

ABSTRACT
The X-linked CDKL5 gene codes for a kinase whose mutations have been associated with a suite of neurodevelopmental disorders generally characterized by early-onset epileptic encephalopathy and severe intellectual disability. The impact of these mutations on CDKL5 functions and brain development remain mainly unknown, although the importance of maintaining the catalytic activity is generally recognized. Since no cure exists for CDKL5 disorders, the demand for innovative therapies is a real emergency. The recent discovery that CDKL5 is dosage sensitive poses concerns on conventional protein and gene augmentative therapies. Thus, RNA-based therapeutic approaches might be preferred. We studied the efficacy of read-through therapy on CDKL5 premature termination codons (PTCs) that correspond roughly to 15% of all mutations. Our results provide the first demonstration that all tested CDKL5 nonsense mutations are efficiently suppressed by aminoglycoside drugs. The functional characterization of the restored full-length CDKL5 reveals that read-through proteins fully recover their subcellular localization, but only partially rescue their catalytic activity. Since read-through can cause amino acid substitution, CDKL5 patients carrying the PTC outside the catalytic domain might benefit more from a nonsense suppression therapy. Eventually, we demonstrate that non-aminoglycoside drugs, such as Ataluren (PTC124) and GJ072, are unable to induce read-through activity on CDKL5 PTCs. Although these drugs might be more effective in vivo, these results question the validity of the Ataluren phase 2 clinical trial that is currently ongoing on CDKL5 patients.
KEYWORDS: CDKL5, encephalopathy, RNA-based therapy, read-through therapy, catalytic activity, aminoglycoside drugs, PTC124, nonsense mutations, PTC
Introduction
Mutations in the X-linked cyclin dependent kinase-like gene (CDKL5) cause a broad spectrum of neuropsychiatric disorders that affect both genders and generally share the common features of severe intellectual disability and early drug-resistant epilepsy, emerging in the first months of life [1].
The CDKL5 gene codes for a serine/threonine kinase with an N-terminal catalytic domain, homologous to that of MAP kinases, and a long C-terminus involved in regulating its catalytic activity and interaction with protein partners [2].
Alternative splicing, which influences mainly the C-terminal region of CDKL5 mRNA, leads to several isoforms; among these, CDKL5_1 is formed by 960 residues and represents the most abundant isoform both in human and mouse [3] (Figure 1a). As main functional features, all CDKL5 isoforms present the ATP binding region and the serine/threonine protein kinase active site (amino acids 19–43 and 131–143, respectively), a TEY motif (amino acids 169–171) whose dual phosphorylation on threonine and tyrosine plays an activating function, and putative nuclear import (NLS) and export (NES) signals located in the C-terminus (Figure 1a). Although the cellular functions of CDKL5 are not fully known and we are still searching for its main role(s) in the brain and how its mutations disrupt neuronal and synaptic functions, we recognize the importance of CDKL5 enzymatic activity. Indeed, the analysis of the distribution of pathogenic missense mutations reveals that they practically all localize in the catalytic domain, while premature stop codons (PTCs) and other truncating mutations can be located anywhere in the gene [2]. Of relevance, most of CDKL5 pathogenic mutations are considered loss of functions; however, the existence of a CDKL5 duplication syndrome suggests that this gene is dosage sensitive [4].
Figure 1.
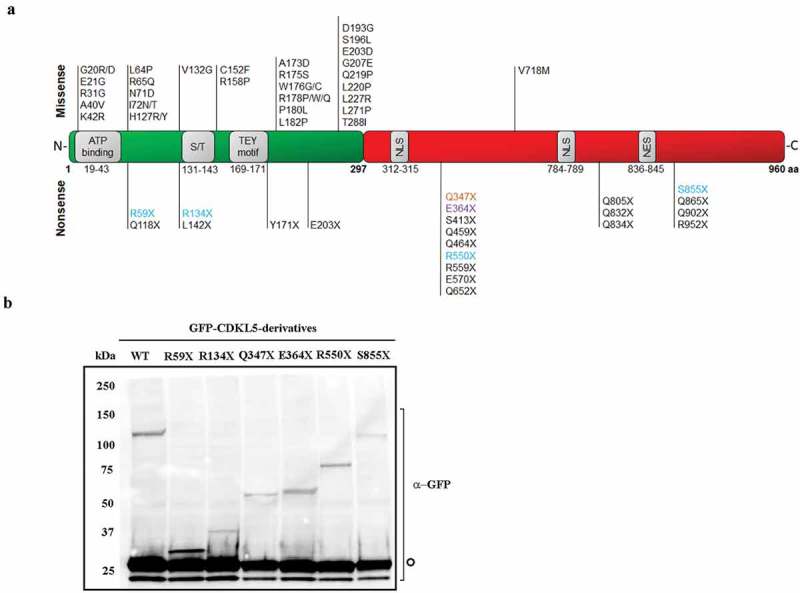
(a) Schematic representation of CDKL5 isoform_1. The catalytic domain is shown in green and the C-terminal domain is in red; functional domains are in grey (ATB binding: ATP binding site; S/T: Ser/Thr kinase active site; TEY motif: Thr/Glu/Tyr motif; NLS: nuclear localization signal; NES: nuclear export signal). Missense pathogenic mutations are indicated above the protein, while nonsense mutations are reported below. PTCs that have been selected for this study are highlighted and distinguished by colour depending on the nonsense codon (UGA: light blue; UAG: orange; UAA: purple). (b) Western blot showing the expression of full length (WT) or truncated CDKL5 of the expected molecular weight in transfected HEK293T cells. The signal of co-transfected GFP served as transfection control in all samples (indicated by an empty circle).
At present, no cure exists for patients with CDKL5 disorder. However, research on Cdkl5-null mice has identified few altered molecular pathways whose restoration improved the animal conditions [5–8], therefore, suggesting the possibility of a pharmaceutical intervention.
While expecting for rationally designed therapies, we found that among pathogenic or likely pathogenic CDKL5 variants (deduced from http://mecp2.chw.edu.au), 15% of patients harbours nonsense mutations and, therefore, might benefit from the treatment with read-through drugs [9]. Indeed, aminoglycosides and other non-aminoglycoside drugs can induce improper recognition of the premature stop codon, favouring the recruitment of near-cognate tRNAs in place of the termination complex. The degree of this ‘mis-reading’ depends upon the sequence of the stop codon itself and the surrounding nucleotide sequence. Importantly, since these drugs do not induce read-through of natural termination codons, their therapeutic potential has been already investigated in preclinical mouse models of genetic diseases and in patients mainly affected by cystic fibrosis and Duchenne muscular dystrophy [10–15]. Of relevance, a phase 2 clinical trial of Ataluren, a non-aminoglycoside drug, has recently started for the treatment of nonsense mutations in CDKL5 deficiency (https://clinicaltrials.gov/ct2/show/NCT02758626). However, to the best of our knowledge, there is no experimental data supporting the clinical investigation. Therefore, we found it important to investigate whether CDKL5 PTCs are sensitive to read-through therapy and to identify the most responsive mutations and/or the most effective drugs. Furthermore, since the amino acid incorporated at the PTC might not necessarily be the authentic one, we compared the activity and subcellular distribution of the read-through products with respect to the WT CDKL5.
