

ABSTRACT
Prokaryotic genomes harbour a plethora of uncharacterized reverse transcriptases (RTs). RTs phylogenetically related to those encoded by group-II introns have been found associated with type III CRISPR-Cas systems, adjacent or fused at the C-terminus to Cas1. It is thought that these RTs may have a relevant function in the CRISPR immune response mediating spacer acquisition from RNA molecules. The origin and relationships of these RTs and the ways in which the various protein domains evolved remain matters of debate. We carried out a large survey of annotated RTs in databases (198,760 sequences) and constructed a large dataset of unique representative sequences (9,141). The combined phylogenetic reconstruction and identification of the RTs and their various protein domains in the vicinity of CRISPR adaptation and effector modules revealed three different origins for these RTs, consistent with their emergence on multiple occasions: a larger group that have evolved from group-II intron RTs, and two minor lineages that may have arisen more recently from Retron/retron-like sequences and Abi-P2 RTs, the latter associated with type I-C systems. We also identified a particular group of RTs associated with CRISPR-cas loci in clade 12, fused C-terminally to an archaeo-eukaryotic primase (AEP), a protein domain (AE-Prim_S_like) forming a particular family within the AEP proper clade. Together, these data provide new insight into the evolution of CRISPR-Cas/RT systems.
KEYWORDS: Abi, archaeo-eukaryotic primase, Cas, CRISPR, group II intron, diversity-generating retroelement, reverse transcriptase, retron
Introduction
Reverse transcriptases (RTs) are RNA-dependent DNA polymerases responsible for converting RNA into cDNA [1,2]. Genes encoding reverse transcriptase are common in prokaryotic genomes, and are generally annotated as ‘retron-type RNA-directed DNA polymerase’ (EC 2.7.7.49), in reference to the first discovery of a prokaryotic RT as a component of a bacterial retroelement called a retron [3,4]. Most prokaryotic RTs are thought to be group-II intron-encoded proteins (IEPs) [5–7], Retron/retron-like sequences [8] and diversity-generating retroelements (DGRs) [9,10]. However, large-scale genomic surveys and phylogenetic analyses have revealed many other predicted RTs that remain uncharacterized [11–13].
Putative RTs phylogenetically related to those encoded by mobile group-II introns were found in association with CRISPR-Cas (clustered regularly interspaced short palindromic repeats and CRISPR-associated proteins) systems, either adjacent or fused at the C-terminus to Cas1 [11,12,14–18]. Some of these RTs had previously been considered to be G2L (group II-like) RT groups [11–14] but had more recently been classified into 13 distinct clades [17,18]. A computational strategy called ‘CRISPRicity’ [19] developed to assess the functional relevance of proteins linked to CRISPR-mediated immunity encoded by genes in CRISPR neighbourhoods, has suggested that RTs may have a relevant function in the CRISPR immune response. CRISPR-Cas systems provide adaptive immunity to invading viruses and plasmids by the sequence-specific targeting of nucleic acids and are classified into two general classes. These two classes are further subdivided into different types based on the composition of their Cas proteins: types I, III, and IV within Class 1, and types II, V, and VI within Class 2, and these types are further subdivided into different subtypes [20–24].
Type III systems are characterized by their targeting mechanism, which results in cotranscriptional cleavage of the target DNA and its transcript [25–27]. Interestingly, the CRISPR-Cas systems encoding RTs usually correspond to a subset of type III systems [15–18] from all known subtypes (III-A, III-B, III-C and III-D). Attention was recently focused on this association by the discovery of RT-mediated spacer acquisition from RNA molecules in a type III-B system from Marinomonas mediterranea [15,28], and the more recent description of an RT-Cas1 fusion from Fusicatenibacter saccharivorans in the so-called ‘Record–seq’ method, making it possible to make transcriptome-scale molecular recordings in cell populations [29].
The origin and evolutionary relationships of RTs functionally linked to CRISPR-Cas systems are currently unknown, and two models have been proposed. The ‘single point of origin’ [16] model suggests that these RTs correspond to a single acquisition event, possibly from the random retrotransposition of a group-II intron into a CRISPR-cas locus, with the various protein domains (RT, RT-Cas1 and Cas6-RT-Cas1 fusions) probably reflecting successive evolutionary episodes. By contrast, the ‘various origins’ model [17,18] suggests that these group II intron-related RTs were recruited by CRISPR-Cas adaptation modules independently on several occasions and that the different RT protein domains also emerged different times in the evolution of these particular CRISPR-Cas systems. Nevertheless, our current knowledge of CRISPR-encoded RTs remains limited and large-scale genomic surveys and phylogenetic analyses are required to improve our understanding of the origin, evolutionary relationships and function of these RTs.
Here, we provide important new insight into RT sequence diversity in prokaryotic genomes by analysing a total of 198,760 predicted RT proteins. We also developed a computational pipeline for identifying unique representative RT sequences encoded by CRISPR-Cas systems. The RTs closely related to group-II introns previously grouped into 13 clades were expanded and novel RTs fused to an archaeo-eukaryotic primase (AEP) domain (AE_Prim_S-like) forming a particular family within the AEP proper clade were identified. We also identified two novel lineages of CRISPR-encoded non-fused RTs, one also associated with type III systems that have evolved from Retron/retron-like sequences (clade 14) and a lineage (clade 15) that has evolved from Abi-P2 RTs associated with type I-C systems. These data provide new insight into the evolution of CRISPR-Cas/RT systems.
