

Abstract
The recent development of plants that overexpress antimicrobial peptides (AMPs) provides opportunities for controlling plant diseases. Because plants employ a broad-spectrum antimicrobial defense, including those based on AMPs, transgenic modification for AMP overexpression represents a potential way to utilize a defense system already present in plants. Herein, using an array of techniques and approaches, we report on VG16KRKP and KYE28, two antimicrobial peptides, which in combination exhibit synergistic antimicrobial effects against plant pathogens and are resistant against plant proteases. Investigating the structural origin of these synergistic antimicrobial effects with NMR spectroscopy of the complex formed between these two peptides and their mutated analogs, we demonstrate the formation of an unusual peptide complex, characterized by the formation of a bulky hydrophobic hub, stabilized by aromatic zippers. Using three-dimensional structure analyses of the complex in bacterial outer and inner membrane components and when bound to lipopolysaccharide (LPS) or bacterial membrane mimics, we found that this structure is key for elevating antimicrobial potency of the peptide combination. We conclude that the synergistic antimicrobial effects of VG16KRKP and KYE28 arise from the formation of a well-defined amphiphilic dimer in the presence of LPS and also in the cytoplasmic bacterial membrane environment. Together, these findings highlight a new application of solution NMR spectroscopy to solve complex structures to study peptide–peptide interactions, and they underscore the importance of structural insights for elucidating the antimicrobial effects of AMP mixtures.
Keywords: nuclear magnetic resonance (NMR), lipopolysaccharide (LPS), antimicrobial peptide (AMP), antibiotic resistance, peptide conformation, synergism, transferred NOESY
Introduction
Despite progress in the field of antimicrobial therapeutics (1, 2), plant production remains profoundly affected by microbial disease outbreaks caused by pathogens of Xanthomonas and Pseudomonas species, leading to considerable losses in crop production and challenges for global food security (3). Finding new approaches for the management of microbe-borne diseases in plants remains a challenge, further complicated by the increasing occurrence of multidrug-resistant pathogens (“superbugs”) that are immune to available treatments (4, 5). Additionally, commonly used bactericides and fungicides pose considerable environmental constraints (6), necessitating the development of sustainable management techniques that can effectively combat such pathogens.
Antimicrobial peptides (AMPs)3 represent the oldest domain of the evolutionary tree, being structurally and functionally conserved across all forms of life (7–10). While having attracted research interest for several decades, peptide-based antimicrobial therapy has seen accelerated attention during the last few years, motivated by its potential applicability against drug-resistant strains (11–15). The direct interaction of AMP with bacterial membranes and membrane components provides broad-spectrum antimicrobial effects (16), and for many AMPs also anti-inflammatory and other such effects (17, 18). Despite such potent host defense functionality, the therapeutic application of AMPs suffers from challenges related to dose-dependent toxicity, as well as scavenging and resulting activity loss by binding to anionic proteins, polysaccharides, and other compounds (19–21), which has motivated recent research on the combination of AMPs with drug-delivery systems (21–23).
Another prospective approach to address challenges with the development of AMP-based therapeutics could be to use a synergistic combination of AMPs for improved potency. The methodology of combination or poly-therapy, involving the rationale of using more than one drug, has attracted recent interest because naturally occurring AMPs are ubiquitously expressed together, and synergism between AMPs can very well explain their success in combating infections (24–27). In combinations, peptide interactions may lead to three primary outcomes, i.e. (i) additivity, (ii) synergism, and (iii) antagonism, in which the interacting peptides exhibit similar, stronger, and weaker activity, respectively, compared with their combined individual profiles at an equivalent dosage. The approach of peptide-based combinatorial therapy has been reported previously in the literature (28). So far, peptide synergism has not been conclusively correlated to the generation of specific structural motifs, which has resulted in other types of descriptions of synergisms, in which the peptides act cooperatively in destabilizing bacterial cell walls.
In previous work, we reported on the synthetically designed peptides VG16KRKP (VARGWKRKCPLFGKGG) and KYE28 (KYEITTIHNLFRKLTHRLFRRNFGYTLR), exhibiting potent activity against devastating plant pathogens of the genus Xanthomonas, including Xanthomonas vesicatoria, Xanthomonas oryzae, and Xanthomonas campestris (29, 30). In doing so, we have also highlighted the biological effects of these peptides ex vivo and provided a structural correlation into the origin of these effects. In this study, we expand on these previously reported findings by combining the two peptides, VG16KRKP and KYE28, to demonstrate synergistic antimicrobial potency of the peptide mixture. In addition, we set out to obtain structural insights into the interaction of these peptides in the presence of bacterial membrane components and mimics and thus to establish the structural basis of the synergism observed in this particular system. As discussed below, the combination of the two peptides promotes killing of several important plant pathogens, as also established by scanning EM (SEM) imaging. Elucidating the molecular origin of these effects, structure–function correlation of the VG16KRKP–KYE28 system was obtained by solving the three-dimensional solution structure of the peptide complex in Escherichia coli total lipid extract bicelles, the major components of the outer membrane of Gram-negative bacteria, lipopolysaccharide (LPS) micelles, and a bacterial cytoplasmic membrane mimic, 3:1 POPE/POPG large unilamellar vesicles (LUVs), using nuclear magnetic resonance (NMR) spectroscopy. Through this, aromatic–aromatic interactions between Trp of VG16KRKP and Phe residues of KYE28, in conjunction with charge clustering, were demonstrated to be instrumental for structural stabilization of the complex formed between these two peptides and for the enhanced biological activity of the peptide combination. These findings were supported by results from site-directed mutagenesis.
Results
Combined antimicrobial effects of VG16KRKP and KYE28
Using binary combinations of the parent peptides, VG16KRKP and KYE28 (Fig. 1A), checkerboard analysis (31) was conducted to calculate the fractional inhibitory concentration index (FICI) (Fig. 1B and see Equation 3 under “Experimental procedures”). As shown in Fig. 1B, for plant pathogens of Xanthomonas and Pseudomonas genus, FICI results demonstrated “partial synergy” to “synergy” between the two parent peptides, with FICI values ranging between 0.32 and 0.60 (Table S1). The combination also showed a synergistic effect against the human fungal pathogen Cryptococcus grubii and a partial synergy against Candida albicans, as well as good plant protease stability (Fig. 1C). In this context, it should be noted that for highly potent peptides, such as VG16KRKP and KYE28, the synergistic factor usually found is considerably smaller as observed in this investigation.
Figure 1.

A, primary amino acid sequences of the parent peptides, VG16KRKP and KYE28, as well as of their mutant analogs. The three-dimensional structure of the individual peptides in LPS is shown on right. The boldface amino acid residues are the mutated ones. B, checkerboard analysis using a modified micro broth dilution assay was performed to calculate the values of MIC99% and FICI. As shown, the parent VG16KRKP and KYE28 peptides interact with each other, demonstrated by the lowering of individual peptide concentration required for microbial killing. All experiments were performed in triplicate. C, proteolytic stability of the respective parent peptides, VG16KRKP and KYE28, in tomato plant extract containing plant proteases was evaluated using HPLC-based analysis. Both peptides displayed a half-life greater than 120 min. The peptides were incubated individually with the plant extract at 310 K, and aliquots were drawn at regular intervals and analyzed using RP-HPLC.
Interaction of VG16KRKP–KYE28 with bacterial membrane components
VG16KRKP–KYE28 interacts and disrupts bacterial membranes
Scanning EM (SEM) analysis of X. vesicatoria cells before and after exposure to the combination of the parent peptides, VG16KRKP and KYE28, at their 0.5× and 1× MIC99%, corresponding to 0.25 and 0.5 μm of each, respectively, demonstrated bacterial cell death to be associated with membrane disruption and leakage of intracellular content after only 1 h of treatment (Fig. 2, A–C). Analogous effects of membrane destabilization were observed for the live X. vesicatoria cells, probed using fluorescence-based experiments. As shown in Fig. 2D, the addition of the peptide combination at 1× and 2× MIC99% concentrations (corresponding to 0.5 and 1 μm of each, respectively) to live X. vesicatoria cells led to immediate depolarization of the bacterial cells as indicated by an increase in the fluorescence intensity of DISC3, a membrane-potential sensitive dye (32). In yet other set of fluorescence experiments, the destabilizing effect of the peptide combination on X. vesicatoria bacterial membranes was confirmed by monitoring the kinetics of bacterial lipopolysaccharide and cytoplasmic membrane permeabilization by measuring the uptake of the fluorescent probes 8-anilino-1-naphthalenesulfonic acid (ANS), N-phenyl-1-naphthylamine (NPN), and propidium iodide (PI), respectively (30, 32, 33). Because dyes are membrane and/or nucleic acid–specific in nature, peptide-induced membrane disruption allows them to bind to their respective counterparts, thus exhibiting enhanced fluorescence. Fig. 2, E–G, displays the increase in fluorescence intensity of these dyes upon treatment of the bacterial cell with 1× and 2× MIC99% concentration, again demonstrating bacterial wall disruption induced by the combination of VG16KRKP and KYE28.
Figure 2.
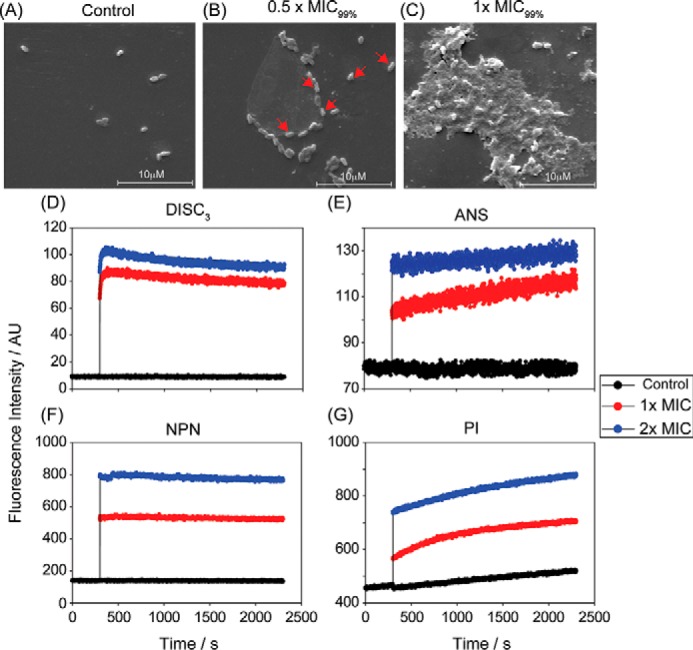
A, SEM images of X. vesicatoria in the absence of both the peptides VG16KRKP and KYE28 (control) showed no visible morphological changes after 1 h of incubation. In contrast, cells exposed to 0.5 × MIC99% (MIC99% = 1 μm each) showed onset of cell distortion (B), and exposure at 1× MIC99% resulted in pronounced membrane perturbation with extensive release of intracellular constituents and loss of cellular morphology (C). The scale corresponds to 10 μm. Additionally, dye-based fluorescence assay with live X. vesicatoria cells displayed that the peptide combination at 1× and 2× MIC99% concentration could effectively interact with bacterial cell membrane bringing about a change in membrane potential as indicated by an increase in DISC3 fluorescence (D), thus disrupting the outer membrane comprising of lipopolysaccharide (E and F), and the inner membrane made of negatively charged lipid components (G). The control cell showed baseline fluorescence under all conditions. It should be noted that ANS and NPN are lipophilic dyes, which fluoresces strongly in nonpolar environments of the cell membrane, which gets exposed upon membrane disintegration. PI, however, binds to nucleic acid components of the dead cell conforming inner membrane disruption. Each experiment was recorded for 30 min at 298 K.
VG16KRKP–KYE28 induces association of bacterial membrane mimics
Fig. 3A shows results from dynamic light-scattering (DLS) measurement performed with anionic LUVs (3:1 POPE/POPG) upon titration with increasing concentrations of the peptide combination. For better representation, the autocorrelation data (Fig. 3A, left) and the particle size distributions (Fig. 3A, right) pertaining to P/L (VG16KRKP–KYE28/LUVs) ratio of 1:1:20 (red), 1:1:10 (blue), 1:1:2 (green), and 1:1:1 (yellow) are shown, all at a constant VG16KRKP–KYE28 ratio of 1:1. Additionally, the same sample caused immediate vesicle flocculation even at the lowest P/L (VG16KRKP–KYE28/LUVs) ratio, leading to the generation of aggregates as indicated by the slowing down of the relaxation decay and increase in the delay time, τ. As can be seen from the figure, the LUVs used for the study displayed a quasi-perfect mono-exponentially decaying correlation graph on the linear-log scale and a low polydispersity index (PDI) of 0.13, indicating a narrow size distribution of spherical LUVs, with the mean hydrodynamic diameter centered around 149 nm. Increasing the total concentration of the 1:1 VG16KRKP–KYE28 peptide mixture resulted in particles ≈1 μm in diameter, and PDI ranging between 0.39 and 0.52 (data not shown). In contrast, the VG16KRKP or KYE28 peptides alone showed much smaller aggregation (Fig. S1).