Results
Aminoglycosides induce read-through of CDKL5 nonsense mutations
Read-through efficacy is highly dependent on the identity of the stop codon (UAG, UGA or UAA) and the nucleotide sequence surrounding the premature stop codon [16,17]. Further, this approach can be considered therapeutically effective only when the activity of the recoded protein is restored. Considering that read-through might favour the incorporation of a wrong amino acid and that the vast majority of the pathogenic missense mutations alters the primary sequence of the catalytic domain of CDKL5 (Figure 1a), we hypothesized that nonsense mutations occurring in the C-terminal regions might represent preferable candidates for a therapeutic read-through. Thus, by screening the database collecting human CDKL5 pathogenic mutations (http://mecp2.chw.edu.au/cdkl5/index.php), we selected four mutations representing the premature UGA codon located either in the N-terminal catalytic domain (R59X and R134X) or in the C-terminus (R550X and S855X), and two C-terminal mutations Q347X and E364X, respectively, representing the UAG and UAA PTCs (Figure 1a). Selected mutations were introduced into a vector coding for a GFP fused to a CDKL5 cDNA encoding for the most abundant human CDKL5 isoform expressed in the brain (hCDKL5_1; 107 kDa [3];) and by western blot we verified that all vectors synthesize truncated proteins of the expected size (Figure 1b). The absence of the full-length GFP-CDKL5 in cells transfected with the selected mutants demonstrated that detectable physiological read-through does not occur in untreated cells. Of note, by comparing the expression levels of the mutants with respect to the transfection efficiency in several experiments (tested through a co-transfected GFP expressing vector) (see also Figure 3b), we always observed that mutant R134X is consistently less expressed.
Figure 3.
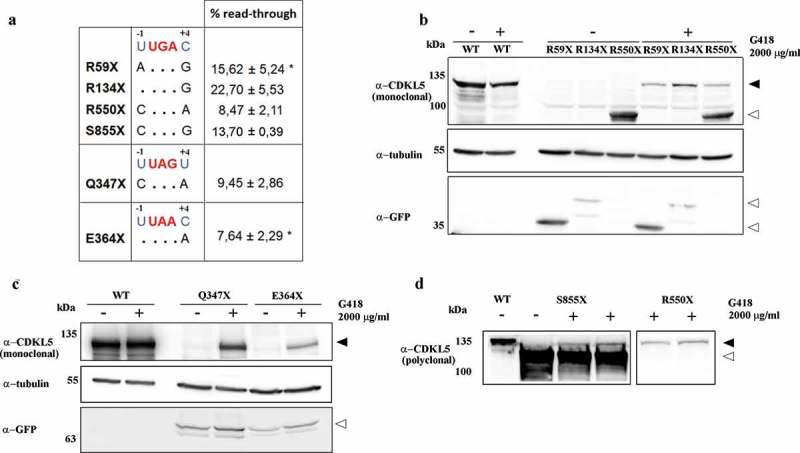
(a) The surrounding sequence of each selected PTCs is compared to the most favourable surrounding sequence for read-through (indicated in blue). Each dot represents identical residue to the consensus sequence. The efficacy of read-through for all PTCs tested is reported as mean ± SEM (n > 4) and calculated by using the anti-GFP antibody. For the R59X and E364X mutants, the asterisk indicates that the calculated efficiency is the cumulative effect of read-through and mRNA stabilization. (b, c, d) Representative western blot of full-length GFP-CDKL5 (α-CDKL5; black arrowheads) or truncated proteins (white arrowheads) in transfected HEK293 cells exposed to vehicle (-) or to 2000 μg/ml of G418 (+). In c and d, the loaded volume of WT samples was halved to avoid signal saturation.
We then assayed whether the suppression of CDKL5 PTCs could be induced by aminoglycosides. HEK293 cells transfected with WT, R59X and R134X CDKL5 constructs were grown for 24 h in the presence of increasing concentrations of gentamicin or geneticin (G418; ranging from 0 to 2000 μg/ml), and read-through was verified by western blots. In particular, full-length GFP-CDKL5 synthesis was revealed using a commercial polyclonal serum raised against a peptide spanning residues 636–758 of the human CDKL5 (Figure 2a), while truncated mutants were recognized using an antiserum against GFP. Of note, a commercial monoclonal antibody raised against a 222–520 human CDKL5 peptide is also available and used in Figure 3b and, in this study, it can recognize only mutants R550X and S855X (Figure 2a). As shown in Figure 2b,c, both drugs were able to restore the expression of full-length GFP-CDKL5 in cells transfected with the mutant cDNAs. Although the effect was dose dependent for both drugs, G418, but not gentamicin, was efficient also at low doses (50–100 μg/ml). The absence of any band exceeding the molecular weight of the WT CDKL5 proved that extension beyond the natural stop codon did not occur.
Figure 2.
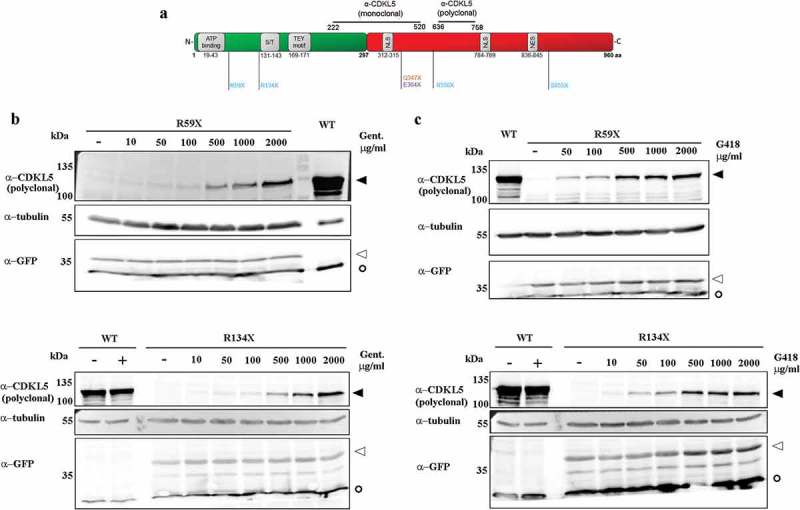
(a) Schematic representation of hCDKL5 isoform_1, the selected PTCs and the peptides used to develop the commercial polyclonal and monoclonal anti-CDKL5 antibodies used in this study. (b, c) Representative western blots of full-length GFP-CDKL5 (α-CDKL5) or truncated proteins (α-GFP) showing the dose-dependent efficacy of gentamicin (Gent; b) or geneticin (G418; c). HEK293 cells were transfected with plasmids expressing the WT, R59X and R134X CDKL5 derivatives and exposed to increasing concentrations of drugs ranging from 0 to 2000 μg/ml or the corresponding vehicle (-) for 24 h. WT transfected cells were treated with 2000 μg/ml for both drugs (+). α-tubulin was used as a loading control. To avoid signal saturation, the WT loaded volume was halved. Black arrowheads indicate full-length GFP-CDKL5; white arrowheads indicate truncated proteins; empty circles indicate co-transfected GFP.
