

ABSTRACT
Noncoding RNA (ncRNA) modulation of gene expression has now been ubiquitously observed across all domains of life. An increasingly apparent role of ncRNAs is to coordinate changes in gene expressions in response to environmental stress. Salmonella enterica, a common food-born pathogen, is known for its striking ability to survive, adapt, and thrive in various unfavourable environments which makes it a particularly difficult pathogen to eliminate as well as an interesting model in which to study ncRNA contributions to cellular stress response. Mounting evidence now suggests that small RNAs (sRNAs) represent key regulators of Salmonella stress adaptation. Approximately 50–500 nucleotides in length, sRNAs regulate gene expression through complementary base pairing with molecular targets and have recently been suggested to outnumber protein-coding genes in bacteria. In this work, we employ small RNA transcriptome sequencing to characterize changes in the sRNA profiles of Salmonella in response to desiccation. In all, we identify 102 previously annotated sRNAs significantly differentially expressed during desiccation; and excitingly, 71 novel sRNAs likewise differentially expressed. Small transcript northern blotting and qRT-PCRs confirm the identities and expressions of several of our novel sRNAs, and computational analyses indicate the majority are highly conserved and structurally related to characterized sRNAs. Predicted sRNA targets include several proteins necessary for desiccation survival and this, in part, suggests a role for desiccation-regulated sRNAs in this stress response. Furthermore, we find individual knock-outs of two of the novel sRNAs identified herein, either sRNA1320429 or sRNA3981754, significantly impairs the ability of Salmonella to survive desiccation, confirming their involvements (and suggesting the potential involvements of other sRNAs we identify in this work) in the Salmonella response to desiccation.
KEYWORDS: Bacteria, desiccation, ncRNA, noncoding RNA, osmoprotectant, prokaryotic, ProP, Salmonella, sRNA, stress, transcriptome
Introduction
Novel noncoding RNA (ncRNA) discovery and the elucidation of defined roles for ncRNAs in gene coordination, transposable element silencing, and normal cellular metabolism continues to accelerate across bacteria and eukaryotes alike. Similar to transcription factors, the majority of ncRNAs are thought to be regulatory and frequently act as master gene regulators [1,2]. That said, cells and organisms are subjected to an array of challenges and perturbations, or stresses, in their environment throughout their life cycles, and the molecular responses to such changes involve the coordinated modulation of gene expressions. Although research into the cellular stress survival has classically focused on protein involvements, ncRNAs, which may ultimately be found to outnumber protein-coding genes in prokaryotes and especially eukaryotes, are increasingly implicated in the molecular mechanisms driving these responses [1,2]. Numerous forms of long and short ncRNAs (e.g. circRNAs [3], lncRNAs [4,5], and miRNAs [6,7] in eukaryotes and CRISPR [8], 6S [9], and sRNAs [10] in prokaryotes) now have well-characterized roles in immediate cellular and organismal stress protections as well as in more persistent adaptions.
The ability of certain species (e.g. nematodes [11]), vascular plants [12], and bacteria [13]) to survive desiccation (or extreme dehydration) for extended periods is a particularly striking example of the ability of cells to adapt to and survive even the most drastic environmental fluctuations. As bacterial small non-coding RNA (sRNA) expressions are frequently associated with stress [14–16], we recently hypothesized likely contribute to Salmonella survival during desiccation. When facing a low water environment, Salmonella adapt primarily by the intracellular accumulation of osmoprotectants (largely through the action of proline channel ProP) preventing intracellular water loss. Interestingly, ProP is regulated by ProQ, an osmoregulatory protein that has been shown to be a global sRNA-binding molecule [17,18], suggesting the likely importance of sRNAs to the desiccation response and cell survival. Understanding the regulatory processes that lead to resistance and adaptation is essential to establishing new control strategies for this pathogen.
Bacterial sRNAs are typically 50 to 500 nucleotides in length, originate from ‘empty’ intergenic regions of chromosomes, and are often induced or repressed by specific stimuli during environmental stress or virulence [19]. SRNAs typically modulate metabolic functions and stress-related responses, such as nutrient deprivation, by binding target mRNAs and inhibiting their expression [14]. The majority of functionally characterized sRNAs are known to bind the 5′ UTRs of multiple mRNAs [20–22] and coordinate their expressions. As an example, the bacterial sRNA OxyS is markedly induced by oxidative stress during which it directly binds and regulates at least eight target mRNAs cumulatively altering the expression of ~40 genes [23].
Until quite recently, it was widely believed that the S. enterica genome was almost entirely annotated and that virtually all its genes had been identified. However, next generation transcriptome sequencing has quickly undermined this paradigm. In just the last 5 years, studies have identified over 350 previously undescribed ncRNA genes dynamically expressed in S. enterica [15,24,25]. That said, in this report, we describe 173 sRNAs significantly differentially expressed in response to desiccative stress (including 71 previously undescribed sRNAs) and outline a detailed methodology for their characterization. Excitingly, we find the loss of either sRNA1320429 or sRNA3981754, significantly impairs the ability of Salmonella to survive low water environments confirming their involvements (and suggesting the potential involvements of other sRNAs identified in this work) in the Salmonella response to desiccation.
Results
Salmonella sRNAs differentially expressed during desiccation
To identify and examine the expression patterns of Salmonella sRNAs during desiccation, Salmonella enterica serovar Typhimurium (strain SL1344) cultures were collected under three sample conditions (stationary phase (SP), 24 h desiccated, and 72 h desiccated), and their small RNA transcriptomes were subsequently isolated and sequenced, resulting in over 40 million raw sequencing reads for each. After mapping the RNA-seq reads to the intergenic regions of the SL1344 reference genome and all known Salmonella sRNAs using the CLC Genomics Workbench, we initially identified 149 chromosomal sequences differentially expressed (≥2.0x) between SP and either 24 h or 72-h desiccated samples (work flow summarized in Supplementary File 1). We found 102 of these differentially expressed sequences corresponded to previously characterized sRNAs (Table 1) and 47 mapped to broader ‘gene-empty’ or intergenic regions suggesting they contain novel sRNAs. In addition to 102 previously annotated sRNAs, we also identified 71 unique, novel sRNAs (Table 2) mapping to these 47 intergenic regions bringing the total number of sRNAs we find differentially expressed during desiccation to 173 (Tables 1 and 2, Supplementary Files 2,3).
Table 1.
Known sRNAs implicated in desiccation stress response in Salmonella enterica. Previous characterisations and expressions in three sample conditions (stationary phase growth, 24 hr desiccated, and 72 hr desiccated) are indicated in transcripts per million.
| sRNA | Previous Characterization | Exp. In Stationary | Exp. In 24-hr Desiccation | Exp. In 72-hr Desiccation |
|---|---|---|---|---|
| RyhB‑1 | Ellermeier & Slauch, 2008; Padalon-Brauch et al., 2008 | 27.9 | 0.0 | 110.3 |
| STnc3830 | Ramachandran et al., 2012 | 6.5 | 0.0 | 115.1 |
| IsrQ | Padalon-Brauch et al., 2008 | 15.9 | 0.0 | 157.6 |
| sRNA697722 | Amin et al., 2016 | 15.9 | 505.4 | 282.4 |
| sRNA1318601 | Amin et al., 2016 | 69.0 | 1865.0 | 430.6 |
| sRNA1170414 | Amin et al., 2016 | 33.9 | 595.6 | 134.1 |
