

ABSTRACT
The facultative intracellular pathogen Listeria monocytogenes can persist and grow in a diverse range of environmental conditions, both outside and within its mammalian host. The alternative sigma factor Sigma B (σB) plays an important role in this adaptability and is critical for the transition into the host. While some of the functions of the σB regulon in facilitating this transition are understood the role of σB-dependent small regulatory RNAs (sRNAs) remain poorly characterized. In this study, we focused on elucidating the function of Rli47, a σB-dependent sRNA that is highly induced in the intestine and in macrophages. Using a combination of in silico and in vivo approaches, a binding interaction was predicted with the Shine-Dalgarno region of the ilvA mRNA, which encodes threonine deaminase, an enzyme required for branched-chain amino acid biosynthesis. Both ilvA transcript levels and threonine deaminase activity were increased in a deletion mutant lacking the rli47 gene. The Δrli47 mutant displayed a shorter growth lag in isoleucine-depleted growth media relative to the wild-type, and a similar phenotype was also observed in a mutant lacking σB. The impact of the Δrli47 on the global transcription profile of the cell was investigated using RNA-seq, and a significant role for Rli47 in modulating amino acid metabolism was uncovered. Taken together, the data point to a model where Rli47 is responsible for specifically repressing isoleucine biosynthesis as a way to restrict growth under harsh conditions, potentially contributing to the survival of L. monocytogenes in niches both outside and within the mammalian host.
KEYWORDS: Listeria monocytogenes, Rli47, Sigma B, Isoleucine biosynthesis, ilvA, sRNA, Threonine deaminase
Introduction
The Gram-positive bacterium Listeria monocytogenes is a facultative intracellular pathogen widely found in the environment [1]. It is capable of colonizing the mammalian host following ingestion by susceptible individuals. Following a successful transit through the gastrointestinal tract, it can pass through the epithelial layer and replicate intracellularly in several organs, causing the life-threatening infection listeriosis [2,3]. Although the number of cases is small, the high mortality rate amongst infected individuals (typically 20%) makes listeriosis a significant public health concern [4–6]. The stress-activated sigma factor σB plays a pivotal role in allowing L. monocytogenes to modulate its transcriptional response to a variety of harsh environments both outside and within the host [7].
While transitioning from a saprophytic to a host-associated state, L. monocytogenes relies on sensing host-specific signals that affect the transcription regulators CodY and PrfA and triggers the initiation of its virulence program [8–12]. In addition to these important regulatory proteins, it is now clear that small regulatory RNAs (sRNAs), non-coding transcripts of about 50 to 500 nucleotides long, can contribute to the control of virulence gene expression at the post-transcriptional level [13,14]. The genome of L. monocytogenes includes about 300 small non-coding RNAs, of which 154 are proposed to be trans-acting sRNAs [15]. Trans-acting sRNAs are located distally from the genes encoding their target RNAs and typically have only limited complementarity. The transcription of six sRNAs (SbrA, Rli47, Rli33-1, Anti-LhrC-5, Anti2270 and Rli95) of L. monocytogenes depends on the stress-inducible sigma factor σB [16]. This group includes the sRNA Rli47, a ~ 500 nucleotide-long sRNA located in the intergenic region between lmo2141 and lmo2142. Transcriptomic studies identified a σB-dependent promoter upstream the rli47 transcript, also referred to as sbrE (sigma-B-dependent RNA) in the literature, and a Rho-dependent terminator at its 3ʹ-end [17–19]. It is expressed at higher levels in L. monocytogenes stationary phase cells [18–20], in the intestinal lumen [18], in macrophages [17] and in soil environments in mutants lacking the AgrA regulator [21]. Although studies on Rli47 function have identified altered transcription of a number of genes including lmo0636, lmo0637 (methyltransferase) and lmo2094 (L-fuculose-phosphate aldolase) in a mutant lacking this sRNA [20], no growth or survival phenotype under a variety of different environmental conditions has been identified and its function has therefore remained undefined.
Here we report predicted binding targets for Rli47 using an in silico and in vivo analysis of its secondary structure, and we confirmed its binding in vitro to a target mRNA associated with branched-chain amino acid (BCAA) biosynthesis. The data presented show that Rli47 can base-pair through a single-stranded CU-rich loop with the Shine-Dalgarno (SD) region of the ilvA mRNA, which encodes threonine deaminase (TD), the first key enzyme on the L-isoleucine (ile) biosynthetic pathway that catalyzes the deamination of L-threonine to produce alpha-ketobutyrate [22,23]. This interaction negatively regulates the expression of ilvA, reducing the threonine deaminase enzymatic activity. Global transcriptional profiling confirmed the role of Rli47 in modulating amino acid metabolism and in particular influencing the CodY regulon. Together, our data support a model where σB, CodY and Rli47 form a regulatory network that serves to repress isoleucine biosynthesis even in conditions where isoleucine is depleted from the environment.
Results
Defining the sequence and structure of rli47 sRNA
The rli47 gene is located on the intergenic region between lmo2141 and lmo2142 of L. monocytogenes EGD-e (Figure 1(a)). Nucleotide BLAST searches [24] against other Listeria species for which whole genome sequence is available revealed that rli47 is present in all Listeria sensu stricto species (L. monocytogenes, L. innocua, L. seeligeri, L. ivanovii, L. welshimeri and L. marthii) but was not found in the genomes of species of the Listeria sensu lato group (L. weihenstephanensis, L. fleschmannii, L. floridensis, L. aquatica, L. newyorkensis, L. cornellensis, L. grandensis, L. riparia, L. booriae, L. rocourtiae).
Figure 1.
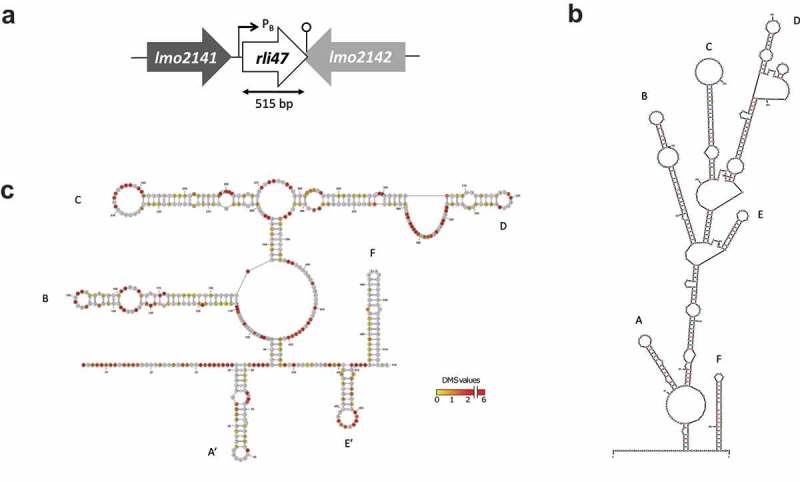
Mapping and folding structure of Listeria monocytogenes Rli47. (a) Mapping rli47 on the genome of Listeria monocytogenes EGD-e. A σB-dependent promoter (PB) is represented, and the lollipop structure denotes a hypothetical transcription terminator structure shared by rli47 and lmo2142. (b) Folding structure was predicted in silico by mfold. The six stem-loop structures were labelled from A to F. (c) Secondary structure of Rli47 small RNA modelled using in vivo DMS-MapSeq structure probing. ‘DMS values’ represent DMS mediated methylation of adenine (a) and cytosine (c) nucleotides, respectively. A DMS value lower than one indicates a base-paired A or C whereas a DMS value higher than one indicates an unpaired A or C. The obtained DMS values were used as guides for the modelling of the Rli47 secondary structure. The six stem-loop structures were labelled based on their position relatively to the in silico structure, as derivative structures were named A’ and E’.
The rli47 gene belongs to the σB regulon and a σB-dependent promoter has been identified upstream from the predicted first nucleotide (Fig. S1A) [17,19]. Earlier studies claimed different sizes for this sRNA; 314 nt [17], 446 nt [25], 509 nt [18] and 514 nt [19]. Additionally, a Rho-independent terminator was previously identified at the 3ʹ-end of the transcript. This terminator is shared with the flanking gene lmo2142 located in the opposite strand [19]. Besides the differences in length, the reported transcription start and stop sites also differed between research groups. Therefore, in order to proceed with the construction of a deletion mutant lacking the entire transcript we mapped rli47 on the genome of L. monocytogenes EGD-e to help better assign its start and stop sites.
Using northern blot analysis (Fig. S1B), primer extension (Fig. S1C) and secondary structure predictions (Figures 1(b and c)), Rli47 was estimated to be 515 nt in length (from nucleotides 2,226,036A to 2,226,550A in the genome of L. monocytogenes EGD-e). Computational (Figure 1(b)) and in vivo (Figure 1(c)) predictions of the secondary structure of Rli47 showed that it is likely to fold as a six stem-loop structure. These predicted six stem-loop structures were labeled from A to F; (A) 42G – 74U, (B) 112C – 185U, (C) 201C – 268G, (D) 279G – 382C, (E) 401U – 424A and (F) 474G – 513U (Figure 1(b)). The six stem-loop structures produced by the DMS-MaPseq in vivo mapping approach (Materials and Methods) showed a similar assembly, although the first and fifth stem-loops were structured in different stretches; (A’) 39U – 81G, (E’) 441U – 470A (Figure 1(c)). Thus, the secondary structures predicted by both the in vivo and in silico approaches are in good agreement.
Regulatory sRNAs of Gram-positive bacteria often contain single-stranded CU-rich motifs, which are important for target mRNA interactions by base-pairing with the SD sequences [26–31]. Analysing the in silico and in vivo generated Rli47 stem-loop structures in detail identified a CU-rich motif on the single-strand of the stem-loop C, making this a promising region for base-pairing with target mRNAs. Furthermore, Rli47 is unlikely to interact with the RNA-binding protein Hfq since the level of Rli47 was not affected by its absence [18].