The efficacy of PTC suppression depends on the location and identity of the stop codon and the surrounding nucleotides. Although some studies have indicated that UGA is the easiest stop codon to be suppressed [16,17], Floquet and collaborators proposed that an uracil immediately upstream of the PTC (−1 position) and a cytosine in +4 promote optimal gentamicin-induced read-through independently of the specific stop codon [18]. The importance of the fourth position was also reported by Manuvakhova and colleagues; however, the authors noticed that we still lack full comprehension of the susceptibility of stop mutations to aminoglycoside-mediated suppression in mammalian cells [19]. We thus tested whether and to what extent premature stop codon read-through occurs at different CDKL5 PTCs. Notably, although none of the selected CDKL5 premature stop codons has both the favourable surrounding −1 and +4 nucleotides (Figure 3a), they were all suppressed by high doses of G418 [9]. Read-through efficacy was estimated using the anti-GFP antibody and measuring the ratio between rescued CDKL5 and the total exogenously expressed kinase (obtained by adding the expression of the full-length protein to that one of the prematurely truncated derivatives). As expected, we observed a different efficacy of PTC suppression among the diverse mutants (Figure 3a). In particular, the R134X mutant exhibited the highest read-through efficacy, a result that can be easily explained considering that it presents the highly relevant −1 uridine upstream of the UGA premature stop codon that is often ranked as the most permissive PTC for read-through (Figure 3a [20];). Complementary, read-through efficiency of the Q347X and E364X mutants appears to well-correlate with the predicted efficacy of their specific PTC; indeed, the least read-through proficient protein appeared the E364X mutant harbouring a UAA codon that is often considered the least permissive for a read-through. We are aware that read-through efficiency is affected by changes in mRNA abundance; we thus used quantitative RT-PCR to investigate whether the addition of G418 could influence the levels of the investigated CDKL5 transcripts. Results were normalized to the transfection levels, obtained analysing the expression of the neomycin gene. We found that the stability of R59X and E364X mRNAs was increased (data not shown). These results suggest that the corresponding read-through efficiency (Figure 3a) is the resultant of two different molecular effects: mRNA stabilization and read-through proficiency.
Additional studies were performed to assess the read-through activity of non-aminoglycoside compounds, such as Ataluren (PTC124; Figure 4a, c) and GJ072 (Figure 4b, d). Of note, GJ072 was found capable of restoring the expression of full-length functional ATM protein, while the efficacy of PTC124 to suppress nonsense mutations both in vitro and in vivo is still highly debated [9]. As shown in Figure 4, in our experimental conditions, both drugs were unable to induce any detectable read-through on the aminoglycoside responsive R59X and R134X nonsense mutants, therefore questioning the opportunity to use these compounds for the treatment of CDKL5 disorder.
Figure 4.
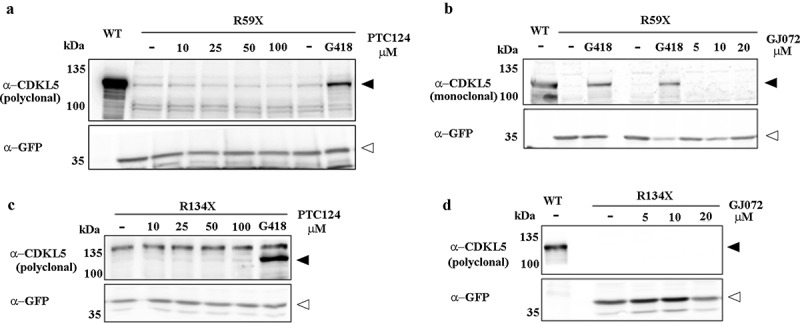
(a, c) Representative western blots of full-length GFP-CDKL5 in HEK293 cells transfected with WT, R59X or R134X mutant constructs and treated or not (-) with increasing doses of Ataluren (PTC124). G418 treated cells served as positive controls. (b, d) Representative western blots of full-length GFP-CDKL5 in HEK293 cells transfected with WT, R59X or R134X mutant constructs and treated with increasing doses of GJ072, or 2000 μg/ml G418 as a comparison. Black arrowheads indicate full-length GFP-CDKL5; white arrowheads indicate truncated proteins. In a and d, 1/3 and 1/5 volume of WT samples were loaded to avoid signal saturation.
The read-through therapy does not lead to a fully functional CDKL5
We then proceeded characterizing at the functional level the restored CDKL5. We have previously demonstrated that the catalytic activity of CDKL5 can be verified measuring the autophosphorylation of its TEY motif enclosed within the activation loop [21]. Indeed, no TEY phosphorylation can be observed when cells express the kinase-dead CDKL5 K42R mutant [21,22].
Thus, we measured the phosphorylation level of the TEY motif of WT and G418-induced read-through CDKL5 derived from R59X, R134X and R550X PTCs, dividing this signal to the total amount of full-length CDKL5.
As shown in Figure 5a, G418 did not affect the catalytic activity of the WT protein, while it partially restored the catalytic activity associated with all the three read-through products. However, quantification of the obtained results demonstrated that all read-through products were significantly hypomorphic (Figure 5a).
Figure 5.
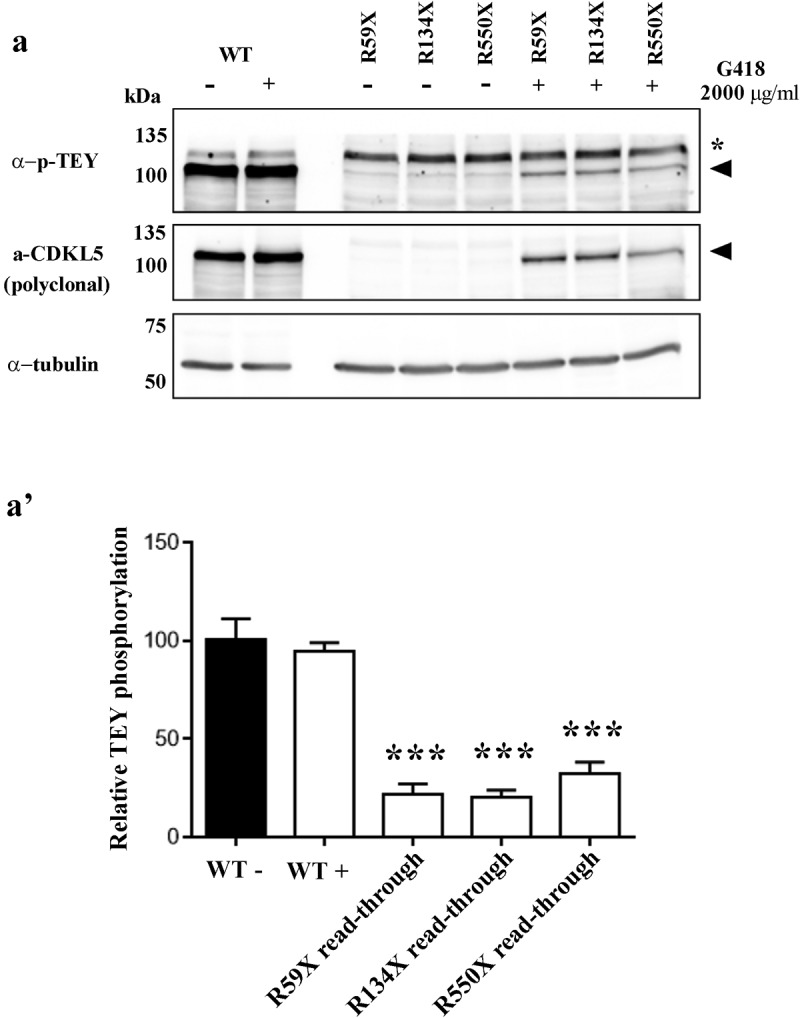
Analysis of the phosphorylation level of the TEY motif of WT and read-through CDKL5 derived from R59X, R134X and R550X PTCs, after treatment with G418 (2000 μg/ml) (+) or the corresponding vehicle (-). (a) Representative western blot of phospho-TEY, CDKL5 and α-tubulin. Black arrowheads indicate full-length GFP-CDKL5; asterisk denotes an aspecific signal. (a’) The histogram shows the mean ± SEM of the percentages of TEY phosphorylation of read-through products with respect to untreated WT (***p < 0.001 by one-way ANOVA followed by Bonferroni post hoc test).