| STnc3150 | Ramachandran et al., 2012 | 137.0 | 1900.5 | 655.2 |
| STnc3420 | Kroger et al., 2013 | 80.6 | 923.8 | 265.5 |
| tpke11 | Rivas et al., 2001 | 63.0 | 707.4 | 103.8 |
| IsrL | Padalon-Brauch et al., 2008 | 614.4 | 6679.4 | 3233.8 |
| STnc3680 | Kroger et al., 2013 | 74.4 | 10.9 | 117.7 |
| STnc3240 | Kroger et al., 2013 | 1001.6 | 9779.0 | 3160.0 |
| MicC | Pfeiffer et al., 2009 | 13.7 | 83.9 | 129.6 |
| C0664 | Hershberg et al., 2003 | 24.8 | 12.1 | 114.4 |
| STnc3500 | Ramachandran et al., 2012 | 124.3 | 1159.3 | 368.4 |
| STnc3920 | Kroger et al., 2013 | 17.6 | 47.5 | 160.5 |
| STnc380 | Pfeiffer et al., 2007; Sittka et al., 2008 | 16.5 | 60.5 | 130.8 |
| STnc790 | Sittka et al., 2009 | 1964.0 | 15245.0 | 2846.0 |
| SroC | Vogel et al., 2003 | 17043.7 | 129573.2 | 40843.5 |
| STnc1590 | Kröger et al., 2012 | 508.5 | 3833.2 | 1477.7 |
| STnc1690 | Kröger et al., 2012 | 579.2 | 4361.7 | 978.6 |
| STnc200 | Pfeiffer et al., 2007; Sittka et al., 2008 | 142.4 | 1022.9 | 368.4 |
| sRNA294324 | Amin et al., 2016 | 35.6 | 142.1 | 249.4 |
| SraA | Argaman et al., 2001 | 102.9 | 720.7 | 231.0 |
| STnc3310 | Kroger et al., 2013 | 3845.6 | 26697.9 | 7451.2 |
| STnc4120 | Kroger et al., 2013 | 425.0 | 2814.5 | 569.4 |
| STnc1540 | Kröger et al., 2012 | 65.1 | 408.5 | 275.8 |
| sRNA10 | Sridhar et al. 2010 | 384.5 | 2314.6 | 643.6 |
| STnc1460 | Kröger et al., 2012 | 2580.2 | 15380.1 | 12827.6 |
| STnc1420 | Kröger et al., 2012 | 494.9 | 2674.8 | 1461.3 |
| STnc1300 | Kröger et al., 2012 | 47.1 | 28.2 | 152.4 |
| STnc1400 | Kröger et al., 2012 | 59.5 | 166.4 | 321.0 |
| RyfA | Wassarman et al., 2001 | 3993.8 | 21215.8 | 20672.9 |
| STnc980 | Kroger et al., 2013 | 3168.7 | 15643.5 | 6274.0 |
| STnc4160 | Kroger et al., 2013 | 353.0 | 102.7 | 504.4 |
| STnc1030 | Kroger et al., 2013 | 23607.0 | 113313.5 | 47610.3 |
| sRNA3405375 | Amin et al., 2016 | 40.6 | 193.1 | 100.3 |
| STnc4040 | Kroger et al., 2013 | 31.2 | 84.3 | 142.4 |
| sRNA3551252 | Amin et al., 2016 | 961.8 | 4237.3 | 1241.5 |
| STnc3110 | Kroger et al., 2013 | 829.7 | 3632.0 | 1157.6 |
| RyeF | Zhang et al., 2003; Chao et al., 2012, Kröger et al., 2012 | 227116.3 | 979720.1 | 306817.5 |
| STnc3850 | Kroger et al., 2013 | 24.7 | 61.9 | 104.5 |
| STnc1650 | Kröger et al., 2012 | 1422.4 | 6009.0 | 1826.6 |
| STnc4220 | Kroger et al., 2013 | 50.1 | 206.4 | 130.1 |
| STnc4250 | Kroger et al., 2013 | 228.3 | 933.6 | 530.9 |
| STnc4190 | Kroger et al., 2013 | 37.2 | 25.1 | 101.8 |
| STnc3640 | Ramachandran et al., 2012 | 243.7 | 116.1 | 463.5 |
| SgrS | Wadler & Vanderpool, 2009; Papenfort et al., 2012; Papenfort et al., 2013 | 9469.1 | 35888.7 | 10789.4 |
| STnc3180 | Kroger et al., 2013 | 106.5 | 371.7 | 162.0 |
| STnc3290 | Kroger et al., 2013 | 733.9 | 2456.0 | 1394.2 |
| STnc770 | Sittka et al., 2009 | 688.8 | 2281.0 | 819.2 |
| sRNA924744 | Amin et al., 2016 | 11361.8 | 36145.1 | 12879.3 |
| STnc3530 | Ramachandran et al., 2012 | 451.8 | 1428.7 | 1046.8 |
| STnc3220 | Ramachandran et al., 2012 | 201.1 | 590.5 | 611.7 |
| STnc1290 | Kröger et al., 2012 | 372.1 | 556.2 | 1108.0 |
| STnc3460 | Kroger et al., 2013 | 92.7 | 203.6 | 274.9 |
| CsrB | Fortune et al., 2006 | 500191.3 | 224632.9 | 653475.2 |
| RygC | Zhang et al., 2003; Fozo et al., 2008; Rudd, 1999 | 4720.3 | 13314.3 | 5186.2 |
| SraL | Argaman et al., 2001 | 17867.1 | 49604.7 | 46410.3 |
| STnc2010 | Chao et al., 2012; Kröger et al., 2012 | 569.8 | 1552.2 | 882.6 |
| STnc3480 | Kroger et al., 2013 | 101.5 | 275.4 | 182.6 |
| STnc4080 | Kroger et al., 2013 | 119.1 | 313.7 | 197.7 |
| STnc1110 | Kröger et al., 2012 | 6793.7 | 14600.4 | 7425.0 |
| AmgR | Lee & Groisman, 2010 | 1171.8 | 771.2 | 559.0 |
| STnc760 | Sittka et al., 2009 | 391.4 | 603.3 | 283.9 |
| FnrS | Boysen et al., 2010; Durand & Storz, 2010 | 1703.3 | 2314.2 | 1070.7 |
| STnc1270 | Kröger et al., 2012 | 1558.4 | 2263.2 | 1024.3 |
| STnc1830 | Kroger et al., 2013 | 286.9 | 427.8 | 192.6 |
| STnc1760 | Ramachandran et al., 2012 | 389.1 | 660.0 | 297.1 |
| STnc1710 | Ramachandran et al., 2012 | 795.3 | 1708.6 | 756.5 |
| STnc780 | Sittka et al., 2009 | 4740.7 | 5332.0 | 2319.6 |
| GlmZ | Urban & Vogel, 2008 | 95171.3 | 92652.8 | 40338.1 |
| t44 | Rivas et al., 2001 | 1833.4 | 1908.9 | 685.8 |
| GcvB | Sharma et al., 2007 | 51769.7 | 60666.1 | 21402.8 |
| STnc3080 | Kroger et al., 2013 | 180.7 | 396.6 | 133.9 |
| STnc2090 | Chao et al., 2012 | 56052.7 | 99725.5 | 32507.6 |
| STnc1480 | Kröger et al., 2012 | 14667.9 | 27422.5 | 8710.1 |
| STnc1330 | Kröger et al., 2012 | 3604.2 | 7379.1 | 2291.1 |
| STnc1990 | Ramachandran et al., 2012 | 424.4 | 643.8 | 197.6 |
| sRNA176086 | Amin et al., 2016 | 225.7 | 445.3 | 135.8 |
| STnc3010 | Ramachandran et al., 2012 | 233.9 | 70.0 | 115.6 |
| STnc1130 | Kröger et al., 2012 | 519.4 | 1195.9 | 350.5 |
| RybA | Wassarman et al., 2001 | 1906.0 | 4460.3 | 1255.7 |
| IsrI | Padalon-Brauch et al., 2008 | 13155.9 | 41853.4 | 11609.5 |
| STnc3990 | Ramachandran et al., 2012 | 676.9 | 350.1 | 178.6 |
| STnc4150 | Kroger et al., 2013 | 744.3 | 281.5 | 176.5 |
| STnc700 | Sittka et al., 2009 | 9605.0 | 3139.9 | 2238.6 |
| sRNA4130247 | Amin et al., 2016 | 365.5 | 82.7 | 134.1 |
| STnc3760 | Ramachandran et al., 2012 | 137.0 | 311.2 | 70.1 |
| OxyS | Altuvia et al., 1997; Padalon-Brauch et al., 2008 | 97.6 | 208.7 | 44.5 |
| SraB | Argaman et al., 2002 | 494.4 | 731.5 | 145.8 |
| STnc4200 | Ramachandran et al., 2012 | 411.8 | 1056.6 | 176.5 |
| sRNA2759412 | Amin et al., 2016 | 229.3 | 736.2 | 121.7 |
| STnc3700 | Ramachandran et al., 2012 | 595.4 | 1719.2 | 270.4 |
| sRNA1799950 | Amin et al., 2016 | 170.6 | 631.5 | 99.2 |
| STnc1170 | Kröger et al., 2012 | 626.1 | 1482.3 | 218.4 |
| sRNA1548865 | Amin et al., 2016 | 238.8 | 675.1 | 58.2 |
| sRNA1253515 | Amin et al., 2016 | 71.6 | 496.4 | 22.3 |
| sRNA1888744 | Amin et al., 2016 | 17.7 | 186.7 | 0.0 |
| MicA | Papenfort et al., 2006; Bossi & Figueroa-Bossi, 2007 | 20.1 | 106.0 | 0.0 |
| DsrA | Majdalani et al., 2001 | 37.6 | 135.2 | 0.0 |
| STnc4070 | Kroger et al., 2013 | 48.1 | 184.8 | 0.0 |
Table 2.
Novel sRNAs implicated in the desiccation stress response in Salmonella enterica. Known sRNA similarities, sRNA conservation, predicted structural complexity, flanking/overlapping genes, putative targets, and expressions in stationary phase, 24 hr desiccated, and 72 hr desiccated cell samples are indicated.
| sRNAa | Conservation, sRNA Similarityb | Structurec | Flanking Genes & Overlapd | Putative Targets – IntaRNAe | Exp. In Stationaryf | Exp. In 24 hr Df | Exp. In 72 hr Df |
|---|---|---|---|---|---|---|---|
| sRNA294677 | WC, RF00391 | HS | rRNA; tRNAAsp; dkgB-1 | - | 25 | 117.7 | 78.8 |
| sRNA458139 | WC | HS | tsaA-1(3ʹ); yajB-1 | - | 16.2 | 31.4 | 13.6 |
| sRNA466762 | WC | HS | STnc1060(3ʹ); tsx-1 | - | 29.2 | 33.7 | 18.5 |
| sRNA539269 | WC | HS | priC-1 | - | 3.7 | 6.4 | 3.6 |
| sRNA539528 | WC | HS | apt-1 | - | 3.9 | 4.4 | 1.5 |
| sRNA898738 | WC | HS | ybiF-1; ompX-1(5ʹ) | - | 45.6 | 56 | 19 |
| sRNA901248 | WC, RF00110 | HS | ybiP-1; RybA(5ʹ); CBW16908 | - | 154.6 | 210.1 | 90.7 |
| sRNA978535 | WC | HS | cspD-1(3ʹ); clpS-1 | cspD | 32.3 | 19.1 | 7.6 |
| sRNA982124 | WC | HS | tnp-1(3ʹ) | - | 71.5 | 108.9 | 56.5 |
| sRNA982481 | WC, RF02902 | HS | CBW16982 | - | 1.9 | 16.7 | 17.2 |
| sRNA997533 | WC | HS | ftsK-1(3ʹ) | - | 2.7 | 9.3 | 14.3 |
| sRNA997685 | WC | HS | ftsK-1(3ʹ) | oadA1 | 5.6 | 15.8 | 7.9 |
| sRNA998116 | WC | HS | ftsK-1; lolA-1(5ʹ) | - | 9.1 | 10.1 | 3.6 |
| sRNA1000215 | - | HS | ycaJ-1; serS-1 | - | 9.8 | 9.8 | 4 |
| sRNA1118334 | WC | HS | ompA-1(3ʹ); sulA-1 | ompA | 24.9 | 28.3 | 15.2 |
| sRNA1227667 | WC | HS | rne-1(3ʹ) | ribonuclease E | 7 | 6 | 2.7 |
| sRNA1227716 | WC, RF00040 | HS | rne-1 | ribonuclease E | 16 | 13.5 | 6 |
| sRNA1290392 | WC, RF02076 | HS | tRNA-Arg; STnc520 | RatA | 42 | 19.8 | 11.1 |
| sRNA1291223 | - | HS | pliC-1(3ʹ) | lysozyme inhibitor | 26.8 | 37.1 | 11.8 |
| sRNA1291436 | - | HS | pliC-1 | - | 27 | 25.3 | 10.6 |
| sRNA1291652 | - | HP | CBW17281 | SsaE; Dcp | 32.4 | 18 | 6.5 |
| sRNA1320429 | WC | HS | yeaG-1(3ʹ) | - | 38 | 581.9 | 582.9 |