σB-dependent transcription of rli47 is induced during exponential growth in DM
To investigate the conditions that lead to Rli47 transcription, the levels of the sRNA were determined via northern blot analysis on total RNA purified from L. monocytogenes EGD-e in both complex medium TSB (Tryptic Soy Broth) and a minimal medium DM (chemically Defined Medium). To further investigate σB-dependency of the rli47 transcription, a sigB deletion mutant strain (ΔsigB) was included in this analysis. An rli47 deletion mutant strain (Δrli47) was constructed that lacked the whole rli47 coding sequence except the hypothetical terminator structure shared with the downstream gene lmo2142 (Fig. S1). This deletion mutant was used as a negative control for the transcript measurements. RNA samples used for northern blots were extracted from cells harvested at exponential and stationary phase cells growing in DM and TSB, which were loaded in a gel per growth stage and probed for Rli47 and rRNA 16S (Fig. S2). Rli47 RNA was detected in the wild-type (WT) in both exponential and stationary phase in both growth media, DM (Figure 2) and TSB (Fig. S3). As expected, Rli47 was not detected in the Δrli47 mutant. Transcription was highly σB-dependent in both media although some σB-independent transcription was detected in TSB at exponential phase (Fig. S3). The deletion of rli47 had minimal impact on the transcription of the upstream (lmo2141) and downstream (lmo2142) genes, as determined by RT-PCR on cells grown in either DM to exponential phase or TSB to stationary phase (Fig. S4).
Figure 2.
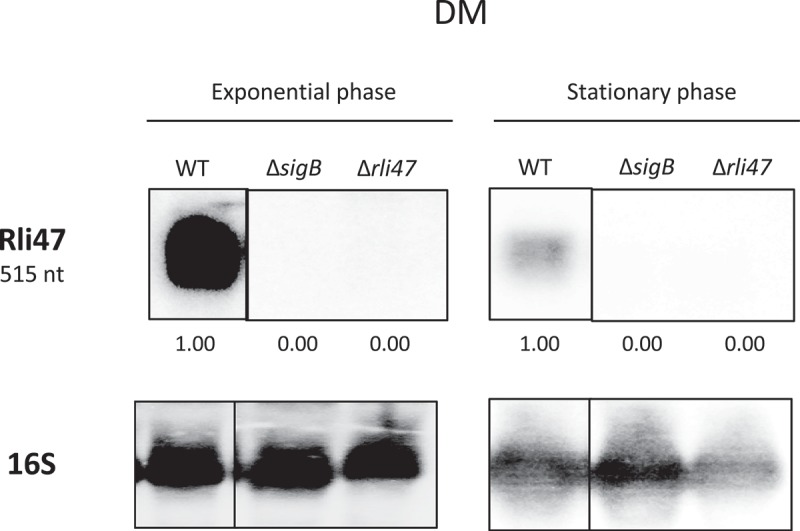
Northern blot analysis of Rli47 expression on a σB-dependent manner. Samples were taken from L. monocytogenes EGD-e wild-type (WT), ΔsigB and Δrli47 cultures at exponential and stationary phases in DM + 0.4% Glucose at 37°C. Northern blot was probed for Rli47 and 16S rRNA as a loading control. Relative levels of Rli47 transcripts were normalized to 16S and are shown below each lane. Vertical lines between bands represent a non-sequential loading of the samples of interest. Representative of two independent technical replicates of two biological samples per condition.
Rli47 binds to the SD region of the ilva mRNA
We hypothesized that this Rli47 might act in trans, via direct binding to the target mRNAs, leading to inhibition of translation initiation and/or influencing transcript stability. To explore this possibility, we first performed in silico analyses of the potential base-pairing between the sRNAs and mRNAs. Several mRNAs were computationally predicted to bind to Rli47 using IntaRNA [32–34]. In order to narrow the list to the most plausible targets, the following criteria were applied to the predicted binding regions: (1) the interaction should involve a single-stranded region of the sRNA; (2) it should include the SD sequence of the target mRNA; (3) the interaction should have a binding energy greater than – 16 kJ mol−1. Based on these constraints, copB, ribCF, ilvA and addB mRNAs were identified as putative targets of Rli47 (Table 1).
Table 1.
Interactions between target mRNAs and Rli47 predicted in silico. Predictions were obtained by the IntaRNA software [32–34]. Top four hypothetical targets which binding prediction fulfilled the selection criteria: (i) Energy > −16 KJ mol−1; (ii) Single-strand region on Rli47; (iii) Overlap the Shine-Dalgarno region on the target mRNA. No σB promoter was found in silico in any of the target genes [25]. TSS – transcription start site.
| Target gene | Energy (kJ mol−1) | mRNA binding region (nt from start codon) | Rli47 binding region (nt from TSS) | Molecular function | Biological process |
|---|---|---|---|---|---|
| copB | (-) 19.74 | −13 to 18 | 259 to 292 | Adenosylcobinamide-phosphate guanyliltransferase | Cobalamin biosynthetic process |
| ribCF | (-) 17.57 | −11 to 14 | 262 to 286 | Bifunctional flavokinase, FAD synthetase | Riboflavin biosynthetic process |
| ilvA | (-) 16.49 | −9 to 17 | 219 to 241 | Threonine deaminase | Isoleucine biosynthetic process |
| addB | (-) 16.26 | −25 to −2 | 379 to 411 | ATP-dependent nuclease, subunit B | Double-strand break repair |
To further investigate the binding of Rli47 to these mRNAs, Electrophoretic Mobility Shift Assays (EMSAs) were performed (Figure 3), where full-length labelled sRNA was incubated with increasing concentrations of 5ʹ-end unlabeled RNA of ilvA (99 nt), copB (75 nt), ribCF (77 nt) or addB (185 nt). For this, RNA fragments corresponding to the region of interest were synthesized, including the entire intergenic region plus at least 20 to 30 nt of the coding region. The electrophoretic mobility of Rli47 was clearly shifted when unlabeled ilvA RNA was used in the assay, suggesting their interaction (Figure 3). However, no binding was detected between Rli47 and copB, ribCF or addB under the same experimental conditions (Figure 3). These results demonstrate a likely interaction between Rli47 and the ilvA mRNA.
Figure 3.
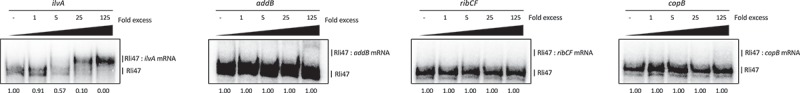
Rli47 and hypothetical target mRNAs base-pairing by Electrophoretic Mobility Shift Assay. Labelled Rli47 full-size transcripts were incubated with increasing concentrations of unlabeled ilvA, addB, ribCF or copB mRNA. The fraction of unbound Rli47 is shown below each lane.
Interestingly, the SD sequence of ilvA mRNA was predicted to bind to the CU-rich single-stranded region of the third stem-loop (stem-loop C) of the sRNA (Fig. 1BC and 4A). To investigate which part of Rli47 was responsible for this interaction, a truncated version of Rli47, from the TSS (transcription start site) to nucleotide 276C, which lies after the third predicted stem-loop structure was synthesized. In silico predictions of the structure of truncated Rli47 showed that the folding of stem-loop C was probably maintained. EMSAs between ilvA RNA and the full (Figure 3) and truncated versions of Rli47 (Figure 4(b)) showed the same pattern of binding (mRNA–sRNA interaction).
Figure 4.
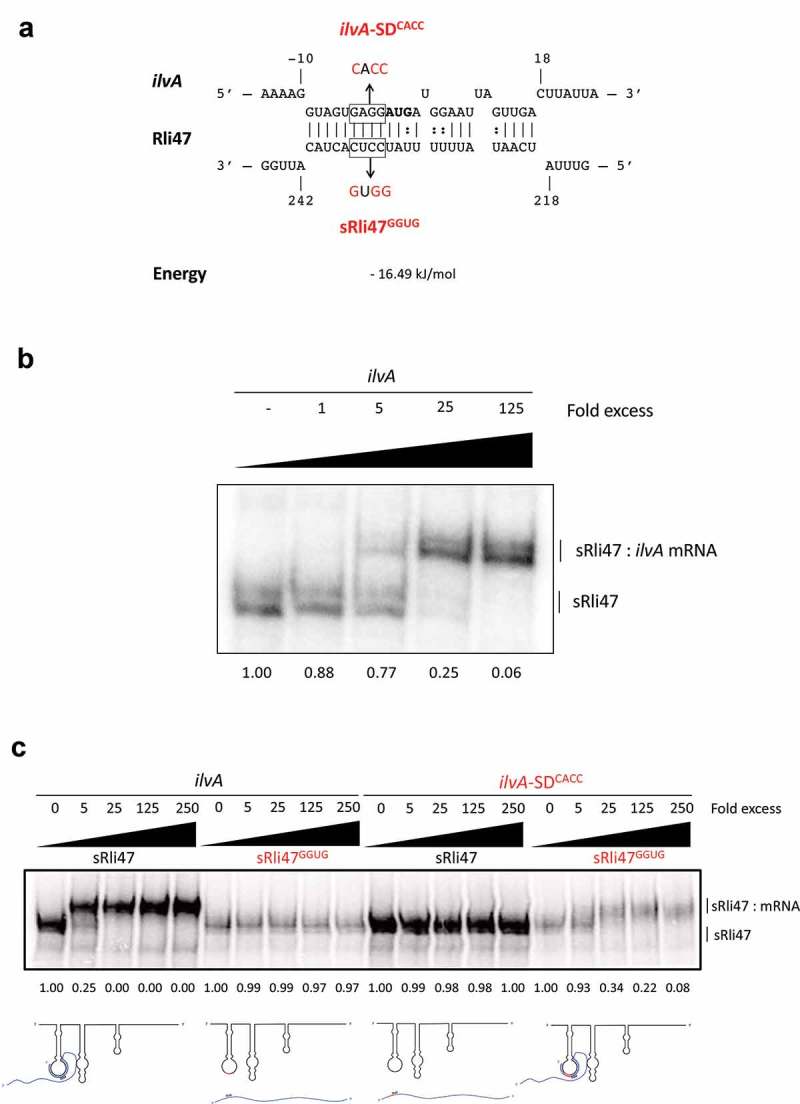
Mutational Analysis of truncated Rli47 and ilvA mRNA base pairing by Electrophoretic Mobility Shift Assay. (a) Predicted basepairing between the SD region of ilvA mRNA and Rli47. The mutated nucleotides are shown in red and the sequences of the minimal mutant variants ilvA-SDCACC and sRli47GGUG are indicated. (b) Electrophoretic mobility shift assays (EMSAs) of the interaction between ilvA mRNA and Rli47 truncated transcripts (sRli47). Truncated (sRli47) transcripts of Rli47 were incubated with increasing concentrations of unlabeled ilvA mRNA. (c) Secondary structure denaturation step was performed previous to binding incubation of labelled sRli47 and sRli47GGUG with increasing concentrations of unlabeled ilvA RNA or the mutant ilvA-SDCACC. Similar results were shown even without the denaturation of the secondary structures (Fig. S5). The fraction of unbound Rli47 is shown below each lane. AUG denotes the start codon of ilvA.
