

Abstract
Benzylisoquinoline alkaloids (BIAs) are a structurally diverse class of plant-specialized metabolites that have been particularly well-studied in the order Ranunculales. The N-methyltransferases (NMTs) in BIA biosynthesis can be divided into three groups according to substrate specificity and amino acid sequence. Here, we report the first crystal structures of enzyme complexes from the tetrahydroprotoberberine NMT (TNMT) subclass, specifically for GfTNMT from the yellow horned poppy (Glaucium flavum). GfTNMT was co-crystallized with the cofactor S-adenosyl-l-methionine (dmin = 1.6 Å), the product S-adenosyl-l-homocysteine (dmin = 1.8 Å), or in complex with S-adenosyl-l-homocysteine and (S)-cis-N-methylstylopine (dmin = 1.8 Å). These structures reveal for the first time how a mostly hydrophobic L-shaped substrate recognition pocket selects for the (S)-cis configuration of the two central six-membered rings in protoberberine BIA compounds. Mutagenesis studies confirm and functionally define the roles of several highly-conserved residues within and near the GfTNMT-active site. The substrate specificity of TNMT enzymes appears to arise from the arrangement of subgroup-specific stereospecific recognition elements relative to catalytic elements that are more widely-conserved among all BIA NMTs. The binding mode of protoberberine compounds to GfTNMT appears to be similar to coclaurine NMT, with the isoquinoline rings buried deepest in the binding pocket. This binding mode differs from that of pavine NMT, in which the benzyl ring is bound more deeply than the isoquinoline rings. The insights into substrate recognition and catalysis provided here form a sound basis for the rational engineering of NMT enzymes for chemoenzymatic synthesis and metabolic engineering.
Keywords: X-ray crystallography, biosynthesis, secondary metabolism, enzyme mechanism, enzyme structure, substrate specificity, stereoselectivity, benzylisoquinoline alkaloids, morphine, N-methyltransferase, structure–function relationship
Introduction
Benzylisoquinoline alkaloids (BIAs)4 are a class of plant-specialized metabolites with a diversity of useful and potent pharmacological properties. Common BIAs include the analgesic morphine, the antitussive and anticancer agent noscapine, and the antibacterial compound sanguinarine (1, 2). Whereas over 2500 BIA substructures have been characterized, a much smaller number of functionally distinct enzymes has been shown to catalyze steps in BIA biosynthesis (3). Elucidation of the major biochemical pathways of BIA biosynthesis in planta is ongoing, with most enzymatic steps of the process determined by substrate turnover assays and/or gene silencing experiments. Because of the importance and utility of BIA compounds, several groups are actively exploring the use of metabolic engineering and synthetic biology approaches to reconstitute portions of the BIA biosynthetic pathways in microbial systems to produce novel or rare BIA (4–6). However, most attempts to date have resulted in modest yields, likely due to the production of off-target side products resulting from the apparent promiscuity of many naturally occurring enzymes. To address this central problem in metabolic engineering, fundamental molecular, structural, and functional studies of BIA enzymes are needed. A deeper understanding of enzyme structure and function will lead to an improved understanding of how BIA diversity is generated. In turn, these insights will provide a basis for the rational design of more efficient microbial systems for the production of a wider range of pharmacologically useful BIA compounds.