Results and discussion
The diversity of prokaryotic RTs
We performed a large survey of CRISPR-encoded RTs, by analysing prokaryotic proteins from complete and draft bacterial and archaeal genomes annotated as RNA-directed DNA polymerases (145,379) or reverse transcriptases (52,684) in databases (hereafter referred to as RTs), and performing phylogenetic analyses, to improve our understanding of the evolutionary history and relationships between prokaryotic reverse transcriptases and CRISPR-Cas systems. Using a previously published dataset of 558 RT sequences, including 137 type III CRISPR RT/RT-Cas1 proteins closely related to group-II intron-encoded RTs [18], we analysed a total of 198,063 sequences, to which 133 new RT-Cas1 entries carrying both a Cas1 domain (pfam01867) and an RT domain (pfam00087) were added. Finally, six additional proteins from Streptomyces species annotated as hypothetical proteins, but predicted to be RTs linked to type I-E CRISPR-Cas systems [16,19] were also included in the analysis. This large dataset (198,760 sequences) was processed by selecting an RT domain (RT0-7) of at least 200 amino acids and unique sequences by multiple-step clustering at 85% sequence identity to remove closer relatives. This procedure yielded a final dataset of 9,141 predicted RT sequences (Figure 1 and Supplementary Table 1).
Figure 1.

Compilation of RTs from databases and generation of the dataset. The procedure depicted yielded 9,141 predicted unique sequences representative of the current diversity of RTs in prokaryotes.
The phylogenetic tree constructed from a multiple sequence alignment (MSA) of the above RT sequences with the FastTree program [30] is shown in Figure 2. Most of the RTs in this set are group-II introns (47%), Retron/retron-like sequences (25%) and DGRs (12%), the remaining 16% clustering into distinct groups including RTs previously reported to be linked to type III CRISPR-Cas systems. The inferred phylogenetic tree also includes other mostly uncharacterized RTs, some of which may be involved in antiphage abortive infection systems (AbiA, AbiK and Abi-P2 groups), together with many others of unknown function (Figure 2 and Supplementaries File 1 and Table 1) expanding the previously defined G2L and UG groups [11–13].
Figure 2.
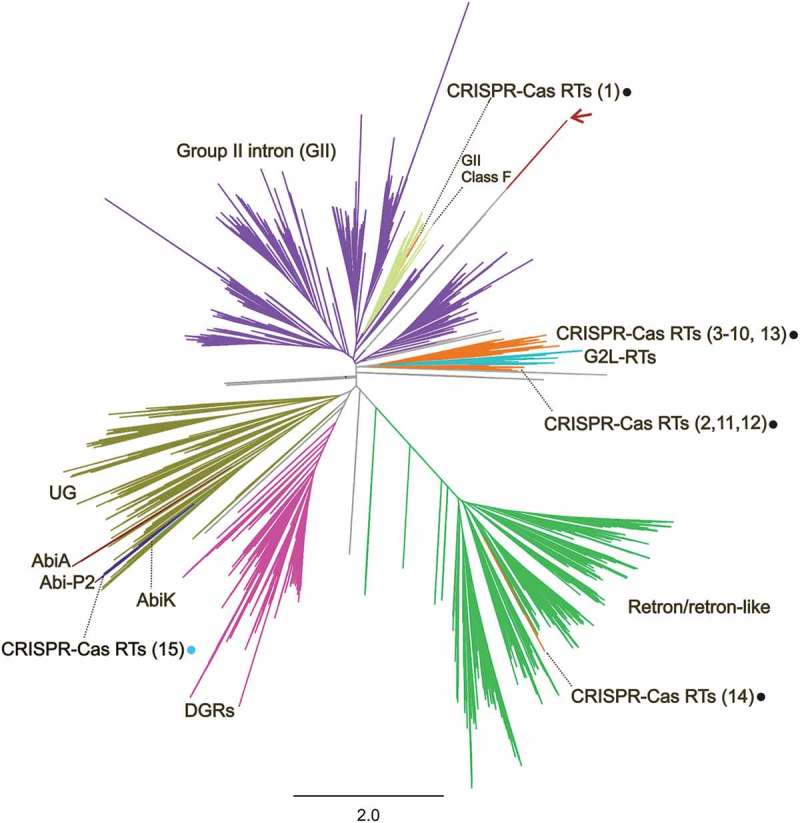
Phylogeny of prokaryotic RTs. The unrooted tree was constructed from an alignment of 9,141 unique predicted RT protein sequences obtained with the FastTree program. The corresponding RT protein sequences, accession number and species names are provided in Supplementary Table 1 and the tree newick file is provided as Supplementary File 1. The branches corresponding to group-II introns (GII), GII class F, Retron/retron-like, DGRs, CRISPR-Cas, G2L, Abi and UG RTs are indicated and highlighted with distinct colours. The numbers of the CRISPR-Cas encoded RT clades are indicated in brackets and the dots indicate the type of system with which they are associated: type III (black) or type I-C (blue). The red arrow indicates the branches corresponding to the putative RTs linked to type I-E CRISPR-Cas systems described by Silas et al. [16]. Relevant subtrees are provided in Figure 4 and Supplementaries Figures 2–4.
Identification of RT genes in the neighbourhood of CRISPR-Cas systems
For the identification of RTs in the neighbourhood of CRISPR-Cas systems, we constructed a computational pipeline for the analysis of the 9,141 RT entries of the dataset. Briefly, we analysed all genes located 30 kb up- and downstream from the RT gene and checked for the presence in the neighbourhood of a CRISPR-Cas adaptation module or CRISPR effector module (for details see the Materials and Methods). In addition to spurious RTs associated with CRISPR-Cas systems related to the retrotransposition of mobile genetic elements that show a recognizable group-II intron structure [6], the computational pipeline confirmed and expanded set of the CRISPR-encoded RTs and RT-Cas1 proteins closely related to group-II introns previously grouped into 13 clades. Remarkably, the analysis also revealed the existence of other lineages with a different origin that evolved from Retron/retron-like (clade 14) and Abi-P2 group (clade 15) RT sequences, the former associated with type III and the latter associated with type I-C systems (Figures 3 and 4, and Supplementaries Table 2 and 3).
Figure 3.