Rli47 CU-rich loop is responsible for binding to SD regulatory region of ilvA mRNA
To further define the base-pairing between the sRNA Rli47 and ilvA mRNA we focused on the CU-rich region of the predicted third stem-loop as a likely location of the interaction. In order to investigate the importance of the CU-rich region of the third stem-loop in the interaction with ilvA mRNA, a mutated version of Rli47 (sRli47GGUG) was synthesized, where nucleotides 233 to 236 were altered from CCUC to GGUG in the truncated version of Rli47. This alteration was predicted to prevent the interaction of the Rli47 CU-rich loop with the SD region of the ilvA transcript (Figure 4(a)). The Rli47 derivative was labeled, mixed with increasing concentrations of ilvA RNA and binding was analyzed in an EMSA (Figure 4(c)). Since Rli47 migrated as two bands in gel electrophoresis, even in the unbound state (Figure 4), it suggests that the sRNA can adopt two alternate structures under the conditions used in the binding experiments. Nonetheless, the possibility that the presence of the two bands could also result from an abortive transcription cannot be ruled out. To further confirm this interaction, a complementary mutation was introduced into the ilvA transcript changing the predicted SD region from GAGG to CACC, and the construct was designated ilvA-SDCACC (Figure 4(a)). Differences in the labelling efficiencies between sRli47 and sRli47GGUG were consistently observed (Figure 4 and S5). In silico analysis predicted several RNA structures with similar folding patterns for both sRli47 and sRli47GGUG (∆G ranging from −66.10 to −63.30 kcal/mol). The single-stranded loop, where the mutated region resides, was preserved in all of the proposed mfold structures, implying that the differences in labelling efficiencies are most likely due to changes in the tertiary structure of the sRNA species. Further secondary structure predictions on ilvA and ilvA-SDCACC mRNAs revealed that the mutation caused differences in folding. Thereafter, to minimize the putative effect of such differences in secondary structures, both RNAs were heated and cooled together for the mutational analysis, ruling out any concerns regarding structural differences on the mutant RNA secondary structures relative to the WT versions (Figure 4(c)). The results revealed that mutations in the CU-rich sequence of the loop prevented the interaction with ilvA RNA, suggesting that this region is crucial for the binding of Rli47 to ilvA transcripts (Figure 4(c)). Similar results were observed in non-denatured RNAs (Fig. S5). The mutated version of ilvA (ilvA-SDCACC) did not interact with the wild-type version of Rli47 (Figure 4(c)). When the complementary mutated versions of ilvA RNA (ilvA-SDCACC) and Rli47 mutated CU-rich loop sequence (sRli47GGUG) were mixed, the interaction was restored (Figure 4(c)). These results confirmed that the CU-rich sequence of the third stem-loop of Rli47 is required for base-pairing to the SD region of ilvA mRNA and thus identified the region that is critical for the interaction.
Deletion of sigB and Rli47 increases ilvA mRNA
The in vitro interaction of Rli47 with the SD region of the ilvA mRNA suggested a regulatory role of the sRNA and we sought to investigate this in vivo. To this end, we performed RT-qPCR on total RNA purified from L. monocytogenes EGD-e wild-type, ΔsigB and Δrli47 exponential phase cells grown in DM and stationary phase cells grown in TSB, conditions where the levels of Rli47 were previously shown to be highest (Figure 2 and S3). The levels of Rli47 and ilvA mRNA were quantified in the knockout cells relative to WT cells and normalized to 16S rRNA transcript levels. In both growth conditions, Rli47 levels were lower in the ΔsigB mutant (log2FC = −7.45 in DM and log2FC = −10.94 in TSB) and essentially undetectable in the Δrli47 strain (log2FC = −16.67 in DM and log2FC = −18.21 in TSB) relative to the wild-type. In contrast, the ilvA transcript was present at an elevated level in both ∆sigB (log2FC = 2.46 in DM and log2FC = 5.57 in TSB) and ∆rli47 (log2FC = 1.05 in DM and log2FC = 7.44 in TSB) strains (Figure 5 and S6). However, in DM, the difference in the ilvA transcript levels in the ∆rli47 strain relative to the WT was not statistically significant (p-value = 0.67). Although Rli47 transcript levels were lower in both knockout mutants, a significant difference in Rli47 mRNA level was shown, suggesting that some transcripts were still being produced in the ΔsigB strain. This provided further evidence that rli47 transcription is largely but not solely dependent on σB. The difference in the ilvA relative transcription between mutant strains was not significant (p-value > 0.05) for cells grown in DM (Figure 5), however in TSB there was significant difference (Fig. S6), suggesting that σB has rli47-independent effects on ilvA mRNA levels. Together these data indicate that Rli47 has a negative effect on the level of ilvA transcripts in vivo, at least in the complex medium TSB, suggesting that the interaction detected in vitro is physiologically significant in vivo.
Figure 5.
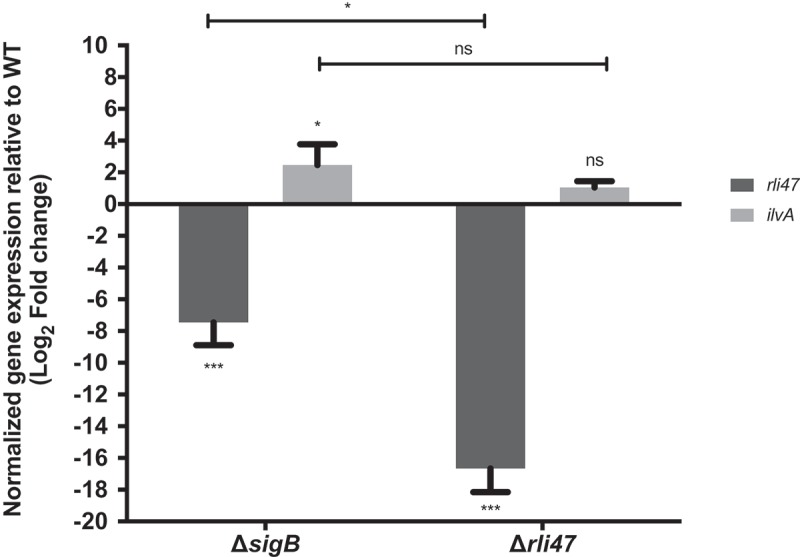
Impact of sigB or rli47 deletion on ilvA expression in exponential growth phase cells in DM. Quantification of rli47 and ilvA in the ∆rli47 or ∆sigB relative to the wild-type was assessed by RT-qPCR and normalized to 16S RNA. Logarithmic fold change values on the mutant strains are relative to the wild-type. The data represent three biological replicates with three technical independent repetitions. Error bars indicate standard deviation. Asterisks represent p-values (* = p-value <0.05, ** = p-value <0.001, *** = p-value <0.0001) calculated using a Two-way ANOVA with Sidak’s multiple comparisons test. Ns: not significant.
Both rli47 and σB cause L-isoleucine auxotrophy by decreasing TD activity
The ilvA gene encodes threonine deaminase (TD), which is the first key enzyme on the L-isoleucine biosynthetic pathway that catalyzes the deamination of threonine to produce alpha-ketobutyrate, water and ammonia [22,23]. To investigate the contribution of Rli47 to L-isoleucine biosynthesis, L. monocytogenes EGD-e wild-type, ΔsigB and Δrli47 strains were grown in DM either with (DM) or without (DM-ile) isoleucine supplementation (Figure 6(a)). No differences in growth rate or lag time were observed among strains growing in DM (Table S3). However, the Δrli47 strain had a significantly shorter lag phase than the WT during growth in DM-ile (Figure 6(a)), as well as in media with reduced levels of isoleucine (Table S3 and Fig. S7). This phenotype was reversed when the Δrli47 deletion was complemented in trans with a native copy of the rli47 (rli47-c) gene including its own promoter region (Figure 6(a)). The rli47-complemented strain shows a longer lag time than the WT in DM-ile medium. In these growth experiments, the ΔsigB mutant displayed a phenotype similar to the Δrli47 mutant but had an even shorter lag time (Figure 6(a)). These data suggest that σB and Rli47 activities delay the onset of growth under isoleucine limiting conditions, consistent with the hypothesis that Rli47 negatively regulates expression of ilvA.
Figure 6.
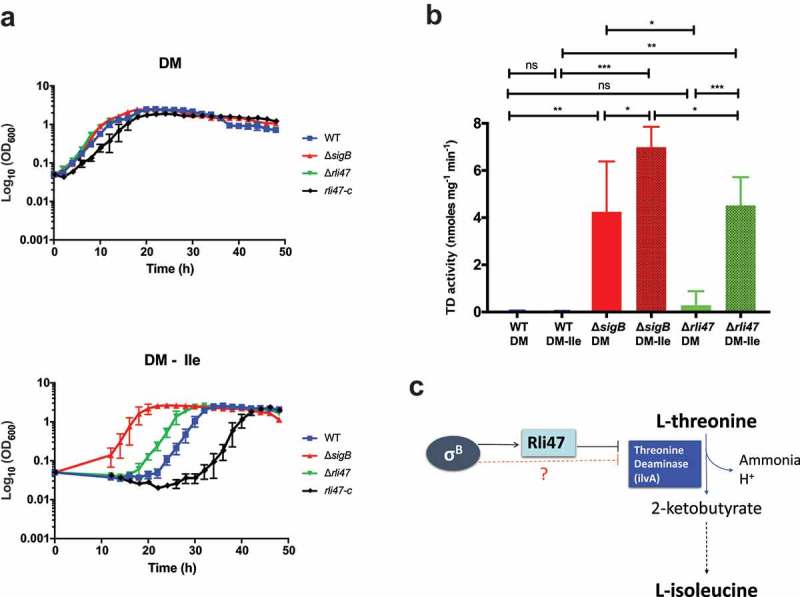
Assessing the effect of Rli47 on the Branched-chain amino acids isoleucine pathway under isoleucine depletion. (a) Growth of L. monocytogenes EGD-e wild-type (WT), ΔsigB, Δrli47 and rli47-complement in chemically defined medium (DM) with and without L-isoleucine. Bacterial growth at 37°C was monitored until all cultures reached stationary phase. The average of three biological replicates is shown, and error bars represent standard deviation. Doubling and lag times are represented in Table S3. (b) Threonine deaminase activity in WT, ΔsigB and Δrli47 cells grown in DM and L-isoleucine depleted medium (DM-ile). Representation of average and standard deviation of two technical replicates of three biological samples per condition. Error bars indicate standard deviation. Asterisks represent p-values (* = p-value <0.05, ** = p-value <0.001, *** = p-value <0.0001) calculated using a Student’s t-test, unpaired with Welch’s correction. (c) Representation of the branched-chain amino acid pathway of isoleucine biosynthesis from L-threonine in L. monocytogenes. Adapted from MetaCyc (22).