Figure 3.

A, dynamic light-scattering experiment was performed with monodisperse population of spherical 3:1 POPE/POPG LUV. The concentrations stated indicate equimolar absolute concentrations of the two peptides individually, in μm. Upon titration with VG16KRKP and KYE28 mixtures, the vesicles showed significant changes in shape and size, as indicated by the increased PDI value and the increase in delay time (black broken line), respectively (left side). Each experimental set was done in triplicate. B, 31P solid-state NMR studies further supported VG16KRKP–KYE28–induced vesicle deformation and aggregation (right) as depicted by the change in intensity of the parallel (∼30 ppm) and perpendicular (∼−15 ppm) edges and an increased spectral span. The inset shows immediate vesicle flocculation upon addition of the peptides, whereas the control vesicles remain unchanged. In contrast, as seen on the left side, the peptide combination could only cause flocculation but no morphological changes in the bicelles prepared from E. coli total lipid extract bicelle.
To further extend our understanding of LUV aggregation induced by the VG16KRKP–KYE28 combination, we next employed 31P solid-state NMR and spectral-shape analysis for model bacterial membranes (Fig. 3 and Fig. S2) (34). The 31P spectrum of 3:1 POPE/POPG LUV or E. coli total lipid extracts showed a typical powder pattern spectrum (Fig. 3B, left) (35). Nonetheless, upon exposure to the peptide combination at 2 and 4 mol %, obvious and immediate changes were observed in the overall spectral shapes, indicating relative population changes of the orientations of phospholipid molecules. Careful comparisons of the spectra of 3:1 POPE/POPG vesicles showed the following: (i) a change in the intensity of the parallel (∼26 ppm) and perpendicular (∼−15 ppm) edges in the 31P spectra, and (ii) an increase in the spectral spans of the powder pattern (40 ± 0.5 ppm for 0 mol % and 44 ± 0.5 ppm for 4 mol %) upon peptide exposure. The relative intensity changes in the spectra implied that upon exposure to the VG16KRKP–KYE28 mixture, the vesicles underwent morphological changes that converted them from spherical LUVs to deformed membranes.
In addition, the increase in the spectral span observed reports on peptide-mediated vesicle aggregation, which retarded the motional averages of the vesicles due to the larger size. Such changes were expected as immediate aggregates were observed after peptide addition to the vesicles for all the samples during the sample preparation step for the NMR experiments (Fig. 3B, inset). This observation is consistent with the DLS data and SEM images.
Comparison of the 31P spectra of 3:1 POPE/POPG LUVs with that of E. coli total lipid extract bicelle showed that the latter underwent similar spectral shapes, but with a slightly increased spectral span (38 ± 0.5 ppm for 0 mol % and 44 ± 0.5 for 4 mol %), implying that the bicelles undergo aggregation without any apparent change in the spherical shape when treated with the peptide combination, as also visible from the NMR samples remaining comparatively clear (Fig. 3B, inset). Such an observation highlighted an important aspect of peptide action through synergism, indicating that, in case of bicelles, the two peptides can bind to the surface of the bicelles and network with other such complexes, hence facilitating aggregate growth.
Probing peptide interaction with bacterial membrane components using NMR spectroscopy to identify the binding “hotspot”
Interaction of VG16KRKP–KYE28 with bacterial lipopolysaccharide
As a major component of the outer membrane in Gram-negative bacteria, LPS acts as an inevitable barrier for AMPs reaching the inner membrane (36, 37). Considering this, we next set out to obtain insights into the structural properties of the VG16KRKP–KYE28 combination, including any complex formed between these, by determining its three-dimensional LPS-bound structure.
LPS form high-molecular-weight aggregates at concentrations above its critical micelle concentration (1.3 μm) (38). As a result of peptide binding to LPS, as well as the large size and slow diffusion of LPS aggregates, successive addition of small aliquots of LPS to an equimolar solution of VG16KRKP and KYE28 led to line-broadening of the peptide proton resonances, indicating a fast–to–intermediate conformational exchange on the NMR time scale (Fig. S3A). Additionally, the binding of the individual VG16KRKP and KYE28 peptides to LPS was in the micromolar range, as determined from isothermal titration calorimetry experiment of the individual peptides with LPS. This suggests that transferred NOESY (tr-NOESY) experiments can be used to determine the three-dimensional structure of the peptide combination after LPS binding (39–41). In aqueous solution, the VG16KRKP–KYE28 displayed only intra-residual (NOEs between residues of individual peptide) αN(i,i + 1) backbone/side-chain NOEs, suggesting these to be unfolded and highly dynamic (Fig. S4). As shown in Fig. S5A, spectral overlap was not observed (contrary to larger peptides), and the spectra could be assigned completely and unambiguously. A large number of intra-residual medium αN(i,i + 2/i + 3) NOEs, such as Ala-2/Gly-4, Arg-3/Trp-5, Arg-3/Lys-6, and Cys-9/Leu-11 in VG16KRKP and Tyr-2/Ile-4, Tyr-2/Thr-5, Ile-4/Ile-7, Ile-7/Asn-9, His-8/Phe-11, Leu-10/Arg-12, Arg-17/Phe-19, Arg-17/Arg-20, Phe-19/Asn-22, Arg-20/Phe-23, Asn-22/Gly-24, Phe-23/Thr-26, Gly-24/Thr-26, Gly-24/Leu-27, and Thr-26/Arg-28 in KYE28 were observed in the presence of LPS Fig. 4A, left. These provided a clear indication that the two peptides adopt combined and well-defined secondary structures when simultaneously present as a complex in LPS. In addition, several intra-molecular NN (i,i + 2) trNOEs between the residues Gly-4/Lys-6, Trp-5/Arg-7, Cys-9/Leu-11, Leu-11/Gly-13, and Gly-13/Gly-15 of VG16KRKP and Thr-6/His-8, Ile-7/Asn-9, His-8/Leu-10, Leu-10/Arg-12, Asn-22/Gly-24, and Phe-23/Tyr-25 of KYE28 (Fig. 4A, right, and Table S4) were also observed, indicating the occurrence of partial helical regions in both the peptides after LPS binding. Nevertheless, neither VG16KRKP nor its shorter analogs alone adopt α-helical conformation in the presence of LPS (29, 42), a change that could be therefore attributed to the interaction with KYE28. The most striking observation was the inter-molecular (inter-peptide NOEs) long-range (i,i + 4/i + 5) side-chain/side-chain NOEs between residues Trp-5VG16KRKP/Phe-23KYE28, Tyr-2KYE28/Trp-5VG16KRKP, Thr-5KYE28/Trp-5VG16KRKP, and Thr-6KYE28/Trp-5VG16KRKP that gave a clear indication that the two peptides interact with each other after simultaneous LPS binding. This interaction between VG16KRKP and KYE28 after LPS binding may therefore be responsible for stabilizing them and generating a unique orientation upon interaction with LPS (Figs. 4B and Fig. S6A).
Figure 4.

A, two-dimensional 1H-1H trNOESY spectra of VG16KRKP–KYE28 complex in the LPS micelle, displaying the NOE contacts in the fingerprint region for CαH-NH and NH-NH resonances important for structure stabilization. B, parent peptide complex also displayed intra- and inter-molecular long-range NOE contacts, illustrating the importance of aromatic residues in the formation of a stabilized structure and justifying the strong synergistic interaction observed. The experiments were performed by titrating 0.6 mm of the total peptides with 10 μm LPS (LPS: each peptide = 1:30) at pH 4.5, using a Bruker Avance III 700 MHz NMR spectrometer (NOESY mixing time = 150 ms) and at 298 K (residues marked in blue are from VG16KKRP and in red are from KYE28).
Interaction of VG16KRKP–KYE28 with bacterial membrane mimics
The majority of antimicrobial peptides act via membranolytic mechanisms, interfering with the integrity of both bacterial cell wall and cell membrane. The inability to do so generally results in the loss of activity, a fact well-established by the example of the cyclic lipopeptide, daptomycin (43). Thus, to obtain in-depth insights into the mechanism of peptide synergism, to conclusively demonstrate the generation of a functional complex between the two peptides, as well as its importance for bacterial cell wall disruption, we set out to probe similar interactions with the cytoplasmic membrane. As in the case of LPS, tr-NOESY experiments were used to determine the three-dimensional structure of the complex when bound to bacterial cytoplasmic membrane mimic, 3:1 POPE/POPG LUVs, and E. coli total lipid extract bicelles. Analogous to LPS, the peptide combination in 3:1 POPE/POPG LUVs displayed unambiguous and explicit intra-residual αN (i,i + 1) and an almost similar intra-residual medium αN (i,i + 2/i + 3) NOEs (Fig. 5A, left, and Fig. S5B). Additionally, intra-molecular NN (i,i + 2) and inter-molecular long-range (i,i + 4/i + 5) side-chain/side-chain trNOEs between the residues remained largely conserved (Fig. 5B and Table S4). However, the long-range interaction between the Hα/β of Thr-5 and Thr-6 and the aromatic ring proton of Trp-5 was lost, implying a structural re-organization of the VG16KRKP–KYE28 complex upon crossing the outer membrane.
Figure 5.

A, two-dimensional 1H-1H trNOESY spectra of VG16KRKP–KYE28 in a 3:1 POPE/POPG vesicle, displaying the NOE contacts in the fingerprint region for CαH-NH and NH-NH resonances important for structure stabilization. VG16KRKP–KYE28 upon interaction with 3:1 POPE/POPG vesicles exhibited several trNOEs hinting toward the adoption of a rigid and well-converged structure. B, parent peptide complex, also displayed intra- and inter-molecular long-range NOE contacts. The experiments were performed by titrating 0.6 mm of the total peptides with 12 μm of 3:1 POPE/POPG LUV (LUV: each peptide = 1:25) at pH 4.5, using a Bruker Avance III 700 MHz NMR spectrometer (NOESY mixing time = 150 ms) and at 298 K (residues marked in blue are from VG16KKRP and in red from KYE28).
These results obtained for VG16KRKP–KYE28 simultaneously present in bound complex with either LPS or 3:1 POPE/POPG suggest that the aromatic residues, because of their innate propensity to exclude water, play a central role in structural stabilization via generation of an aromatic zipper and a hydrophobic hub, effectively defining a structural hotspot for synergism in the VG16KRKP–KYE28 system. Also, this results in energetically-favorable salt-bridge interactions that have a substantial overall contribution in structure stabilization and hence activity. Of note, the indole ring proton (NϵH), resonating at ∼10.2 ppm, did not show any NOE contacts with other peptide residues in the complex. This is consistent with the observation for the individual peptide VG16KRKP interacting with LPS. Apart from that, the Cys–Pro bond maintained a trans-conformation in LPS as indicated by the presence of C9CαH/P10CδH NOE in all the three complexes.
In the case of E. coli total lipid extract bicelles, interference from CHAPSO, the detergent used for bicelles preparation, structural destabilization, and the lack of unambiguous peaks were observed, thus impeding identification of a well-defined structure (Figs. S6C and S7, A–E). The latter finding suggests that the VG16KRKP–KYE28 complex is relatively subtle, sensitive to surfactant exposure, in line with well-established conformational changes in peptides and proteins observed by a wide range of surfactants (44–46).
Aromatic cluster stabilizes the bound three-dimensional structure of the peptide complex
Interaction of VG16KRKP–KYE28 with bacterial lipopolysaccharide
The 15-ensemble structures of VG16KRKP–KYE28 in LPS, 3:1 POPE/POPG LUVs, and E. coli total lipid extract bicelle were generated using CYANA (47), at an equimolar peptide ratio and an LPS/peptide mixture of 1:30, based on the NOE distance constraints obtained from the tr-NOESY spectrum. In LPS, good convergence upon superposition of the backbone (Cα, N, and C′) atoms was obtained, as expected due to the higher number of NOEs obtained (Fig. 6, A and B), as also demonstrated by the low value of root mean square deviation for backbone and heavy side-chain atoms (Table S6).
Figure 6.

A and B, 15 ensemble LPS-bound structure of the VG16KRKP–KYE28 complex showed a well-defined conformation with good convergence of the backbone atoms (Protein Data Bank code 6KBO). C, cartoon representation of the complex indicated adoption of an amphipathic structure stabilized by an inter-molecular aromatic–aromatic hub, with the agreement of an overall “helix-loop-helix” structure. Interestingly, both the peptides showed a marked difference from the original structure in LPS, which could give a plausible justification for their enhanced activity. D, hydrophobic hub of VG16KRKP–KYE28 complex was characterized by energetically favorable cluster of aromatic–aromatic interactions, especially T-shaped geometries, formed between aromatic residues, which maintained a spatial distribution of ∼3.5–7 Å between them. E, two positively charged clusters with an end-to-end distance of 29 Å, corroborating closely with the thickness of the LPS bilayer, giving subtle hints toward the mode of action of the peptide complex. F, electrostatic surface potential of the LPS-bound complex at an angle of 180° further depicted the clear demarcation of the surface charge distribution, generated using MOLMOL software.