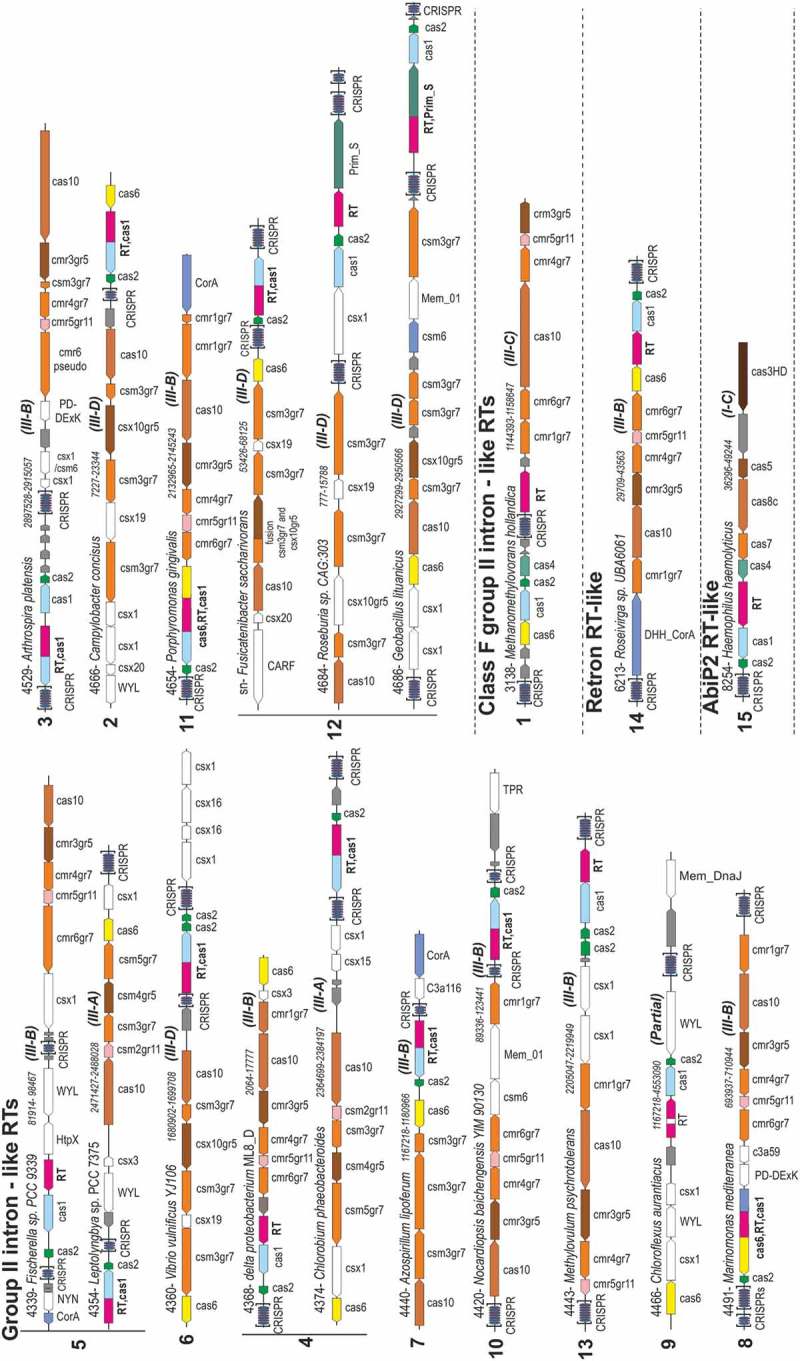
Architectures of genomic loci for the representative subtypes of CRISPR-Cas systems associated with RTs. Group-II intron-like RTs (ancient, clades 2–13; and recent, clade 1), Retron RT-like (clade 14) and Abi-P2 RT-like (clade 15). For each locus, the node number (Fusicatenibacter saccharivorans was not included in the 9,141 entries), species, respective nucleotide coordinates and CRISPR-Cas system subtype (derived from the respective effector genes) are indicated. Genes are shown roughly to scale; CRISPR arrays are indicated in brackets and are not to scale. The genes within each locus are denoted as in Supplementary Table 3. Homologous genes are colour-coded, with the exception of most of the ancillary genes, which are shown in white; unknown proteins are shown in grey.
Figure 4.
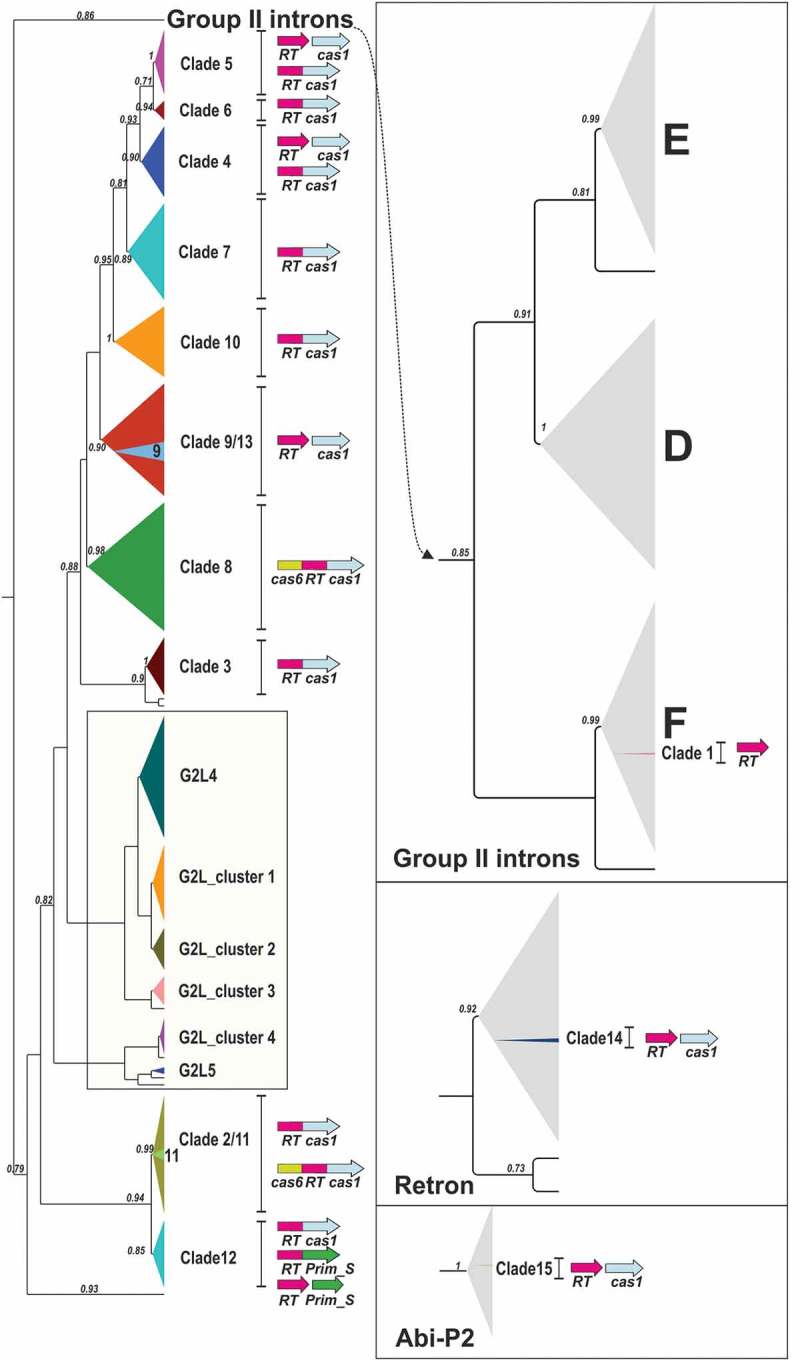
Phylogeny of CRISPR-Cas encoded RTs. The identified lineages of CRISPR-Cas RTs, three evolving from group-II introns, one from Retron/retron-like and one from Abi-P2 RTs, are shown. The CRISPR-Cas RTs and neighbouring group-II intron classes (F, D and E); G2L; Retron and Abi-P2 clades are depicted schematically, with collapsed branches (FastTree support ≥0.85). For the CRISPR-Cas RT clades, the most common RT domains or gene organizations are indicated. Prim_S indicates an archaeo-eukaryotic primase AE_Prim_S-like domain.
The origin and phylogenetic relationships of the predicted RTs from Streptomyces species reported to be linked to type I-E systems [16,19] remain uncertain. These protein sequences form a distinct long branch in the RT phylogeny (Figure 2), with a large number of substitutions per site (2.4; Supplementary File 1). However, its placement in the tree, sharing a common ancestral node with group-II intron RT sequences, is not consistently supported by other phylogenetic analyses, as previously reported [18].
CRISPR-associated RTs closely related to group-II intron-encoded RTs
The 13 previously reported clades of RTs that may have evolved from an ancestral group-II intron retrotransposition event linked to type III CRISPR-Cas systems of different subtypes are not monophyletic, and three distinct clusters can be distinguished. One comprises clade 1 branching within class F introns; this clade is the most recent (Supplementary Figure 1) and arose in methanogenic archaea (Methanosarcina and Methanomethylovorans species). The