cited 2019 June11];70:589–603. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16622074 [DOI] [PubMed] [Google Scholar]
- [50].Chiu H-C, Soni S, Kulp SK, et al. Eradication of intracellular Francisella tularensis in THP-1 human macrophages with a novel autophagy inducing agent. J Biomed Sci [Internet] 2009. [cited 2017 May4];16:110 Available from: http://www.ncbi.nlm.nih.gov/pubmed/20003180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Park MA, Yacoub A, Rahmani M, et al. OSU-03012 stimulates PKR-like endoplasmic reticulum-dependent increases in 70-kDa heat shock protein expression, attenuating its lethal actions in transformed cells. Mol Pharmacol [Internet] 2008. [cited 2019 June11];73:1168–1184. Available from: http://molpharm.aspetjournals.org/cgi/doi/10.1124/mol.107.042697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Booth L, Roberts JL, Tavallai M, et al. OSU-03012 and viagra treatment inhibits the activity of multiple chaperone proteins and disrupts the blood-brain barrier: implications for anti-cancer therapies. J Cell Physiol [Internet] 2015. [cited 2017 May4];230:1982–1998. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25736380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hamdani S, Thiolat A, Naserian S, et al. Delayed and short course of rapamycin prevents organ rejection after allogeneic liver transplantation in rats. World J Gastroenterol [Internet] 2017. [cited 2019 June11];23:6962–6972. Available from: http://www.wjgnet.com/1007-9327/full/v23/i38/6962.htm [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kahan BD. Efficacy of sirolimus compared with azathioprine for reduction of acute renal allograft rejection: a randomised multicentre study. The rapamune US study group. Lancet [Internet] 2000. [cited 2019 June11];356:194–202. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10963197 [DOI] [PubMed] [Google Scholar]
- [55].Johnson SC, Yanos ME, Kayser E-B, et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of leigh syndrome. Science [Internet] 2013. [cited 2019 June11];342:1524–1528. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24231806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kolosova NG, Vitovtov AO, Muraleva NA, et al. Rapamycin suppresses brain aging in senescence-accelerated OXYS rats. Aging (Albany NY) [Internet] 2013. [cited 2019 June11];5:474–484. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23817674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sarkar S, Ravikumar B, Floto RA, et al. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ [Internet] 2009. [cited 2019 June11];16:46–56. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18636076 [DOI] [PubMed] [Google Scholar]
- [58].Ozcelik S, Fraser G, Castets P, et al. Rapamycin attenuates the progression of Tau pathology in P301S Tau transgenic mice. Feany MB, editor PLoS One [Internet] 2013. [cited 2019 June11];8:e62459 Available from: http://www.ncbi.nlm.nih.gov/pubmed/23667480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chen J, Long Z, Li Y, et al. Alteration of the Wnt/GSK3β/β catenin signalling pathway by rapamycin ameliorates pathology in an Alzheimer’s disease model. Int J Mol Med [Internet] 2019. [cited 2019 June11];44:313–323. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31115485 [DOI] [PubMed] [Google Scholar]
- [60].Caughey B, Raymond GJ. Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J Virol [Internet] 1993. [cited 2019 June12];67:643–650. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7678300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Farquhar C, Dickinson A, Bruce M. Prophylactic potential of pentosan polysulphate in transmissible spongiform encephalopathies. Lancet [Internet] 1999. [cited 2019 June12];353:117 Available from: http://www.ncbi.nlm.nih.gov/pubmed/10023899 [DOI] [PubMed] [Google Scholar]
- [62].Fischer M, Appelhans D, Schwarz S, et al. Influence of surface functionality of poly(propylene imine) dendrimers on protease resistance and propagation of the scrapie prion protein. Biomacromolecules [Internet] 2010. [cited 2019 June12];11:1314–1325. Available from: https://pubs.acs.org/doi/10.1021/bm100101s [DOI] [PubMed] [Google Scholar]
- [63].Solassol J, Crozet C, Perrier V, et al. Cationic phosphorus-containing dendrimers reduce prion replication both in cell culture and in mice infected with scrapie. J Gen Virol [Internet] 2004. [cited 2019 June12];85:1791–1799. Availabe from: http://www.ncbi.nlm.nih.gov/pubmed/15166465 [DOI] [PubMed] [Google Scholar]
- [64].Ryou C, Titlow WB, Mays CE, et al. The suppression of prion propagation using poly-l-lysine by targeting plasminogen that stimulates prion protein conversion. Biomaterials [Internet] 2011. [cited 2019 June12];32:3141–3149. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0142961211000305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lim Y, Mays CE, Kim Y, et al. The inhibition of prions through blocking prion conversion by permanently charged branched polyamines of low cytotoxicity. Biomaterials [Internet] 2010. [cited 2019 June12];31:2025–2033. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0142961209013295 [DOI] [PubMed] [Google Scholar]
- [66].Supattapone S, Wille H, Uyechi L, et al. Branched polyamines cure prion-infected neuroblastoma cells. J Virol [Internet] 2001. [cited 2019 June12];75:3453–3461. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11238871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Cortes CJ, La Spada AR. The many faces of autophagy dysfunction in Huntington’s disease: from mechanism to therapy. Drug Discov Today [Internet] 2014. [cited 2016 November3];19:963–971. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1359644614000762 [DOI] [PMC free article] [PubMed] [Google Scholar]