| sRNA1320654 | WC | HS | yeaG-1 | PrkA | 43.7 | 250 | 82.1 |
| sRNA1440272 | WC, RF00391 | HS | ssaE-1; sseA-1 | CpxP-like | 120.1 | 252.1 | 186.4 |
| sRNA1440474 | - | HP | ssaE-1; sseA-1(5ʹ); sseBa-1 | - | 31.6 | 18.5 | 6.2 |
| sRNA1448803 | - | HS | ssaK-1(3ʹ); ssaL-1; STnc1220 | - | 11.1 | 14 | 5.6 |
| sRNA1866912 | - | HS | CBW17833(3ʹ); ycgN-1 | - | 547.2 | 1420.8 | 603 |
| sRNA1886080 | WC | HS | CBW17853(5ʹ) | - | 23.7 | 25.2 | 6.6 |
| sRNA1922930 | - | HS | CBW17896; CBW17897 | endonuclease | 1.7 | 6.5 | 2.4 |
| sRNA1954465 | WC | HS | aspS-1; yecD-1 | aspS | 8.7 | 11.5 | 4 |
| sRNA1954754 | - | HS | yecD-1(3ʹ) | - | 7.2 | 10.3 | 3.4 |
| sRNA1996414 | WC | HS | sirA-1 (3ʹ) | dbrr | 22.9 | 8.1 | 2.8 |
| sRNA1996681 | WC | HS | yecF-1; sirA-1 | - | 4.8 | 2.8 | 1.9 |
| sRNA2111563 | WC, RF00174 | HS | cbiA-1; pag | ProP effector protein | 10.9 | 3.6 | 1.8 |
| sRNA2111772 | - | HS | cbiA-1; pocR-1 | - | 7.2 | 1.3 | 0.9 |
| sRNA2437186 | WC | HS | nuoA-1(3ʹ) | nuoA | 15.8 | 17.6 | 6.1 |
| sRNA2437382 | WC | HP | nuoA-1; CBW184000 | lytictransglycosylase; patatin family protein |
26.3 | 14.8 | 7.6 |
| sRNA2495587 | WC | HS | sixA-1(3ʹ); fadJ-1 | sixA | 100 | 162.4 | 82.9 |
| sRNA2499697 | WC | HS | yfcZ-1(3ʹ) | - | 24.9 | 21.3 | 7.9 |
| sRNA2499837 | WC | HS | fadL-1; yfcZ-1 | - | 5.6 | 18.3 | 9.6 |
| sRNA2522653 | WC | HS | nupC-1; mntH-1(3ʹ) | mntH | 4.7 | 24.3 | 7.4 |
| sRNA2522758 | - | HS | nupC-1; mntH-1 | treR; CysZ | 6.3 | 23.1 | 6.7 |
| sRNA2522977 | WC | HS | nupC-1(5ʹ) | - | 3.6 | 23.2 | 8 |
| sRNA2828025 | WC, RF00391 | HS | rimM-1; rps16-1(3ʹ); ffh-1 | 30S ribosomal protein S16 | 7.9 | 21.7 | 10.2 |
| sRNA2828147 | WC | HS | rps16-1; ffh-1 | ABC transporter; RecF; pncA |
3.9 | 13.7 | 5.5 |
| sRNA2828280 | WC | HS | rps16-1; ffh-1(5ʹ) | - | 8.6 | 26.1 | 10.3 |
| sRNA2948961 | - | HS | pipB2-1 | glgX; Kdul; HyaE; nrdD | 19.4 | 73.8 | 37.3 |
| sRNA2950404 | - | HP | pipB2-1(3ʹ) | pipB2 | 7.3 | 9.6 | 3.3 |
| sRNA2950578 | WC | HP | pipB2-1 | murC; SDR | 20.6 | 7.6 | 4.4 |
| sRNA2969551 | WC | HS | ygaP-1; stpA-1 | - | 9.8 | 14 | 9.3 |
| sRNA3393373 | - | HS | ygiH-1; gcp-1 | - | 26 | 63.5 | 38.3 |
| sRNA3395090 | WC, RF02818 | HS | rpsU-1; dnaG-1(5ʹ) | - | 7 | 25.3 | 9 |
| sRNA3417448 | WC | HS | yqjC-1(3ʹ); yqjE-1 | ferredoxin | 80 | 405.6 | 151.6 |
| sRNA3417670 | WC | HS | yqjC-1; yqjE-1 | ybbK; oadA1 | 64.7 | 484 | 218 |
| sRNA3435592 | WC, RF00010 | HP | gark-1 | - | 4254.2 | 2399.9 | 1744.1 |
| sRNA3435639 | WC, RF00010 | HS | gark-1 | - | 11178.2 | 7799 | 5212.6 |
| sRNA3758077 | WC | HS | rpoH-1(3ʹ); ftsX-1 | rpoH | 123.6 | 132.5 | 58.9 |
| sRNA3857685 | WC, RF01766 | HS | yiaG-1; cspA-1(5ʹ) | - | 452.4 | 1577 | 563.7 |
| sRNA3981754 | WC | HS | slsA-1(3ʹ); cigR-1 | - | 187.6 | 75.8 | 21.6 |
| sRNA3981945 | WC, RF00391 | HS | sciZ-1; yafV-1 | - | 106.6 | 250.6 | 166.6 |
| sRNA4066303 | WC | HS | dnaA-1(3ʹ) | DnaA | 12.6 | 19.9 | 7.7 |
| sRNA4066490 | - | HS | dnaA-1 | UmuD | 15.5 | 13.5 | 6.4 |
| sRNA4318787 | - | HS | yiiU-1(3ʹ); menG-1 | ilvD; NirC; kup; manY |
64.7 | 114.2 | 50.4 |
| sRNA4433610 | WC | HS | metH-1; yjbB-1(5ʹ) | - | 21.7 | 72.7 | 34 |
| sRNA4534936 | WC | HS | yjcH-1; acs-1(5ʹ) | rtcA | 3.7 | 26.9 | 13.5 |
| sRNA4626941 | WC | HS | hflX-1; hflK-1(5ʹ) | CreC; rpiA | 12.9 | 47.7 | 20.6 |
| sRNA4662099 | - | HS | cpdB-1; cysQ-1 | cpdB | 10.1 | 16.6 | 7.9 |
| sRNA4662318 | WC | HS | cpdB-1; cysQ-1(5ʹ) | - | 7.2 | 25.3 | 10.2 |
| sRNA4663946 | WC | HS | ytfK-1(5ʹ); ytfL-1 | - | 83 | 98.2 | 50.6 |
| sRNA4837237 | WC | HS | osmY-1; yjjU-1 | ndk; epmA | 43.8 | 203.2 | 108.5 |
| sRNA4876851 | WC | HS | arcA-1; yjjY-1(3ʹ) | - | 19.7 | 29.6 | 11.2 |
asRNA – name refers to the starting nucleotide position of the putative sRNA sequence on the SL1344 chromosome.
bConservation, sRNA Similarity – (WC), ‘well-conserved’ sRNAs are at least 80% identical over their entire length to a genomic sequence found within at least one other distinct bacterial genus. RF number refer to RFAM sequence family significantly aligning to indicated sRNA.
cStructure – Hairpin (HP), simple sRNA secondary structure containing one hairpin predicted with Mfold; Highly structured (HS), possesses more than one hairpin or simple structural element within predicted secondary structure.
dFlanking genes and Overlap determined by alignment in Ensembl – (5′) indicates that sRNA overlaps 5′-end of CDS; (3′) indicates that sRNA overlaps 3′-end of CDS.
ePutative Targets determined by IntaRNA, assuming a fdr cut-off of 0.1.
fExp. In Stationary – normalized number of transcripts per million (TPM) at sRNA location during normal stationary-phase growth; Exp. In 24 hr D – normalized TPM at sRNA location after 24 h of desiccation; Exp. In 72 hr D – normalized TPM at sRNA location after 72 h of desiccation.
102 differentially expressed sRNAs have been previously identified
In brief, we identified 173 putative sRNAs differentially expressed after desiccation for 24 and/or 72 h as compared to controls (fold change ≥ 2.0). Of these, we found 102 (~59%) correspond to previously annotated sRNAs (Table 1). Interestingly, most of the previously reported sRNAs we found under- or overexpressed during desiccation were also differentially expressed under nutrient limitation conditions (63%) (including 14 we previously identified as being involved in the carbon starvation-stress response [15]), followed by peroxide shock (41%), bile shock (33%), and NaCl shock (31%) [24,25] (Table 1). Notably, we find RyhB-1, an sRNA which acts on at least four genes related to iron homoeostasis (sodB, acnA, acnB, ftn) in Salmonella, significantly overexpressed after 72 h of desiccation. Previous experiments have shown that RyhB-mediated regulation is responsible for phenotypic changes related to acid and peroxide resistance, carbon source utilization, tricarboxylic acid cycle and sensitivity to antibiotics [26,27]. As such, our results further suggest the importance of RyhB as a general regulator of the cellular stress response system.
71 putative sRNAs are novel
While we found 102 of our differentially expressed sRNA sequences corresponded to known Salmonella sRNAs, we also identified differential expressions from 47 broader regions containing no annotated sRNAs or other known genes. To more precisely define sRNA sequences from these 47 intergenic regions, we employed a peak-calling strategy utilizing visualization of expression data on the SL1344 chromosome via the Integrative Genomics Viewer (IGV) and identified 71 distinct putative sRNA sequences expressed from the 47 broader intergenic regions (Table 2, Supplementary File 2). While we intially identified each of our novel sRNAs by examining intergenic regions, peak-calling of these broader regions along with their flanking sequences did result in the identification of 40 of our novel sRNAs partially overlapping the 5ʹ or 3ʹ ends of protein-coding genes. That said, overlaps were typically <25% of total sRNA lengths, and putative sRNA expressions were confirmed to significantly differ from those of size matched sequences selected from both a central internal location and the opposing terminus. Importantly, each of the 71 consensus sequences resulting from peak-calling within these broad regions were realigned to the full body of known SL1344 sRNAs and again confirmed to not correspond to any previously annotated sRNAs. Significant differential expression (>1.5-fold) for each of the 71 putative sRNAs was independently validated in our sequencing data with 65 of these shorter, better defined sequences exhibiting over a two-fold change in expression (Table 2, Supplementary Files 3,4). Unsupervised hierarchical clustering of the expressions of these 71 novel sRNAs identified four principle clusters. Nearly 2/3 of the putative sRNAs make up the largest cluster characterized by expression almost exclusively at 24 h of desiccation. In contrast, 13 genes are predominately expressed during normal conditions then turned off during desiccation whereas only 3 sRNAs are the most highly expressed after 72 h of desiccation (Figure 1).
Figure 1.