other two clusters seem to be older and arose in bacteria: a group comprising clades 3–10 and 13, and a separate group comprising clades 2, 11 and 12 (Supplementary Figure 2). These bacterial CRISPR-RT clades and group-II intron RTs are closely related to other uncharacterized RTs that also lack a group-II intron ribozyme RNA structure but are not encoded by sequences in the vicinity of CRISPR-Cas systems. These RTs cluster with other RTs previously described as members of the G2L4 and G2L5 groups [11–13], and within four new additional G2L groups (Figure 4). Three of these G2L groups, together with those of the G2L4 class, appear to form a well-supported larger clade (Supplementary Figure 2). These G2L RTs and CRISPR-RT clades (2–13) branch off from a common node, suggesting that these CRISPR-associated RTs may have evolved from ancestral G2L RT-like sequences (Figure 4 and Supplementary Figure 2).
The cluster comprising clades 3–10 and 13 branches off from a common node, consistent with previous observations [17,18], which suggests that they may be descended from a common ancestor (Figure 4 and Supplementary Figure 2). Clades 9 and 13 cluster into a single clade, referred to hereafter as clade 9/13. This clade is also characterized by the frequent presence of two different cas2 loci (Supplementary Table 3), a feature common to clade 6 CRISPR-RT systems. Unlike the other RTs of clade 9/13, the RT sequences of clade 9 have an insertion of ~105 to 145 amino-acid residues upstream from the RT4 domain (insertion removed in the MSA used to construct the phylogenetic tree shown in Figure 2). These RTs are from bacterial species belonging to class Chloroflexi [31], with the exception of Poribacteria bacterium WGA-4CII, which does not branch from the same internal node (Supplementary Figure 2) and may correspond to a lateral transfer event.
Some of these clades are relatively homogeneous, harbouring RTs alone, as in clades 9 and 13, RT-Cas1 fusions, as in clades 3, 6, 7, 10, or Cas6-RT-Cas1 fusions, as in clade 8 (Figure 4). By contrast, other clades, such as clades 4 and 5, contain both RTs and RT-Cas1 fusions (Supplementary Table 2). Furthermore, the RT and RT-Cas1 fusions belonging to these clades branch off from different single nodes (Supplementary Figure 2). Together, these traits may be consistent with single RT-Cas1 fusion events occurring within the clades, rather than fission events.