Closer inspection of the three-dimensional LPS-bound structure of VG16KRKP–KYE28 complex highlighted a marked change from their native LPS-bound structure (Fig. 6A and Fig. S8). In the binary peptide complex, VG16KRKP exhibited a more prominent structural change than KYE28 after LPS binding (Fig. S8, top). Notably, a helix Pro-10–Gly-16 was obtained at the C-terminal end of VG16KRKP to adopt a triad “Trp-5–Leu-11–Phe-12,” as opposed to a turn structure when present individually (Fig. 6C). In contrast, KYE28 in the peptide complex conserved its native “helix–loop–helix” motif with a minor (Glu-3–Ile-7) and a major (Leu-18–Leu-27) helix, also adopted when this peptide was present alone in LPS (Fig. S8, bottom). Importantly, the structural contribution of each residue in KYE28 in the VG16KRKP–KYE28 complex was not the same as in the case of the KYE28 peptide alone in LPS. A key structural change was observed in the central region of both VG16KRKP and KYE28, which together formed a dimer, characterized by a tight and bulky aromatic cluster stabilized by residues Trp-5 and Phe-12 of VG16KRKP and Tyr-2, Phe-19, and Phe-23 of KYE28, together forming a hotspot (Fig. 6D). Additionally, a smaller and loosely arranged hydrophobic hub formed by Leu-10–Leu-14–Leu-18–Phe-19 of KYE28 was also observed.
It is worth mentioning that the orientation of aromatic residues within a distance of 4.5–7.0 Å plays an essential role in structural stabilization (48, 49). Among the three probable aromatic–aromatic orientations, i.e. T-shaped or edge–to–face, face–to–face, and parallel-displaced, the obtained distance constraints allowed the VG16KRKP–KYE28 complex to adopt an energetically favorable aromatic cluster geometry by forming a T-shaped interaction (50) between Phe-12VG16KRKP–Trp-5VG16KRKP–Phe-23KYE28 (Fig. 6D). An additional cluster between Tyr-2KYE28–Phe-19KYE28 was also observed between the helix–loop–helix structure of KYE28, allowing correct folding and providing scaffold stability (51). Although these interactions provide weak contributions individually, together they helped to maintain the VG16KRKP–KYE28 complex in a globally favorable conformation in LPS to provide stability and rigidity.
Opposing the hydrophobic hub, the positively charged Lys and Arg residues of both peptides in the LPS-bound VG16KRKP–KYE28 complex remained disseminated toward the solvent front, maintaining a distance of 10–15 Å between the headgroup, being almost identical to the distance between two-phosphate groups of the LPS (Fig. 6E) (39). An interesting observation emerging here was the presence of two distinct clusters of concentrated positive charges at the two opposite faces of the adopted complex structure.
Collectively, the LPS-bound complex displayed an amphipathic structure, with a hydrophobic hub with well-separated charge distribution at the two termini, similar to some previously reported designed antimicrobial peptides, upon interaction with the bacterial membrane (39, 52). The electrostatic potential map confirmed the adoption of an amphipathic rigid structure in LPS that helped the two peptides to interact cooperatively with each other (Fig. 6F).
Interaction of VG16KRKP–KYE28 with bacterial membrane mimics
The three-dimensional structure of VG16KRKP–KYE28 bound to a negatively charged bacterial cytoplasmic membrane mimic, 3:1 POPE/POPG LUV, exhibited marked changes from their native as well as their complex LPS-bound structure (Fig. 7). Although the distorted helix at the C terminus remains conserved, the “Trp-5–Leu-11–Phe-12” triad was lost (Fig. 7, A–C). Additionally, KYE28 adopted a helix–loop–helix orientation, with two short helices, between residues Ile-4–Leu-10 and Leu-18–Thr-26, distinct from the major and minor helices adopted in LPS, both when alone and in the complex (Fig. 7C). Of note, the hydrophobic cluster formed by Trp-5 and Phe-12 of VG16KRKP and Tyr-2, Phe-19, and Phe-23 of KYE28 in LPS remained conserved upon interaction with the bacterial cytoplasmic membrane mimic, thus forming a hotspot helping the peptide complex to penetrate deeper into the membrane bilayer (Fig. 7C). Despite this conservation of the hydrophobic hub, local reorientation of aromatic residues was observed, with the generation of three T-shaped or edge–to–face interactions, between residues Phe-12VG16KRKP–Trp-5VG16KRKP–Phe-23KYE28, Trp-5VG16KRKP–Tyr-2KYE28–Phe-12VG16KRKP, and Phe-19KYE28–Trp-5VG16KRKP–Phe-23KYE28, respectively (Fig. 7D).
Figure 7.

A and B, 15 ensemble 3:1 POPE/POPG LUV-bound structure of the VG16KRKP–KYE28 complex displayed a well-defined conformation that, when compared with the LPS-bound structure, was more flexible (Protein Data Bank code 6KBV). C and D, one molecule cartoon representation of the complex indicated adoption of an amphipathic “helix-loop-helix” structure with a clear separation of charge with the hydrophobic network formed by the bulky aromatic groups as well as aliphatic residues involved in intra- and inter-molecular aromatic–aromatic and aromatic–aliphatic interaction via three T-shaped interactions between the residues Phe-12VG16KRKP–Trp-5VG16KRKP–Phe-23KYE28, Trp-5VG16KRKP–Tyr-2KYE28–Phe-12VG16KRKP, and Phe-19KYE28–Trp-5VG16KRKP–Phe-23KYE28, whereas E, the cationic face remained exposed to the solvent front, forming a single tight cluster of positive charges, which aids in initial electrostatic interaction with the negatively charged phosphate headgroups of the lipid molecules. F, electrostatic surface potential of the vesicle-bound complex at an angle of 180° further depicted the clear demarcation of the surface charge distribution, generated using MOLMOL software.
The positively charged residues Lys and Arg remained flexible and oriented toward the solvent, causing the complex to adopt an overall amphipathic structure. Here again, the anticipated re-distribution in residue assembly was observed when compared with the LPS-bound complex. Thus, the two positively charged clusters found in the presence of LPS was disrupted, and instead a single face of high-charge density was observed, located opposite to the hydrophobic hub (Fig. 7E). Such local changes could be attributed to the difference in mechanism adopted by the peptide to establish an initial interaction with the microbial cell and thus leading onto the killing of the same.
On the contrary, the three-dimensional structure of the peptide complex bound to E. coli total lipid extract bicelles adopted a very loose and open structure, consisting of two very short helices, Thr-6–Asn-9 and Leu-18–Gly-24, generating a nearly helix–loop–helix motif for KYE28, whereas the helix at the C terminus of VG16KRKP remained conserved, with the “Trp-5–Leu-11–Phe-12” triad lost (Fig. S7F). The overall structure as a whole resonated more with the complex structure bound to 3:1 POPE/POPG membrane mimic, having a distinct dual-face residue alignment, a bulky hydrophobic hub, and a highly-flexible hydrophilic face consisting of positively charged amino acids.
As mentioned above, interference of the surfactant CHAPSO used in the bicelles destabilized the VG16KRKP–KYE28 dimer and precluded structure determination. Considering this, further studies with this system were not undertaken, but the findings are nevertheless interesting in that they indicate that the VG16KRKP–KYE28 dimer is a restrained structure, sensitive to the presence of surfactants, in line with observations in the literature for a wide range of features in the structure of proteins and peptides (44–46).
Structural characterization and validation of the hotspot region in lipopolysaccharide
Site-directed mutagenesis studies
To validate the structure of VG16KRKP–KYE28 after LPS and membrane mimic binding, as well as the hotspot responsible for the same, mutants of the parent peptides (VG16KRKP and KYE28) were generated, viz. VG16W5A (VARGAKRKCPLFGKGG), VG16F12A (VARGWKRKCPLAGKGG), VG16WFA (VARGAKRKCPLAGKGG), and KYE28A (KYEITTIHNLARKLTHRLARRNAGATLR, respectively (Fig. 1A). Here, it should be noted that the three VG16KRKP mutant analogs were generated via a single-point mutation, whereas KYE28A bore four individual point mutations. In addition to providing further insight into the structural motifs forming the hotspot, these analogs can also be used for investigating the consequences of the observed structural features for antimicrobial activity, and synergy in this system, in particular. Considering this, FICIs were calculated for the parent and mutated analogs in pairs. The mutated peptides, VG16W5A and VG16F12A, at a concentration of 10 μm individually, displayed synergistic interaction with the parent peptide, KYE28 against Xanthomonas species (Tables S1–S3). This is in striking contrast to the finding that VG16F12A, either alone or in combination with other mutated analogs, lacked any activity. The same, however, was not observed for a combination of VG16KRKP with the mutated analog KYE28A.
These findings indicate that if two peptides, in the presence of microbes, interact with one another and undergo favorable structural transition, different from when they act alone, the complex generated can enhance their potency or, in some cases, induce potency in the nonactive peptide partner. This also implicated a structural re-organization that leads to the generation of an aromatic hub. Addressing the structural basis for this, the trNOESY profiles revealed medium-range αN (i,i + 2/i + 3) intra-molecular NOEs between the residues Tyr-2/Thr-6, Ile-4/Ile-7, Ile-7/Asn-9, Leu-10/Arg-12, Arg-17/Phe-19, Arg-17/Arg-20, Phe-19/Phe-23, Arg-20/Phe-23, and Phe-23/Thr-26 of KYE28 (Figs. S9A and S10B and Table S5). Once again, it was clear that aromatic–aromatic, inter-residual, and long-range interactions between Trp-5VG16F12A/Phe-11KYE28 and Trp-5VG16F12A/Phe-19KYE28 are the major players for the observed synergistic interaction, allowing VG16F12A to participate in the formation of an aromatic hub with KYE8 (Fig. S9B). The similar intra- and inter-molecular long-range NOEs were absent in VG16W5A-KYE28 or VG16WFA-KYE28 complexes in LPS (data not shown), suggesting that the aromatic–aromatic interaction plays an important role in structural stabilization and hence the antimicrobial activity. Additionally, checkerboard analysis using VG16KRKP and KYE28A indicated that the low antimicrobial activity displayed by the mutant peptide KYE28A remains unaltered even upon the inclusion of the potent VG16KRKP. This observation was also evident in their trNOESY profile, lacking inter-molecular medium- and long-range contacts (Fig. S10A).
Next, the three-dimensional LPS-bound structure of the mutants in complex with either of the parent peptides was calculated (Table S7). The VG16F12A–KYE28 complex adopted a comparatively relaxed structure, being held together by the aromatic tetrad formed by residue Trp-5 of VG16F12A with Phe-11, Phe-19, and Phe-23 of KYE28, which may explain its biological activity (Fig. S9, C and D, and Fig. S11, A and B, left). Apart from this, the overall structure of the complex-forming peptides remained conserved, with VG16F12A remaining in an extended conformation and KYE28 adopting a helix–loop–helix structure via formation of a major (Glu-3–Leu-14) and a minor (Phe-F23–Leu-27) helix (Fig. S9, C and D).
Although the helical regions of KYE28 remain conserved in both the VG16KRKP–KYE28 and VG16F12A–KYE28 complexes in LPS, a slight change in the locus and overall orientation allows conserving the variants in their bioactive conformation, resulting in partially maintained antimicrobial potency. This structural re-arrangement could also justify the relatively relaxed and dynamic orientation of the VG16F12A–KYE28 complex structure, as peptides and proteins have evolved to prioritize functional aspects by compensating in terms of structural stability (53). Exemplifying this, the mutation of the Phe-12 residue in this complex generated a change in the overall geometry and orientation of aromatic residues, so that the inter-helical interaction between Tyr-2KYE28 and Phe-19KYE28 was lost to maintain the T-shaped or face–to–edge interaction between Phe-xKYE28 (x = 11, 19, and 23) and Trp-5VG16KRKP (Fig. S9C) in order to retain the synergism and the activity of VG16F12A. In line with this, the lack of a well-folded three-dimensional structure is consistent with the inactivity of the VG16WFA–KYE28A complex against the plant pathogens and the apparent lack of synergism.
Additionally, as predicted from the lack of inter-molecular NOE contacts in the VG16KRKP–KYE28A complex and the tendency of protein/peptide folding toward functional conservation, these peptides adopt their native secondary structure mutually exclusive of the other, conforming with the low antimicrobial activity displayed (Fig. S11, right). Circular dichroism (CD) studies supported the adoption of an extensive α-helical conformation for the VG16KRKP–KYE28, VG16F12A–KYE28, and VG16KRKP–KYE28A complexes in LPS (Fig. S12). Intriguingly, VG16F12A–KYE28, and to some extent VG16KRKP–KYE28, displayed a diffuse CD spectral pattern, which could be corroborated to their interaction with the other peptide partner in the complex, the same being absent in VG16KRKP–KYE28A.