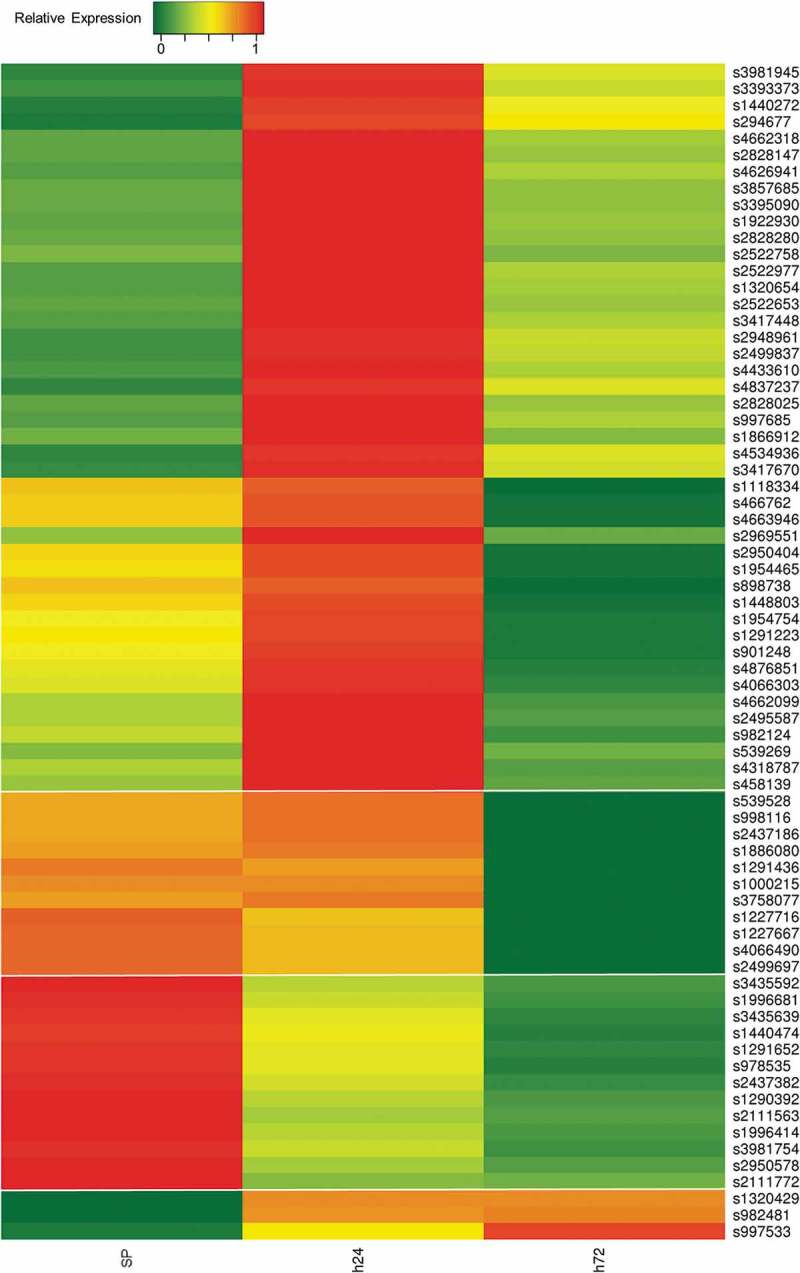
Gene expression analysis of novel desiccation-related sRNA genes. Heatmap showing relative expressions of novel desiccation-related sRNA genes. SP, stationary phase; h24, 24 h desiccated; h72, 72 h desiccated. Four gene clusters (demarcated by breaks) were defined in accordance with unsupervised hierarchical clustering [28].
Next, to determine similarities between our novel sequences and other known bacterial sRNAs, we aligned our putative sRNAs to the non-coding RNA databases Rfam and sRNATarbase [29,30] and also examined their probable secondary structures via Mfold [31]. Notably, we found 55 of our putative sRNA gene loci are well conserved (at least 80% identical over their entire length) in the genomes of related bacterial genera (Figure 2), and that 13 of our novel sRNAs correspond to annotated sRNAs or other noncoding RNA species in related bacteria (Table 2). In addition, we found another of our putative sRNAs (sRNA901248) shares an identical 23 base pair sequence with RybA, a known SL1344 sRNA we also find differentially expressed during desiccation [32] (Table 1). Furthermore, Mfold analyses indicate over 90% of our candidate sRNAs likely adopt complex secondary structures consisting of a series of short stem loops similar to structures of characterized bacterial sRNAs [33] (Figure 3, Table 2, Supplementary File 2).
Figure 2.

Multiple alignments of candidate sRNAs with genomic sequences from other bacterial genera. (a) sRNA294677. (b) sRNA1118334. (c) sRNA978535. Genomic sequences were retrieved from NCBI using BLASTN [55] search for bacterial RNA. Alignments were generated using ClustalW [56]. *, 100% nucleotide identity.
Figure 3.
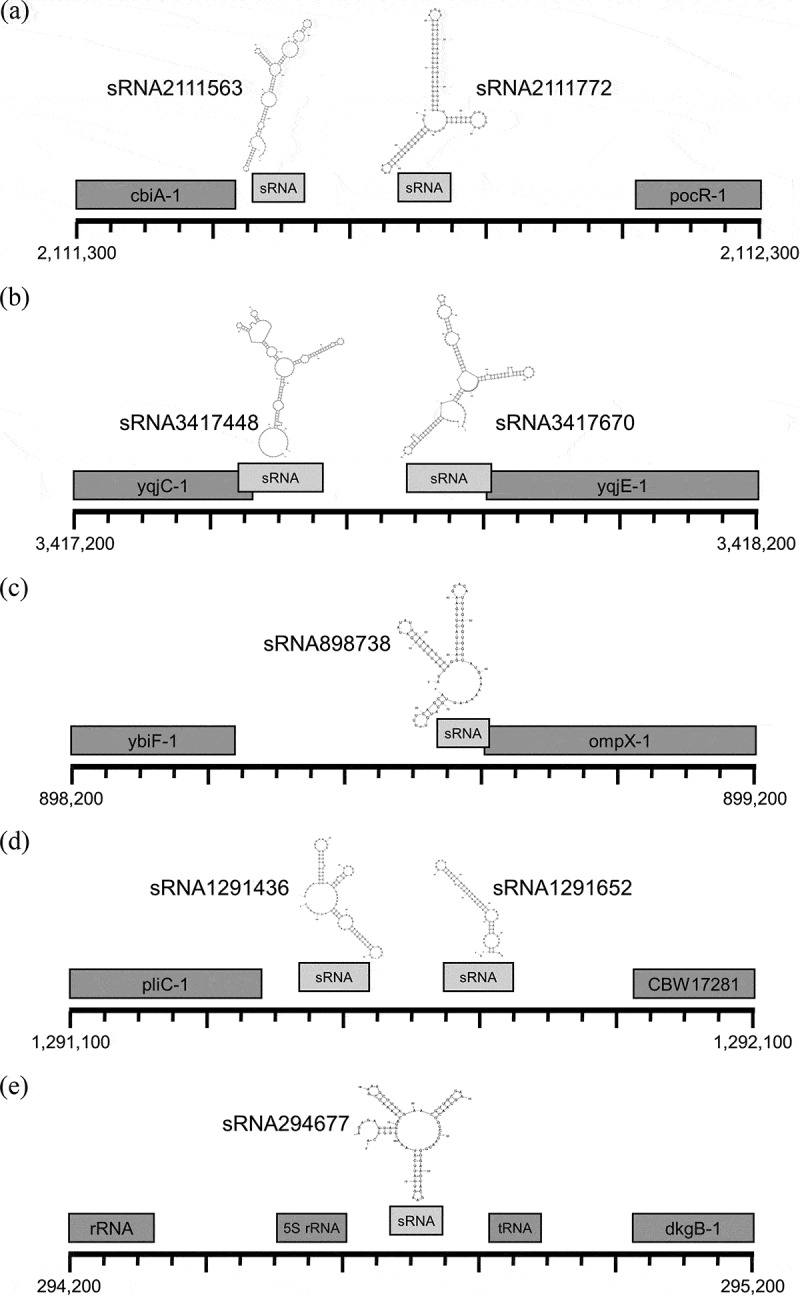
Select sRNA loci. Cartoons depicting 1kb genomic regions roughly centred on 1 or 2 sRNA sequences are illustrated. The most thermodynamically stable secondary structures of individual sRNAs (as predicted by Mfold [31]) are included above respective sRNA sequences (light grey). Names and positions of neighbouring genes occurring within the 1kb region as defined in the current Ensembl build [44] are also indicated (dark grey). SL1344 chromosomal positions are shown (bottom left and right). (a) sRNA2111563 and sRNA2111772 locus. (b) sRNA3417448 and sRNA3417670 locus. (c) sRNA898738 locus. (d) sRNA1291436 and sRNA1291652 locus. (e) sRNA294677 locus.
Confirmation of sRNA expressions
Differential expressions of five annotated and five putative sRNAs were verified by qRT-PCR (Figure 4(a,b)). Importantly, all qPCR-based sRNA expression trends largely agreed with RNA-seq-based expressions (Tables 1 and 2, Figure 1, Supplementary File 3). The magnitude of changes in expression, however, did at times differ notably between techniques. For example, the RNA-seq-based expression of STnc3920 at 72 h was determined to be ~10x its expression in SP controls (Table 1) whereas its qRT-PCR -based expression at 72 h was calculated as a more modest 3x that of controls (Figure 4(a)). That said, the disparity between STnc3920 changes in expression was by far the most pronounced with sRNA relative expression changes typically agreeing within 50% between methods (Tables 1 and 2, Figure 4(a,b)).
Figure 4.
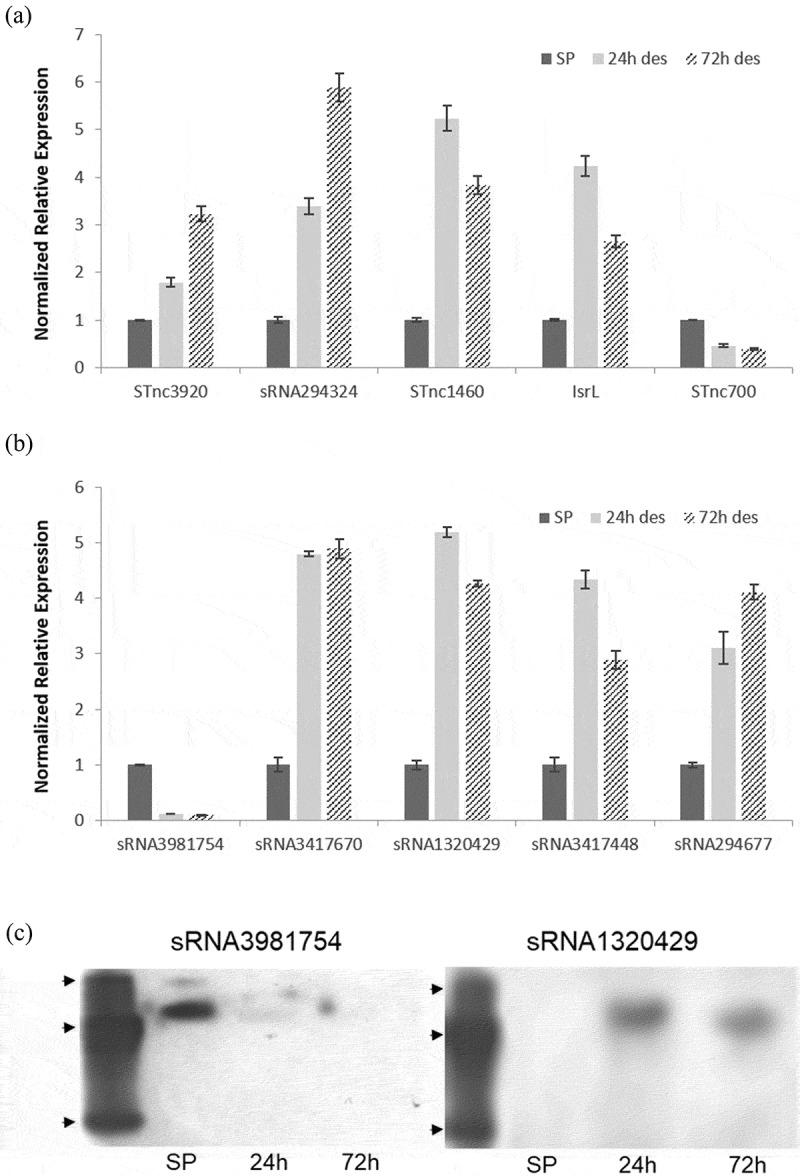
qRT-PCR and small transcript northern validation of sRNA differential expressions. (a) Annotated sRNA qPCR expressions. (b) Putative sRNA qPCR expressions. The specificity of each amplification was verified via melting curves, and a control without reverse transcriptase was included in parallel. Gene expressions were calculated via the Delta-Delta cycle threshold method [56]. SP, stationary phase; 24 h des, 24 h desiccated; 72 h des, 72 h desiccated. Expressions were normalized to SP. Error bars in A and B indicate SD (n = 3). (c) sRNA3981754 (left) and sRNA1320429 (right) small transcript northern blot. Arrowheads indicate 25, 50 and 200 nt oligonucleotide bands in custom ssDNA 5ʹ biotinylated ladder. SP, stationary phase. 24 h, 24 h desiccated. 72 h, 72 h desiccated.