Although these results put some concerns on the potential efficacy of read-through therapy for the treatment of CDKL5 disorder, we decided to corroborate them analysing whether read-through proteins might also manifest a non-canonical subcellular distribution. Since CDKL5 localization has usually been tested in HeLa cells [21,23], we transfected WT GFP-CDKL5 and its truncated derivatives R59X, R134X and R550X in these cells and compared their distribution between nucleus and cytoplasm including GFP as control (Figure 6a, a'). However, since we evaluated read-through therapy in HEK cell lines, we performed the same assay also in these cells (Figure 6b). As shown, all mutants exhibited a significantly different subcellular distribution with respect to the WT kinase. In particular, the R550X derivative manifested the highest accumulation in the nucleus and its distribution diverged both from the WT and GFP protein. On the contrary, and in apparent good accordance with GFP, representing the prevalent moiety of the recombinant proteins, GFP-CDKL5 R59X and R134X chimaeras behaved similarly to the fluorescent protein, therefore diverging only from WT CDKL5.
Figure 6.
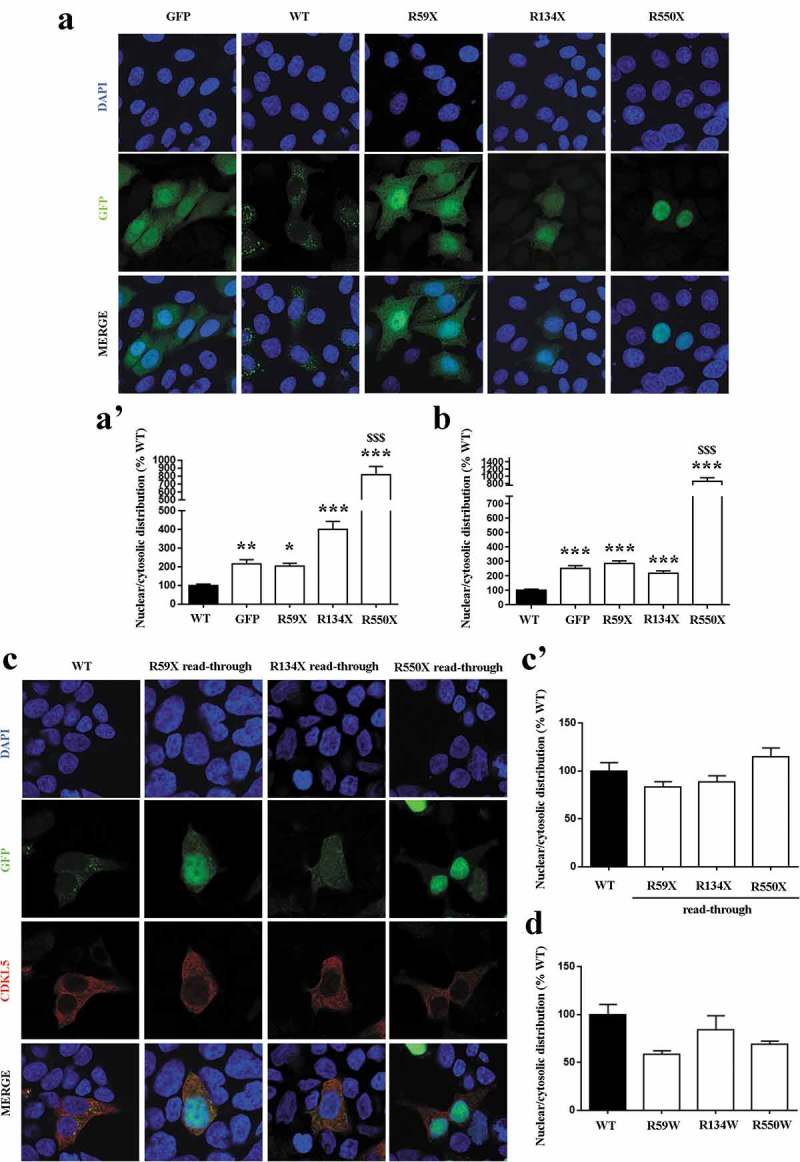
(a, b) Analysis of the nuclear/cytosolic distribution of WT and mutant CDKL5 in transfected HeLa cells (a, a’) and HEK293T (b). Representative immunofluorescence of CDKL5 in cells transfected with WT GFP-CDKL5 and its truncated derivatives R59X, R134X and R550X. GFP transfected cells served as control. Histograms depict the analysis of nuclear/cytosolic distribution of CDKL5, calculated by measuring the integrated density of GFP fluorescent signal in the two cellular compartments and expressed as the mean ± SEM of the percentages with respect to WT (*p < 0.05, **p < 0.01, ***p < 0.001vsWT; $$$p < 0.001vsGFP, by one-way ANOVA, followed by Dunn post hoc test). (c, c’) Analysis of the nuclear/cytosolic distribution of mutant CDKL5 following read-through treatment. Representative immunofluorescence of nuclei (blue), GFP-CDKL5 (green) and full-length CDKL5 (red) in transfected HEK293T cells. Histogram depicts the analysis of the nuclear/cytosolic distribution of full-length CDKL5 after exposure with G418 (2000 µg/ml for 24 h), reporting the mean ± SEM of the percentages of calculated integrated density with respect to WT. (d) Analysis of the nuclear/cytosolic distribution of full-length CDKL5 derivatives containing a tryptophan residue at the level of the selected PTCs. Histogram depicts the mean ± SEM of the percentages of calculated integrated density respect to WT.
Next, we asked whether read-through CDKL5 proteins rescued their subcellular distribution. To this purpose, we exploited the polyclonal α-CDKL5 serum that can only detect the full-length protein. Interestingly, we found that read-through R59X, R134X and R550X derivatives normalized their subcellular distribution, therefore phenocopying the untreated WT CDKL5 (Figure 6c, c’). These results well match with the demonstration that full-length CDKL5 derivatives containing a recoded tryptophan residue at the level of the selected PTCs did not manifest any phenotype with respect to the WT protein (Figure 6d).
The incorporation of a wrong amino acid at PTC sites might have been responsible for the observed reduced catalytic activity. It has been suggested that both in yeast and mammals read-through of UGA PTCs leads to the production of either WT proteins or recoded variants containing more frequently a tryptophan (W) or a cysteine (C) [24,25]. Considering all the above, we mutagenized the GFP-CDKL5 cDNA in order to obtain two different probable recoded full-length proteins and transfected them in HEK293 cells. Interestingly and in good accordance with our initial working hypothesis, we found that the introduction of a missense mutation in the catalytic domain of CDKL5 (CDKL5R59W/C; CDKL5R134W/C) significantly impaired its enzymatic activity, while the introduction of a diverse amino acid in the C-terminal domain (CDKL5R550W/C) did not or only slightly affected the catalytic activity of the kinase (Figure 7).
Figure 7.