Several key steps in BIA biosynthesis involve the methylation of the central amino group (1, 7). Several BIA-specific N-methyltransferases (NMTs) have been identified from different species within the order Ranunculales. BIA NMTs can be organized into three main groups based on sequence conservation and substrate specificity: the coclaurine NMTs (CNMTs), tetrahydroprotoberberine NMTs (TNMTs), and pavine/reticuline NMTs (PavNMT/RNMTs). The CNMTs are involved in the generation of (S)-reticuline, a common intermediate in the biosynthesis of most BIA metabolites, including the morphinans (morphine and codeine) (8–10). The PavNMTs/RNMTs and TNMTs appear to have a broader substrate specificity, acting on a wider range of substrates in multiple distinct pathways downstream of (S)-reticuline. Both PavNMT and RNMT have been shown to efficiently produce quaternary 1-benzylisoquinolines in vitro (including (S)-tembetarine) (11–13). In planta evidence suggests that PavNMT is involved in the biosynthesis of quaternary pavinans (including N-methyleschscholtzidine), and RNMT is implicated in the biosynthesis of quaternary aporphines (including magnoflorine). TNMT plays a key role in pathways leading to the formation of protoberberines (including tetrahydrocolumbamine), protopines (including protopine), benzo[c]phenanthridines (including sanguinarine), and phthalideisoquinolines (including noscapine) (Fig. 1) (14, 15). Notably, TNMTs have been reported previously to produce only (S)-cis-N-methyl stereoisomers in contrast to chemical N-methylation, which predominantly yields (S)-trans products (16–18). The stereospecificity of the N-methylation reactions catalyzed by TNMTs appears to be crucial for furnishing protoberberine substrates with the correct stereochemistry for downstream enzymes that do not accept substrates with (S)-trans stereochemistry (19).
Figure 1.

Contributions of tetrahydroprotoberberine N-methyltransferases to BIA biosynthesis in plants. The central BIA biosynthetic pathway yields the core intermediate (S)-reticuline from which branch pathways diverge. TNMT catalyzes the N-methylation of (S)-stylopine in the pathway leading to protopines and benzo[c]phenanthridines (e.g. sanguinarine). TNMT also acts on (S)-canadine in a different pathway that leads to the production of phthalideisoquinolines (e.g. noscapine). Dashed arrows represent multiple enzymatic conversions. The carbon numbering and ring labeling schemes used in the text are shown using (S)-stylopine as a representative protoberberine.
Two previous structural studies on BIA NMTs have been performed for Thalictrum flavum PavNMT (11) and Coptis japonica CNMT (8). These studies have established the overall architecture for NMT enzymes involved with the biosynthesis of BIA compounds, including the identification of a conserved active-site region containing several residues important for substrate binding and catalysis. These recent studies have also raised questions relating to the specific mechanisms utilized by BIA NMTs for differentiating between structurally similar BIAs, as well as the underlying mechanism of catalysis. To further elucidate the central issues of substrate specificity and catalytic mechanism, we report the first structural and functional characterization of an enzyme from the TNMT subclass, GfTNMT from Glaucium flavum (yellow horned poppy). The structure–function relationships seen in GfTNMT reveal novel insights into the mechanisms used by BIA NMT enzymes for defining substrate specificity and catalysis that will be invaluable for future efforts in metabolic engineering.
Results
Functional characterization of GfTNMT
G. flavum TNMT (GfTNMT; originally reported as GFLNMT2) was one of six BIA NMT enzymes previously cloned and characterized from G. flavum (20). A preliminary assessment of substrate specificity suggested that GfTNMT was functionally most similar to the tetrahydroprotoberberine NMTs characterized from opium poppy, T. flavum, and Eschscholzia californica (12, 21, 22). The amino acid sequence of GfTNMT shows 47–53% identity to CNMTs, RNMTs, and PavNMTs although it shares greater than 80% identity with the three functionally characterized TNMTs (Fig. S1) (12, 21). Phylogenetic analysis of functionally characterized BIA NMT sequences from Ranunculales placed it firmly in a well-supported clade along with known TNMTs. Based on these analyses, the enzyme was considered representative of a canonical TNMT and was renamed GfTNMT.
The natural GfTNMT translation product is 40.9 kDa and 358 amino acids in length, whereas the recombinant protein includes a 25-amino-acid N-terminal linker and hexahistidine tag, giving a calculated molecular mass of 43.5 kDa. As with all known BIA NMTs, the polypeptide does not appear to contain any canonical subcellular targeting signals (23, 24). Both Papaver somniferum CNMT and TNMT were shown to localize to the cytoplasm of cultured opium poppy cells, and this feature is thought to be conserved across BIA NMTs, including GfTNMT (25).