Paramagnetic relaxation enhancement (PRE)–NMR studies
To corroborate our findings further, PRE NMR experiments were performed next. Because a Cys residue was present at the central region of VG16KRKP, 1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl methanethiosulfonate (MTSL) was tagged to VG16KRKP via disulfide bonding. On the NMR timescale, the T2 relaxation rate of nuclei in the vicinity of a paramagnetic group is increased, leading to peak broadening and loss of peak intensity for the corresponding nearby nuclei (Fig. S13C). Therefore, MTSL conjugation can be applied to infer proximity of any one nucleus with the other, within a distance of 25 Å (54). Employing this method, MTSL-conjugated VG16KRKP was found to result in peak broadening in the Cα-H of the N-terminal residues of VG16KRKP and KYE28 peptides in complex with LPS, because of their distal proximity (Fig. 8A). Most notably, residues in KYE28, which were interacting with or were in close juxtaposition to Cys-9 of VG16KRKP and its nearby amino acid groups, also experienced faster T2 relaxation of the Cα-H. This mainly includes residues in the extreme N- and C-terminal of KYE28, which underwent complete relaxation.
Figure 8.

A, PRE NMR was performed using MTSL-tagged VG16KRKP with KYE28 in LPS. The trNOESY experiment was conducted under similar conditions with the unlabeled peptide to compare residue-specific T2 relaxation, depicted as I/I0. The residues in the vicinity of the labeled Cys-9 residue exhibited almost complete relaxation as shown by broadened Hα/NH trNOESY peaks. B, similarly, H/D experiments performed throughout 5 h corroborated with the solved structure with residues involved in the formation of the hydrophobic hub and those engaged in π-cation interaction remaining shielded from the exchange kinetics. C, FRET experiment showed a linear change in the emission intensity of the Trp residue of the donor peptide, VG16KRKP upon titration with increasing concentrations of dansylated acceptor KYE28, upto 0.5 mol fraction. The FRET experiment was performed at 298 K in 10 mm potassium phosphate buffer (pH 7.4).
In comparison, the residues at further distances experienced a suppressed effect. The same was found true for the Cβ/γ/δ-H, which because of the tapered relaxation effect was not affected to a similar extent as Cα-H (Fig. S14A). Nonetheless, the observation that the end–to–end distance from MTSL-C9 is ∼25 Å (Fig. S14B) helps to justify the phased out quenching pattern observed for all the residues and hence corroborates well with the complex three-dimensional structure discussed above.
Hydrogen/deuterium (H/D) exchange studies
NMR-based H/D exchange experiment helped to assess the degree of protection of the LPS-bound VG16KRKP–KYE28 complex. Careful examination of the 1H-1H TOCSY spectra, recorded through 5 h of exchange, demonstrated that the backbone amide protons of several residues, notably Ala-2/Trp-5/Phe-12 (from VG16KRKP) and Phe-11/Phe-19/Phe-23 (from KYE28), undergo slow solvent exchange, as evident from their estimated protection factors (Fig. 8B and Fig. S15A). These residues play a central role in forming the α-helix and are involved in structure stabilization and folding through aromatic–aromatic and aromatic–aliphatic interaction, as well as through π–cation interactions, as also indicated by the protection factor (Fig. S15A). Moreover, the solvent-accessible surface area (SASA), calculated using the POPS* software (55), indicated that the residues involved in the peptide–peptide and the peptide–LPS and peptide–peptide–LPS interactions are characterized by an overall decrease in SASA on complexation. In contrast, the cationic residues in both the peptides forming the complex demonstrated an enhanced value of the same (Figs. S15B and S16). Collectively, these results show that such interactions play a vital role in maintaining the conformational stability of the peptide complex.
FRET experiments indicate complex formation
The interaction between the parent peptides and their mutant analogs to come together and generate synergistic antimicrobial activity was further investigated by fluorescence-based FRET analysis (56). Interaction between the Trp of donor peptide (VG16KRKP) and dansyl of N-terminal labeled acceptor peptide (KYE28) was probed by monitoring the quenching of Trp emission intensity upon titration with the acceptor dansyl in the presence of E. coli LPS, cytoplasmic membrane mimic 3:1 POPE/POPG, and E. coli total lipid-extract bicelles. The VG16KRKP–KYE28 complex showed highly quenched Trp emission upon addition of increasing concentrations of dansylated KYE28 (dans-KYE28), giving a linear relationship between the quenching of donor and concentration of the acceptor molecule (Fig. 8C). The interaction between VG16KRKP and the parent KYE28 exhibited a strong FRET effect amounting to a maximum quenching of 0.56, corroborating to a distance of 16.5 Å, in satisfactory agreement with the 3D structure (Fig. S17, A and F). The slight deviation from the NMR-derived structure could be attributed to the motional freedom and difference in orientation of the fluorophore groups attached. In contrast to these findings for LPS-bound peptide complexes, FRET analysis in 3:1 POPE/POPG and E. coli total lipid-extract bicelles did not reveal a strong interaction between VG16KRKP and KYE28.
The same could be true for VG16F12A–KYE28, which exhibited a modest FRET effect surmounting to a distance of 15.6 Å, in satisfactory agreement with the 3D structure (Fig. S17, E and F). Strikingly, the VG16KRKP–KYE28A complex did not show any FRET effect, in line with the absence of any significant interaction observed on the NMR timescale (Fig. S17B).
Discussion
Matsuzaki et al. (57) did the first pioneering work to study peptide synergism and attempted to reach an understanding of the mechanism of the same by trying to elucidate the molecular and structural basis of peptide synergism, using the two amphibian antimicrobial peptides, magainin 2 and PGLa. Initial studies with the two peptides displayed maximum synergy at a 1:1 molecular ratio, hinting toward a probable heterodimer complex formation (57, 58). The three-dimensional structure of the hybrid peptides generated in the presence of SDS and DPC micelles, however, failed to demonstrate any stable heterosupramolecular structure, underlying the synergism observed (59). Following this early work, studies of the structural aspect of synergism remained largely unexplored because of the limitations associated with the available biophysical techniques. Therefore, Matsuzaki et al. (57) also highlighted the importance of well-defined NMR experiments required for obtaining unambiguous NOE peaks, which can make structure determination more precise. Later, Bhunia et al. (56) studied the synergistic interaction between two amphibian peptides, temporin 1Tl (TL) and temporin 1Tb (TB), using NMR in conjunction with other biophysical experiments, showing that the peptide TL affects the LPS-mediated oligomerization state of TB, while maintaining its native antiparallel dimer helix. However, the group did not report on the complex structure or its implications.
Also addressing the structural origin of AMP synergy, Ulrich and co-workers (60), working on PGLa–MAG2 peptide combination, highlighted that the GXXXG motif plays an important role in synergy. These authors proposed the formation of a functionally active PGLa–MAG2 complex, in which an anti-parallel PGLa dimer stabilized by the GXXXG motif was generated and interacted with MAG2 monomers via its C terminus (60). In addition to studying the structural features underlying synergy, various groups have highlighted the importance of intrinsic membrane curvature for AMP synergy (61–63). Here, Bechinger and co-workers (64) have performed breakthrough work, associating peptide synergism with the “Soft Membranes Adapt and Respond, also Transiently” ('SMART') model to gain mechanistic insights for peptide synergism. This highlights the importance of both the peptide and the interacting lipid partners in deciding the mode of action. Additionally, work from these authors has identified the importance of individual peptides, which do not form complexes but do interact at the membrane interface to generate peptide synergism. For example, through their work on the PGLa–MAG2 system, Bechinger and co-workers (64) highlighted that magainin displays stable membrane surface alignment, whereas PGLa undergoes re-alignment when in the presence of magainin, but only in the unsaturated lipid membrane, adopting a mesophase arrangement.
In this study, probing the structural basis of synergism via three-dimensional structure calculation in bacterial outer and inner membrane components highlighted the underlying key aspect of observed synergism to be lying in its aromatic core, which formed the hotspot region for peptide activity. Studies have associated overly high hydrophobicity of small peptides with an enhanced ability to self-associate and precipitate, thus negating availability and decreasing the antimicrobial efficacy (65). Nevertheless, the fact that VG16KKRP–KYE28 complex generated an aromatic packing cluster that remained globally conserved while incorporating local changes in all three structures calculated, as well as in the mutant complex studied, highlighted the importance of these hotspot residues. Experimental and structural analyses have previously established the role of such interaction and the resulting higher-order clusters in generating a super-secondary structure in α-helices of the type “helix–loop–helix” in proteins (66) as observed for our peptide combination.
A comparison between the LPS-bound and the bacterial membrane mimic-bound structure of the VG16KKRP–KYE28 complex further indicated defined differences between the two, which might help to deduce the mode–of–action of the peptide complex. A key change observed was in the segmentation of positive charges in the three-dimensional LPS-bound structure. Although the complex maintained its amphipathicity, the hydrophilic, cationic residues generated two clusters of mild positive charges when bound to LPS as opposed to a highly-concentrated charged region adopted in a bacterial membrane mimic. The end–to–end distance between the two clusters was ∼29 Å, closer to the LPS bilayer thickness of 32 Å, implying that the complex as a whole could mount itself into the bilayer and generate transient pores and traverse further to reach the inner membrane. Such charge segmentation could be looked upon as an approach to help prevent excess peptide oligomerization (60), so that higher numbers of peptide complexes could be generated, attacking the Gram-negative bacteria's first line of defense, the LPS. Upon reaching the inner membrane, composed of a 3:1 mixture of bulky POPE and smaller POPG, the peptide undergoes structural reorganization as explained above. Such clustering of positive charges helps the peptide complex to interact with a negatively charged phosphate headgroup of the lipid, by launching themselves into the spatial voids. Such voids are generated due to the difference in size of the lipid headgroup constituting the inner membrane, and peptide interaction at these voids would cause further membrane incompatibility leading to increased mobility and stress through generation of positive curvature, thus affecting lipid alignment in the membrane, membrane thinning, and manifesting in a detergent-like mechanism of action (67). This structural attribute correlated very well with the 31P NMR data, which indicated similar deformation of the membrane mimics.
An important point to note here is the fact that the electrostatic interaction observed emerging from the positive charge cluster and their orientation in different membrane mimics, although important for launching an initial assault on the membrane, remained secondary in importance, contributing indirectly to synergy. This could be attributed to the observation that the mutant peptides, lacking critical aromatic residues, displayed diminished or no synergy.
VG16KRKP and KYE28 individually are capable of forming membrane defects and killing cells by a detergent-like or toroidal-pore mechanism (29, 30, 68). As clearly demonstrated in this investigation, the VG16KRKP–KYE28 complex similarly destabilizes both bacterial cell walls and model membrane mimics. Strikingly, this is paralleled by the generation of defined complexes, involving VG16KRKP–KYE28 dimer formation. Although some features of these binary peptide complexes are the same in the LPS and plasma membrane environment, notably in relation to the overall symmetry and the aromatic hub, there are also distinct differences between these environments, as is evident by the changed orientation of particular residues, especially the polar cationic ones which differ locally, upon interaction with a different region of the bacterial membrane. When added together to a bacterial suspension, the two peptides first interact with the bacterial outer membrane-forming heteropeptide complex that disrupts LPS assembly, followed by diffusion deeper into the bacterial cell wall to reach the inner membrane. Here, the complex disturbs membrane fluidity as found by 31P NMR experiments, causing membrane deformation and ultimately lysis, in turn contributing to the observed synergism. Such a scenario is also compatible with findings of both structure, membrane disruption, and antimicrobial effects of combinations of native and mutated VG16KRKP and KYE28 analogs.
Although synergy between VG16KRKP and KYE28 thus seems to involve formation of a well-defined structure in the LPS region of the bacterial wall, which is partly retained and partly transformed when reaching the cytoplasmic membrane environment, a key question is that of the generality of this mechanism of AMP synergy. Here, it should be noted that aromatic–aromatic interactions have been observed to be widely important for structural stabilization and hydrophobic domain formation in AMPs, e.g. the peptide combination used in this study, VG16KRKP and KYE28 as well as their shorter analogs, when present individually in LPS, exhibit similar structural characteristics (29, 30, 52, 68). One should also note that previous work on AMP synergy has only partly succeeded in identifying structural features associated with synergy, which in turn has led to other approaches, such as that based on local curvature (62, 64). Also, as shown for the results obtained for the E. coli total lipid extract bicelles, the presence of the surfactant CHAPSO was sufficient to destabilize the dimer formed by VG16KRKP–KYE28 in either LPS or cytoplasmic membrane mimic environments. This, in turn, suggests a relatively low stability of the binary structure formed. Adding these considerations together, further work is therefore needed to assess whether the structure formed in this study is a rare exception or whether related aromatic–aromatic hotspot formation through dimerization can also be found in other AMP systems.
