

ABSTRACT
The omics technologies provide an invaluable opportunity to employ a global view towards human diseases. However, the appropriate translation of big data to knowledge remains a major challenge. In this study, we have performed quality control assessments for 91 transcriptomics datasets deposited in gene expression omnibus database and also have evaluated the publications derived from these datasets. This survey shows that drawbacks in the analyses and reports of transcriptomics studies are more common than one may assume. This report is concluded with some suggestions for researchers and reviewers to enhance the minimal requirements for gene expression data generation, analysis and report.
KEYWORDS: Big data, data analysis, differentially expressed gene, transcriptomics, quality control
Introduction
The recent emergence of high-throughput technologies has provided an invaluable opportunity to generate holistic views towards complex biological processes. However, in spite of considerable generation and availability of omics data, translation of big biological data to clinically valuable knowledge remains a major challenge. It is not only due to the insufficiency of current data processing and integration approaches but also inappropriate application of existing analysis methods. To provide an estimation of the quality of transcriptomics datasets, we here have performed a comprehensive assessment of microarray data on kidney disorders as a representation of complex disorders. In addition, the publications emerged from these studies were manually reviewed to investigate the accuracy of analysis techniques. This survey reveals common pitfalls in quality control, statistical methods and down-stream processing steps in genome-wide expression profiling studies.
Methods
Microarray data was retrieved from Gene Expression Omnibus (GEO) database [1]. We focused on datasets related to kidney diseases as an example of complex disorders. In search strategy, gene expression profiling array deposited until September 2017 were chosen and the type of organism was limited to Homo sapiens or Mus musculus. The search terms were ‘Diabetic nephropathy’ OR ‘Polycystic Kidney Disease’ OR ‘PKD’ OR ‘Chronic Kidney Disease’ OR ‘CKD’ OR ‘Acute Kidney Injury’ OR ‘AKI’ OR ‘End-Stage Renal Disease’ OR “ESRD. For quality control assessment, principal component analysis (PCA) was carried out using ggplot2 package [2] and prcomp function of R software [3]. Then, the publications derived from the datasets were assessed for accuracy of data analysis.
Results and discussion
Surveying Gene Expression Omnibus (GEO) database, 118 microarray mRNA expression datasets were retrieved (Supplementary table 1) and studies with inappropriate design or incomplete deposited data were excluded (Supplementary table 2). Eighty-nine datasets remained that corresponded to 94 platforms (Figure 1). Remarkably, among these experiments, 50 studies (53%) had unsatisfying quality according to principle component analysis (PCA) as more than 10% of the samples were not segregated based on experimental groups (Figure 1a, Supplementary figure 1). Although an inappropriate PCA may indicate wrong wet-lab steps, batch effect, poor pre-processing analysis, or mild differences between conditions, we here have tolerated up to 10% deviation from an ideal PCA plot to avoid the stringency of quality control assessments.
Figure 1.
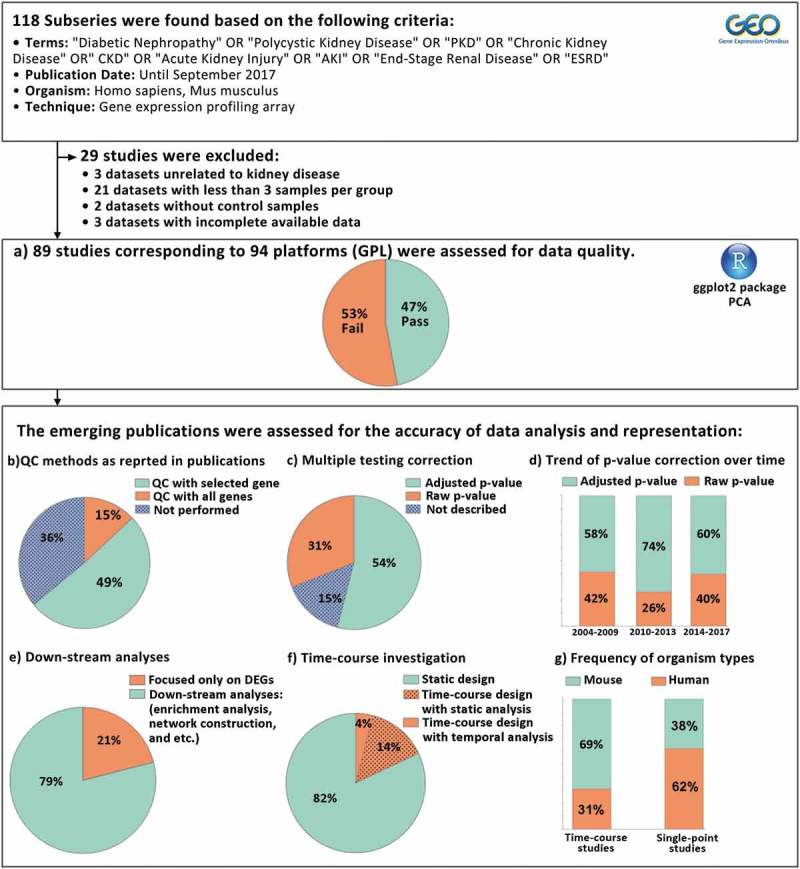
The framework followed in this study is depicted. For all datasets quality control assessment was performed using PCA (a). The publications derived from the datasets were inspected for the accuracy of data analysis and representation (b–g). QC: quality control, PCA: principal component analysis. DEGs: differentially expressed genes.