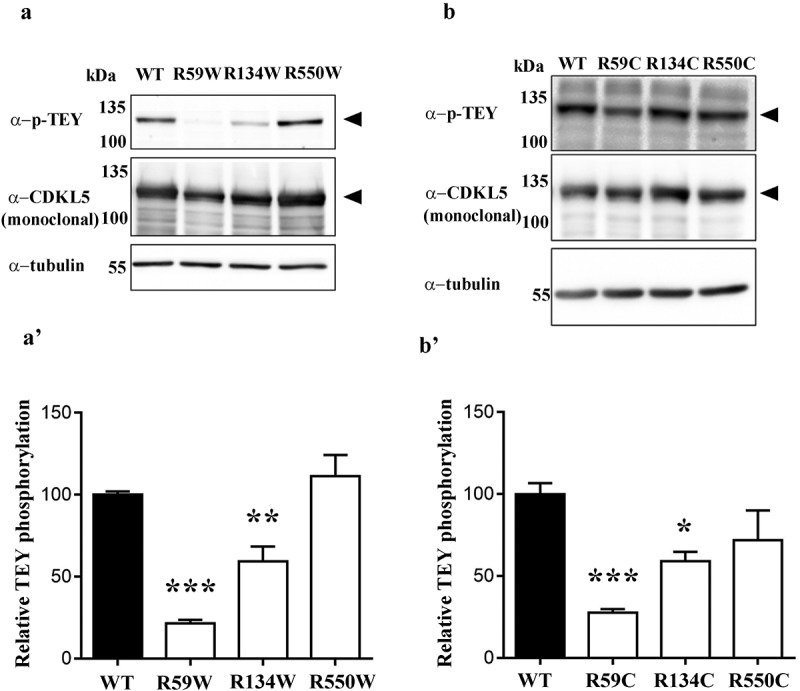
(a-b) Phosphorylation level of the TEY motif of CDKL5 derivatives harbouring a missense mutation in the catalytic domain (CDKL5R59W/C; CDKL5R134W/C) or in the C-terminus (CDKL5R550W/C). Black arrowheads indicate full-length GFP-CDKL5. (a’-b’) The histograms show the mean ± SEM of the percentages of TEY phosphorylation of mutant CDKL5 compared to WT (*p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA followed by Bonferroni post hoc test).
All in all, the obtained results suggest that read-through therapy can restore the subcellular distribution of CDKL5, while only partially rescuing its catalytic activity. Further, as expected, they indicate that missense mutations in the catalytic domain are more detrimental than in the C-terminal portion.
Discussion
Nonsense suppression therapy generally exploits small molecules to suppress translation termination at in-frame premature termination codons. The concept of this therapeutic approach was introduced more than 20 years ago [26] and since then many studies have proposed to use the read-through approach to restore deficient protein functions. However, read-through efficiency varies among genes and depends on the sequence context, in particular, the type of stop codon and surrounding nucleotides. Further, the amino acid incorporated at the PTC might differ from the one encoded by the wild type protein, therefore leading to a missense mutation. For these reasons, the potential validity of read-through therapy should be carefully investigated for every gene and mutation. The main aim of our study was thus to explore whether CDKL5 disorder might benefit from a nonsense suppression therapy. In particular, we investigated the efficacy of read-through on different CDKL5 nonsense mutations and tested the compatibility of the amino acid inserted at the PTC with CDKL5 kinase activity and subcellular localization. In this light, we assumed that an effective read-through of PTCs located in the CDKL5 C-terminal domain should be favoured because very few, if any, certain missense pathogenic mutations have been identified in this region.
The importance of investigating the effectiveness of the read-through approach for CDKL5 disorder is highlighted by several aspects. First, although CDKL5 disorder represents a devastating condition affecting kids since the very early weeks of life, no cure exists to combat the primary pathology of these patients and current treatments are geared only to ameliorate secondary symptoms, in particular, infantile seizures. Further, although the still incomplete knowledge of CDKL5 functions in the brain limits the identification of therapies, it has become clear that CDKL5 activity depends on its subcellular localization, synthesis and degradation. Accordingly, neuronal activity quickly induces CDKL5 expression by two main molecular mechanisms: an increase in transcription and the activation of localized protein synthesis in dendritic fractions. Importantly, this activation is immediately followed by proteasomal degradation, therefore returning to basal levels within few minutes [27]. Moreover, sustained glutamate stimulation of mature neurons determines a massive degradation of CDKL5 that occurs through NMDA receptors [28]. These observations, together with the spatiotemporal regulation of CDKL5 expression in the brain [21], indicate that CDKL5 levels have to be strictly modulated. Accordingly, although the CDKL5 disorder is generally associated with loss of function mutations, patients carrying CDKL5 duplications have recently been reported [4]. These evidences cause concerns on the possibility to use gene- and protein-augmentative therapies for the treatment of CDKL5 patients; in fact, both these approaches will not reproduce the subtle regulation of CDKL5. Further, the gene therapy approach suffers from the difficulty in targeting a significant percentage of neurons expressing the mutated X-linked allele, while avoiding cells expressing the WT allele.
On the contrary, read-through therapy exerts its effects on mature mRNA, that, importantly, maintains all the regulative sequences affecting its localization and translation. Further, since it is not an irreversible approach, it offers the advantage to be easily interrupted if negative effects occur or in case a more efficient therapy might become available for the same patient. However, since read-through will lead to limited amounts of functional CDKL5 protein, it remains to be established whether the obtained levels are sufficient to rescue clinical symptoms.
A number of small molecules have been discovered or specifically designed to induce read-through; among these, several in vitro studies have reported that certain aminoglycosides own this activity. Although their capability to restore functional protein was also proved in short-term studies using mouse models of diseases, results from pilot clinical trials are often unclear [9].
This study demonstrates for the first time that read-through occurs quite efficiently on CDKL5 and that several nonsense mutations, independently of the type of stop codon, can be suppressed by aminoglycosides; indeed, all tested mutations could be efficiently suppressed.
Since a long-term administration of traditional aminoglycosides is not feasible because of their toxic side effects [29,30], we assessed, in the same experimental setting, the activity of the safer Ataluren (PTC124) and GJ072 [9]. Importantly, both drugs were unable to manifest any appreciable read-through activity on all tested CDKL5 PTCs. Although the ability of PTC124 to suppress nonsense mutations has already been questioned few times [9,31], we find these results quite important considering that a phase 2 clinical trial for the treatment of CDKL5 patients with Ataluren is currently ongoing. Our positive results obtained with aminoglycosides but not Ataluren suggest that in case the clinical trial will fail, the prospect of using read-through therapy for CDKL5 patients might not be abandoned; the capability of small molecules to enhance read-through might be also tempted [32]. However, the functional characterization of the restored full-length CDKL5 causes some concerns on the potential efficacy of a CDKL5 nonsense suppression therapy. Indeed, our results demonstrate that although all the analysed read-through CDKL5 proteins manifest a full-recovery of the subcellular distribution, they are unable to fully recover the catalytic activity that remains significantly hypomorphic. Furthermore, the analysis of the catalytic activity of two probable recoded CDKL5 products suggests that the observed defect is not only caused by the introduction of missense mutations. Indeed, while the full-length CDKL5 recovered from the R550 PTC is highly hypomorphic, no significant impairment of the TEY phosphorylation was observed analysing its R550W/C mutant derivatives, that represent the more probable full-length proteins produced by read-through. It has recently been discovered that the structure and function of some proteins are affected by the rate at which protein synthesis elongation occurs [33]. Since ribosomal toeprinting studies led to propose protracted ribosomal pausing at PTCs [34], we speculate that during read-through a prolonged ribosomal pausing might affect CDKL5 folding and activity. In the future, we will thus investigate whether CDKL5 kinase activity suffers from the alteration in the speed of its synthesis.