Furthermore, a useful understanding of the interplay in mixtures of AMP will require wider considerations than addressed here, e.g. relating to pharmacodynamic aspects of such interactions. Synergism between two to three peptides, including naturally occurring AMP, as well as between AMP and either antibiotics or nanoparticles, has been reported (69, 70). Exemplifying this, interaction studies between peptides of the dermaseptin family, between β-defensins and the cationic protein BPI, and between the α-helical peptides magainin and PGLa have all been reported before (64, 71, 72). In none of these cases, however, has a well-defined structure of the type reported here been observed.
Within the context of plant protection, it should also be noted that transgenic plants with individual defense genes have been found to display only partial resistance against key plant pathogens (73–75). Therefore, combining heterologous defense genes from various sources offers a more promising alternative for enhanced resistance in transgenic plants. Simultaneous expression of chitinase and glucanase is one such first example of synergism-induced enhanced resistance against fungus infection in the transgenic tomato (76). Similarly, using a synergistic combination of Fusarium-specific antibody linked to antifungal peptides was found to boost resistance against Fusarium oxysporum f. sp. matthiola in Arabidopsis (77).
In conclusion, the studies above demonstrate that the synergistic antimicrobial effects observed for VG16KRKP and KYE28 in combination are related to the formation of a well-defined amphiphilic dimer in the presence of LPS, which is partially retained upon interaction of this peptide pair in the cytoplasmic membrane environment. In addition, the adoption of such an aromatic zipper motif in the three-dimensional LPS-bound structure was demonstrated to induce antimicrobial potency in an inherently inactive peptide VG16F12A. Although only one peptide pair was demonstrated, both the structural requirements and the effects of peptide amino acid changes suggest these effects to be of potentially wider importance, applicable also to other peptide combinations. That said, additional work is required to elucidate the potential generalization of the effects observed in this investigation.
Experimental procedures
Reagents
E. coli O111:B4 LPS and polymyxin B were purchased from Sigma. 3-(Trimethylsilyl)propionic-2,2,3,3-d4 acid (TSP) sodium salt and deuterium oxide were purchased from Cambridge Isotope Laboratories, Inc. (Tewksbury). MTSL was purchased from Santa Cruz Biotechnology, Inc. (Dallas). DISC3, ANS, NPN, and PI were purchased from Thermo Fisher Scientific (Waltham, MA). All buffer components, including salts, were purchased from Merck (Kenilworth, NJ). CHAPSO was purchased from Sigma. POPE and POPG were obtained from Avanti Polar Lipids (Alabaster, AL).
Bacterial and fungal media components were purchased from Himedia Laboratories Pvt. Ltd. (Mumbai, India). VG16KRKP and its analogs were obtained from GL Biochem (Shanghai, China), and KYE28 and its variant were from Biopeptide Co. (San Diego), both with ≥95% purity. Peptide purity was confirmed by RP-HPLC, MS, and NMR spectroscopy (78). Peptide stock solutions were prepared either in autoclaved water or phosphate buffer (pH 4.5 and 7.4, respectively) and filter-sterilized unless stated otherwise.
Bacterial and fungal strains and reagents
Plant pathogenic strain X. campestris pv. vesicatoria was a kind gift from Dr. Christian Lindermayr, Institute of Biochemical Plant Pathology, Germany. X. campestris pv. campestris was isolated locally from fields of Kalyani, West Bengal, India, and X. oryzae pv. oryzae was kindly provided by Professor Sampa Das, Bose Institute, India. Pseudomonas syringae DCM 300 was a gift from Dr. Pallob Kundu, Bose Institute, India. Fungal strains C. albicans SC5314 and Cryptococcus neoformans var. grubii H99 were the kind gift from Prof. Kaustuv Sanyal, JNCASR, India.
For microbroth dilution and checkerboard activity assay, X. vesicatoria, X. campestris, and X. oryzae were grown in Nutrient Broth, PS broth (1% peptone and 1% sucrose), and PSP broth (0.5% peptone, 2% sucrose dissolved in starch obtained from boiling 200 g of potato/liter of media), respectively, and maintained at 301 K under shaking. P. syringae DCM 300 was grown in King's Broth supplemented with 50 μg/ml rifampicin and kanamycin, respectively, and maintained at 301 K under shaking. C. albicans and C. grubii were grown in YPDU broth (1% yeast extract, 1% peptone, 2% dextrose, and 20 μg/ml uridine) and maintained at 301 K under shaking.
Microbroth dilution assay
To assay for antimicrobial activity of the peptides, standard microdilution broth assay was used, as described previously (79). Overnight-grown cultures of the respective plant pathogens, X. vesicatoria, X. campestris, X. oryzae, and P. syringae DCM 300, were used to obtain mid-log–phase cultures The cell suspensions were centrifuged at 6000 rpm for 5 min, after which cell pellets were washed three times with 10 mm phosphate buffer (pH 7.4), followed by resuspension in the same buffer to 105 CFU/ml. The reactions were set in a 96-well flat-bottom plate by incubating 50 μl of the cell suspension with 2-fold increasing concentrations of the respective peptides (ranging from 1 to 100 μm) prepared from 1 mm peptide stock in phosphate buffer (pH 7.4) and incubated at 301 K, 150 rpm shaking for 4 h. For negative and positive controls, samples containing only cell suspension and 10 μm polymyxin B–treated cell suspension, respectively, were included. Next, 150 μl of suitable media was added to each well and incubated for 48 h with shaking at 301 K. Bacterial growth was monitored by measuring the absorbance at 630 nm. All absorbance readings were normalized using the negative control polymyxin B. The peptide concentration at which 99 ± 0.5% growth inhibition was observed served as its MIC99%. All experiments were performed in triplicate.
Checkerboard dilution assay
A modified version of a standard microdilution broth assay was used to analyze the possible synergistic interaction between the parent peptides (VG16KRKP and KYE28) and their mutant variants using two-dimensional checkerboard dilution assay (80). Mid-log phase cultures of each organism were obtained by inoculating the overnight grown cultures of respective pathogens in their respective growth medium. The cell suspensions were centrifuged at 6000 rpm for 5 min, followed by washing three times with 10 mm phosphate buffer (pH 7.4) and resuspension in the same buffer to 105 CFU/ml for bacteria and 104 CFU/ml for fungi. In a 96-well plate format, 50 μl of this suspension was incubated with varying concentrations of VG16KRKP against a 2-fold dilution range of KYE28 to obtain MIC99% of the combination by incubating at their respective temperatures for 4 h. A negative control containing only cell suspension, as well as a positive control containing 10 μm polymyxin B with cell suspension, was included as well. 150 μl of suitable media was added to each well and incubated overnight with shaking at their respective temperatures. All experiments were performed in triplicates. Peptide interaction was assessed by calculating the fractional inhibitory concentrations of the two-peptide combinations, according to Equations 1–3,
| (Eq. 1) |
| (Eq. 2) |
| (Eq. 3) |
FICI, calculated as the sum of each individual fractional inhibitory concentration, was interpreted as follows: FICI < 0.5 synergy; 0.5 ≤ FICI < 1 partial synergy; 1 ≤ FICI < 4 additive effect or indifference; and 4 < FICI antagonism.
Plant extract stability assay
Stability of VG16KRKP and KYE28 against degradation by plant proteases obtained from plant leaf extract was investigated using a modified version of a previously reported experiment (81). For this, 1 g of a 1-month-old tomato (Pusa Ruby) leaf was ground in liquid nitrogen, followed by extraction with 2 ml of 10 mm phosphate buffer (pH 7.4). The mixture was centrifuged at 13,500 rpm (5 min, 277 K), and the supernatant was collected and saved in aliquots at −80 °C until further use. To analyze peptide stability, 1 mm of the respective peptide was incubated with 20 μg/ml of the leaf extract (concentration determined using BSA assay) at 310 K, and aliquots of 100 μl were withdrawn at the respective time points. The reaction was stopped by adding trifluoroacetic acid (TFA) to a final concentration of 1%. Water and plant extract (20 μg/ml) were used as blank and negative control, respectively. The reaction mixture was next loaded onto a Phenomenix C18 column (Dimension 250 × 10.0-mm, pore size 100 Å, particle size 5 μm), at room temperature, to be analyzed by an LC-20AT reverse phase-HPLC system (SHIMADZU, Japan) using dual solvent (acetonitrile and water in 0.1% TFA) linear gradient elution at a flow rate of 1 ml/min. To determine the remaining peptide in percentage (%), the peak obtained at 220 nm was integrated using the SPINCHROME CFR software, and the area obtained was compared with that of a free peptide in the absence of plant extract.
Scanning Electron Microscopy
X. vesicatoria cells were cultured in nutrient broth (NB) to mid-logarithmic phase and were harvested by centrifugation at 6000 rpm for 5 min, washed twice with phosphate buffer (10 mm (pH 7.4), and resuspended in the same to a final concentration of 106 cells per ml. 50 μl of this suspension was incubated with 0.5× and 1× MIC99% concentration of the peptide combination, VG16KRKP and KYE28, for 1 h at 310 K. Untreated cells were set as control. After treatment, the cells were fixed with 4% (v/v) glutaraldehyde, and 15 μl of the same was spotted on clean poly-l-lysine–coated glass slides and dried overnight. Next, the slides were washed twice with phosphate buffer, followed by dehydration with a graded series of ethanol (30, 50, 70, 90, and 100%), each for 15 min. The sample slides were then air-dried, followed by gold coating, and observed by SEM (FEI, QUANTA 200).
Fluorescence spectroscopy
All studies were conducted with X. vesicatoria live cell. The cells were grown and maintained as mentioned above, and a final concentration of 106 or 107 cells per ml was used for experimental purposes until unless specified otherwise. Measurements were performed at 298 K by monitoring the emitted fluorescence, at the excitation wavelength of the dye used, upon peptide treatment of the cells using a Hitachi F-7000 FL spectrometer.
Cytoplasmic membrane depolarization assay
Mid-logarithmic phase X. vesicatoria cells were washed in 5 mm HEPES buffer (pH 7.4) supplemented with 5 mm glucose and resuspended in the same along with 100 mm potassium chloride in 1:1:1 (v/v) ratio to a final concentration of 106 cells per ml. Next, 1 μm of the membrane potential sensitive dye, DISC3, was added to the cell suspension and incubated in the dark for an hour. Fluorescence from DISC3 was recorded for 30 min at an excitation wavelength of 622 nm, an emission wavelength of 670 nm, and a slit width of 10 and 5 nm, respectively. The uptake of the dye by the cell resulting in self-quenching properties are dependent on the potential of the membrane. Upon maximum uptake, indicated by a decrease in the inherent fluorescence of the dye, the peptides were added to the cells at 1× and 2× MIC99% concentration, and the decrease in membrane potential as indicated by an increase in dye fluorescence was recorded.
Outer-membrane disruption assay
The experiment for outer membrane interaction and disruption by the peptide combination was performed using ANS and NPN dyes. 10 μm of the respective dye was added to the cell suspension and incubated in the dark for 60 and 15 min, respectively, to allow equilibration. Next, the cells were titrated with 1× and 2× MIC99% concentration of the parent peptides, and the increase in fluorescence intensity was recorded for 30 min.
Fluorescence from ANS was recorded at an excitation wavelength of 380 nm, an emission wavelength of 470 nm, and a slit width of 1 nm. Fluorescence from NPN was recorded at an excitation wavelength of 350 nm, an emission wavelength of 400 nm, and a slit width of 5 nm.
Inner-membrane permeabilization assay
PI dye, which binds DNA/RNA of dead cells, was used to probe disruption of the inner membrane. 10 μm of the dye was added to the cell suspension (107 cells/ml) and incubated in the dark for 15 min for equilibration. Next, the cells were titrated with 1× and 2× MIC99% concentration of the parent peptides, and the increase in fluorescence intensity of PI was recorded for 30 min. Fluorescence from PI was recorded at an excitation wavelength of 535 nm, an emission wavelength of 617 nm, and a slit width of 10 nm.
Liposome preparation
Approximately, 1 mg of POPE and POPG was weighed and dissolved in chloroform to obtain stock solutions and further mixed to obtain the model liposomes, a 3:1 molar ratio of anionic POPE/POPG, which is reported to be used extensively as a model system for negatively charged bacterial membranes in the literature (82). Next, uniform lipid films were obtained by drying the lipid suspension of model liposomes in a nitrogen stream, followed by overnight lyophilization. The film was resuspended in 10 mm sodium phosphate buffer at pH 7.4 by vigorous vortexing for 30 min, followed by five freeze-thaw cycles in liquid nitrogen to obtain vesicles. To isolate unilamellar vesicles, the suspension was passed through a mini-extruder (Avanti Polar Lipids, Alabaster, AL) using stacked 100-nm pore-size polycarbonate membrane filters. Samples were subjected to 23 passes through the filter. To avoid contamination by vesicles that may not have passed through the first filter, we performed an odd number of passages. The LUVs so generated were used for DLS experiments.