To determine if the elevated ilvA mRNA levels found in the ΔsigB and Δrli47 mutant backgrounds resulted in a corresponding effect on the levels of TD, its activity was measured in total protein extracts from L. monocytogenes EGD-e wild-type, ΔsigB and Δrli47 grown to stationary phase in both DM and DM-ile (Figure 6(b)). While low levels of TD were detected in the WT grown in DM, significantly higher activity was recorded in the ΔsigB mutant, but the increase in the Δrli47 was not significant. Isoleucine depletion did not affect TD activity in the WT, but it induced a significant increase of TD activity in both ΔsigB and Δrli47 mutants (p-value = 0.024 and p-value < 0.0001, respectively, for ΔsigB and Δrli47 mutant strains) (Figure 6(b)). Consistent with the growth lag data in DM-ile (Figure 6(a)), the activity of TD was significantly higher in the ΔsigB mutant than in the Δrli47 mutant strain. Altogether, these results strongly suggest that Rli47 plays a role in the impairment of isoleucine biosynthesis through repression of ilvA expression, even in media where this amino acid is absent. They further suggest that additional σB-dependent mechanisms leading to TD repression are also present (Figure 6(c)).
Transcriptomic analysis shows that amino acid transport and metabolism is affected by rli47 deletion
To complement the in silico and in vitro data described above, and investigate the overall impact of Rli47 on gene transcription, a transcriptomic analysis of L. monocytogenes Δrli47 mutant and the parental strain EGD-e was performed using RNA-seq during growth at 37°C in DM, a condition where Rli47 is strongly expressed in the WT (Figure 2). Significant changes in the transcript levels of 155 genes were detected between the two strains (Figure 7(a)). The relative abundance of each COG category in the set of differentially transcribed genes was determined. Overall, genes involved in amino acids transport and metabolism were highly represented in both upregulated and downregulated sets of genes, suggesting a direct and/or indirect role of rli47 in modulating amino acid metabolism (Figure 7(b)). A set of 105 genes showed higher transcript levels in the Δrli47 mutant. The gene with the highest fold change was lmo1634 (Fold-change = 29.9). It encodes a bifunctional acetaldehyde-CoA/alcohol dehydrogenase involved in pyruvate metabolism. Three pyruvate-formate lyase encoding genes were found upregulated as well (pflA, pflC and pflB). Transcripts of genes encoding transcription regulators (lmo2365, lmo2364 and lmo2447), in ferrous iron transport (feoA and feoB), (R)-2-hydroxyglutaryl-CoA dehydratase (yjiL), pheromones (lmo2637), inosol degradation into acetyl-CoA (iolI), anaerobic bacterial respiration (lmo0355), arginine biosynthesis (argG) and valine/isoleucine biosynthesis (ilvD) were all found in the top 20 most highly expressed genes (Table S4 and Figure 7(a)). Thirteen sRNAs were in the set of genes with higher transcript level, of which Rli60 (Fold-change = 2.8) was the only figuring in the top 20 most upregulated genes in the Δrli47 mutant strain. Interestingly, rli60 belongs to the ilv-leu BCAA biosynthesis operon (rli60, ilvD, ilvB, ilvN, ilvC, leuA, leuB, leuC, leuD and ilvA) This sRNA has recently been identified as a riboregulator regulating the transcription of the ilv-leu operon depending on isoleucine levels [12]. Four of the genes (rli60, ilvD, ilvB and ilvN) of this operon were in the set of genes with higher transcript levels (Fold-changes of 2.8, 2.7, 2.00, 1.8, respectively), but the increased levels detected for the last six genes of the operon (ilvC, leuA, leuB, leuC, leuD and ilvA) did not reach the significance threshold (Fold-change = 1.4, 1.3, 1.2, 1.1, 1.1 and 1.2, respectively). Fifty genes including four sRNAs showed lower transcript levels (Table S4). The most downregulated gene in the Δrli47 mutant was copB (Fold-change = −3.4), one of the in silico predicted Rli47 targets (Table S4) and which encodes adenosylcobinamide-phosphate guanylyltransferase, a bifunctional cobalamin biosynthesis protein. Another gene that was significantly downregulated was cbiD (cobalt percorrin-6A synthase), which is also involved in cobalamin biosynthesis. In the top 20 most downregulated were genes encoding proteins involved in late competence (lmo1341), secretion (esaC), propanediol dehydratase reactivation (lmo1157), ABC transporter systems (lmo0861 and lmo0135), glycine cleavage (gcvT and gcvPA), phosphate transport system (lmo2497), uroprophyrinogen-III methyltransferase/synthase (lmo1201) and de novo purine biosynthesis (purH, purN and purM) (Figure 7(a) and Table S4). However, the function of some other genes (lmo2807, lmo2161, lmo0702 and lmo0461) remains unknown as all encode hypothetical proteins. Four sRNAs were in the set of genes with lower transcript levels in the Δrli47 mutant strain. The role of Rli89 is undetermined while SbrA is a highly σB-dependent sRNA that is likely to be involved in the fine-tuning expression of genes involved in stress response, metabolism and virulence [18,19,35]
Figure 7.
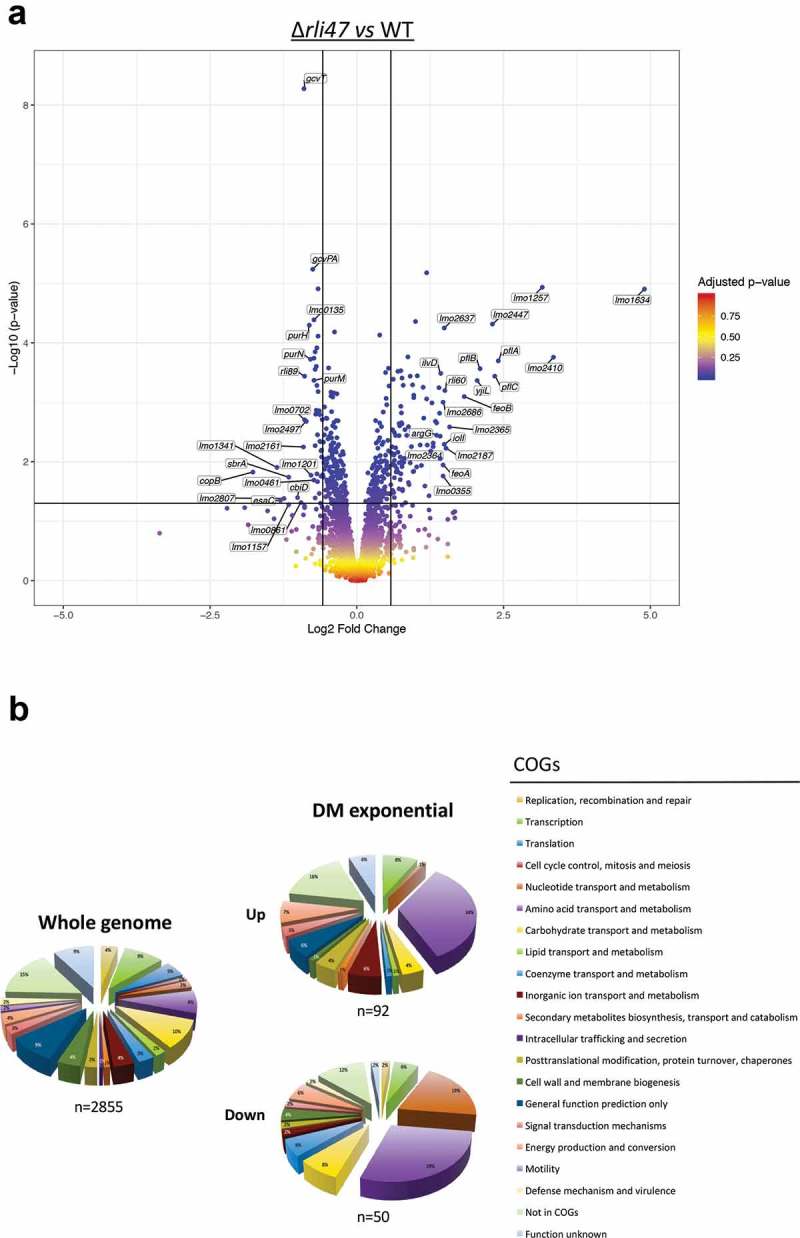
Expression analysis of genes differentially transcribed by L. monocytogenes rli47 deletion mutant grown to exponential phase in defined media, summarizing RNA sequencing results. (a) Volcano plots displaying differentially expressed genes in exponential phase DM. The top 20 most upregulated and downregulated significantly expressed genes are labelled. The negative log10 of p-value is plotted on the Y-axis, and the log2FC is plotted on the X-axis. The red points on this graph represent sRNAs and mRNAs that are significantly differently expressed in the rli47 deletion mutant (p-value <0.05), the blue points represent sRNAs and mRNAs with p-value >0.05. The horizontal line represents the p-value >0.05 cutoff. The horizontal lines represent a − 0.58> log2FC > 0.58 cutoffs. (b) Relative abundance of categories of genes differentially expressed by L. monocytogenes rli47 deletion mutant comparatively to the wild-type EGD-e. Left pie chart, percentage of genes belonging to each COGs in the whole genome of L. monocytogenes EGD-e (NC_003210.1). Right upper pie chart, genes upregulated compared with wild-type; right down pie chart, genes downregulated compared with wild-type. Colours correspond to categories in the COGs database.
Some overlap with AgrA, σB, CodY and PrfA regulons was recorded (Table S5). The overlap with CodY regulon was the largest (n = 42 genes), and most of the overlapping genes belong to the amino acids transport and metabolism functional category. Indeed, in L. monocytogenes CodY controls the transcription of genes involved in metabolism, stress responses, motility and virulence in response to the availability of BCAA [10]. Since isoleucine serves as ligand for modulating CodY activity, deregulation of ilvA expression in the Δrli47 mutant likely accounts for the high proportion of CodY-regulated genes in this RNA-seq dataset.