Next, we elected to confirm the existence and the changes in expression of two of the sRNAs described here by small transcript northern blotting, a more direct and definitive method. This allowed us to confirm that proposed sRNAs form discrete species rather than being part of longer transcripts. We found northern analysis of the first of these, sRNA3981754, largely confirmed our predicted length of 87nts (Figure 4(c)) and in agreement with our expression analyses. That said, we found northern blotting more closely agreed with our qRT-PCR -based expression analyses than with our RNA-seq-based expressions. RNA-seq-based expressions for sRNA3981754 were 187.6, 75.8, and 21.6 transcripts per million during SP, 24 h, and 72 h of desiccation, respectively (Table 2). In contrast, while our qRT-PCR likewise found sRNA3981754 to be most highly expressed during SP, it determined expression decreased by 88% by 24 h of desiccation then decreased even further by 72 h (Figure 4(b)) closely paralleling our northern analyses. Similarly, northern analysis of sRNA1320429 generally confirmed our predicted length of 78 nts (Figure 4(c)) and in agreement with our expression analyses suggesting sRNA1320429 expression increases by 5 to 15 fold during desiccation (Figure 4(b), Table 2).
Deletion of novel sRNAs can significantly impair the Salmonella desiccation response
To explore potential functional roles of novel sRNAs during the desiccation response, we selected sRNA1320429 and sRNA3981754 for deletion analysis, as we found both significantly and dynamically expressed in response to desiccation and were able to successfully confirm their expressions and identitites by northern (Figure 4). The Lambda-Red recombinase method was employed to delete each of these sRNAs [34] resulting in the creation of two mutant Salmonella strains: ∆13204 and ∆3981 for subsequent phenotypic testing. Deletions were verified by PCR confirmation of sRNA (~100–200 bp) replacement with a chloramphenicol resistance cassette (1,034 bp) (Figure 5(a)).
Figure 5.
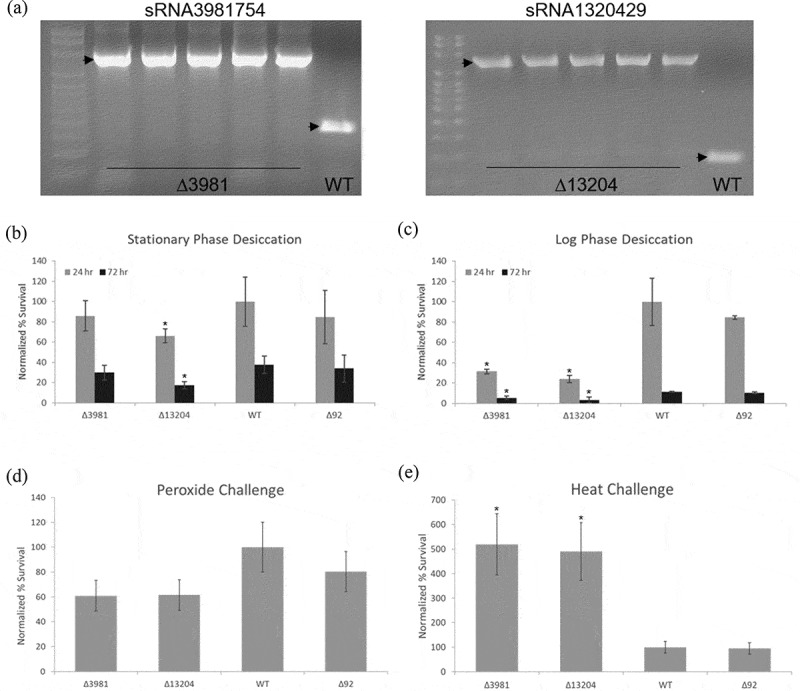
Effects of silencing sRNA1320429 and sRNA3981754. (a) PCR confirmation of sRNA genomic replacement with chloramphenicol resistance cassette. Arrowheads indicate amplicons generated utilizing primers flanking indicated sRNA loci. Lane 1, l00 bp ladder. Lanes 2-6, mutant colonies (harbouring ~1,000 bp insertions at indicated loci). Lane 7, wild type sRNA amplicons (<200 bp). (b) Desiccation of stationary phase deletion mutants and wild type Salmonella. Survival determined after desiccation for 24 and 72 h by comparison to wild type samples maintained under the same conditions. (c) Survival of log phase deletion mutants and wild type Salmonella subjected to desiccation for 24 and 72 h. (d) Survival of peroxide-challenged mutants and wild type Salmonella. (e) Survival of mutant and wild type Salmonella after heat exposure (55°C) for 45 min. Error bars in B-E indicate SD (n = 5). Survivals were normalized to wild type (WT). * indicates p ≤ 0.05; p-values determined by unpaired two-tailed t-test. ∆13204, sRNA1320429 deletion mutant; ∆3981, sRNA3981754 deletion mutant; ∆92, sRNA deletion mutant previously generated using the same methodology [15].
Since these sRNAs were identified by their dynamic expression in response to desiccation, we subjected deletion mutant strains to conditions identical to those used during our initial desiccation response sequencing analysis. When SP Salmonella were subjected to these same desiccation conditions, we found ∆13204 survival was significantly lower, ~34% less than controls after 24 h of desiccation (p = 0.0067) and ~55% less than wild type Salmonella after 72 h (p = 0.0017) as compared to controls. Similarly, reduced but less pronounced effects on survivability were observed for ∆3981 (Figure 5(b)). More strikingly, when log phase Salmonella mutants were subjected to these same desiccation conditions, both deletion mutants showed roughly a 70% to 80% lower survival rate than wild type Salmonella or a control sRNA deletion mutant (∆92) [15] at 24 h (p = 0.000297 and p = 0.000271) and similarly significant lower survivals at 72 h (Figure 5(c)). This is expected as bacteria in an exponential phase of growth are generally thought to be more susceptible to various environmental stresses due to the inherent vulnerability of highly prolific and metabolically active systems [35–37]. Importantly, effects of sRNA overexpression on survival of log phase deletion mutants and wild type Salmonella subjected to desiccation for 24 and 72 h was also determined. We find transformation of sRNA deletion mutants with complementary sRNA expression vectors fully corrects impaired survivability during desiccation. In addition, we find transformation of a sRNA3981754 expression vector similarly conveys enhanced desiccation survivability to control strains (Supplementary File 5).
Deletion of novel sRNAs can significantly impair the Salmonella response to other stresses
To determine whether deletions of these sRNAs similarly affected survivability in response to other stresses, we next evaluated the effects of heat and peroxide exposure. While both desiccation sRNA deletion mutants did consistently exhibit approximately 20% to 40% lower survivability after hydrogen peroxide exposure depending on controls, observed decreases were not statistically significant (Figure 5(d)). In contrast, we found deletion of either sRNA conferred a marked effect on survivability during heat challenge. After exposure to 55°C for 45 min, we observed significantly enhanced survivability for both mutants (∆13204, p = 0.0282 and ∆3981, p = 0.0158) with each surviving over five times greater than wild type Salmonella or a control sRNA mutant (∆92) [15] (Figure 5(e)).
Prediction and analysis of potential targets
Many sRNAs have been found to regulate their neighbouring transcripts [38]. Therefore, as an initial means of identifying potential regulatory targets of our novel sRNAs, we determined the proximity of our sequences to neighbouring genes in the SL1344 annotated reference genome and found that all 71 of our sequences were located within 300 bp of an annotated SL1344 gene, including ribosomal RNA, transfer RNA, and protein-coding loci (Table 2, Supplementary File 2). However, while some sRNAs do target neighbouring genes, proximity is often not a requirement for regulation, and as the majority of sRNAs have been shown to regulate genes from distant loci in trans, IntaRNA [39] was principally employed to predict potential sRNA-target interactions based on the thermodynamic energy requirements for hybridization and site accessibility resulting in a total of 144 predicted targets (Table 2).
We next mapped 87 of 130 predicted target gene GIs (GenInfo Identifiers) to 1507 UniProtKB IDs, and mapped an additional 32 of the remaining GIs to the UniParc sequence archive. Target genes were then clustered according to gene ontology (molecular function) and enzyme class (Table 2, Supplementary File 6). Interestingly, ~89% of the predicted targets of sRNAs differentially expressed in desiccation are associated with catalytic, binding or transcription regulator activities, and ~75% of the predicted enzyme targets correspond to transferases, oxidoreductases and lyases (Supplementary File 6).