Dynamic light scattering
The experiment was performed on a Malvern ZetasizerNano S (Malvern Instruments) provided with a 4-milliwatt He-Ne laser (λ = 633 nm) and a back-scattering angle of 173°. DLS experiments were performed using Malvern ZetasizerNano S (Malvern Instruments) provided with a 4-milliwatt He-Ne laser (λ = 633 nm) and a back-scattering angle of 173°. LUVs (pH 7.4) were titrated with increasing concentrations of the peptide solution, prepared in the same buffer (phosphate buffer (pH 7.4)), and DLS measurement was performed in triplicate and three independent experimental sets were recorded. All samples were filtered using Millipore 0.45-μm polycarbonate membrane filters, degassed before use, and measured at 298 K in low-volume disposable sizing cuvettes. For data analysis, the viscosity and refractive index of 10 mm Tris buffer containing 100 mm NaCl buffer (pH 7.4) were taken to be 0.89 and 1.33, respectively. The particle size distributions were generated using the autocorrelation data from the non-negative least-squares fit algorithm provided by the instrument manufacturer (83).
Circular dichroism spectroscopy
CD spectra of the peptide combination were obtained using a JASCO 814 spectrometer (Japan). For this, 25 μm of both peptides, prepared in 10 mm phosphate buffer (pH 7.4), was titrated with increasing concentrations of LPS, prepared in the same buffer. The spectra were recorded in a 0.2-cm cuvette, at 298 K with three scans accumulating at a speed of 100 nm/min, scanning from 190 to 260 nm, at a data interval of 1 nm. The spectra generated were corrected using a baseline spectrum for each combination of peptide, and the ellipticity in milli-degrees was plotted against wavelength in nanometers.
Solid-state NMR spectroscopy
NMR sample preparation
Sample preparation for 31P solid-state NMR was done by dissolving 3:1 POPE/POPG in chloroform. The mixed samples were dried under a stream of nitrogen followed by vacuum-dry oven (30 °C) overnight to completely remove any residual solvent. 10 mm Tris buffer (pH 6.0) was added to the samples to hydrate the lipid film. The hydrated samples were next vortexed for 2 min above the lipid-phase transition temperature and freeze-thawed for a minimum of four times using liquid nitrogen, ensuring a generation of a single lamellar vesicle. To obtain LUVs with a uniform size of 1 μm in diameter, the sample was extruded through polycarbonate filters (pore size of 1 μm, Nuclepore®, Whatman, NJ) mounted on a mini-extruder (Avanti Polar Lipids, AL) fitted with two 1.0-ml Hamilton gastight syringes (Hamilton, NV). Samples were typically subjected to 23 passes through the filter. Such an odd number of passages helps to avoid contamination of the sample by vesicles that have not passed through the first filter. The VG16KKRP or KYE28 or both peptide solutions, dissolved in similar buffer, were added to the LUVs to prepare the appropriate final peptide concentration/lipid molar ratio, making a final sample volume of 120 μl.
Solid-state NMR spectroscopy
NMR experiments were performed on an Agilent NMR spectrometer (DD2) operating at the resonance frequency of 699.88 MHz for 1H and 283.31 MHz for 31P nuclei and a 4-mm MAS HXY solid probe (Agilent). 31P NMR experiments were performed using a 90° pulse and 24 kHz TPPM proton decoupling. Pulse length for the 31P experiment was 5.5 μs. In all cases, a total of 512 scans were acquired for each sample with a relaxation delay time of 2.0 s. LUVs were put in a 4-mm Pyrex glass tube, which was cut to fit into the MAS probe. 31P NMR spectra of LUVs without peptide were first recorded, and next an appropriate amount of peptide for first molar ratio (2 and 4 mol %) from a stock solution in buffer was added; the mixture was gently shaken, and the sample was placed back into the magnet for acquiring 31P NMR spectra at the ambient temperature. All 31P NMR spectra were processed using 150 Hz line broadening referenced externally to 85% phosphoric acid (0 ppm).
Solution-state NMR spectroscopy
All NMR experiments were carried out at 298 K using a Bruker Avance III 700 MHz NMR spectrometer, equipped with 5-mm cryogenic cooled triple resonance probe. NMR samples were prepared in 10% deuterated water (pH 4.5), and TSP was used as an internal standard (0.0 ppm). Two-dimensional 1H-1H nuclear Overhauser effect spectroscopy (2D NOESY) with a sweep width of 12 ppm in both directions was recorded for the peptides VG16KRKP and KYE28 at a 1:1 molar ratio in aqueous solution. The number of scans was fixed at 16 per t1 increment with 16 dummy scans. Next, to probe the synergistic interaction of the two peptides with LPS, 1D proton NMR spectra of 0.6 mm VG16KRKP and KYE28 was recorded alone and upon titration with increasing concentrations of E. coli LPS. The 1D spectra were acquired using excitation-sculpting scheme for water suppression and the States-TPPI for quadrature detection in the t1 dimension (84). The molar ratio of LPS/VG16KRKP/KYE28 of 1:30:30 was used for two-dimensional 1H-1H total correlation spectroscopy (2D TOCSY) (mixing time = 80 ms) and two-dimensional 1H-1H trNOESY (mixing times = 100, 150, and 200 ms) experiments, keeping all other parameters the same. The number of scans was fixed to 16 and 72, respectively, for TOCSY and trNOESY experiments. 512 increments in t1 and 2048 data points in t2 dimension along with States TPPI for quadrature detection in t1 dimension and excitation-sculpting scheme for water suppression were used to record the experiments. The experiments were processed and analyzed using Bruker TopSpinTM 3.1 and Sparky (85). Similar TOCSY and trNOESY NMR experiments were also recorded for VG16KRKP-KYE28A, VG16F12A-KYE28, and VG16WFA-KYE28 to study the role of aromatic residues in the interaction of peptides with lipopolysaccharide using Bruker Avance III 700 MHz NMR spectrometer.
Calculation of NMR-derived structures
The LPS-bound structure of the individual peptides VG16KRKP, KYE28, and KYE28A have already been reported with Protein Data Bank acquisition codes of 2MWL, 2NAT, and 2NAU, respectively (29, 30). To get structural insights into their mode of interaction and the combination effects, we calculated the LPS-bound three-dimensional conformation of the complexes VG16KRKP-KYE28, VG16KRKP-KYE28A, VG16F12A-KYE28, and VG16WFA-KYE28, generated by linking the two peptides via 10 poly-glycine residues, using CYANA version 2.1 software (47). It could be noted here that the choice of glycine poly-linker merely aids in structure calculation by helping to comprehend the influence of the peptides on their interaction abilities with each other, as well as with LPS, and to ensure that the peptide complex remains flexible and unbiased. For this, the inter-proton upper bound distances were calculated initially by sorting trNOESY cross-peaks based on their intensities into strong (3.0 Å), medium (4.0 Å), and weak (5.0 Å). The lower-bound distance constraint was kept fixed at 2.0 Å. The backbone dihedral angles, φ (between −30 and 120° and/or −80 and −60°) and ψ (120° and −120° and/or −40° and −20°), were kept flexible for all nonglycine residues so as to limit the conformational space. The distance constraints were then used for iterative refinement at each round of structure calculation. Hydrogen bonding and stereo-specificity were excluded from the structure calculation. The ensemble structure was generated from the 15 lowest-energy structures, which were analyzed using PyMOL, MOLMOL, and Chimera software. PROCHECK server was used to authenticate the backbone stereochemistry (47, 86–88).
PRE NMR
To conduct PRE NMR, site-directed spin labeling was done using a paramagnetic quencher, MTSL, attached to the thiol group of the cysteine residue present in VG16KRKP. This was done by incubating a 10:1 molar ratio of the peptide VG16KRKP and MTSL in 50% acetone overnight in the dark at 298 K under a mild shaking condition. The resulting solution was diluted and subjected to RP-HPLC (LC-20AT Phenomenix C-18 column, SHIMADZU, Kyoto, Japan) to purify labeled VG16KRKP from unlabeled peptide and free MTSL and characterized using MS in a Bruker autoflex speed TOF/TOF in a positive reflector mode. MTSL-labeled VG16KRKP was then dissolved in deuterated water in an equimolar ratio with KYE28 (0.6 mm (pH 4.5)) and 1H-1H TOCSY, and trNOESY experiments were recorded, keeping all the recording parameters similar as described previously. NMR data were analyzed using Bruker TOPSPINTM 3.1 and SPARKY (85). The Hα/NH cross-peak intensities of MTSL-labeled and unlabeled complex in LPS were calculated, and the remaining amplitudes (I/I0) in percentage was calculated using Equation 4,
| (Eq. 4) |
where IHα/NH(MTSL-labeled) and IHα/NH(unlabeled) indicate the Hα/NH peaks intensities of each amino acid in the unlabeled and MTSL-labeled peptide complexes, respectively. No attempts were made to back-calculate theoretical I/I0 values due to uncertainties related to R2 relaxation effects and INEPT transfer calculations from 2D trNOSEY spectra.
Hydrogen/deuterium exchange NMR
To gain insight into the stability and solvent accessibility of the VG16KRKP–KYE28 peptide complex, amide proton exchange was monitored. For this, 0.5 mm VG16KRKP/KYE28 was suspended in deuterium oxide (pD 4.5), and a series of 2D TOCSY (80-ms mixing time) was recorded until 5 h with an interval of 1 h each. The number of scans and dummy scans were kept fixed at 8 and 16, respectively, with 256 and 2048 data point increments in t1 and t2 dimensions, respectively. NMR data were analyzed using Bruker TOPSPINTM 3.1 and SPARKY (85). The hydrogen exchange rates (kex) were obtained by fitting to a single-exponential decay equation, and the protection factor was determined by taking a ratio of the calculated intrinsic exchange rate and the experimentally determined exchange rate.
SASA calculations
Online POPS* algorithm was used to calculate the SASA value of each peptide, individually as well as the peptide complex in LPS (55).
FRET analysis
FRET experiment between the Trp of donor peptide and dansyl of the N-terminally–labeled acceptor peptide was carried out by monitoring the quenching of Trp emission intensity upon titration with the acceptor dansyl in the presence of E. coli LPS. 5 μm of the donor peptide, VG16KRKP, in the presence of LPS, 3:1 POPE/POPG vesicle, or E. coli total lipid extract bicelle was titrated with increasing concentrations of dansyl-KYE28, and the emission was recorded at a 300–550-nm range after exciting at 280 nm wavelength in a Hitachi F7000 instrument (Tokyo, Japan) at room temperature. The normalized quenching value (F/F0) was plotted as a function of mole fraction (Pa) of the acceptor molecule. F and F0 represent the intensity of fluorescence emission of Trp in the presence and absence of the dansyl group, and Pa was calculated by Equation 5,
| (Eq. 5) |
This quenching graph was extrapolated linearly to obtain the value of E (1 − Q), with Q being the maximum quenching value. Next, using Förster equation, the inter-chromophore distance between Trp and the dansyl group was calculated by Equation 6,
| (Eq. 6) |
where r = distance between the two chromophores (Trp and dansyl in this case), R0 = Föster distance (23 Å for Trp-dansyl) and E = efficiency of FRET obtained for the quenching plot. The same experiment was carried out for a combination of donor–acceptor pairs composed of the mutant variants of the two peptides, and in all cases the inter-chromophore distance between Trp and the dansyl group was calculated.
Statistical analysis
All biological experiments were repeated three times. Data were analyzed by using SigmaPlot 12.0 (Copyright 2011 Systat Software, Inc., Germany) and Origin Pro 2018 (Northampton, MA).
Author contributions
H. I., J. K., D. L., and A. B. data curation; H. I., D. L., M. M., and A. B. formal analysis; H. I. and A. B. methodology; H. I. and A. B. writing-original draft; M. M. and A. B. conceptualization; M. M. and A. B. funding acquisition; M. M. and A. B. validation; M. M. and A. B. investigation; M. M. and A. B. writing-review and editing; A. B. supervision; A. B. project administration.
Supplementary Material
This work was supported by the India-Swedish (DST-VR) Programme of Cooperation in Science & Technology Project 2015-06720 or DST/INT/SWD/VR/P-02/2016 (to M. M. and A. B.), Science and Engineering Research Board File No. EMR/2017/003457 (to A. B.) from the Government of India, and LEO Foundation Center for Cutaneous Drug Delivery Grant 2016-11-01 (to M. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S17 and Tables S1–S7.