The cluster comprising clades 2, 11 and 12 branches off from a well-supported common node, suggesting that these clades descended from a common ancestor (Figure 4 and Supplementary Figure 2). In previous phylogenetic analyses [18], this group traditionally comprised only RT-Cas1 (clades 2 and 12) and Cas6-RT-Cas1 fusions (clade 11). Cas6-RT-Cas1 fusions occur in Porphyromonas species and were considered to have evolved independently of the acquisition of a Cas6 domain in clade 8, which was used to support the ‘various origins’ hypothesis [18]. Nevertheless, as it forms a distinct long branch, with 0.91 substitutions per site (Supplementary File 1), that is taxonomically constrained with no stand-alone closely related Cas6 sequences, it has been argued that this clade may be incorrectly positioned in the RT phylogeny [28]. Interestingly, the Cas6 domain of the Cas6-RT-Cas1 fusions of clade 11 also forms a separate branch (Branch 17) different from that of clade 8 (Branch 11), indicating that both the RT and Cas6 domains have high rates of amino-acid replacement [28]. In an analysis of a larger dataset, increasing the representativeness of taxon sampling, clade 11 also branched within clade 2 (Supplementary Figure 2), but no other taxa in the clade had obvious long or short branches that might explain this attraction. Moreover, the addition of a new member at the base of the clade (Bacteroidetes bacteria, PID94761.1) did not influence the topology. The larger linker [28] between the RT domain and the Cas1 domain that differentiates these fusions from those of clade 8 located downstream from the RT7 domain was removed from the MSAs and did not, therefore, influence the topology of clade 11 in the RT phylogenies constructed ([18], and this work). The position of the Cas6-RT-Cas1 fusions of clade 11 away from the major Cas6-RT-Cas1 fusions (clade 8) does not necessarily involve a long branch attraction (LBA) effect, and it remains possible that this clade has a faster rate of molecular evolution within clade 2.
The previously reported members of clades 2/11 and 12 included only RTs fused to Cas1 [17,18]. However, the phylogenetic analysis based on the larger dataset revealed new members at the base of the clade in which the RTs are adjacent to or fused at the C-terminus to an archaeo-eukaryotic primase (AEP) domain (AE_Prim_S_like) similar to the small catalytic subunit PriS (Supplementaries Table 2 and 3). The NCBI database contains 16 (14 unique) protein sequences with this characteristic RT-Prim_S (AE_Prim_S_like) domain architecture, including some of the above RTs. AEPs have been classified into 13 distinct families, and 12 can be clustered into three major clades: the AEP proper clade, the NCLDV-herpesvirus primase, and the Prim-Pol proper family. All these families have three conserved motifs (I, II and III) essential for catalysis [32,33] in common. The AE_Prim_S_like domain of the CRISPR-associated RTs (truncated in Armatimonadetes species) contains the three conserved motifs (Supplementary File 2). Phylogenetic reconstruction with known AEPs indicated that the AEP domain of the CRISPR-associated RTs formed a new group of primases related to archaeal and eukaryotic PriS proteins, NHEJ primases, and Lef-1-like primases of baculoviruses within the AEP proper clade (Figure 5). Thus, these CRISPR-associated RTs may have both reverse transcriptase and primase activity, and it is tempting to speculate that this primase activity could be used to convert RNA molecules into cDNA in the absence of a primer, facilitating the acquisition of spacers by the CRISPR array, which should be further explored.
Figure 5.
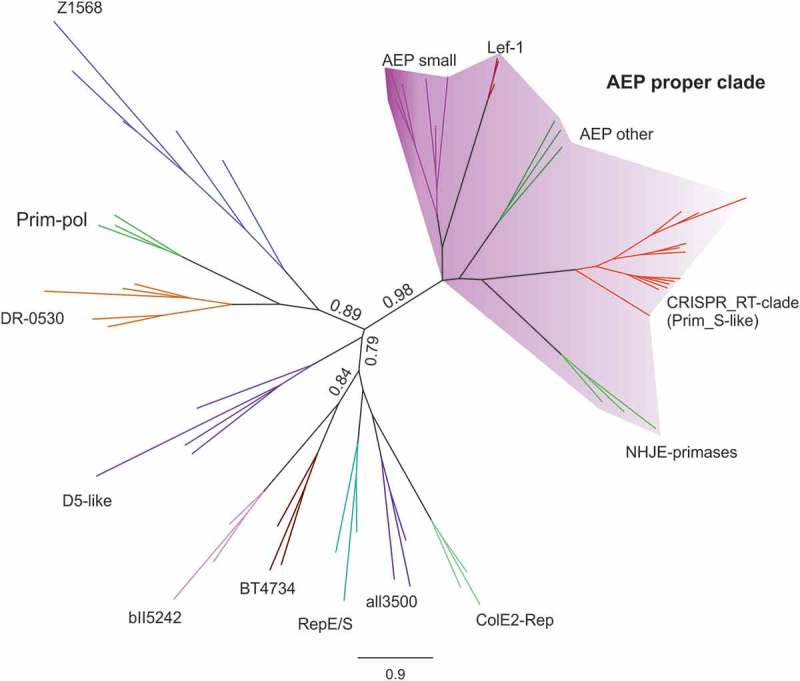
Phylogeny of CRISPR-Cas encoded RT AE_Prim_S_like domains. The tree was constructed with FastTree, from an alignment of 62 protein sequences, including members of the AEP proper clade (AEP small_PriS proteins, NHEJ primases, Lef-1-like primases of baculoviruses and other related sequences), Prim-Pol clade (Z1568-like family, DR0530-like family, all3500-like family, bll5242-like family, ColE2 Rep-like family, RepE/RepS family), BT4734-like family, and the AE_Prim_S_like domain of 14 unique RT proteins with this architecture (NCBI database). All the clades except the all3500-like family (FastTree support 0.65) have a FastTree support ≥0.85. Other relevant FastTree support values are indicated. The corresponding tree newick file (Supplementary File 3) and the subalignment of the AE_Prim_S_like domain of 14 unique RT proteins with the three conserved motifs (Supplementary File 2) is provided in the Supplementary material.
In clade 12, all the RT-Cas1 fusions branched from a single node and the Cas1 proteins were more similar to those found in CRISPR-Cas type I-B systems, which was not the case for the Cas1 proteins adjacent to the RT-Prim_S fusions in the other members of the clade. It, therefore, seems plausible that the acquisition of a former type I-B Cas1 protein was the event triggering the generation of the RT-Cas1 fusion with the AE_Prim_S_like domain loss (Supplementary Table 3).