Discussion
Taken together, the data presented here suggest a model where the regulatory RNA Rli47 exerts tight control of isoleucine biosynthesis by repressing expression of the threonine deaminase gene ilvA (Figure 8). This repression is likely achieved primarily through the repression of translation by occlusion of the ilvA ribosome binding site. There may also be a negative effect of Rli47 on ilvA mRNA levels since there was a significant increase in the ilvA transcript in the Δrli47 mutant strain cells grown in TSB (Fig. S6). This effect was not detected in stationary growth phase cells grown in DM (Figure 5), but the transcriptomic data further suggested a role of Rli47 in negatively affecting ilv-leu operon mRNA levels. Differences in the magnitude of the effect between growth media could be partly caused by differences in the abundance of amino acids or other nutrients in the media. Transcripts of the first four genes of the of the ilv-leu operon, rli60 and ilvDBN, were found at significantly elevated levels in the Δrli47 mutant, suggesting that the levels of the entire ilv-leu operon transcript are lower in the presence of Rli47. It seems unlikely that this effect occurs at the transcriptional level since transcription is controlled by the combined actions of CodY and the transcriptional attenuator Rli60 which is located upstream from ilvD and acts as a ribosome-dependent transcriptional attenuator of the operon [12]. More likely the binding of Rli47 to the SD region of ilvA, negatively affects the overall stability of the mRNA. Thus, the recent model presented by Brenner and co-workers [12] describing the regulation of the ilv-leu operon in L. monocytogenes has acquired an additional layer, with Rli47 providing another level of negative control that operates to restrict isoleucine biosynthesis even when this amino acid is absent from the growth medium.
Figure 8.
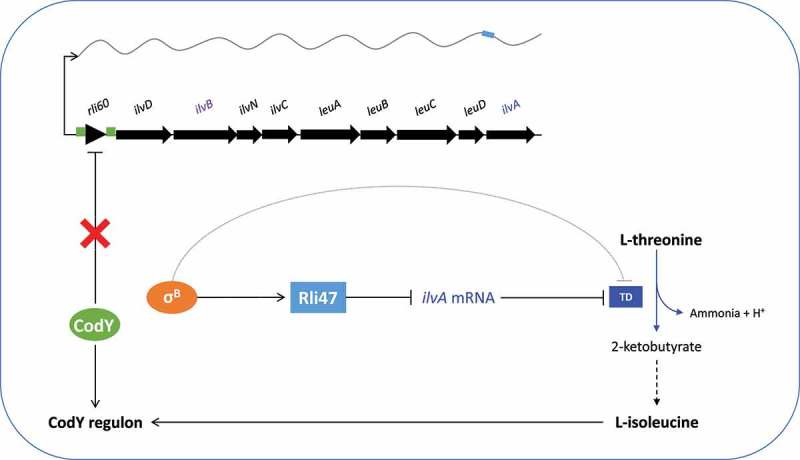
Proposed model of the regulatory effect of Rli47 on isoleucine biosynthesis in L. monocytogenes. Transcription of rli47 is under σB control and when it is expressed it interacts directly with mRNA from the ilv-leu operon at the predicted Shine-Dalgarno sequence upstream from the ilvA start codon. The Rli47-ilv interaction blocks the first step of isoleucine biosynthesis by preventing the translation of ilvA to produce threonine deaminase (TD), and also by affecting the stability of the ilv-leu transcript. σB negatively influences the activity of TD through an Rli47-independent route which remains to be identified. CodY represses transcription of the ilv-leu operon in a manner that depends on the availability of isoleucine (ile) through an interaction with two binding sites, shown in green [10,12]. When σB is active Rli47 influences the CodY regulon through an effect on the cytoplasmic pool of isoleucine.
The question of why L. monocytogenes would repress the biosynthesis of an amino acid it requires for growth is not easily answered. It has been known for many years that L. monocytogenes behaves as a partial auxotroph for BCAA, requiring supplementation of these amino acids into the growth medium in order to achieve optimal growth [12,36,37], despite having the genes required for their biosynthesis [38]. Here we provide evidence suggesting that Rli47 contributes to the repression of the ilv-leu operon, which encodes the genes necessary for BCAA biosynthesis, and show that growth is improved in a defined medium lacking isoleucine when this sRNA is absent (Figure 6). Interestingly, a similar phenotype was reported by Brenner et al. (2018) for L. monocytogenes 10403S when the rli60 was deleted from the genome. It encodes a leader peptide that is rich in ile/val/leu codons which serves to control transcriptional termination in a manner that is dependent on the availability of BCAA in the medium, a response that is mediated by the translation rate of the leader peptide [12]. The authors of this study proposed that this regulation of BCAA might represent a host adaptation strategy, whereby the pathogen could couple virulence gene expression with the levels of BCAA encountered in micro-niches within the host. In this model, the link between BCAA and virulence gene expression is made by CodY which positively regulates PrfA expression in response to low BCAA levels [9,12,39]. While this model is certainly worth exploring further it is not yet clear how an additional layer of negative regulation, mediated by the σB–dependent sRNA Rli47, can be incorporated into this view. One possibility is that the stresses encountered within the host, particularly in the gastrointestinal tract where σB is known to be active [18], trigger this additional layer of negative control. This Rli47-mediated effect could serve as an early stimulus to prime cells for virulence by inducing prfA transcription in response to σB activation, but further studies will be needed to explore this idea fully.
An alternative model that seems worth considering is the possibility that restricted BCAA biosynthesis may serve to limit the growth of L. monocytogenes when it is under stress, thereby enhancing its survival. This possibility is suggested by a number of observations, the first of which is that rli47 transcription is under the control of σB, the regulator of the general stress response [7,40–42], and so it would be unsurprising if it played a role in stress survival. Secondly, a number of studies have highlighted the fact that mutants lacking sigB grow at a faster rate than the WT under some culture conditions [43–46], suggesting that σB acts to limit growth under some conditions. A recent study on the intracellular behaviour of L. monocytogenes reports that a subpopulation can exist in a non-growing state within vacuoles in mammalian cells [47], although the mechanism for this phenomenon has yet to be identified. In several other bacterial species, the presence of dormant persister cells within the population has been shown to contribute to increased antibiotic and stress resistance, and a variety of different mechanisms contribute to this persister state. Indeed, this phenomenon appears to be ubiquitous amongst bacteria although in many cases the underlying mechanisms are unknown [48–50]. Thus, we speculate that tight negative control of BCAA biosynthesis under conditions where cells are subject to stress might serve to block growth and confer an increased resistance that is associated with non-growing cells, even under conditions when the supply of BCAA in the medium is limiting. This effect could contribute to survival in both terrestrial and host-associated niches and is consistent with the finding that this sRNA is present in non-pathogenic species of Listeria.
Interestingly the effect of the Δrli47 deletion on growth in DM with limiting BCAA was apparently confined to the lag phase (Figure 6 and S7). Once cells began to grow the growth rates were not significantly different in this medium (Table S3), whether this sRNA was present or not. Since Rli47 is expressed strongly under conditions where σB is highly active (Figures 2 and S3) [18,19], it is possible that Rli47 serves to limit the exit from lag phase under stressful conditions in order to allow a period of protection and repair prior to renewed growth. The question of how the levels of Rli47 are modulated during this transition from lag to active growth will require further investigation, but it could simply occur when σB activity drops due to the presence of more benign prevailing conditions and subsequent loss of Rli47 through normal RNA degradation processes.
The size of Rli47 and the complexity of its secondary structure points to other possible roles for this sRNA other than its involvement in regulating BCAA biosynthesis. The transcriptomic analysis presented here was undertaken to complement the more targeted approaches that we used to identify binding targets for Rli47. Strikingly one of the potential binding targets predicted in silico, copB, was found to be significantly affected in the Δrli47 mutant, where it was the transcript showing the largest reduction compared to the WT (Figure 7 and Table S4). While the appearance copB in the RNA-seq data set does not provide evidence of a direct interaction between it and Rli47 this certainly remains a distinct possibility. It could be that the in vitro binding conditions used for the EMSA analysis fail to emulate the conditions within the cytoplasm in some crucial aspect, for example, if additional factors such as RNA chaperones are required to permit successful stable interactions, or if the interactions occur very transiently.
In any case, it is interesting to observe that copB and cbiD, both involved in cobalamin (vitamin B12) biosynthesis, as well as genes involved in the B12-dependent pathway for 1,2-propanediol utilisation (iolI and lmo1157) were all significantly affected in the absence of Rli47. These genes are only present in Listeria species that can survive within the gastrointestinal tract, the so-called Listeria sensu stricto species that include both the pathogenic species L. monocytogenes, L. ivanovii [51], and commensal species L. innocua, L. marthii, L. welshimeri and L. seeligeri [52]. Interestingly Rli47 is also confined to the genomes of these species and is not found in the Listeria sensu lato species, a clade that includes species only isolated from non-host-associated environments (data not shown) [52,53]. These observations lead us to speculate that Rli47 may contribute to modulating the expression of genes that are required for survival within the environment of the gastrointestinal tract.
Overall this study identifies a defined role for an sRNA that is known to be under σB control and induced within the mammalian gastrointestinal tract but whose function has remained elusive. It is now one of the small number of sRNAs in L. monocytogenes where a clear function has been established. The finding that it acts to repress BCAA biosynthesis implicates it as a global regulator of transcription in this pathogen because of its influence on isoleucine levels, the ligand that determines the activity of the global regulator CodY. The evolutionary rationale for supressing the biosynthesis of isoleucine remains to be fully determined, but we favour a model that involves growth restriction under harsh conditions, contributing to the survival of this adaptable pathogen in niches both outside and within its mammalian host.