The atomic coordinates and structure factors (codes 6KBO and 6KBV) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The three-dimensional LPS and 3:1 POPE/POPG vesicle-bound structures of the complex VG16KRKP–KYE28 have been submitted to Biological Magnetic Resonance Data Bank under codes 36265 for LPS-bound and 36266 for 3:1 POPE/POPG-bound.1
- AMP
- antimicrobial peptide
- SEM
- scanning electron microscopy
- LPS
- lipopolysaccharide
- LUV
- large unilamellar vesicle
- FICI
- fractional inhibitory concentration index
- DLS
- dynamic light scattering
- PDI
- polydispersity index
- TOCSY
- total correlation spectroscopy
- trNOESY
- transferred nuclear Overhauser effect spectroscopy
- PRE
- paramagnetic relaxation enhancement
- SASA
- solvent accessible surface area
- MIC99%
- minimal inhibitory concentration for 99% killing
- TSP
- 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt
- POPE
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- POPG
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- CHAPSO
- 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonic acid
- MTSL
- 1-S-(1-oxy-2,2,5,5-tetramethyl-2, 5-dihydro-1H-pyrrol-3-yl) methyl methanesulfonothioate
- DISC3
- 3,3′-dipropylthiodicarbocyanine iodide
- ANS
- 8-anilino-1-naphthalenesulfonic acid
- NPN
- N-phenyl-1-naphthylamine
- PI
- propidium iodide
- H/D
- hydrogen–deuterium
- dansyl
- 5-dimethylaminonaphthalene-1-sulfonyl
- CFU
- colony-forming unit
- P/L
- VG16KRKP–KYE28/LUVs.
References
- 1. Chiang C. Y., Uzoma I., Moore R. T., Gilbert M., Duplantier A. J., and Panchal R. G. (2018) Mitigating the impact of antibacterial drug resistance through host-directed therapies: current progress, outlook, and challenges. MBio 9, e01932–17 10.1128/mBio.01932-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Spaulding C. N., Klein R. D., Schreiber H. L. 4th, Janetka J. W., and Hultgren S. J. (2018) Precision antimicrobial therapeutics: the path of least resistance? NPJ Biofilms Microbiomes 4, 4 10.1038/s41522-018-0048-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gustavsson J., Cederberg C., Sonesson U., Otterdijk V. R., and Maybeck A. (2011) Global Food Losses and Food Waste–Extent, Causes, and Prevention. Food and Agriculture Organization of the United Nations, New York: ISBN 978-92-5-107205-9 [Google Scholar]
- 4. Levy S. B., and Marshall B. (2004) Antibacterial resistance worldwide: causes, challenges and responses. Nat. Med. 10, S122–S129 10.1038/nm1145 [DOI] [PubMed] [Google Scholar]
- 5. Strange R. N., and Scott P. R. (2005) Plant disease: a threat to global food security. Annu. Rev. Phytopathol. 43, 83–116 10.1146/annurev.phyto.43.113004.133839 [DOI] [PubMed] [Google Scholar]
- 6. McManus P. S., Stockwell V. O., Sundin G. W., and Jones A. L. (2002) Antibiotic use in plant agriculture. Annu. Rev. Phytopathol. 40, 443–465 10.1146/annurev.phyto.40.120301.093927 [DOI] [PubMed] [Google Scholar]
- 7. Zasloff M. (2002) Antimicrobial peptides of multicellular organisms. Nature 415, 389–395 10.1038/415389a [DOI] [PubMed] [Google Scholar]
- 8. Strominger J. L. (2009) Animal antimicrobial peptides: ancient players in innate immunity. J. Immunol. 182, 6633–6634 10.4049/jimmunol.0990038 [DOI] [PubMed] [Google Scholar]
- 9. Hancock R. E., and Sahl H. G. (2006) Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551–1557 10.1038/nbt1267 [DOI] [PubMed] [Google Scholar]
- 10. Mahlapuu M., Håkansson J., Ringstad L., and Björn C. (2016) Antimicrobial peptides: an emerging category of therapeutic agents. Front. Cell Infect. Microbiol. 6, 194 10.3389/fcimb.2016.00194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hancock R. E., and Patrzykat A. (2002) Clinical development of cationic antimicrobial peptides: from natural to novel antibiotics. Curr. Drug Targets Infect. Disord. 2, 79–83 10.2174/1568005024605855 [DOI] [PubMed] [Google Scholar]
- 12. Hancock R. E. (2001) Cationic peptides: effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis. 1, 156–164 10.1016/S1473-3099(01)00092-5 [DOI] [PubMed] [Google Scholar]
- 13. Hancock R. E., Brown K. L., and Mookherjee N. (2006) Host defence peptides from invertebrates–emerging antimicrobial strategies. Immunobiology 211, 315–322 10.1016/j.imbio.2005.10.017 [DOI] [PubMed] [Google Scholar]
- 14. Brogden K. A. (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250 10.1038/nrmicro1098 [DOI] [PubMed] [Google Scholar]
- 15. Sahl H. G. (2006) Optimizing antimicrobial host defense peptides. Chem. Biol. 13, 1015–1017 10.1016/j.chembiol.2006.10.001 [DOI] [PubMed] [Google Scholar]
- 16. Shai Y. (2002) Mode of action of membrane active antimicrobial peptides. Biopolymers 66, 236–248 10.1002/bip.10260 [DOI] [PubMed] [Google Scholar]
- 17. Kalle M., Papareddy P., Kasetty G., van der Plas M. J., Mörgelin M., Malmsten M., and Schmidtchen A. (2014) A peptide of heparin cofactor II inhibits endotoxin-mediated shock and invasive Pseudomonas aeruginosa infection. PLoS ONE 9, e102577 10.1371/journal.pone.0102577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhattacharjya S. (2010) De novo designed lipopolysaccharide binding peptides: structure-based development of antiendotoxic and antimicrobial drugs. Curr. Med. Chem. 17, 3080–3093 10.2174/092986710791959756 [DOI] [PubMed] [Google Scholar]
- 19. Wimley W. C., and Hristova K. (2011) Antimicrobial peptides: successes, challenges and unanswered questions. J. Membr. Biol. 239, 27–34 10.1007/s00232-011-9343-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aoki W., and Ueda M. (2013) Characterization of antimicrobial peptides toward the development of novel antibiotics. Pharmaceuticals 6, 1055–1081 10.3390/ph6081055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nordström R., Nyström L., Andrén O. C. J., Malkoch M., Umerska A., Davoudi M., Schmidtchen A., and Malmsten M. (2018) Membrane interactions of microgels as carriers of antimicrobial peptides. J. Colloid. Interface Sci. 513, 141–150 10.1016/j.jcis.2017.11.014 [DOI] [PubMed] [Google Scholar]
- 22. Biswaro L. S., da Costa Sousa M. G., Rezende T. M. B., Dias S. C., and Franco O. L. (2018) Antimicrobial peptides and nanotechnology, recent advances and challenges. Front. Microbiol. 9, 855 10.3389/fmicb.2018.00855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kovalainen M., Mönkäre J., Riikonen J., Pesonen U., Vlasova M., Salonen J., Lehto V.-P., Järvinen K., and Herzig K.-H. (2015) Novel delivery systems for improving the clinical use of peptides. Pharmacol. Rev. 67, 541–561 10.1124/pr.113.008367 [DOI] [PubMed] [Google Scholar]
- 24. Yu G., Baeder D. Y., Regoes R. R., and Rolff J. (2016) Combination effects of antimicrobial peptides. Antimicrob. Agents Chemother. 60, 1717–1724 10.1128/AAC.02434-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yan H., and Hancock R. E. (2001) Synergistic interactions between mammalian antimicrobial defense peptides. Antimicrob. Agents Chemother. 45, 1558–1560 10.1128/AAC.45.5.1558-1560.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Siavoshi M. A., Prokopczuk F. I., Del Rosario N.-A., Abasi L., and Taheri-Araghi S. (2018) Combination therapies with antimicrobial peptide LL-37 and conventional antibiotics. Biophys. J. 114, 417 10.1016/j.bpj.2017.11.2310 [DOI] [Google Scholar]
- 27. Grassi L., Maisetta G., Esin S., and Batoni G. (2017) Combination strategies to enhance the efficacy of antimicrobial peptides against bacterial biofilms. Front. Microbiol. 8, 2409 10.3389/fmicb.2017.02409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Choi H., and Lee D. G. (2012) Synergistic effect of antimicrobial peptide arenicin-1 in combination with antibiotics against pathogenic bacteria. Res. Microbiol. 163, 479–486 10.1016/j.resmic.2012.06.001 [DOI] [PubMed] [Google Scholar]
- 29. Datta A., Ghosh A., Airoldi C., Sperandeo P., Mroue K. H., Jiménez-Barbero J., Kundu P., Ramamoorthy A., and Bhunia A. (2015) Antimicrobial peptides: insights into membrane permeabilization, lipopolysaccharide fragmentation and application in plant disease control. Sci. Rep. 5, 11951 10.1038/srep11951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Datta A., Bhattacharyya D., Singh S., Ghosh A., Schmidtchen A., Malmsten M., and Bhunia A. (2016) Role of aromatic amino acids in lipopolysaccharide and membrane interactions of antimicrobial peptides for use in plant disease control. J. Biol. Chem. 291, 13301–13317 10.1074/jbc.M116.719575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bremmer D. N., Bauer K. A., Pouch S. M., Thomas K., Smith D., Goff D. A., Pancholi P., and Balada-Llasat J. M. (2016) Correlation of checkerboard synergy testing with time-kill analysis and clinical outcomes of extensively drug-resistant Acinetobacter baumannii respiratory infections. Antimicrob. Agents Chemother. 60, 6892–6895 10.1128/AAC.00981-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yarlagadda V., Akkapeddi P., Manjunath G. B., and Haldar J. (2014) Membrane active vancomycin analogs: a strategy to combat bacterial resistance. J. Med. Chem. 57, 4558–4568 10.1021/jm500270w [DOI] [PubMed] [Google Scholar]
- 33. Thennarasu S., Lee D. K., Tan A., Prasad Kari U., and Ramamoorthy A. (2005) Antimicrobial activity and membrane selective interactions of a synthetic lipopeptide MSI-843. Biochim. Biophys. Acta 1711, 49–58 10.1016/j.bbamem.2005.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schiller J., Muller M., Fuchs B., Arnold K., and Huster D. (2007) 31P NMR spectroscopy of phospholipids: from micelles to membranes. Curr. Anal. Chem. 3, 283–301 10.2174/157341107782109635 [DOI] [Google Scholar]
- 35. Bechinger B., and Salnikov E. S. (2012) The membrane interactions of antimicrobial peptides revealed by solid-state NMR spectroscopy. Chem. Phys. Lipids 165, 282–301 10.1016/j.chemphyslip.2012.01.009 [DOI] [PubMed] [Google Scholar]
- 36. Snyder D. S., and McIntosh T. J. (2000) The lipopolysaccharide barrier: correlation of antibiotic susceptibility with antibiotic permeability and fluorescent probe binding kinetics. Biochemistry 39, 11777–11787 10.1021/bi000810n [DOI] [PubMed] [Google Scholar]
- 37. Rosenfeld Y., Barra D., Simmaco M., Shai Y., and Mangoni M. L. (2006) A synergism between temporins toward Gram-negative bacteria overcomes resistance imposed by the lipopolysaccharide protective layer. J. Biol. Chem. 281, 28565–28574 10.1074/jbc.M606031200 [DOI] [PubMed] [Google Scholar]
- 38. Yu L., Tan M., Ho B., Ding J. L., and Wohland T. (2006) Determination of critical micelle concentrations and aggregation numbers by fluorescence correlation spectroscopy: aggregation of a lipopolysaccharide. Anal. Chim. Acta 556, 216–225 10.1016/j.aca.2005.09.008 [DOI] [PubMed] [Google Scholar]
- 39. Bhunia A., Domadia P. N., Torres J., Hallock K. J., Ramamoorthy A., and Bhattacharjya S. (2010) NMR structure of pardaxin, a pore-forming antimicrobial peptide, in lipopolysaccharide micelles: mechanism of outer membrane permeabilization. J. Biol. Chem. 285, 3883–3895 10.1074/jbc.M109.065672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ghosh A., Datta A., Jana J., Kar R. K., Chatterjee C., Chatterjee S., and Bhunia A. (2014) Sequence context induced antimicrobial activity: insight into lipopolysaccharide permeabilization. Mol. Biosyst. 10, 1596–1612 10.1039/C4MB00111G [DOI] [PubMed] [Google Scholar]
- 41. Chowdhury R., Ilyas H., Ghosh A., Ali H., Ghorai A., Midya A., Jana N. R., Das S., and Bhunia A. (2017) Multivalent gold nanoparticle–peptide conjugates for targeting intracellular bacterial infections. Nanoscale 9, 14074–14093 10.1039/C7NR04062H [DOI] [PubMed] [Google Scholar]
- 42. Datta A., Jaiswal N., Ilyas H., Debnath S., Biswas K., Kumar D., and Bhunia A. (2017) Structural and dynamic insights into a glycine-mediated short analog of a designed peptide in lipopolysaccharide micelles: correlation between compact structure and anti-endotoxin activity. Biochemistry 56, 1348–1362 10.1021/acs.biochem.6b01229 [DOI] [PubMed] [Google Scholar]