CRISPR-associated RTs that have evolved from Retron/retron-like RT sequences
The ‘single-point’ and ‘various origins’ models consider all the RTs that can be predicted to be evolutionary and functionally linked to CRISPR-Cas systems to have branched off from a node common to group-II introns [15–18]. However, the analytical pipeline in this study revealed a novel clade (clade 14) of RTs in our dataset, with adjacent Cas1 linked to subtype III-B and III-D CRISPR-Cas systems that branched off from Retron/retron-like RT sequences (nodes 6211–6220 in Supplementaries Tables 1–3, Figures 2 and 4). This clade is constrained to bacterial species of the phylum Bacteroidetes within the classes Flavobacteria, Cytophagia and Sphingobacteriia and branched off from a node common to a taxonomically similar clade grouping RTs encoded by genes not in the neighbourhood of CRISPR-Cas systems (Supplementary Figure 3), suggesting the existence of a common ancestor within this phylum. These RTs have the characteristic signatures of retron RTs, with the conserved VTG motif within RT domain 7 in the ‘Y’ region crucial for specific recognition and binding to the template-primer RNA used for the synthesis of msDNA, and the conserved AXXH motif in region ‘X’, located between domains 2 and 3 [34]. However, no retron msr and msd genes (the primer-template region) were identified in the vicinity. Outside clade 14, but within the retron lineage, we found another RT harbored by Caloramator australicus RC3 (node 6869 in Supplementaries Tables 1–3, accession: fig|857293.11.peg.1034) that is fused to an AE_Prim_S_like domain phylogenetically related to those of clade 12 (not shown), with adjacent cas1 and cas2 loci, with this entire set of genes flanked by CRISPR arrays (Supplementary Table 3). These findings suggest that, like the RTs closely related to group-II introns, Retron/retron-like RTs were captured relatively recently by type III CRISPR-Cas systems.
CRISPR-associated RTs that evolved from AbiP2-like RTs
The computational pipeline designed here highlighted two closely related RT sequences in the Abi-P2 group linked to the second most abundant type of CRISPR-Cas systems (type I-C) in prokaryotes [21]. These RTs (Figure 4 and nodes 8253, 8254 in Supplementaries Tables 1–3) are harboured by bacterial species of the order Pasteurellales (Basfia succiniciproducens and Haemophilus haemolyticus strain HK386) and share a node common to RTs from Haemophilus influenza (strain 723), Salmonella enterica subsp. enterica (serovar Oranienburg str. 701) and Erwinia tasmaniensis, encoded by genes, not in the vicinity of any CRISPR-Cas system (Supplementary Figure 4). A search for close relatives to B. succiniciproducens and H. haemolyticus led to the identification of other RTs linked to type I-C systems in Mannheimia varigena and various Neisseria (Neisseriales) species. A phylogenetic analysis of these RTs (Figure 6) revealed that they split into two differentiated subgroups corresponding to different ecological niches. The B. succiniciproducens group comprises members isolated from livestock- and animal-associated habitats, whereas the H. haemolyticus group appears to be restricted to the human microbiome. The relatively low level of representation of these RTs, which were harbored by only two bacterial phylae in current databases, and their clustering by host/environmental niche rather than by vertical inheritance in microbial species suggest that they probably spread recently, by lateral transfer, in each habitat, possibly between bacteria occupying the same microniches.
Figure 6.
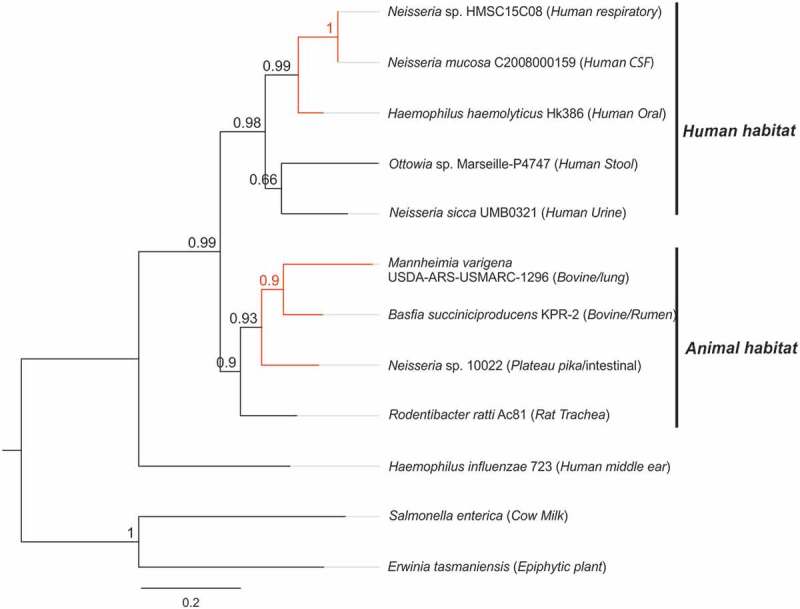
Phylogeny of RTs linked to CRISPR-Cas systems type I-C. The tree was constructed with FastTree, from an alignment including the Abi-P2 RTs from bacterial species of the order Pasteurellales included in the 9,141 entries: Basfia succiniciproducens and Haemophilus haemolyticus strain HK386. Other close relatives identified by Blast searches of the NCBI database also associated with type I-C systems were included. The RTs linked to type I-C systems correspond to the red branches. The host and environment of the isolates are indicated. The tree newick file is provided (Supplementary File 4).
Conclusions and perspectives
We performed a large survey of RTs in databases and constructed a large dataset of unique sequences representative of prokaryotic RTs. The combined phylogenetic reconstruction and identification of RT genes in the vicinity of CRISPR adaptation and effector modules identified sequences that may be evolutionary and functionally linked to CRISPR-Cas systems. These RTs have at least three different origins: the largest lineage evolved from group-II intron RTs, and two smaller lineages, one probably recently arising from retron RT-like sequences and to the other apparently emerging from Abi-P2 RTs.
Most of the RTs studied were associated with type III systems, but those evolving from Abi-P2 RTs were linked to type I-C systems. Our data also suggest that the acquisition of the various protein domains (RT, RT-Prim_S, RT-Cas1 and Cas6-RT-Cas1) may have occurred independently several times during evolution. Nevertheless, these results should be interpreted with care due to the difference in the branch lengths and branching patterns, because the RT sequences have evolved at different rates.