Materials and methods
Bacterial strains, plasmids and primers
In this study, Listeria monocytogenes EGD-e was used as the wild-type strain [38]. The isogenic mutant derivatives of this strain were constructed using standard techniques for DNA manipulation in L. monocytogenes [54]. An rli47 deletion mutant was obtained by allelic replacement of the wild-type gene by homologous recombination using the pMAD shuttle-suicide plasmid as previously described [55]. The gene deletion construct was synthesised by Invitrogen GeneArt Gene Synthesis (Thermo Fisher Scientific). The deletion insert (552 bp) spanning 258 bases upstream of the TSS of rli47 to 209 bases downstream of the hypothetical terminator structure (Fig. S1), containing a deletion of 439 bp (from base 18 to 456) in the rli47 gene, was sub-cloned in the pMAD vector and confirmed by DNA sequencing. Alternatively, a sigB knockout mutant, including a 561 bases deletion (from base 64 to 624) was constructed using the pMAD shuttle plasmid [55], and confirmed by DNA sequencing. Whole-genome sequencing was performed by Ilummina sequencing (MicrobesNG) and genomes analyzed by Breseq [56]. Sequencing indicated additional point mutations in lmo2761 (G1063→T) in the sigB deletion mutant and two in the rli47 deletion mutant, lmo1255 (G1156→A) and lmo2142 (T527→A). A complement of rli47 deletion was constructed by cloning a PCR fragment, spanning approximately 91 bases upstream of the TSS of rli47 to a region lying just downstream of the hypothetical terminator, into the pMK4 vector [57], and transform into Listeria monocytogenes Δrli47 competent cells by electroporation. Primers used for in-frame deletions are listed in Table S1. All plasmids and strains used in this study are listed in Table S2.
Growth conditions
L. monocytogenes was routinely grown at 37°C with aeration in tryptic soy broth (TSB, Conda) as a rich medium or in a chemically defined medium (DM) for L. monocytogenes. Alternatively, strains were grown in brain-heart infusion broth (BHI, Lab M) or Luria-Bertani broth (LB, Sigma Aldrich) supplemented with 50 mM glucose, when stated. DM was prepared as described previously [58]. When appropriate, cultures were supplemented with chloramphenicol (10 μg ml−1), ampicillin (100 μg ml−1), kanamycin (50 μg ml−1), erythromycin (5 μg ml−1) or L-threonine (0.1 μg ml−1). For growth under limiting concentrations of isoleucine (ile), DM was freshly made with 10-fold less of isoleucine (resulting in a final concentration of 10 μg ml−1 or 80 μM), 100-fold less of isoleucine (resulting in a final concentration of 1 μg ml−1 or 8 μM), or completely depleted of isoleucine. For growth curves, bacteria from overnight DM cultures were washed 3 times with PBS and adjusted to OD600 of 0.05 in fresh DM without ile or supplemented with 100, 10 or 1 μg ml−1 of ile (800, 80 or 8 μM ile, respectively). OD600 measurements were taken every 2 h. Lag and Doubling times were calculated by GrowthRates 3.0 [59].
Primer extension
Primer extension experiments were performed as previously described [60]. A ΔsigB strain was used as a negative control, given the σB-dependency of rli47 transcription [17], in opposite to a ΔagrA strain where Rli47 was expected to be highly expressed [21]. RNA was extracted from stationary phase cells grown at 37°C in both complex media BHI and LB + Glucose.
In silico predictions
The IntaRNA software [32–34] was used for predicting interactions between target mRNAs and Rli47. RNA–RNA interaction search was performed using the full-length Rli47 sRNA sequence as a query against the RNA sequences from L. monocytogenes EGD-e genome (NC_003210). The following levels of stringency were applied; (1) sequences spanning 75 nt upstream to 75 nt downstream the start codon of target genes, (2) one (sub)optimal interaction output overlap in query, (3) minimum of 7 basepairs in seed, (4) output including only interactions with a delta energy ≤100 kcal mol−1, (5) folding computational temperature of 37°C with the (6) energy parameter set (Vienna package) Turner model, 2004. To narrow the list to the most plausible targets, the following criteria were applied to the predicted binding regions: (1) the interaction should involve a single-stranded region of the sRNA; (2) it should include the SD sequence of the target mRNA; and (3) the interaction should have a binding energy greater than −16 kJ mol−1. The secondary structure prediction of Rli47 sRNA folding form was obtained using mfold [61]. A folding temperature of 37°C and a flat exterior loop type criterion were applied to the default parameters.
Modelling of rli47 secondary structure in vivo
The data on dimethyl sulphate (DMS) reactivity of Rli47 RNA were imported from NCBI’s Gene Expression Omnibus (GEO Series accession number GSE118387). In that study, DMS reactivity of L. monocytogenes non-coding RNAs was probed using the DMS-MaPseq protocol during mid-exponential growth at 37°C in BHI medium [62]. DMS methylates unpaired adenines (A) or cytosines (C) but not A or C engaged in pairing. In brief, DMS (3%) was added to growing cells for 3 min before quenching and RNA-preparation. cDNA of the RNA were synthesized using TGIRTIII reverse transcriptase which incorporates a random base if encountering a methylated A or C. After sequencing and aligning of the cDNA, any mismatch from the reference sequence was indicative of an unpaired A or C in the RNA at the time of DMS addition. The DMS values represent the normalized measure of DMS reactivity of adenines (A) and cytosines (C) and thus reflect their base-pairing status. A or C engaged in base-pairing have low DMS values (no methylation of A or C), whereas unpaired A or C have high DMS values since such bases are accessible to DMS. The secondary structure of Rli47 small RNA was predicted with RNA fold algorithm [63] and manually corrected to minimize discrepancy between DMS values and base-pairing. The model of Rli47 secondary structure and DMS values of its nucleotides were visualized with VARNA applet [64].
RNA extractions
For primer extension and northern blot analysis, L. monocytogenes was grown to exponential or stationary phases. Cultures where then split, stressed with the indicated stressor concentration, and samples were taken (10 ml) at the indicated time points. Cells were harvested by centrifugation at 11,000 x rcf for 3 min at 4°C, and snap-frozen in liquid nitrogen. Cells were disrupted by the FastPrep-24 instrument (MP Biomedicals). Total RNA was extracted using TRI Reagent (Molecular Research Center, Inc.) as previously described [65]. Alternatively, total RNA was extracted using the RNeasy Mini Kit (QIAGEN). The integrity, concentration and purity of the RNA were confirmed by agarose gel electrophoresis, NanoDrop 2000 and Bioanalyzer 2100 (Agilent).
Northern blot
Total RNA (10 μg) was resolved on a 6% or 8% polyacrylamide 8 M urea gel as previously described [65]; alternatively, 20 μg of total RNA were separated on a formaldehyde agarose gel for 3 h and 15 min prior to capillarity blotting on a Zeta-Probe membrane (Bio-Rad) [66]. Membranes were hybridized with 32P-labeled DNA single-stranded probes listed in Table S1. RNA bands were visualized using a Typhoon FLA9000 (GE Healthcare) and analyzed with IQTL 8.0 quantification software (GE Healthcare). For mapping the 3ʹ-end of Rli47, three probes were used. The first probe used annealed in the middle of Rli47 (Rli47_NB), the second one Rli47(2)_NB annealed before the predicted stop site, and the third probe Rli47(3)_NB annealed after this predicted stop site.
RNA integrity, quantification and DNase I treatment
Three independent biological replicates of RNA samples extracted from L. monocytogenes EGDe and Δrli47 were stored at −80°C before treatment. The quality of RNA was assessed using Labchip GX II bioanalyzer (Perkin Elmer). To remove DNA contamination, total RNA was incubated with Baseline-Zero DNase (Epicentre) in the presence of RiboLock RNase inhibitor (40 U µl−1) (Thermo Fisher Scientific) for 30 min at 37°C followed by purification using Zymo-Spin column (ZymoResearch). Briefly, sample was mixed with 2 volumes of RNA-Binding Buffer and added to an equal volume of ≥99.8% ethanol. The mixture was vortexed and transferred to Zymo-Spin™ IC Column and centrifuged at 12,000 x g. The RNA bound to column was washed twice with RNA Wash Buffer then RNA was eluted in DNase/RNase free water. Concentration of RNA was measured using fluorescence-based Qubit™ RNA HS Assay (Thermo Fisher Scientific).
Ribosomal RNA depletion and library preparation
To enrich mRNA and remove ribosomal RNA (rRNA) from total RNA, total RNA was treated with Ribo-Zero rRNA removal kit (Illumina). Briefly, beads were washed twice and hybridized with probes at 68°C for 10 min. Total 500 ng RNA was added to the mixture and incubated at RT and 50°C for 5 min each followed by separation of mRNA from rRNA which was bound to the beads using magnetic stand. Enriched mRNA was purified by Zymo-Spin column (ZymoResearch) and run on Labchip GX II bioanalyzer (Perkin Elmer) to confirm depletion of rRNA. Preparation of cDNA fragment libraries was performed using the NEBNext® Ultra™ II Directional RNA Library Prep Kit for Illumina® (Illumina) with slight modifications. Briefly, the enriched mRNA was fragmented for 15 min at 94°C and reverse transcribed to synthesize the first-strand cDNA followed by second strand cDNA synthesis. Double-stranded cDNA (ds cDNA) was purified using NucleoMag (Macherey nagel) SPRI selection. End repair was performed on the ds cDNA library followed by ligation of adaptors. After purification using NucleoMag SPRI beads, test RT-qPCR (Applied Biosystems) was performed using KAPA Hifi polymerase (Roche) with EvaGreen® (Biotium) to determine appropriate cycle numbers for PCR. Using NEBNext Multiplex Oligos for Illumina (Dual Index Primers), high fidelity PCR was performed using KAPA Hifi polymerase to selectively enrich library fragments. The PCR products were purified twice using NucleoMag (Macherey nagel) SPRI beads and the quality of the final library was assessed on Labchip GX II bioanalyzer (Perkin Elmer).
RNA sequencing and data analysis
Indexed and purified libraries were loaded together onto a flow cell, sequencing was carried out on the Illumina NextSeq 500 platform (paired-end, 2 × 75 bp per read). Sequencing quality was assessed using FastQC, and Illumina adapter sequences and low-quality base pairs were removed using cutadapt version 1.9 [67]. Reads were mapped to the complete sequenced genome of reference strain EGD-e (ENSEMBL ASM19603v1) using Bowtie 2 v2.2.4 with standard parameters and sensitive-local [68]. BAM alignment files were used as input for read counting using htseq-count version 0.6.0. Differential expression (DE) analyses were performed using DESeq2 in R v3.2.2 [69], and the DE was reported as log2 fold changes. p-values were adjusted by the DESeq2 default Benjamini-Hochberg (BH) adjustment method and genes with a >2-fold (>1 log2) change in expression and an adjusted p-value <0.05 were considered as DE. The transcriptomic data have been deposited on the Sequence Read Archive (SRA) database and is accessible through the SRA accession SUB5067488. Volcano plots were drawn by R software, with the integration of the ggplot2 and ggrepel graphical packages for data analysis [70]. The top 20 most significantly upregulated and downregulated expressed genes in the deletion mutant were labelled. The negative log10 of p-value was plotted on the Y-axis, and the log2FC was plotted on the X-axis. The blue points on this graph represent sRNAs and mRNAs that are significantly differently expressed in the rli47 deletion mutant (p-value <0.05), the red points represent sRNAs and mRNAs with p-value >0.05. The horizontal line represents a p-value >0.05 cutoff. The horizontal lines represent a − 0.58> log2FC > 0.58 cutoff. Genes were categorized into Cluster Orthologous Groups (COGs) and pie charts were plotted representing the transcript levels of up- and downregulated genes grouped by COGs.