Discussion
Salmonella is one of the most challenging bacteria for food manufacturers and is a leading cause of enterocolitis worldwide. As the food processing chain commonly involves dehydration as a means of long-term food storage, desiccation is an important environmental stress encountered by Salmonella [40]. As such, a better understanding of the desiccation stress response of this pathogen is therefore of great practical importance. Such knowledge is critical for the development of effective strategies for commercial application in control of potential bacterial contaminants. Although low water activity has been associated with an increase of Salmonella resistance to heat and other severe conditions, often contributing to foodborne outbreaks, there is a lack of information regarding regulatory genes that may act on these specific pathways [40]. As such, to gain further insight into the mechanisms underlying Salmonella responses to desiccation, in this work we have identified 173 intergenic sRNAs differentially expressed during desiccation. While 102 of these sRNAs have been previously annotated, to our knowledge, this is the first work describing changes in their expressions during (and potential to act as regulators of) this pathogen’s desiccation response system. Notably, in addition to these 102 previously annotated sRNAs our transcriptomic analyses have simultaneously identified 71 novel, putative sRNAs likewise differentially expressed in response to desiccation. While qRT-PCRs of five of these strongly agree with RNA-seq-based expressions, the lengths and identities of only two were directly confirmed by small transcript northern blotting (Figure 4), and similar experiments will ultimately be required to confirm that each of the remaining putative sRNAs truly represent bona fide sRNAs and definitively exclude the possibility that they represent RNA-seq/qRT-PCR bias artefacts.
That said, unsupervised hierarchical clustering of the expressions of the 71 novel sRNAs described here identified four principle clusters. Nearly 2/3 of the putative sRNAs make up the largest cluster characterized by expression almost exclusively at 24 h of desiccation (Figure 1). Notably, clustering of the expressions of the 102 annotated sRNAs we also find differentially expressed during desiccation (Table 1) closely resembles that of our novel sRNAs in that 72 of the 102 annotated sRNAs group together into a single predominant cluster likewise characterized by expression almost exclusively at 24 h of desiccation (Supplementary File 7). Of note, the two sRNAs examined in our deletion analyses were in large part selected based on their opposing expression profiles. We find sRNA1320429 levels to be negligible during SP and that it is primarily expressed during desiccation. In contrast, sRNA3981754 is most highly expressed during SP, and its expression is decreased by at least 60% after 24 h of desiccation (Figure 4). As such, we initially hypothesized that a loss of sRNA1320429 would impair desiccation survival whereas a loss of sRNA3981754 would likely have no effect. Much to our surprise, whereas we found deletions of other unrelated sRNA loci had little to no effect on desiccation survivability, we found knocking out either of these sRNAs significantly impairs the ability of cells to survive desiccation while conversely conferring an enhanced survivability during heat challenge (Figure 5). While the mechanisms responsible for these phenotypic observations remain unclear, we feel it suggests both of these sRNAs are necessary for initial survivability during desiccation and that these sRNAs are likely involved in regulating the same or related genetic pathways.
Importantly, recent reports have demonstrated the association of several distinct sRNAs in regulatory stress responses and virulence in Salmonella. Salmonella is quickly becoming a model organism for RNA-mediated regulation, with hundreds of novel sRNAs recently identified by transcriptomic studies [15,24,25]. Of note, our previous work identified uncharacterized sRNAs that are differentially expressed under starvation stress response (SSR). Remarkably, we found that 58 of the 63 sRNAs we identified under SSR conditions had not been previously characterized in Salmonella or other Enterobacteriaceae [15]. These results clearly suggest that sRNA regulation in Salmonella remains largely unexplored and that the discovery of novel sRNAs is an essential first step to understanding how sRNAs control pathogenesis and adaptation processes in response to environmental changes.
While Salmonella sRNA discovery is proceeding robustly, the identification of targets now becomes a critical bottleneck for further progress in this field. In this report, we used a combinational strategy taking both proximity and hybridization kinetics into consideration in order to identify the most likely targets of desiccation-induced sRNAs. Of note, predicted primary targets of all differentially expressed sRNAs were clustered according to their gene ontology (GO) terms, focusing on their molecular function. Interestingly, we find the targets of sRNAs differentially expressed in desiccation appear to present mainly catalytic, binding and transcription regulator activities (Supplementary File 6), and the enzymes predicted to be the most affected mostly correspond to transferases, oxidoreductases and lyases. That said, one sRNA differentially expressed in 24 and 72-h desiccated cells, sRNA2111563, was predicted to interact with and potentially repress proP, which encodes an osmoprotectant proline/betaine transporter crucial for osmoprotection and survival of desiccated Salmonella enterica Serovar Typhimurium [41]. We find this novel sRNA significantly repressed after 72 h of desiccation, suggesting a higher reciprocal expression of proP. In fact, proP is known to be overexpressed in low-moisture environments, and mutant ∆proP shows significant reduction in viability when compared to wild type strain in the same conditions [41]. In addition, we find another of our putative sRNAs (sRNA1448803) is predicted to regulate ssaL-1 a type III secretion system apparatus protein previously shown to be necessary for surviving desiccation [42]. Although experimental assays will be required to confirm the role of these sRNAs (and several of our other putative sRNAs) in regulating the desiccation stress response, we find it exciting that several of our predicted targets are associated with characterized desiccation responses in Salmonella (Table 2). As such, we now suggest that these logical target associations, taken with our demonstration that a loss of specific sRNAs identified in this study can significantly impair the ability of Salmonella to survive desiccation, strongly indicate that many of the sRNAs described in this report represent uncharacterized, critical regulators of desiccation [25,43].
Of note, the 71 novel sRNAs described here bring the total number of Salmonella enterica serovar Typhimurium sRNAs to over 450 [15,24,25,41,44–48]. Excitingly, mounting evidence suggests there are many additional Salmonella sRNAs yet to be described. Although our work only considered intergenic sRNAs, recent reports suggest that functional ncRNAs can also be derived from gene untranslated regions (UTRs), and that some sRNAs are entirely embedded within protein-coding regions [49]. As such, it is tempting to speculate that sRNAs may actually outnumber protein-coding genes in this and perhaps many additional prokaryotic genomes. That said, the functional dissection of the specific roles for each of these novel genetic regulators represents the next major hurdle to realizing a more thorough understanding of the genetic networks driving bacterial biology.
Materials and methods
Strain and growth conditions
Salmonella enterica serovar Typhimurium strain SL1344 was grown and maintained on LB agar (1% w/v tryptone/1% w/v NaCl/0.5% w/v yeast extract/1.5% w/v agar). To prepare the cell suspension, a fresh overnight-grown culture was streaked onto 2 LB Agar plates and grown overnight at 37ºC. On the next day, a sterile cell scraper was used to harvest the cells. The scraped cells were washed 3 times with sterile distilled water for 5 min at 5,000 g. At the end of the last wash, water was added up to reach OD600 = 1.0 (~ 8 × 108 cells/ml).
Salmonella desiccation and small RNA sequencing
Cells were desiccated following a protocol used in a previous work examining the Salmonella transcriptome under dehydration [50], with minor modifications. Petri dishes containing 15 mL of the cell suspension were left open inside of a laminar flow hood (relative humidity of 40%) for up to 72 h at room temperature. Control samples were maintained at the same conditions but in water suspension in a 50 mL conical tube. 24 and 72-h cells were harvested from Petri dishes by adding 10 mL RNA storage solution (25 mM sodium citrate, 10 mM EDTA, 70% w/v ammonium sulphate, pH 5.2). Cells were pelleted (4,500 rpm for 10 min at room temperature) and washed two times with a sterile PBS solution. The final pellet was resuspended in 1 mL PBS solution, then samples frozen in liquid nitrogen and stored at −80ºC. Final frozen samples were shipped to the Genomic Services Lab at HudsonAlpha (HudsonAlpha Institute for Biotechnology, Huntsville, AL, USA) where RNA isolation and small RNA-seq using an Illumina HiSeq v4 genome sequencer were performed (paired ends, 100 bp, 25M reads). HudsonAlpha performed RNA isolations following standard Trizol (Invitrogen) protocols then employed the NEBNext Multiplex Small RNA Library Prep Set for Illumina (NEB) coupled with automated agarose gel size selection (30 to 200 nt) using the Pipin Prep Instrument (Sage Science) for small RNA library prep. Adapter sequences were removed with cutadapt [51], and reads were mapped against S. Typhimurium SL1344 genome using BWA-MEM [52].
Identification and analysis of known small RNAs (sRNAs)
Reads from the RNA-seq library were mapped against the intergenic regions of SL1344 reference genome and known sRNAs from Salmonella using the CLC Genomics Workbench (CLC bio – v9.0; Finlandsgade, Dk) with the following parameters: mapping settings (minimum length fraction = 0.9, minimum similarity fraction = 0.8, and maximum number of hits for a read = 15) and paired settings (minimum distance = 180 and maximum distance = 250, including the broken pairs counting scheme). Expression values were reported in reads per kilobase of exon model per million mapped reads (RPKM), and the normalized value for each sample was calculated in transcripts per million (TPM). The expression data were log2 transformed and normalized to identify sRNAs exhibiting a fold change ≥2.0 during desiccation.
Identification of sRNA consensus sequences
To define consensus sRNA sequences with inconclusive mapping from RNA-seq data, Integrative Genomics Viewer (IGV) was employed to refine the sRNA locations within the larger intergenic regions of the SL1344 chromosome [53,54]. Indexed BAM files for each culture sample (SP, 24 h desiccated, and 72 h desiccated) and a BED file containing chromosomal positions for the intergenic regions were uploaded to IGV to determine expression levels within these regions along the SL1344 chromosome. Chromosomal position of peak expression within each intergenic region was determined and chromosomal positions (upstream and downstream) where peaks fell below 75% of the maximum were identified in each sample. Start and stop positions in each of the three samples where a putative sRNA was expressed were averaged resulting in the final sRNA consensus sequence calls.
Computational analyses of novel sRNAs
After definition of sRNA, consensus sequences through peak calling in IGV, the newly identified sRNA sequences were aligned to the original raw RNA-seq read data for each sample (SP, 24 h desiccated, and 72 h desiccated) using BLAST+ (2.2.27) with best hit parameters and an e-value of 1e−1 [55]. Alignments were also required to have percent identity ≥95% and length ≥28 bp (-evalue 1e-1 -best_hit_score_edge 0.05 -best_hit_overhang 0.25 -perc_identity 95 -max_target_seqs 1). Resulting alignments were then counted for each unique sequence read (sRNA) in each of the three samples, and counts were normalized using the total reads from the raw RNA-seq data for each sample to allow comparison of expression across all three sample conditions. Candidate sRNA sequences were aligned to Rfam, sRNATarBase, and SalCOM databases to determine similarity to known sRNAs [24,25,29,30]. NCBI BLAST megablast tool (word size: 16) was used to search for homologous sequences in all available bacterial genomes (excluding Salmonella). SRNAs were considered conserved if genomic sequences with ≥80% identity to the full length of a novel sRNA were identified in at least one other bacterial genera [56]. All RNA and protein-coding genes located within 300 bp upstream or downstream of the sRNA in Ensembl were also identified to analyse the expressions of flanking and overlapping genes [44], and putative sRNA expressions required to differ from overlapping genes by ≥200%. The computational tool ‘IntaRNA’ (http://rna.informatik.uni-freiburg.de) was used to predict potential, primary sRNA targets, and target genes were further analysed using Blast2GO and UniProtKB (https://www.uniprot.org/) [39,55,57]. Predicted secondary structures for each sRNA were generated using Mfold [31].