However, we note that our functional studies confirm that missense mutations outside of the catalytic domain are better tolerated, therefore suggesting which CDKL5 patients might gain more from a nonsense suppression therapy.
We are aware that our in vitro assays have limitations. Indeed, PTCs can affect splicing, possibly inducing exon skipping [35,36]. Further, it is well known that the level of nonsense transcripts can significantly affect read-through efficiency. PTC-bearing transcripts can be degraded by the NMD pathway that is affected by several parameters, including splicing and the length of the 3ʹUTR [37,38]. Further, NMD efficiency might vary considerably among patients carrying the same gene mutation and even among cells derived from the same patient [39]. Importantly, all these mechanisms occur simultaneously in vivo, therefore affecting the specific responsiveness to read-through therapy. Being based on transfected cDNAs, our assays could not take into account all these parameters. However, recent publications demonstrated that NMD occurs in transfected cells regardless of the presence or absence of an exon junction complex (EJC) downstream of the PTC. The authors suggested that EJC might serve as an 0NMD enhancer [40]. Notably, in our experimental conditions mutant CDKL5 mRNAs did not appear significantly affected by NMD (data not shown).
To conclude, our results suggest that read-through therapy might be an opportunity for a selected cohort of CDKL5 patients, although indicating some limitations. We believe that further studies involving genetically modified human cells and mouse models will be fundamental to understand whether read-through therapy permits to obtain sufficient levels of functional CDKL5 to induce relevant neurological rescue.
Materials and methods
Antibodies
The following antibodies were used throughout the experiments: mouse monoclonal anti-α-tubulin (T6074; Sigma-Aldrich, St Louis, MO, USA), mouse monoclonal anti-GFP (1,814,460; Roche Diagnostics Ltd, Switzerland), rabbit polyclonal anti-phospho-ERK1/2 (Thr 202/Tyr 204, sc-16,982 used to detect the phosphorylated TEY motif; Santa Cruz Biotechnology, Inc., Texas, USA), rabbit polyclonal anti-CDKL5 (HPA002847, a.a. 636–758; Sigma-Aldrich), mouse monoclonal anti-CDKL5 (sc-376,314, clone D-12, a.a. 222–520; Santa Cruz Biotechnologies). HRP-conjugated anti-mouse and anti-rabbit secondary antibodies were purchased from KPL (Gaithersburg, MD, USA). Secondary Alexa anti-mouse 488 and anti-rabbit 568 antibodies were purchased from Life Technologies (Carlsbad, CA, USA).
Read-through drugs
Geneticin (50mg/ml in H2O; Life Technologies), gentamicin (50mg/ml in H2O; Sigma Aldrich), PTC124 (200mM in DMSO; Selleck Chemicals, Munich, Germany) and GJ072 (10mM in DMSO; Ambinter, Orleans, France) were properly prepared and stored until use.
Generation of mutated CDKL5 vectors
Pathological nonsense mutations (http://mecp2.chw.edu.au/cdkl5/index.php) were generated by site-directed mutagenesis of the pEGFPC1-hCDKL5107 plasmid codifying for the human CDKL5_1 isoform, weighing 107 kDa and fused to an EGFP at the N-terminus [41]. Such plasmid, herein defined as WT, was used as a template for generating R59X (175 C > T), R134X (400 C > T), R550X (1648 C > T), S855X (2564 C > G), Q347X (1039 C > T), E364X (1090 G > T) nonsense mutants or R59W (175–177 CGA>TGG), R134W (400–402 CGA>TGG), R550W (1648–1650 CGA>TGG), R59C (175–177 CGA>TGT), R134C (400–402 CGA>TGT), R550C (1648–1650 CGA>TGT) missense constructs.
Site-directed mutagenesis was performed using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA, USA) as instructed by the manufacturer and using the following specific primers: R59X sense 5ʹ-AACGACTTTATGAGAGCTTAAAATG-3ʹ, antisense 5ʹ- TCTTTGACTTCTTCATTTTCTTC-3ʹ; R134X sense 5ʹ-TATTGTCCATTGAGATATAAAACCAG-3ʹ, antisense 5ʹ-TCATTCTTATGGCACCAG-3ʹ; R550X 5ʹ-AAGAAATAACTGAAATGAGGGAACG-3ʹ, antisense 5ʹ-CCAGAAGGGCTGAGCAAAG-3ʹ; S855X sense 5ʹ- CCGGCTTCCTGAGATCCCCGC-3ʹ, antisense 5ʹ-GTGATTTGAGGCCGAAGAGAGATG-3ʹ; Q347X sense 5ʹ-CAAGGACATCTAGAACCTGAG-3ʹ, antisense 5ʹ-CTGTTAGATCTGTGGTGAG-3ʹ; E364X sense 5ʹ-CCCTGCCAATTAAAGCTTCCTAAATGG-3ʹ, antisense 5ʹ-AGACCTTCGTCAGCCCGG-3ʹ; R59W sense 5ʹ-AACGACTTTATGGGAGCTTAAAATG-3ʹ, antisense 5ʹ-TCTTTGACTTCTTCATTTTCTTC-3ʹ; R134W sense 5ʹ-TATTGTCCATTGGGATATAAAACCAG-3ʹ, antisense 5ʹ-TCATTCTTATGGCACCAG-3ʹ; R550W sense 5ʹ-AAGAAATAACTGGAATGAGGGAACGC-3ʹ, antisense 5ʹ-CCAGAAGGGCTGAGCAAAG-3ʹ; R59C sense 5ʹ-AACGACTTTATGTGAGCTTAAAATG-3ʹ, antisense 5ʹ- TCTTTGACTTCTTCATTTTCTTC-3ʹ; R134C sense 5ʹ-TATTGTCCATTGTGATATAAAACCAGAAAATC-3ʹ, antisense 5ʹ-TCATTCTTATGGCACCAG-3ʹ; R550C sense 5ʹ- AAGAAATAACTGTAATGAGGGAACGCTG-3ʹ, antisense 5ʹ- CCAGAAGGGCTGAGCAAA-3ʹ. All PCR-generated constructs were verified by sequencing.
Cell cultures and transfection
HEK293T, HEK293 and HeLa cells were maintained in DMEM (Dulbecco’s modified Eagle’s medium, Sigma Aldrich) supplemented with 10% FBS (Life Technologies), 1% L-glutamine (Sigma Aldrich), 1% penicillin/streptomycin (Sigma Aldrich) at 37°C with 5% CO2 in T75 flasks. Cells were seeded in 12-well dishes and transfected when they reached 70–80% of confluence. The cell lines were transiently transfected with 1.6 μg of DNA using LipofectamineTM 3000 (Life Technologies; 2μl) or 2 μg of DNA for calcium phosphate method. The amount of transfected DNA could be modified according to the number of cells.