CDNA synthesis and reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR)
Fifty micrograms of total RNA were DNase-treated with TURBO DNA-free kit, according to the manufacturer’s instructions (InvitrogenTM by Thermo Fischer Scientific). First-strand cDNA from purified total RNA was synthesized using SuperScript™ IV First-Strand Synthesis System (InvitrogenTM by Thermo Fischer Scientific) according to the manufacturer’s instructions. The cDNA quantity was determined by Qubit™ fluorometer (Invitrogen™ by Thermo Fischer Scientific) following manufacturer’s recommendations. RT-qPCR was performed using the QuantiTectTM SYBR Green PCR Kit (Qiagen) and specific primer sets for the genes of interest (Table S1). Primers for rli47, ilvA and 16s genes were tested with genomic DNA prior to analysis. Three sets of cDNA were analyzed, originating from three sets of RNA extracted as described above. Primers efficiency was calculated prior to sample runs, in the same system. The relative expression ratio was used to analyze RT-qPCR results. The samples were run on a LightCycler® 480 System (Roche) with an initial step at 95°C for 15 min, 45 cycles of 15 s at 95°C, 15 s at 53°C and 30 s at 72°C, a melting curve was drawn for 5 s at 95°C, 1 min at 55°C followed by increases of 0.11°C s−1 until 95°C, and a cooling for 30 s at 40°C. Cycle quantification values were calculated by the software LightCycler® 480 Software version 1.5.1 (Roche) and the Pfaffl relative expression formula [71,72]. The 16s rDNA gene served as a reference gene. The experiment was carried out in three biological replicates, each in technical triplicates.
Electrophoretic mobility shift assays (EMSAs)
Templates for in vitro transcription carried a T7 RNA polymerase binding site at their 5′-end and were generated by PCR, as described in Lillebæk and Kallipolitis (2018) [73]. Templates for ilvA were obtained by PCR from chromosomal DNA and sRNA transcripts were made using overlapping primers (Table S1). In vitro transcription, RNA purification, de-phosphorylation and labelling were performed as described as previously described [65]. Briefly, 40 fmol of 5′-end-labelled RNA were incubated with a molar excess of unlabeled RNA in a total volume of 10 μL in the presence of non-specific competitor (tRNA) for 1 h at 37°C followed by 10 min on ice. Samples were separated on a 5% non-denaturing gel at 4°C. RNA bands were visualized and analyzed as described for the northern blotting experiments.
Protein extraction and TD colorimetric assay
Ten μg ml−1 chloramphenicol were added to each of the 25 ml cell cultures at stationary growth phase. Samples were centrifuged for 15 min at 4°C at 9,000 x g and resuspended in 2 ml sonication buffer (10 mM Tris- HCl, 0.1 mM EDTA and 5 mM MgCl2, prepared in dH2O and autoclaved, pH 7.4) with 2 mg ml−1 lysozyme (Sigma-Aldrich). Suspensions were incubated at 37°C for 10 min before centrifugation at 9,000 x g for 15 min at 4°C. Pellets were resuspended in 0.3 ml sonication buffer containing 1% (v/v) protease inhibitor cocktail (P2714, Sigma-Aldrich). Cell disruption was performed by FastPrep-24 (MP Biomedicals). Samples were centrifuged at 13,000 x g for 30 min to remove cell debris. Protein extracts were quantified by Bradford Protein assay (Bio-Rad).
Threonine deaminase colorimetric assay
The activity of TD was assayed using the method of Harris (1981) [74] with some modifications. Eight hundred μl of assay buffer consisting of 0.1 M Tris-HCl (pH 8.0), 0.1 M NH4Cl, 0.1 mM pyridoxal phosphate and 50 mM L-threonine were placed into each 15 ml tube. One hundred microlitres of cell extract was added to the tubes and incubated in a water bath at 37°C for 20 min. One hundred microlitres of 50% [v/v] trichloroacetic acid was then added, followed by 3 ml of 0.025% [w/v] 2,4. dinitrophenylhydrazine (Sigma-Aldrich) in 0.5 M HCl. After 15 min incubation at 25°C, 1 ml of 10 M NaOH was added. Absorbance at 540 nm was then measured and recorded for each sample. The amount of product formed was determined from a standard curve prepared using known concentrations of α-ketobutyrate (Sigma – Aldrich).
Funding Statement
This work was supported by the European Union’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreements No. 641984 and No. 721456..
Acknowledgments
The authors wish to acknowledge members of the Bacterial Stress Response Group for helpful discussions and ideas, and BoHyung Lee for technical support.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed here.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- [1].Gray MJ, Freitag NE, Boor KJ.. How the bacterial pathogen Listeria monocytogenes mediates the switch from environmental Dr. Jekyll to pathogenic Mr. Hyde. Infect Immun. 2006;74(5):2505–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cossart P, Lebreton A. A trip in the “New Microbiology” with the bacterial pathogen Listeria monocytogenes. FEBS Lett. 2014;588(15):2437–2445. [DOI] [PubMed] [Google Scholar]
- [3].NicAogain K, O’Byrne CP. The role of stress and stress adaptations in determining the fate of the bacterial pathogen Listeria monocytogenes in the food chain. Front Microbiol. 2016;7:1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].PoBHB EFSA. Listeria monocytogenes contamination of ready-to-eat foods and the risk for human health in the EU. Efsa J. 2017;16:(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].CDC Listeria (Listeriosis). 2018. [cited 2019 Jan 10]. Available from: https://www.cdc.gov/listeria/index.html
- [6].WHO Listeriosis, fact sheet https://www.who.int/mediacentre/factsheets/listeriosis/en/: WHO (World Health Organization); 2018. [cited 2019 Jan 10]. Available from: https://www.who.int/mediacentre/factsheets/listeriosis/en/.
- [7].O’Byrne CP, Karatzas KA. The role of sigma B (sigma B) in the stress adaptations of Listeria monocytogenes: overlaps between stress adaptation and virulence. Adv Appl Microbiol. 2008;65:115–140. [DOI] [PubMed] [Google Scholar]
- [8].Freitag NE, Port GC, Miner MD. Listeria monocytogenes – from saprophyte to intracellular pathogen. Nat Rev Microbiol. 2009;7(9):623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lobel L, Sigal N, Borovok I, et al. Integrative genomic analysis identifies isoleucine and CodY as regulators of Listeria monocytogenes virulence. PLoS Genet. 2012;8(9):e1002887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lobel L, Herskovits AA. Systems level analyses reveal multiple regulatory activities of cody controlling metabolism, motility and virulence in Listeria monocytogenes. PLoS Genet. 2016;12(2):e1005870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sonenshein AL. CodY, a global regulator of stationary phase and virulence in Gram-positive bacteria. Curr Opin Microbiol. 2005;8(2):203–207. [DOI] [PubMed] [Google Scholar]
- [12].Brenner M, Lobel L, Borovok I, et al. Controlled branched-chain amino acids auxotrophy in Listeria monocytogenes allows isoleucine to serve as a host signal and virulence effector. PLoS Genet. 2018;14(3):e1007283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thorsing M, Dos Santos PT, Kallipolitis BH. Small RNAs in major foodborne pathogens: from novel regulatory activities to future applications. Curr Opin Biotechnol. 2018;49:120–128. [DOI] [PubMed] [Google Scholar]
- [14].Oliva G, Sahr T, Small BC. RNAs, 5ʹ UTR elements and RNA-binding proteins in intracellular bacteria: impact on metabolism and virulence. FEMS Microbiol Rev. 2015;39(3):331–349. [DOI] [PubMed] [Google Scholar]
- [15].Becavin C, Koutero M, Tchitchek N, et al. Listeriomics: an interactive web platform for systems biology of Listeria. mSystems. 2017;2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dorey A, Marinho C, Piveteau P, et al. Role and regulation of the stress activated sigma factor sigma B (σB) in the saprophytic and host-associated life stages of Listeria monocytogenes. Adv Appl Microbiol. 2019;106:1–48. [DOI] [PubMed] [Google Scholar]
- [17].Mraheil MA, Billion A, Mohamed W, et al. The intracellular sRNA transcriptome of Listeria monocytogenes during growth in macrophages. Nucleic Acids Res. 2011;39(10):4235–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Toledo-Arana A, Dussurget O, Nikitas G, et al. The Listeria transcriptional landscape from saprophytism to virulence. Nature. 2009;459(7249):950–956. [DOI] [PubMed] [Google Scholar]
- [19].Oliver HF, Orsi RH, Ponnala L, et al. Deep RNA sequencing of L. monocytogenes reveals overlapping and extensive stationary phase and sigma B-dependent transcriptomes, including multiple highly transcribed noncoding RNAs. BMC Genomics. 2009;10:641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mujahid S, Bergholz TM, Oliver HF, et al. Exploration of the role of the non-coding RNA SbrE in L. monocytogenes stress response. Int J Mol Sci. 2012;14(1):378–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vivant AL, Garmyn D, Gal L, et al. Survival of Listeria monocytogenes in soil requires AgrA-mediated regulation. Appl Environ Microbiol. 2015;81(15):5073–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Caspi R, Altman T, Billington R, et al. The metacyc database of metabolic pathways and enzymes and the bioCyc collection of pathway/genome databases. Nucleic Acids Res. 2014;42(Databaseissue):D459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yu X, Li Y, Wang X. Molecular evolution of threonine dehydratase in bacteria. PLoS One. 2013;8(12):e80750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].NCBI BLASTn: national center for biotechnology information; 2018. [cited 2016 Jan 06]. Available from: https://blast.ncbi.nlm.nih.gov/.