Real-time quantitative qRT-PCR analysis
Total RNA was isolated from control (24-h water-suspended) and desiccated samples (24 and 72 h) using the RNeasy PowerMicrobiome Kit (Qiagen, Cat No.:26000–50). qPCR was performed using the iTaqTM Universal SYBR® Green One-Step Kit (Bio-Rad, Cat No.: 172–5150) according to the manufacturer’s instructions. Five annotated genes were selected to validate the small RNA-Seq data: STnc3920, STnc1460, STnc700, IsrL and sRNA294324. Five previously unannotated genes were also selected for validation: sRNA3981754, sRNA3417670, sRNA1320429, sRNA3417448, and sRNA294677. The gene rpoD was used as a control to normalize the values as it has shown not to be differentially expressed under these conditions. The reactions were performed in duplicate in a 384-well plate containing 5 μL of iTaq universal SYBR® Green reaction mix, 0.125 μL of the iScript reverse transcriptase, 300 nM of each forward and reverse primers, 45 ng of RNA and nuclease-free water to a total reaction mix volume of 10 μL. Primers are listed in Supplementary File 8. qRT-PCR was conducted in the CFX384 Touch Real-Time PCR Detection System (Bio-Rad). The real-time PCR program was as follows: initial reverse transcription reaction at 50ºC for 10 min, polymerase activation and DNA denaturation at 95°C for 1 min, followed by 40 cycles of amplification at 95ºC for 10 sec and 60ºC for 15 sec. All the PCR amplifications were performed in triplicates. DNase-treated RNA was used in all sample extractions, and a control without reverse transcriptase was included. Specificity of amplifications was verified using melting curves. Gene expression was calculated via the Delta-Delta cycle threshold method [58].
Deletion and over expression of select sRNAs
Select sRNAs were deleted from the SL1344 genome to analyse the phenotypic effects of their removal under various conditions. The sRNAs chosen for deletion were sRNA1320429 and sRNA3981754, and deletion mutants were generated employing the previously described Lambda-Red recombinase method [17,34]. Briefly, lambda-red recombinase genes from the pKD46 plasmid were induced with arabinose in wild type SL1344 cells. The pKD3 plasmid was used to amplify the chloramphenicol resistance cassette flanked by sequences corresponding to the sRNAs selected for knockout. Resulting SL1344 mutant strains with chloramphenicol resistance in place of either sRNA3981754 or sRNA1320429 were confirmed by colony PCR before use in further analysis. ∆92, an sRNA deletion mutant we previously generated using the same methodology [15] was utilized as an sRNA deletion control as it had previously been identified by both our group (as sRNA924744) and independently (as STnc1200) [24].
Following Lambda Red protocol, colonies having incorporated the antibiotic resistance cassette (selected by growth on chloramphenicol plates) were re-suspended in 10 μL of 1x PBS and boiled for 15 min at 95°C. Samples were then pulse centrifuged to remove cell debris and 2 μL of resulting supernatant used for colony PCR amplifications. Prior to visual confirmation by being run on a 1.5% agarose gel and imaged with EtBr, colony PCR amplifications were performed in 25 μL reactions at standard concentrations (2.5 μL of 10x NH4 PCR buffer, 1.25 μL of 50mM MgCl2, 1 μL of 10mM dNTP, 0.2 U Taq (Bioline USA, Inc., Randolph, MA), 0.5 µM each primer) and using standard cycling parameters (94°C – 2 min (94°C – 45 s, 56°C – 30 s, 72°C – 45 s) × 30 cycles, 72°C – 2 min), using the following primers:
sRNA13204F – CGCAGGATAGCGAGCAATAACC
sRNA13204R – CGCAGTGCTTATGCCAACGC
sRNA3981F – GCAGAATGGGCGGATGTATATAC
sRNA3981R – CTAACGCGTTTTGGCGAGCAC.
PCR amplifications of expression construct inserts were performed in 40 μl reactions at standard concentrations (1.5 mM MgCl2, 0.2 mM dNTP, 1x Biolase PCR buffer, 0.5 U Taq (Bioline), 0.5 uM each primer) and using standard cycling parameters (94°C – 3 min (94°C – 30 s, 55°C – 30 s, 72°C – 60 s) x 30 cycles, 72°C – 3 min). sRNA expression vector sRNA insert primers 5ʹ to 3ʹ were: s132f – CAGCGAGCGATGATTTG; s132r – GCGAAAAAACGAAAGGATGG; s398f – GTACCAATGGATACCG; s398r – GAGCGTACACGTAGTACG. Resulting amplicons were separated on a 1% agarose gel and a band excised from the appropriate lane at ~350 bp. Gel extractions were then cloned into Topo TA PCR 2.1 and sequenced. Resultant amplicons were cloned into Topo PCR 2.1 and sequenced. RT-PCR reactions were performed to confirm expression using the Tetro cDNA synthesis kit (Bioline) and random hexamer primers per standard manufacturer protocols followed by PCR amplifications (as above) with the following primers:
S132rtF53 – GTCGGGAAAGTCGGG with
S132rtR53 – GGCAGAGCGGCTATTAATG and
S398rtF53 – ATCAGCTACTGATTGAAAGTTATACC with
S398rtR53 – TTATTTATGCGCGTTGAGAATCC.
For sRNA add-back expression experiments, deletion mutants and controls were grown to mid-log phase and subsequently washed 2x with sterile ddH2O and 2x with sterile 10% (v/v) glycerol solution to allow for electro-competency. Using Bio-Rad E. coli Pulser (1.8kV, 0.1 cm E cuvette), 60uL aliquot of each EC mutant or control was immediately electroporated with 850 ng of TOPO vector sRNA expression construct or empty TOPO control and plated on Ampicillin-selective agar plates.
Desiccation survival
To assess the effects of removing and/or over expressing selected sRNAs from the SL1344 genome, mutant and wild type strains were desiccated under the same conditions as described previously [42]. After 24 and 72 h, desiccated cells were harvested from Petri dishes using a sterile cell scraper and resuspended in 10 mL sterile distilled water. Desiccated resuspensions and control samples were serially diluted (1:5) in sterile distilled water, spread onto LB agar plates, and incubated overnight at 37°C. Colony-forming units (CFUs) were determined for countable dilutions, and CFUs between 30 and 300 were used to calculate desiccation survival for each sample. The same procedure was also repeated using Salmonella cells in log-phase growth.
Heat and peroxide challenge
To assess tolerance to heat and hydrogen peroxide challenges, deletion mutants and wild type Salmonella were exposed to these conditions under the following parameters. Cultures were grown overnight in LB broth with shaking at 37°C. The next morning, overnight cultures were inoculated 1:100 into fresh LB broth and grown to an OD600 of approximately 0.3–0.4 to generate log-phase cells. Aliquots of log-phase culture were serially diluted (1:5) in sterile distilled water and plated on LB agar to serve as control. For challenge with hydrogen peroxide, 10 µL of H2O2 (1M) and 10 µL of log-phase culture were added to 980 µL of fresh LB broth and incubated at 37°C with shaking for 45 min. For heat tolerance assessment, log-phase cultures were incubated at 55°C with shaking for 30 min. Following each treatment, aliquots from each sample were serially diluted and plated in the same manner as control, and all plated samples were incubated at 37°C overnight. CFUs were then determined and used to calculate survival under each condition.
Small transcript northern blots
Total RNA from SL1344 cultures was isolated with Trizol® (Life Sciences) per standard manufacture protocol. A 15% acrylamide/bis-acrylamide (29:1) gel containing 8 M urea (48% (w/v)) and 1X TBE was prerun for 30 min at 100 V in a vertical mini-PROTEAN tank (Bio-Rad). Gels were flushed and loaded with 10 µg of total RNA in 2X TBE/Urea sample buffer (Bio-Rad), then run at 200 V until the bromophenol blue dye front reached the gel bottom. As a size reference, 1 µl of pooled, commercially synthesized biotin 5ʹ end–labelled DNA oligonucleotides (25, 50 and 200 bp each at 1 µM) was also loaded in 2X TBE/Urea sample buffer. After removal from the electrophoresis plates, gels were gently rinsed with water then washed in 0.5X TBE for 5 min on an orbital shaker. After electrophoresis, RNA was electro-transferred (Mini Trans-Blot Electrophoretic Transfer Cell apparatus, Bio-Rad) to Biodyne B Pre-cut Modified Nylon Membranes 0.45 µm (Thermo Scientific) for 2 h at 20 V in 0.5X TBE. After removal from the transfer stack, membranes were gently washed in 1X TBE for 15 min on an orbital shaker, then UV cross-linked at 1200 mJ for 2 min (Stratalinker, Stratagene). Prehybridization was performed in North2South® Hybridization Buffer (Thermo Scientific) at 42°C for 30 min, after which 30 ng (per millilitre of hybridization buffer) of each appropriate biotin 5ʹ end–labelled oligonucleotide was added directly to the hybridization buffer as probe.
Probe1320429 5pBio- CAGTGTCGACCATATTAGGCTCGCCGATAG
Probe3981754 5pBio- AGCTACTGATTGAAAGTTATACCAAAGCGC
Probe1320429ctl 5pBio- CTATCGGCGAGCCTAATATGGTCGACACTG
Probe3981754ctl 5pBio- GCGCTTTGGTATAACTTTCAATCAGTAGCT
Blots were hybridized overnight with gentle rotation at 23°C. Hybridization buffer was removed the following day, and membranes washed and developed using the Thermo Scientific™ North2South® Chemiluminescent Hybridization and Detection Kit per manufacturer instructions then imaged on a LI-COR C-DiGit Chemiluminescent Blot Scanner.
Funding Statement
Funding was provided in part by NSF CAREER grant 1350064 (GMB) awarded by Division of Molecular and Cellular Biosciences (with co-funding provided by the NSF EPSCoR program). Graduate funding was also provided in part by Alabama Commission on Higher Education ALEPSCoR grants 150380 (JTR),160330 (VMK), and 180435 (DH). Postdoctoral funding was provided by São Paulo Research Foundation (FAPESP) Grants # 2014/17387-8 and # 2015/19400-4 (AC). A.S.Sant’Ana acknowledges the support of ‘Conselho Nacional de Desenvolvimento Cientifico e Tecnológico’ (CNPq) (Grants #302763/2014-7; #305804/2017-0); Alabama Commission on Higher Education [180435]; Alabama Commission on Higher Education [160330]; Alabama Commission on Higher Education [150380]; Conselho Nacional de Desenvolvimento Científico e Tecnológico [305804/2017-0]; Fundo de Apoio ao Ensino, à Pesquisa e Extensão, Universidade Estadual de Campinas [2015/19400-4]; Fundo de Apoio ao Ensino, à Pesquisa e Extensão, Universidade Estadual de Campinas (BR) [2014/17387-8]; Conselho Nacional de Desenvolvimento Científico e Tecnológico (BR) [302763/2014-7].