- 43. Li J., Koh J. J., Liu S., Lakshminarayanan R., Verma C. S., and Beuerman R. W. (2017) Membrane active antimicrobial peptides: translating mechanistic insights to design. Front. Neurosci. 11, 73 10.3389/fnins.2017.00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Loo R. R., Dales N., and Andrews P. C. (1994) Surfactant effects on protein structure examined by electrospray ionization mass spectrometry. Protein Sci. 3, 1975–1983 10.1002/pro.5560031109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sjögren H., Ericsson C. A., Evenäs J., and Ulvenlund S. (2005) Interactions between charged polypeptides and nonionic surfactants. Biophys. J. 89, 4219–4233 10.1529/biophysj.105.065342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mcdonnell P. A., and Opella S. (1993) Effect of detergent concentration on multidimensional solution NMR spectra of membrane proteins in micelles. J. Magn. Reson. Series B 102, 120–125 10.1006/jmrb.1993.1073 [DOI] [Google Scholar]
- 47. Güntert P., Mumenthaler C., and Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 10.1006/jmbi.1997.1284 [DOI] [PubMed] [Google Scholar]
- 48. Burley S. K., and Petsko G. A. (1986) Amino-aromatic interactions in proteins. FEBS Lett. 203, 139–143 10.1016/0014-5793(86)80730-X [DOI] [PubMed] [Google Scholar]
- 49. Anjana R., Vaishnavi M. K., Sherlin D., Kumar S. P., Naveen K., Kanth P. S., and Sekar K. (2012) Aromatic-aromatic interactions in structures of proteins and protein-DNA complexes: a study based on orientation and distance. Bioinformation 8, 1220–1224 10.6026/97320630081220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Samanta U., Pal D., and Chakrabarti P. (1999) Packing of aromatic rings against tryptophan residues in proteins. Acta Crystallogr. D Biol. Crystallogr. 55, 1421–1427 10.1107/S090744499900726X [DOI] [PubMed] [Google Scholar]
- 51. Serrano L., Bycroft M., and Fersht A. R. (1991) Aromatic-aromatic interactions and protein stability. Investigation by double-mutant cycles. J. Mol. Biol. 218, 465–475 10.1016/0022-2836(91)90725-L [DOI] [PubMed] [Google Scholar]
- 52. Datta A., Kundu P., and Bhunia A. (2016) Designing potent antimicrobial peptides by disulphide linked dimerization and N-terminal lipidation to increase antimicrobial activity and membrane perturbation: structural insights into lipopolysaccharide binding. J. Colloid. Interface Sci. 461, 335–345 10.1016/j.jcis.2015.09.036 [DOI] [PubMed] [Google Scholar]
- 53. Madhusudan Makwana K., and Mahalakshmi R. (2015) Implications of aromatic–aromatic interactions: from protein structures to peptide models. Protein Sci. 24, 1920–1933 10.1002/pro.2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cabrita L. D., Waudby C. A., Dobson C. M., and Christodoulou J. (2011) Solution-state nuclear magnetic resonance spectroscopy and protein folding. Methods Mol. Biol. 752, 97–120 10.1007/978-1-60327-223-0_7 [DOI] [PubMed] [Google Scholar]
- 55. Cavallo L., Kleinjung J., and Fraternali F. (2003) POPS: a fast algorithm for solvent accessible surface areas at atomic and residue level. Nucleic Acids Res. 31, 3364–3366 10.1093/nar/gkg601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bhunia A., Saravanan R., Mohanram H., Mangoni M. L., and Bhattacharjya S. (2011) NMR structures and interactions of temporin-1Tl and temporin-1Tb with lipopolysaccharide micelles: mechanistic insights into outer membrane permeabilization and synergistic activity. J. Biol. Chem. 286, 24394–24406 10.1074/jbc.M110.189662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Matsuzaki K., Mitani Y., Akada K. Y., Murase O., Yoneyama S., Zasloff M., and Miyajima K. (1998) Mechanism of synergism between antimicrobial peptides magainin 2 and PGLa. Biochemistry 37, 15144–15153 10.1021/bi9811617 [DOI] [PubMed] [Google Scholar]
- 58. Zerweck J., Strandberg E., Bürck J., Reichert J., Wadhwani P., Kukharenko O., and Ulrich A. S. (2016) Homo- and heteromeric interaction strengths of the synergistic antimicrobial peptides PGLa and magainin 2 in membranes. Eur. Biophys. J. 45, 535–547 10.1007/s00249-016-1120-7 [DOI] [PubMed] [Google Scholar]
- 59. Haney E. F., Hunter H. N., Matsuzaki K., and Vogel H. J. (2009) Solution NMR studies of amphibian antimicrobial peptides: linking structure to function? Biochim. Biophys. Acta 1788, 1639–1655 10.1016/j.bbamem.2009.01.002 [DOI] [PubMed] [Google Scholar]
- 60. Zerweck J., Strandberg E., Kukharenko O., Reichert J., Bürck J., Wadhwani P., and Ulrich A. S. (2017) Molecular mechanism of synergy between the antimicrobial peptides PGLa and magainin 2. Sci. Rep. 7, 13153 10.1038/s41598-017-12599-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Marquette A., and Bechinger B. (2018) Biophysical investigations elucidating the mechanisms of action of antimicrobial peptides and their synergism. Biomolecules 8, E18 10.3390/biom8020018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Leber R., Pachler M., Kabelka I., Svoboda I., Enkoller D., Vácha R., Lohner K., and Pabst G. (2018) Synergism of antimicrobial frog peptides couples to membrane intrinsic curvature strain. Biophys. J. 114, 1945–1954 10.1016/j.bpj.2018.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Harmouche N., and Bechinger B. (2018) Lipid-mediated interactions between the antimicrobial peptides magainin 2 and PGLa in bilayers. Biophys. J. 115, 1033–1044 10.1016/j.bpj.2018.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Marquette A., Salnikov E. S., Glattard E., Aisenbrey C., and Bechinger B. (2016) Magainin 2-PGLa interactions in membranes–two peptides that exhibit synergistic enhancement of antimicrobial activity. Curr. Top. Med. Chem. 16, 65–75 [DOI] [PubMed] [Google Scholar]
- 65. Chen Y., Guarnieri M. T., Vasil A. I., Vasil M. L., Mant C. T., and Hodges R. S. (2007) Role of peptide hydrophobicity in the mechanism of action of α-helical antimicrobial peptides. Antimicrob. Agents Chemother. 51, 1398–1406 10.1128/AAC.00925-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lanzarotti E., Biekofsky R. R., Estrin D. A., Marti M. A., and Turjanski A. G. (2011) Aromatic-aromatic interactions in proteins: beyond the dimer. J. Chem. Inf. Model 51, 1623–1633 10.1021/ci200062e [DOI] [PubMed] [Google Scholar]
- 67. Strandberg E., Tiltak D., Ehni S., Wadhwani P., and Ulrich A. S. (2012) Lipid shape is a key factor for membrane interactions of amphipathic helical peptides. Biochim. Biophys. Acta 1818, 1764–1776 10.1016/j.bbamem.2012.02.027 [DOI] [PubMed] [Google Scholar]
- 68. Datta A., Yadav V., Ghosh A., Choi J., Bhattacharyya D., Kar R. K., Ilyas H., Dutta A., An E., Mukhopadhyay J., Lee D., Sanyal K., Ramamoorthy A., and Bhunia A. (2016) Mode of action of a designed antimicrobial peptide: high potency against Cryptococcus neoformans. Biophys. J. 111, 1724–1737 10.1016/j.bpj.2016.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Le C. F., Yusof M. Y., Hassan H., and Sekaran S. D. (2015) In vitro properties of designed antimicrobial peptides that exhibit potent antipneumococcal activity and produces synergism in combination with penicillin. Sci. Rep. 5, 9761 10.1038/srep09761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ruden S., Hilpert K., Berditsch M., Wadhwani P., and Ulrich A. S. (2009) Synergistic interaction between silver nanoparticles and membrane-permeabilizing antimicrobial peptides. Antimicrob. Agents Chemother. 53, 3538–3540 10.1128/AAC.01106-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mor A., Hani K., and Nicolas P. (1994) The vertebrate peptide antibiotics dermaseptins have overlapping structural features but target specific microorganisms. J. Biol. Chem. 269, 31635–31641 [PubMed] [Google Scholar]
- 72. Levy O., Ooi C. E., Weiss J., Lehrer R. I., and Elsbach P. (1994) Individual and synergistic effects of rabbit granulocyte proteins on Escherichia coli. J. Clin. Invest. 94, 672–682 10.1172/JCI117384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Brogue K., Chet I., Holliday M., Cressman R., Biddle P., Knowlton S., Mauvais C. J., and Broglie R. (1991) Transgenic plants with enhanced resistance to the fungal pathogen Rhizoctonia solani. Science 254, 1194–1197 10.1126/science.254.5035.1194 [DOI] [PubMed] [Google Scholar]
- 74. Alexander D., Goodman R. M., Gut-Rella M., Glascock C., Weymann K., Friedrich L., Maddox D., Ahl-Goy P., Luntz T., and Ward E. (1993) Increased tolerance to two oomycete pathogens in transgenic tobacco expressing pathogenesis-related protein 1a. Proc. Natl. Acad. Sci. U.S.A. 90, 7327–7331 10.1073/pnas.90.15.7327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhu B., Chen T. H., and Li P. H. (1996) Analysis of late-blight disease resistance and freezing tolerance in transgenic potato plants expressing sense and antisense genes for an osmotin-like protein. Planta 198, 70–77 [DOI] [PubMed] [Google Scholar]
- 76. Jongedijk E., Tigelaar H., van Roekel J. S. C., Bres-Vloemans S. A., Dekker I., van den Elzen P. J. M., Cornelissen B. J. C., and Melchers L. S. (1995) Synergistic activity of chitinases and β-1,3-glucanases enhances fungal resistance in transgenic tomato plants. Euphytica 85, 173–180 10.1007/BF00023946 [DOI] [Google Scholar]
- 77. Peschen D., Li H. P., Fischer R., Kreuzaler F., and Liao Y. C. (2004) Fusion proteins comprising a Fusarium-specific antibody linked to antifungal peptides protect plants against a fungal pathogen. Nat. Biotechnol. 22, 732–738 10.1038/nbt970 [DOI] [PubMed] [Google Scholar]
- 78. Bhattacharyya D., Kim M., Mroue K. H., Park M., Tiwari A., Saleem M., Lee D., and Bhunia A. (2019) Role of non-electrostatic forces in antimicrobial potency of a dengue-virus derived fusion peptide VG16KRKP: mechanistic insight into the interfacial peptide-lipid interactions. Biochim. Biophys. Acta Biomembr. 1861, 798–809 10.1016/j.bbamem.2019.01.011 [DOI] [PubMed] [Google Scholar]
- 79. Blazyk J., Wiegand R., Klein J., Hammer J., Epand R. M., Epand R. F., Maloy W. L., and Kari U. P. (2001) A novel linear amphipathic β-sheet cationic antimicrobial peptide with enhanced selectivity for bacterial lipids. J. Biol. Chem. 276, 27899–27906 10.1074/jbc.M102865200 [DOI] [PubMed] [Google Scholar]
- 80. Odds F. C. (2003) Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 52, 1 10.1093/jac/dkg301 [DOI] [PubMed] [Google Scholar]
- 81. Li Q., Lawrence C. B., Maelor Davies H., and Everett N. P. (2002) A tridecapeptide possesses both antimicrobial and protease-inhibitory activities. Peptides 23, 1–6 10.1016/S0196-9781(01)00572-1 [DOI] [PubMed] [Google Scholar]
- 82. Schmidtchen A., Ringstad L., Kasetty G., Mizuno H., Rutland M. W., and Malmsten M. (2011) Membrane selectivity by W-tagging of antimicrobial peptides. Biochim. Biophys. Acta 1808, 1081–1091 10.1016/j.bbamem.2010.12.020 [DOI] [PubMed] [Google Scholar]
- 83. Twomey S. (2002) Introduction to the Mathematics of Inversion of Remote Sensing and Indirect Measurements. Dover Publications, Mineola, NY [Google Scholar]
- 84. Sklenar V., Piotto M., Leppik R., and Saudek V. (1993) Gradient-tailored water suppression for 1H-15N HSQC experiments optimized to retain full sensitivity. J. Magn. Reson. 102, 241–245 10.1006/jmra.1993.1098 [DOI] [Google Scholar]
- 85. Goddard T. D., and Kneller D. G. (2008) Sparky3, University of California, San Francisco [Google Scholar]
- 86. Koradi R., Billeter M., and Wüthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55. 29–32 10.1016/0263-7855(96)00009-4 [DOI] [PubMed] [Google Scholar]
- 87. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., and Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 88. Schrödinger, LLC. (2017) The PyMOL Molecular Graphics System, Version 2.0, Schrödinger, LLC, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.