Six hours after transfection, a fresh medium devoid of penicillin and streptomycin but containing the read-through drug was added. Cells were treated for 24 or 40 h with the drug or the corresponding vehicle. Of note the capability of aminoglycosides to induce read-through is generally tested in HEK293 cells and, more seldom, in HEK293T cells, that express the aminoglycoside-3ʹ-phosphotransferase together with the large T antigen. We reasoned that by conferring neomycin resistance this gene might affect the efficacy of the aminoglycoside treatments in HEK293T cells [42]. However, by applying the same procedure to the two cell-lines and comparing the level of the read-through, we did not observe any significant difference (data not shown).
Western blotting
Cells were lysed using Laemmli buffer (150 μl/well for 12-well plates) and sonicated for 10 s at 30% amplitude. Protein lysates were heated at 95°C for 5 min (or at 70°C for 10 min to preserve the phosphorylations) and separated on a 10% or 8% SDS-PAGE. Specifically for lysates derived from cells transfected with CDKL5 S855X derivative, the electrophoretic separation was conducted on a 4–20% TGXTM precast gel (Biorad) for x 2 h. Proteins were blotted onto nitrocellulose membrane using a semidry transfer apparatus (Trans-blot SD; Bio-Rad, Hercules, CA, USA). Membranes were incubated 1 h in blocking solution (Tris-buffered saline containing 0.2% Tween-20 [TBS-T] and 5% non-fat dry milk) and then incubated overnight (4°C) with primary antibodies at the proper dilution: anti-CDKL5 (1:1000 in 5% milk in TBS-T), anti-GFP (1:1000 in 5% milk in TBS-T), anti-phospho-TEY (1:500 in 5% bovine serum albumin in TBS-T), anti-α-tubulin (1:30,000 in 5% milk in TBS-T). After three washes in TBS-T, blots were incubated for 1 h at room temperature with the appropriate HRP-conjugated secondary antibody (1:10,000 in 5% milk in TBS-T), and the immunocomplexes were visualized by using the ECL substrate kit (GenSpin, Milan, Italy) and the Uvitec system (Cleaver Scientific Ltd, UK). Band density measurements were performed using the Uvitec software, and the mean value of the control group (untreated WT) was set at 100% and data expressed as a percentage of controls. TEY phosphorylation was measured by dividing the signal intensity of TEY with that of CDKL5.
Immunofluorescence
Twenty-four or 32 h after transfection, cells seeded on poly-L-lysine coated coverslips were fixed with 4% paraformaldehyde in PBS for 20 min (RT) and then washed three times with 10 mM PBS.
For CDKL5 staining, cells were post-fixed in ice-cold methanol for 10 min (−20°C), permeabilized in 10 mM PBS containing 0.1% Tween-20 for 30 min and incubated with 1X DNase (Sigma Aldrich) in PBS for 90 min.
After incubation in a blocking solution (5% horse serum in 10 mM PBS containing 0.2% Triton X-100) for 2 h, immunofluorescence staining was performed incubating the primary anti-CDKL5 antibody (1:500 in blocking buffer) overnight at 4°C. Cells were then incubated with Alexa secondary antibodies for 1 h at room temperature. DNA was stained for 10 min with DAPI solution (1:1000 in PBS; Life Technologies) and slides were mounted using Fluoromount-G reagent (Life Technologies). Images were acquired from at least three coverslips for each experimental group.
For the analysis of the nuclear-cytosolic distribution of proteins, single optical sections were acquired using the confocal laser-scanning microscope Zeiss LSM 800 equipped with a 63X oil immersion objective (Zeiss) and by Zen 2.3 system software. During acquisition, the pinhole size was set to 1 Airy Unit (AU), and the laser percentage was maintained constant while master gain was adjusted to avoid saturated pixels. In all images, the background level of full-length CDKL5 staining obtained after read-through was set deducting the signal from untreated samples.
Image analysis and cell counting were performed using Fiji software (https://imagej.net/Fiji). Fluorescence values were expressed as Integrated Density (mean fluorescence x area measured; IntDen) corrected for the background: InDen (area of selected cell x mean fluorescence of background readings).
RNA isolation, cDNA synthesis and qPCR analysis
To assess whether G418 affects the RNA stability of exogenously expressed CDKL5 mRNAs, HEK293T cells were transfected with vectors expressing the WT or the CDKL5 derivatives, and drug treated as described above.
Total RNA was isolated from transiently transfected cells using TRIzol (Life Technologies). DNA was removed by DNase I digestion (Sigma-Aldrich). RNA was quantified using a NanoDrop spectrophotometer and integrity verified on 1% agarose gel. RNA (500 ng) were reverse-transcribed using the RT2 First Strand Kit (Qiagen, Dusseldorf, Germany) as instructed by the manufacturer, and the resulting cDNA was used as template for real-time qPCR using the SYBR green Master Mix (Life Technologies) and the following primers: human CDKL5 forward 5ʹ- GGGTTGTAGGTGAAGGAGCC-3ʹ, reverse 5ʹ-CCTCCGACGAAATGCTTCCT-3ʹ; human GAPDH forward 5ʹ-CCACATCGCTCAGACACCAT-3ʹ, reverse 5ʹ-CCAGGCGCCCAATACG-3ʹ; Neomycin forward 5ʹ-TTGTCAAGACCGACCTGTCC-3ʹ, reverse 5ʹ-CACTTCGCCCAATAGCAGCC-3ʹ. qPCR was performed in 384-well plates using QuantStudio 5 (Life Technologies) and fold change in gene expression was calculated by the 2(-delta Ct) method, using Neomycin as the internal standard.
Statistical analysis
Data are expressed as means ± standard error (SEM) and analysed by GraphPad Prism 7.0 software. The significance of results was evaluated by one-way or two-way ANOVA followed by Bonferroni’s post hoc test. When a large set of data were analysed, the normality distribution was verified using Kolmogorov–Smirnov, D’Agostino & Pearson and Shapiro-Wilk tests; in case of a non-Gaussian distribution of data, the non-parametric Kruskal Wallis one-way ANOVA with Dunn’s post hoc test was used. A p-value <0.05 was considered significant.
Funding Statement
The project ‘Novel approaches of “personalized medicine” as proof of principle for CDKL5 related pathologies’ granted by Foundation Jérôme Lejeune supported this work. The salary of MF was supported by L’Albero di Greta parents' association.