- [25].Wurtzel O, Sesto N, Mellin JR, et al. Comparative transcriptomics of pathogenic and non-pathogenic Listeria species. Mol Syst Biol. 2012;8:583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Geissmann T, Marzi S, Romby P. The role of mRNA structure in translational control in bacteria. RNA Biol. 2009;6(2):153–160. [DOI] [PubMed] [Google Scholar]
- [27].Chevalier C, Boisset S, Romilly C, et al. Staphylococcus aureus RNAIII binds to two distant regions of coa mRNA to arrest translation and promote mRNA degradation. PLoS Pathog. 2010;6(3):e1000809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Miller EW, Cao TN, Pflughoeft KJ, et al. RNA-mediated regulation in Gram-positive pathogens: an overview punctuated with examples from the group A Streptococcus. Mol Microbiol. 2014;94(1):9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Patenge N, Pappesch R, Khani A, et al. Genome-wide analyses of small non-coding RNAs in streptococci. Front Genet. 2015;6:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mollerup MS, Ross JA, Helfer AC, et al. Two novel members of the LhrC family of small RNAs in Listeria monocytogenes with overlapping regulatory functions but distinctive expression profiles. RNA Biol. 2016;13(9):895–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dos Santos PT, Menendez-Gil P, Sabharwal D, et al. The small regulatory RNAs LhrC1-5 contribute to the response of Listeria monocytogenes to heme toxicity. Front Microbiol. 2018;9:599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wright PR, Georg J, Mann M, et al. CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains. Nucleic Acids Res. 2014;42(WebServer issue):W119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mann M, Wright PR, Backofen R. IntaRNA 2.0: enhanced and customizable prediction of RNA-RNA interactions. Nucleic Acids Res. 2017;45(W1):W435–W9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Busch A, Richter AS, Backofen R. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics. 2008;24(24):2849–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nielsen JS, Olsen AS, Bonde M, et al. Identification of a sigma B-dependent small noncoding RNA in Listeria monocytogenes. J Bacteriol. 2008;190(18):6264–6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Joseph B, Goebel W. Life of Listeria monocytogenes in the host cells’ cytosol. Microbes Infect. 2007;9(10):1188–1195. [DOI] [PubMed] [Google Scholar]
- [37].Premaratne RJ, Lin WJ, Johnson EA. Development of an improved chemically defined minimal medium for Listeria monocytogenes. Appl Environ Microbiol. 1991;57(10):3046–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Glaser P, Frangeul L, Buchrieser C, et al. Comparative genomics of Listeria species. Science. 2001;294(5543):849–852. [DOI] [PubMed] [Google Scholar]
- [39].Lobel L, Sigal N, Borovok I, et al. The metabolic regulator CodY links Listeria monocytogenes metabolism to virulence by directly activating the virulence regulatory gene prfA. Mol Microbiol. 2015;95(4):624–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Becker LA, Cetin MS, Hutkins RW, et al. Identification of the gene encoding the alternative sigma factor sigmaB from Listeria monocytogenes and its role in osmotolerance. J Bacteriol. 1998;180(17):4547–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wiedmann M, Arvik TJ, Hurley RJ, et al. General stress transcription factor sigmaB and its role in acid tolerance and virulence of Listeria monocytogenes. J Bacteriol. 1998;180(14):3650–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hecker M, Pane-Farre J, Volker U. SigB-dependent general stress response in Bacillus subtilis and related gram-positive bacteria. Annu Rev Microbiol. 2007;61:215–236. [DOI] [PubMed] [Google Scholar]
- [43].Chaturongakul S, Boor KJ. RsbT and RsbV contribute to sigmaB-dependent survival under environmental, energy, and intracellular stress conditions in Listeria monocytogenes. Appl Environ Microbiol. 2004;70(9):5349–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Brondsted L, Kallipolitis BH, Ingmer H, et al. kdpE and a putative RsbQ homologue contribute to growth of Listeria monocytogenes at high osmolarity and low temperature. FEMS Microbiol Lett. 2003;219(2):233–239. [DOI] [PubMed] [Google Scholar]
- [45].Abram F, Starr E, Karatzas KA, et al. Identification of components of the sigma B regulon in Listeria monocytogenes that contribute to acid and salt tolerance. Appl Environ Microbiol. 2008;74(22):6848–6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].O’Donoghue B, NicAogain K, Bennett C, et al Blue-light inhibition of Listeria monocytogenes growth is mediated by reactive oxygen species and is influenced by sigmab and the blue-light sensor Lmo0799. Appl Environ Microbiol. 2016;82(13):4017–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bierne H, Milohanic E, Kortebi M. To be cytosolic or vacuolar: the double life of Listeria monocytogenes. Front Cell Infect Microbiol. 2018;8:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lewis K. Persister cells. Annu Rev Microbiol. 2010;64:357–372. [DOI] [PubMed] [Google Scholar]
- [49].Fisher RA, Gollan B, Helaine S. Persistent bacterial infections and persister cells. Nat Rev Microbiol. 2017;15(8):453–464. [DOI] [PubMed] [Google Scholar]
- [50].Maisonneuve E, Gerdes K. Molecular mechanisms underlying bacterial persisters. Cell. 2014;157(3):539–548. [DOI] [PubMed] [Google Scholar]
- [51].Seeliger HP. Modern taxonomy of the Listeria group relationship to its pathogenicity. Clin Invest Med. 1984;7(4):217–221. [PubMed] [Google Scholar]
- [52].Chiara M, Caruso M, D’Erchia AM, et al. Comparative genomics of Listeria Sensu Lato: genus-wide differences in evolutionary dynamics and the progressive gain of complex, potentially pathogenicity-related traits through lateral gene transfer. Genome Biol Evol. 2015;7(8):2154–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Schardt J, Jones G, Muller-Herbst S, et al. Comparison between Listeria sensu stricto and Listeria sensu lato strains identifies novel determinants involved in infection. Sci Rep. 2017;7(1):17821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Monk IR, Gahan CG, Hill C. Tools for functional postgenomic analysis of Listeria monocytogenes. Appl Environ Microbiol. 2008;74(13):3921–3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Arnaud M, Chastanet A, Debarbouille M. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol. 2004;70(11):6887–6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Deatherage DE, Barrick JE. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol. 2014;1151:165–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sullivan MA, Yasbin RE, Young FE. New shuttle vectors for Bacillus subtilis and Escherichia coli which allow rapid detection of inserted fragments. Gene. 1984;29(1–2):21–26. [DOI] [PubMed] [Google Scholar]
- [58].Amezaga MR, Davidson I, McLaggan D, et al. The role of peptide metabolism in the growth of Listeria monocytogenes ATCC 23074 at high osmolarity. Microbiology. 1995;141(Pt 1):41–49. [DOI] [PubMed] [Google Scholar]
- [59].Hall BG, Acar H, Nandipati A, et al. Growth rates made easy. Mol Biol Evol. 2014;31(1):323–328. [DOI] [PubMed] [Google Scholar]
- [60].Christiansen JK, Larsen MH, Ingmer H, et al. The RNA-binding protein Hfq of Listeria monocytogenes: role in stress tolerance and virulence. J Bacteriol. 2004;186(11):3355–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ignatov D, Liu X, Kallipolitis BH, et al. Comparative analysis of RNA structures reveals an mRNA-mRNA interaction controlling listeria virulence factor expression; 2018. [cited 2018 Nov 16]. Available from: https://ssrncom/abstract=3238688
- [63].Lorenz R, Bernhart SH, Honer Zu Siederdissen C, et al. ViennaRNA Package 2.0. Algor Mol Biol. 2011;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Darty K, Denise A, VARNA: PY. Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009;25(15):1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Nielsen JS, Lei LK, Ebersbach T, et al. Defining a role for Hfq in Gram-positive bacteria: evidence for Hfq-dependent antisense regulation in Listeria monocytogenes. Nucleic Acids Res. 2010;38(3):907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sheehan B, Klarsfeld A, Msadek T, et al. Differential activation of virulence gene expression by PrfA, the Listeria monocytogenes virulence regulator. J Bacteriol. 1995;177(22):6469–6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Martin M. Cutadapt removes adapter sequences from high-thoughput sequencing reads. EMBnet J. 2011;17:1. [Google Scholar]
- [68].Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag; 2016. [Google Scholar]
- [71].Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Pfaffl MW, Georgieva TM, Georgiev IP, et al. Real-time RT-PCR quantification of insulin-like growth factor (IGF)-1, IGF-1 receptor, IGF-2, IGF-2 receptor, insulin receptor, growth hormone receptor, IGF-binding proteins 1, 2 and 3 in the bovine species. Domest Anim Endocrinol. 2002;22(2):91–102. [DOI] [PubMed] [Google Scholar]
- [73].Lillebæk EMS, Kallipolitis BH. Mutational analysis of sRNA–mRNA base pairing by electrophoretic mobility shift assay. In bacterial regulatory RNA: methods and protocols. Methods Mol Biol. 2018;1737:165–176. [DOI] [PubMed] [Google Scholar]
- [74].Harris CL. Cysteine and growth inhibition of Escherichia coli: threonine deaminase as the target enzyme. J Bacteriol. 1981;145(2):1031–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Rieu A, Weidmann S, Garmyn D, et al. Agr system of Listeria monocytogenes EGD-e: role in adherence and differential expression pattern. Appl Environ Microbiol. 2007;73(19):6125–6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Garmyn D, Augagneur Y, Gal L, et al. Listeria monocytogenes differential transcriptome analysis reveals temperature-dependent Agr regulation and suggests overlaps with other regulons. PLoS One. 2012;7(9):e43154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Hain T, Hossain H, Chatterjee SS, et al. Temporal transcriptomic analysis of the Listeria monocytogenes EGD-e sigmaB regulon. BMC Microbiol. 2008;8:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bennett HJ, Pearce DM, Glenn S, et al. Characterization of relA and codY mutants of Listeria monocytogenes: identification of the CodY regulon and its role in virulence. Mol Microbiol. 2007;63(5):1453–1467. [DOI] [PubMed] [Google Scholar]
- [79].Milohanic E, Glaser P, Coppee JY, et al. Transcriptome analysis of Listeria monocytogenes identifies three groups of genes differently regulated by PrfA. Mol Microbiol. 2003;47(6):1613–1625. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- CDC Listeria (Listeriosis). 2018. [cited 2019 Jan 10]. Available from: https://www.cdc.gov/listeria/index.html