Acknowledgments
This work was facilitated by the University of South Alabama (USA) College of Arts & Sciences Department of Biology, the USA College of Allied Health Professions Department of Biomedical Sciences, and the USA College of Medicine Department of Pharmacology. This study was also financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental Material
Supplemental data for this article can be accessed here.
References
- [1].Hombach S, Kretz M.. Non-coding RNAs: classification, biology and functioning. Adv Exp Med Biol. 2016;937:3–17. [DOI] [PubMed] [Google Scholar]
- [2].Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014. March;157(1):77–94. [DOI] [PubMed] [Google Scholar]
- [3].Fischer JW, Leung AKL. CircRNAs: a regulator of cellular stress. Crit Rev Biochem Mol Biol. 2017. April;52(2):220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sun X, Zheng H, Sui N. Regulation mechanism of long non-coding RNA in plant response to stress. Biochem Biophys Res Commun. 2018. September;503(2):402–407. [DOI] [PubMed] [Google Scholar]
- [5].Tehrani SS, Karimian A, Parsian H, et al. Multiple functions of long non-coding RNAs in oxidative stress, DNA damage response and cancer progression. J Cell Biochem. 2018. January;119(1):223–236. [DOI] [PubMed] [Google Scholar]
- [6].Hollins SL, Cairns MJ. MicroRNA: Small RNA mediators of the brains genomic response to environmental stress. Prog Neurobiol. 2016. August;143:61–81. [DOI] [PubMed] [Google Scholar]
- [7].Olejniczak M, Kotowska-Zimmer A, Krzyzosiak W. Stress-induced changes in miRNA biogenesis and functioning. Cell Mol Life Sci. 2018. January;75(2):177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Amaral PP, Dinger ME, Mattick JS. Non-coding RNAs in homeostasis, disease and stress responses: an evolutionary perspective. Brief Funct Genomics. 2013. May;12(3):254–278. [DOI] [PubMed] [Google Scholar]
- [9].Cavanagh AT, Wassarman KM. 6S RNA, a global regulator of transcription in Escherichia coli, Bacillus subtilis, and beyond. Annu Rev Microbiol. 2014. September;68(1):45–60. [DOI] [PubMed] [Google Scholar]
- [10].Holmqvist E, Wagner EGH. Impact of bacterial sRNAs in stress responses. Biochem Soc Trans. 2017;45:1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Perry RN. Desiccation survival of parasitic nematodes. Parasitology. 1999;119 Suppl:S19–S30. [PubMed] [Google Scholar]
- [12].Giarola V, Hou Q, Bartels D. Angiosperm plant desiccation tolerance: hints from transcriptomics and genome sequencing. Trends Plant Sci. 2017. August;22(8):705–717. [DOI] [PubMed] [Google Scholar]
- [13].Esbelin J, Santos T, Hébraud M. Desiccation: An environmental and food industry stress that bacteria commonly face. Food Microbiol. 2018. February;69:82–88. [DOI] [PubMed] [Google Scholar]
- [14].Sharma CM, Vogel J. Experimental approaches for the discovery and characterization of regulatory small RNA. Curr Opin Microbiol. 2009. October;12(5):536–546. [DOI] [PubMed] [Google Scholar]
- [15].Amin SV, Roberts JT, Patterson DG, et al. Novel small RNA (sRNA) landscape of the starvation-stress response transcriptome of Salmonella enterica serovar typhimurium. RNA Biol. 2016. March;13(3):331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Deng X, Li Z, Zhang W. Transcriptome sequencing of Salmonella enterica serovar Enteritidis under desiccation and starvation stress in peanut oil. Food Microbiol. 2012. May;30(1):311–315. [DOI] [PubMed] [Google Scholar]
- [17].Spector MP, Kenyon WJ. Resistance and survival strategies of Salmonella enterica to environmental stresses. Food Res Int. 2012. March;45(2):455–481. [Google Scholar]
- [18].Smirnov A, Förstner KU, Holmqvist E, et al. Grad-seq guides the discovery of ProQ as a major small RNA-binding protein. Proc Natl Acad Sci U S A. 2016. October;113(41):11591–11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vogel J, Sharma CM. How to find small non-coding RNAs in bacteria. Biol Chem. 2005. January;386(12):1219–1238. [DOI] [PubMed] [Google Scholar]
- [20].Hoe CH, Raabe CA, Rozhdestvensky TS, et al. Bacterial sRNAs: regulation in stress. Int J Med Microbiol. 2013;303(5):217–229. [DOI] [PubMed] [Google Scholar]
- [21].Oliva G, Sahr T, Buchrieser C. Small RNAs, 5ʹ UTR elements and RNA-binding proteins in intracellular bacteria: impact on metabolism and virulence. FEMS Microbiol Rev. 2015. May;39(3):331–349. [DOI] [PubMed] [Google Scholar]
- [22].Mandin P, Guillier M. Expanding control in bacteria: interplay between small RNAs and transcriptional regulators to control gene expression. Curr Opin Microbiol. 2013. April;16(2):125–132. [DOI] [PubMed] [Google Scholar]
- [23].Altuvia S, Weinstein-Fischer D, Zhang A, et al. A small, stable RNA induced by oxidative stress: role as a pleiotropic regulator and antimutator. Cell. 1997. July;90(1):43–53. [DOI] [PubMed] [Google Scholar]
- [24].Kröger C, Colgan A, Srikumar S, et al. An infection-relevant transcriptomic compendium for Salmonella enterica serovar Typhimurium. Cell Host Microbe. 2013. December;14(6):683–695. [DOI] [PubMed] [Google Scholar]
- [25].Kroger C, Dillon SC, Cameron ADS, et al. The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc Natl Acad Sci. 2012. May;109(20):E1277–E1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Calderón IL, Morales EH, Collao B, et al. Role of Salmonella Typhimurium small RNAs RyhB-1 and RyhB-2 in the oxidative stress response. Res Microbiol. 2014. January;165(1):30–40. [DOI] [PubMed] [Google Scholar]
- [27].Kim JN, Kwon YM. Genetic and phenotypic characterization of the RyhB regulon in Salmonella Typhimurium. Microbiol Res. 2013. January;168(1):41–49. [DOI] [PubMed] [Google Scholar]
- [28].Babicki S, Arndt D, Marcu A, et al. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 2016. July;44(W1):W147–W153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kalvari I, Argasinska J, Quinones-Olvera N, et al. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018. January;46(D1):D335–D342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang J, Liu T, Zhao B, et al. sRNATarBase 3.0: an updated database for sRNA-target interactions in bacteria. Nucleic Acids Res. 2016. January;44(D1):D248–D253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003. July;31(13):3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wassarman KM, Repoila F, Rosenow C, et al. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev. 2001. July;15(13):1637–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Schlüter J-P, Reinkensmeier J, Daschkey S, et al. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genomics. 2010. April;11(1):245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Murphy KC. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J Bacteriol. 1998. April;180(8):2063–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Evans DJ, Brown MR, Allison DG, et al. Susceptibility of bacterial biofilms to tobramycin: role of specific growth rate and phase in the division cycle. J Antimicrob Chemother. 1990. April;25(4):585–591. [DOI] [PubMed] [Google Scholar]
- [36].Mayhall CG, Apollo E. Effect of storage and changes in bacterial growth phase and antibiotic concentrations on antimicrobial tolerance in Staphylococcus aureus. Antimicrob Agents Chemother. 1980. November;18(5):784–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Purdy MA, Tenovuo J, Pruitt KM, et al. Effect of growth phase and cell envelope structure on susceptibility of Salmonella typhimurium to the lactoperoxidase-thiocyanate-hydrogen peroxide system. Infect Immun. 1983. March;39(3):1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vogel J, Wagner EGH. Target identification of small noncoding RNAs in bacteria. Curr Opin Microbiol. 2007. June;10(3):262–270. [DOI] [PubMed] [Google Scholar]
- [39].Busch A, Richter AS, Backofen R. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics. 2008. December;24(24):2849–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Podolak R, Enache E, Stone W, et al. Sources and risk factors for contamination, survival, persistence, and heat resistance of salmonella in low-moisture foods. J Food Prot. 2010. October;73(10):1919–1936. [DOI] [PubMed] [Google Scholar]
- [41].Finn S, Handler K, Condell O, et al. ProP is required for the survival of desiccated salmonella enterica serovar typhimurium cells on a stainless steel surface. Appl Environ Microbiol. 2013. July;79(14):4376–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mandal RK, Kwon YM. Global screening of salmonella enterica serovar typhimurium genes for desiccation survival. Front Microbiol. 2017. September;8:1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hébrard M, Kröger C, Srikumar S, et al. sRNAs and the virulence of Salmonella enterica serovar Typhimurium. RNA Biol. 2012. April;9(4):437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zerbino DR, Achuthan P, Akanni W, et al. Ensembl 2018. Nucleic Acids Res. 2018. January;46(D1):D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ryan D, Mukherjee M, Nayak R, et al. Biological and regulatory roles of acid-induced small RNA RyeC in Salmonella Typhimurium. Biochimie. 2018. July;150:48–56. [DOI] [PubMed] [Google Scholar]
- [46].Srikumar S, Kröger C, Hébrard M, et al. RNA-seq brings new insights to the intra-macrophage transcriptome of Salmonella Typhimurium. PLoS Pathog. 2015. November;11(11):e1005262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lévi-Meyrueis C, Monteil V, Sismeiro O, et al. Expanding the RpoS/σS-network by RNA sequencing and identification of σS-controlled small RNAs in Salmonella. PLoS One. 2014. May;9(5):e96918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Colgan AM, Kröger C, Diard M, et al. The impact of 18 ancestral and horizontally-acquired regulatory proteins upon the transcriptome and sRNA landscape of Salmonella enterica serovar Typhimurium. PLoS Genet. 2016. August;12(8):e1006258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dar D, Sorek R. Bacterial noncoding RNAs excised from within protein-coding transcripts. MBio. 2018. September;9(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gruzdev N, McClelland M, Porwollik S, et al. Global transcriptional analysis of dehydrated Salmonella enterica Serovar Typhimurium. Appl Environ Microbiol. 2012. November;78(22):7866–7875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chen C, Khaleel SS, Huang H, et al. Software for pre-processing Illumina next-generation sequencing short read sequences. Source Code Biol Med. 2014;9:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010. March;26(5):589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011. January;29(1):24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013. March;14(2):178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009. December;10(1):421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protocals Bioinf. 2002 Aug;Chapter 2:Unit 2.3. [DOI] [PubMed] [Google Scholar]
- [57].Götz S, Garcia-Gomez JM, Terol J, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008. June;36(10):3420–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.