The recent acquisition of RTs of different origins by CRISPR-Cas adaptation modules raises questions as to whether the evolutionary change of the adaptation modules and the ecological response mediated by the CRISPR-Cas systems may occur over a similar timescale.
With the increasing diversity of RT annotation due to the growing scale of sequencing projects and more specifically using metagenomics datasets, we expect to see an increase in the known diversity of CRISPR-Cas/RT adaptation and effector modules. Further studies of these hypothetical eco-evolutionary dynamics will shed light on the forces responsible for generating and maintaining diversity in microbial populations.
Methods
Reverse transcriptase dataset
A heterogeneous dataset was built through several different approaches, to encompass the wide diversity of prokaryotic RTs. RT sequences annotated as RNA-directed DNA polymerases (145,379) or reverse transcriptases (52,684) were downloaded from the PATRIC web server [35] and 133 new protein entries (23 February 2018) with RT-Cas1 architecture were retrieved from the Conserved Domain Architecture Retrieval Tool (CDART) [36]. These two datasets were merged and incorporated into a previously analysed dataset of 558 RT sequences, including 137 type III CRISPR RT/RT-Cas1 proteins closely related to group-II intron-encoded RTs [18]. Six additional proteins from Streptomyces species (annotated as hypothetical proteins) predicted to be RTs linked to type I-E CRISPR-Cas systems [16,19] were also included in the analysis. The resulting dataset of 198,760 proteins was then filtered by selecting the RT domain (RT0-7) of the proteins with a length ≥200 amino acids, in multiple-step clustering with a threshold of 85% sequence identity [18]. This procedure resulted in the construction of a set of 9,141 diverse unique RTs for further analysis. The various steps used to compile the final dataset are described in Figure 1.
Identification pipeline for CRISPR/Cas systems
A custom script was written in python 3.7 to predict the CRISPR arrays, to annotate the proteins encoded by genes in the neighborhood of the 9,141 RTs analysed in this study, to obtain the taxonomic information about the carrier organism, to classify the system in which the RT was embedded and to use this information to predict the putative association of an RT with a CRISPR/cas locus. Briefly, the CRISPR arrays were identified by CRT [37], which provided information about the number of repeats, the mean length of the spacers and the mean length of the direct repeats (DRs). Information about the proteins was retrieved by constructing a database of profiles by combining existing datasets [20] with datasets describing novel effector proteins from recent publications [22,38–40], datasets relating to defence systems [41] and recent updates of databases of CRISPR/Cas accessory proteins [19,42]. The resulting database was then used with hmmscan (http://hmmer.org), to identify the proteins encoded by genes in the neighbourhood of the RT sequence (within 30 kb on either side). Two consecutive rounds of analysis with an e-value cutoff of 1e-5 and 1e-2, respectively, were performed. The taxonomic information was downloaded from the NCBI taxonomy database [43]. Finally, the systems were classified (based on the effector proteins of the loci) on the basis of the previously described methodology and profiles [20,22,38–40]. Any RT gene not surrounded by Cas or CRISPR arrays was considered not to be associated with CRISPR/Cas and was discarded from subsequent analyses. The reported clades were defined by revising the rest of the RTs manually and considering them to be associated with CRISPR/Cas if it does not show a recognizable group-II intron RNA structure [6] and: i) it is located within a CRISPR/Cas locus usually closed to an adaptation module, and ii) with at least one other RT matching this criterion branching from the same node in the phylogenetic tree. Other RTs associated with CRISPR-Cas locus were also considered, but they remain unclassified.
Phylogenetic analyses of RT sequences
We used MAFFT software [44] and progressive methods to perform the MSAs. The MSA corresponding to the RT0-7 domain of the 9,141 entries in the final dataset was filtered to remove sites containing gaps in more than 50% of all sequences. The phylogenetic trees were constructed with the FastTree program [30] as previously described [17].
Funding Statement
This work was supported by the Spanish Ministerio de Ciencia, Innovación y Universidades, including ERDF (European Regional Development Funds) research grants [BIO2014-51953-P, BIO2017-82244-P]. A.G-D was supported by a FPU predoctoral fellowship grant from the Ministerio de Economía y Competitividad [FPU15/02714].
Authors contribution
NT conceived the study and performed the phylogenetic analyses. MRM designed the computational pipeline with FM-A and obtained protein sequences from databases. AG-D obtained RT sequences fused to Cas1 from databases. NT wrote the paper with contributions from all the authors.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplementary data for this article can be accessed here.