To investigate the accuracy of data representation, 72 publications derived from the collected datasets were manually reviewed. Importantly, in 36% of the papers quality assessment is not reported at all, 49% of the studies have used selected genes for this analysis, and in only 15% of the reports quality control was appropriately performed by considering the expression values of all genes (Figure 1b).
In big data analysis, adjustment for multiple testing is critical to avoid false positive results. However, we noticed that in 31% of publications, raw p-value is used as a significance cut-off for the identification of differentially expressed genes (DEGs). Also, in 15% of papers details regarding p-value correction are not provided (Figure 1c). Notably, we did not observe a tendency towards multiple testing correction in recent transcriptomics studies (Figure 1d). Furthermore, we explored the details of microarray data processing in publications (Supplementary Table 1). Data processing to detect DEGs is commonly an initial assessment in microarray data analysis in which false positive records can dramatically affect down-stream analyses. Hence, this step should be cautiously performed and comprehensively described.
Microarray technology allows the simultaneous identification of a large number of altered genes in a given biological process. However, the holistic interpretation of all gene expression variations is a challenging task and so some investigators may prefer to focus on most variably expressed genes. We noticed that in 49% of the publications only the top DEGs were analysed and considerable numbers of genes with mild alterations were totally ignored. This reductionist approach can be misleading as correlated slight variations of a bundle of genes can have a significant effect on cell behaviour [4]. Depending on study aims and scopes, DEG identification should be followed by downstream procedures such as gene set enrichment analyses, interaction network construction, data integration, etc. to generate clinically significant hypotheses. However, in 21% of the reviewed publications, microarray data analysis was restricted to DEG identification (Figure 1e).
Biological phenomena are dynamic processes which cannot be appropriately studied by static investigations. Hence, time course assessments are critical to represent temporal alterations. We have previously shown that it is important to follow both treatment and control groups over time to avoid misinterpretations raised by gene expression noise and inherent variations such as circadian rhythms [5]. Notably, only 13 studies (18%) had a time course design with at least three steps and in the remaining experiments all measurements were performed in a single point. Even in most of these time-series studies, data analysis was performed with a static viewpoint (Figure 1f). For instance, the time points were compared one by one instead of inspecting the trajectories as a whole. Indeed, in spite of the availability of specialized tools for time-series big data analysis [6,7], such techniques were employed only in 3 studies. In addition, we explored the distribution of studies based on their model organism. In 9 out of 13 time-course published studies (69%), murine models were employed. This rate is 38% for single-point experiments (Figure 1g). This shows a tendency towards animal models in time-series investigations due to practical limitations for using human tissues in such experiments.
Based on this survey, we propose the following remarks:
The quality of microarray datasets should be scrutinized before any analysis and appropriate tests should be performed to decide whether to exclude specific samples or even all data.
For reporting microarray studies, it is critical to indicate the details of statistical methods such as methods used for multiple testing adjustment and DEG identification.
In order to translate big data to clinically valuable knowledge, it is critical to analyse and interpret microarray data with a hypothesis-free holistic framework rather than a reductionist approach.
Considering the time-dependency of biological phenomena and the scarcity of time-series big data, the generation of such datasets followed by appropriate temporal analysis is highly encouraged.
These suggestions may assist researchers, reviewers, and editors to improve the arena of gene expression profiling.
Author contributions
MA, RF, and KM retrieved the datasets and performed the analyses. MA and RF draughted the manuscript and the other authors critically revised it. YG designed and supervised the whole project. All authors contributed to data analysis, approved the final draught, and agreed to be accountable for all aspects of the work.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material:
Supplemental data for this article can be accessed here.
References
- [1].Clough E., Barrett T. (2016) The Gene Expression Omnibus Database. In: Mathé E., Davis S. (eds) Statistical Genomics. Methods in Molecular Biology, vol 1418. Humana Press, New York, NY. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wickham H. Ggplot2: elegant graphics for data analysis. Springer-Verlag New York 2009. [Google Scholar]
- [3].R Core Team (2014). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/.
- [4].Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003. July;34(3):267–273. [DOI] [PubMed] [Google Scholar]
- [5].Rabieian R, Moein S, Khanahmad H, et al. Transcriptional noise in intact and TGF-beta treated human kidney cells; the importance of time-series designs. Cell Biol Int. 2018. Sep;42(9):1265-1269. [DOI] [PubMed] [Google Scholar]
- [6].Moradzadeh K, Moein S, Nickaeen N, et al. Analysis of time-course microarray data: comparison of common tools. Genomics. 2018. Jul;111(4):636-641. [DOI] [PubMed] [Google Scholar]
- [7].Bar-Joseph Z, Gitter A, Simon I. Studying and modelling dynamic biological processes using time-series gene expression data. Nat Rev Genet. 2012. July 18;13(8):552–564. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.