Acknowledgments
We are grateful to Dr.ssa Federica Miramondi, Dr. Domenico Giorgio, Dr.ssa Eleonora Spiombi, Dr.ssa Alice Fratton and Dr.ssa Anais Serati for helpful discussion and technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Fehr S, Wilson M, Downs J, et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur J Human Genet. 2012;21:266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kilstrup-nielsen C, Rusconi L, La MP, et al. What we know and would like to know about CDKL5 and its involvement in epileptic encephalopathy. 2012. DOI: 10.1155/2012/728267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hector RD, Dando O, Landsberger N, et al. Characterisation of CDKL5 transcript isoforms in human and mouse. PlosOne. 2016;5:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Szafranski P, Golla S, Jin W, et al. Neurodevelopmental and neurobehavioral characteristics in males and females with CDKL5 duplications, 2015; 915–921. DOI: 10.1038/ejhg.2014.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fuchs C, Trazzi S, Torricella R, et al. Neurobiology of disease loss of CDKL5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3 β signaling. Neurobiol Dis. 2014;70:53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Della SG, Putignano E, Chelini G, et al. Archival report dendritic spine instability in a mouse model of CDKL5 disorder is rescued by insulin-like growth factor 1. Biol Psychiatry. 2016;80:302–311. [DOI] [PubMed] [Google Scholar]
- [7].Trazzi S, Fuchs C, Viggiano R, et al. HDAC4 : a key factor underlying brain developmental alterations in CDKL5 disorder. Human Molecul Genet. 2016;25:3887–3907. [DOI] [PubMed] [Google Scholar]
- [8].Barbiero I, Peroni D, Tramarin M, et al. The neurosteroid pregnenolone reverts microtubule derangement induced by the loss of a functional CDKL5-IQGAP1 complex. Human Molecul Genet. 2017;26:3520–3530. [DOI] [PubMed] [Google Scholar]
- [9].Keeling KM, Xue X, Gunn G, et al. Therapeutics based on stop codon readthrough kim. Annu Rev Hum Genet. 2014;15:371–394. Therapeutics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Finkel RS, Flanigan KM, Wong B, et al. . Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation duchenne muscular dystrophy. PlosOne. 2013;8 DOI: 10.1371/journal.pone.0081302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wilschanski M, Miller LL, Shoseyov D, et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respirat J. 2011;59–69 DOI: 10.1183/09031936.00120910. [DOI] [PubMed] [Google Scholar]
- [12].Barton-davis ER, Cordier L, Shoturma DI, et al. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clinic Invest. 1999;104:375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Malik V, Rodino-Klapac LR, Viollet L, et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol. 2010;67:771–780. [DOI] [PubMed] [Google Scholar]
- [14].Wilschanski M, Yahav Y, Yaacov Y, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349:1433–1441. [DOI] [PubMed] [Google Scholar]
- [15].Du M, Jones JR, Lanier J, et al. Aminoglycoside suppression of a premature stop mutation in a Cftr-/- mouse carrying a human CFTR-G542X transgene. J Mol Med. 2002;80:595–604. [DOI] [PubMed] [Google Scholar]
- [16].Bidou L, Allamand V, Rousset J-P, et al. Sense from nonsense: therapies for premature stop codon diseases. Trends Mol Med. 2012;18:679–688. [DOI] [PubMed] [Google Scholar]
- [17].Loughran G, Chou M, Ivanov IP, et al. Evidence of efficient stop codon readthrough in four mammalian genes. Nucl Acid Res. 2014;42:8928–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Floquet C, Hatin I, Rousset J, et al. Statistical analysis of readthrough levels for nonsense mutations in mammalian cells reveals a major determinant of response to gentamicin. PlosOne. 2012;8 DOI: 10.1371/journal.pgen.1002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Manuvakhova M, Keeling KIM, Bedwell DM.. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000;1044–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bonetti B, Fu L, Moon J, et al. The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in saccharomyces cerevisiae. J Mol Biol. 1995;251:334–345. [DOI] [PubMed] [Google Scholar]
- [21].Bertani I, Rusconi L, Bolognese F, et al. Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J Biolog Chem. 2006;281:32048–32056. [DOI] [PubMed] [Google Scholar]
- [22].Lin C, Franco B, Rosner MR. CDKL5/Stk9 kinase inactivation is associated with neuronal developmental disorders. Hum Mol Genet. 2005;14: 3775–3786. 10.1093/hmg/ddi391. [DOI] [PubMed] [Google Scholar]
- [23].Rusconi L, Salvatoni L, Giudici L, et al. CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J Biolog Chem. 2008;283:30101–30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pranke I, Bidou L, Martin N, et al. Factors influencing readthrough therapy for frequent cystic fibrosis premature termination codons. European Respirat Soc. 2017;4:00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Blanchet S, Cornu D, Argentini M, et al. New insights into the incorporation of natural suppressor tRNAs at stop codons in Saccharomyces cerevisiae. Nucl Acid Res. 2014;42:10061–10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Howard M, Frizzel R, Bedwell DM. Aminoglycoside antibiotics restore CFfR function by overcoming premature stop mutations. Nat Med. 1996;2:467–469. [DOI] [PubMed] [Google Scholar]
- [27].La MP, Rusconi L, Locarno A, et al. Synaptic synthesis, dephosphorylation, and degradation. J Biolog Chem. 2015;290:4512–4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rusconi L, Kilstrup-Nielsen C, Landsbergers N. Extrasynaptic N-Methyl-D-aspartate (NMDA) receptor stimulation induces cytoplasmic translocation of the CDKL5 kinase and its proteasomal degradation. J Biol Chem. 2011;286:36550–36558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ali BH, Al M, Blunden G, et al. MiniReview experimental gentamicin nephrotoxicity and agents that modify it : a mini-review of recent research. 2011;109:225–232. DOI: 10.1111/j.1742-7843.2011.00728.x. [DOI] [PubMed] [Google Scholar]
- [30].Huth ME, Ricci AJ, Cheng AG. Mechanisms of aminoglycoside ototoxicity and targets of hair cell protection. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ferrarese M, Testa MF, Balestra D, et al. Secretion of wild-type factor IX upon readthrough over F9 pre-peptide nonsense mutations causing hemophilia B. Human Mutat. 2018;39:702–708. DOI: 10.1002/humu.23404. [DOI] [PubMed] [Google Scholar]
- [32].Baradaran-Heravi A, Balgi AD, Zimmerman C, et al. Novel small molecules potentiate premature termination codon readthrough by aminoglycosides. Nucleic Acids Res. 2016;44:6583–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sharma AK, Brien EPO. ScienceDirect Non-equilibrium coupling of protein structure and function to translation – elongation kinetics. Curr Opin Struct Biol. 2018;49:94–103. [DOI] [PubMed] [Google Scholar]
- [34].Amrani N, Ganesan R, Kervestin S, et al Amrani-2004-A faux 3[prime]-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature. 2004;432:33818–33824. [DOI] [PubMed] [Google Scholar]
- [35].Barny I, Perrault I, Michel C, et al. Basal exon skipping and nonsense-associated altered splicing allows bypassing complete CEP290 loss-of-function in individuals with unusually mild retinal disease. Hum Mol Genet. 2018;27:2689–2702. [DOI] [PubMed] [Google Scholar]
- [36].Peterlongo P, Catucci I, Colombo M, et al. FANCM c.5791C>T nonsense mutation (rs144567652) induces exon skipping, affects DNA repair activity and is a familial breast cancer risk factor. Hum Mol Genet. 2015;24:5345–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Karousis ED, Mühlemann O. Nonsense-mediated mRNA decay begins where translation ends. Cold Spring Harb Perspect Biol. 2019;11 DOI: 10.1101/cshperspect.a032862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Schweingruber C, Rufener SC, Zünd D, et al. Nonsense-mediated mRNA decay - Mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim Biophys Acta - Gene Regul Mech. 2013;1829:612–623. [DOI] [PubMed] [Google Scholar]
- [39].Linde L, Boelz S, Nissim-Rafinia M, et al. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J Clin Invest. 2007;117:683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Metze S, Herzog VA, Ruepp MD, et al. Comparison of EJC-enhanced and EJC-independent NMD in human cells reveals two partially redundant degradation pathways. Rna. 2013;19:1432–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Williamson SL, Giudici L, Kilstrup-Nielsen C, et al. A novel transcript of cyclin-dependent kinase-like 5 (CDKL5) has an alternative C-terminus and is the predominant transcript in brain. Hum Genet. 2012;131:187–200. [DOI] [PubMed] [Google Scholar]
- [42].DuBridge RB, Tang P, Hsia HC, et al. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol Cell Biol. 1987;7:379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]