References
- [1].Baltimore D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature. 1970;226:1209–1211. [DOI] [PubMed] [Google Scholar]
- [2].Temin HM, Mizutani S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature. 1970;226:1211–1213. [DOI] [PubMed] [Google Scholar]
- [3].Lampson BC, Sun J, Hsu MY, et al. Reverse transcriptase in a clinical strain of Escherichia coli: production of branched RNA-linked msDNA. Science. 1989;243:1033–1038. [DOI] [PubMed] [Google Scholar]
- [4].Lim D, Maas WK. Reverse transcriptase-dependent synthesis of a covalently linked, branched DNA-RNA compound in E. coli B. Cell. 1989;56:891–904. [DOI] [PubMed] [Google Scholar]
- [5].Toro N, Jiménez-Zurdo JI, García-Rodríguez FM. Bacterial group II introns: not just splicing. FEMS Microbiol Rev. 2007;31(3):342–358. [DOI] [PubMed] [Google Scholar]
- [6].Lambowitz AM, Zimmerly S. Group II introns: mobile ribozymes that invade DNA. Cold Spring Harb Perspect Biol. 2011;3(8):a003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].McNeil BA, Semper C, Zimmerly S. Group II introns: versatile ribozymes and retroelements. RNA. 2016;7(3):341–355. [DOI] [PubMed] [Google Scholar]
- [8].Lampson BC, Inouye M, Inouye S. Retrons, msDNA, and the bacterial genome. Cytogenet Genome Res. 2005;110(1–4):491–499. [DOI] [PubMed] [Google Scholar]
- [9].Guo H, Arambula D, Ghosh P, et al. Diversity-generating retroelements in phage and bacterial genomes. Microbiol Spectr. 2014;2:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wu L, Gingery M, Abebe M, et al. Diversity-generating retroelements: natural variation, classification and evolution inferred from a large-scale genomic survey. Nucleic Acids Res. 2018;46(1):11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Simon DM, Zimmerly S. A diversity of uncharacterized reverse transcriptases in bacteria. Nucleic Acids Res. 2008;36(22):7219–7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Toro N, Nisa-Martínez R. Comprehensive phylogenetic analysis of bacterial reverse transcriptases. PLoS One. 2014;9(11):e114083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zimmerly S, Wu L. An unexplored diversity of reverse transcriptases in bacteria. Microbiol Spectr. 2015;3(2):MDNA3-0058–2014. [DOI] [PubMed] [Google Scholar]
- [14].Kojima KK, Kanehisa M. Systematic survey for novel types of prokaryotic retroelements based on gene neighborhood and protein architecture. Mol Biol Evol. 2008;25:1395–1404. [DOI] [PubMed] [Google Scholar]
- [15].Silas S, Mohr G, Sidote DJ, et al. Direct CRISPR spacer acquisition from RNA by a natural reverse transcriptase-Cas1 fusion protein. Science. 2016;351(6276):aad4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Silas S, Makarova KS, Shmakov S, et al. On the origin of reverse transcriptase-using CRISPR-Cas systems and their hyperdiverse, enigmatic spacer repertoires. MBio. 2017;8(4):pii: e00897–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Toro N, Martínez-Abarca F, González-Delgado A. The reverse transcriptases associated with CRISPR-Cas systems. Sci Rep. 2017;7(1):7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Toro N, Martínez-Abarca F, González-Delgado A, et al. On the origin and evolutionary relationships of the reverse transcriptases associated with type III CRISPR-Cas systems. Front Microbiol. 2018;9 DOI: 10.3389/fmicb.2018.01317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shmakov SA, Makarova KS, Wolf YI, et al. Systematic prediction of genes functionally linked to CRISPR-Cas systems by gene neighborhood analysis. Proc Natl Acad Sci USA. 2018;115(23):E5307–E5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Makarova KS, Koonin EV. Annotation and classification of CRISPR-Cas systems. Methods Mol Biol. 2015;1311:47–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Makarova KS, Wolf YI, Alkhnbashi OS, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13(11):722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shmakov S, Smargon A, Scott D, et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol. 2017;15(3):169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Makarova KS, Zhang F, Koonin EV. SnapShot: class 1 CRISPR-Cas systems. Cell. 2017;168(5):946–946.e1. [DOI] [PubMed] [Google Scholar]
- [24].Makarova KS, Zhang F, Koonin EV. SnapShot: class 2 CRISPR-Cas systems. Cell. 2017;168(1–2):328–328.e1. [DOI] [PubMed] [Google Scholar]
- [25].Staals RH, Zhu Y, Taylor DW, et al. RNA targeting by the type III-A CRISPR-Cas Csm complex of Thermus thermophilus. Mol Cell. 2014;56:518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tamulaitis G, Kazlauskiene M, Manakova E, et al. Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol Cell. 2014;56:506–517. [DOI] [PubMed] [Google Scholar]
- [27].Pyenson NC, Marraffini LA. Type III CRISPR-Cas systems: when DNA cleavage just isn’t enough. Curr Opin Microbiol. 2017;37:150–154. [DOI] [PubMed] [Google Scholar]
- [28].Mohr G, Silas S, Stamos JL, et al. A reverse transcriptase-Cas1 fusion protein contains a Cas6 domain required for both CRISPR RNA biogenesis and RNA spacer acquisition. Mol Cell. 2018;72(4):700–714.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schmidt F, Cherepkova MY, Platt RJ. Transcriptional recording by CRISPR spacer acquisition from RNA. Nature. 2018;562:380–385. [DOI] [PubMed] [Google Scholar]
- [30].Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Muñoz R, Rosselló-Móra R, Amann R. Revised phylogeny of Bacteroidetes and proposal of sixteen new taxa and two new combinations including Rhodothermaeota phyl. nov. Syst Appl Microbiol. 2016;39(5):281–296. [DOI] [PubMed] [Google Scholar]
- [32].Iyer LM, Koonin EV, Leipe DD, et al. Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-domain proteins: structural insights and new members. Nucleic Acids Res. 2005;33(12):3875–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kazlauskas D, Sezonov G, Charpin N, et al. Novel families of archaeo-eukaryotic primases associated with mobile genetic elements of bacteria and archaea. J Mol Biol. 2018;430(5):737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Inouye S, Hsu M-Y, Xu A, et al. Highly specific recognition of primer RNA structures for 2)-OH priming reaction by bacterial reverse transcriptases. J Biol Chem. 1999;274:31236–31244. [DOI] [PubMed] [Google Scholar]
- [35].Wattam AR, Davis JJ, Assaf R, et al. Improvements to PATRIC, the All-Bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017;45:D535–D542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Geer LY. CDART: protein homology by domain architecture. Genome Res. 2002;12(10):1619–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bland C, Ramsey TL, Sabree F, et al. “CRISPR Recognition Tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007;8:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Burstein D, Harrington LB, Strutt SC, et al. New CRISPR-Cas systems from uncultivated microbes. Nature. 2017;542:237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Harrington LB, Burstein D, Chen JS, et al. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science. 2018;362:839–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yan WX, Hunnewell P, Alfonse LE, et al. Functionally diverse type V CRISPR-Cas systems. Science. 2019;363:88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Doron S, Melamed S, Ofir G, et al. Systematic discovery of antiphage defense systems in the microbial pangenome. Science. 2018;359:eaar4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shah SA, Alkhnbashi OS, Behler J, et al. Comprehensive search for accessory proteins encoded with archaeal and bacterial type III CRISPR-Cas gene cassettes reveals 39 new Cas gene families. RNA Biol. 2019;16:530–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Federhen S. The NCBI Taxonomy Database. Nucleic Acids Res. 2011;D136–D143. DOI: 10.1093/nar/gkr1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.