Expression of GfTNMT in Escherichia coli and purification by immobilized metal ion affinity chromatography allowed for the determination of the enzyme's characteristics in vitro. The activity of GfTNMT was assessed with 35 alkaloids primarily representing the major BIA structural subclasses and a wide range of naturally occurring functional group modifications (Table 1). Reaction products consistent with N-methylation (+14 m/z with respect to the substrate) were detected for 22 substrates spanning seven BIA structural subclasses. Alkaloids belonging to the isoquinoline, indole, β-carboline, phenylalkylamine, and tryptamine classes were not accepted as substrates, suggesting that GfTNMT is relatively specialized in function. The protoberbeine substrates stylopine and tetrahydropalmatine (THP) were most efficiently methylated. Intermediate activities were observed with select morphinan, aporphine, and pavine substrates, and trace activities were present with 1-benzylisoquinoline and phthalideisoquinoline alkaloids. Collision-induced dissociation spectra obtained for the protoberberine products confirmed their identities as (S)-cis-N-methylstylopine (SMS) and N-methyl-THP (Fig. S2). The activity of GfTNMT was reduced to approximately one-half for the substrate with one free hydroxyl group in the C-3 position (i.e. tetrahydrocolumbamine) and further reduced to one-fourth with two free hydroxyl groups at the C-2 and C-9 positions (i.e. scoulerine). In an analogous manner, the O-methylated morphinan alkaloids were turned over, whereas those with free hydroxyl groups (i.e. codeine and morphine) were not. Although GfTNMT exhibited a preference for the nonphenolic tertiary amine substrates, it was also capable of N-methylating substrates with secondary amines (e.g. coclaurine) to a lesser extent. Under initial rate conditions with secondary amine BIA substrates, GfTNMT yielded products with tertiary amines resulting from a single methyl transfer.
Table 1.
Relative substrate turnover for GfTNMT in vitro
Activity values represent the relative percentage of substrate turned over (mean ± standard deviation of three replicates), where the assay with the most turnover was set to 100% and others were scaled proportionally. + and ++ denote substrates where product formation was detected in minor or major trace amounts (less than 2% substrate loss), respectively. nd denotes no detectable activity. R group designations are provided in Table S1.
a Stylopine was at 6.0 pmol s−1 μg−1.
The dependence of GfTNMT activity on temperature and pH displayed features consistent with the ranges previously reported for BIA NMTs (9, 11, 13, 21). The recombinant enzyme's activity was maximal at pH 8.0 and dropped to less than 10% of the total activity at pH 6.0 and 9.5 (Fig. S3). Activity was greatest at 30 °C and dropped to less than 10% maximal activity at 60 °C, but the enzyme retained ∼40% activity at 4 °C. Based on these results, kinetic parameters for a wide range of substrates were measured at pH 8.0 and 30 °C (Fig. S4). Maximal activity was detected at 25 μm stylopine (13.4 pmol s−1 μg−1) and 63 μm THP (6.2 pmol s−1 μg−1), followed by a pronounced inhibition of activity at higher concentrations for both substrates (Fig. S4, B and C). Half-maximal activities were calculated to occur at 13 and 18 μm, respectively. In addition to substrate inhibition, GfTNMT displayed substantial product inhibition. The activity of GfTNMT with 0.1 mm THP was reduced to 50% of the maximum activity in the presence of 2.5 μm SMS (Fig. S4D). Fitting the data to a rapid-equilibrium model of reversible substrate (S) inhibition in which a single E/S/S-inhibited complex forms reveals a poor fit at higher substrate concentrations, possibly because of the confounding effects of product inhibition or the presence of a more complex mechanism of substrate inhibition. As a result, values for kcat (∼0.8 s−1 for stylopine and ∼0.4 s−1 for THP) and Km (∼15 μm for stylopine and ∼30 μm for THP) can only be approximately determined from steady-state measurements. However, values of kcat/Km can be more reliably estimated from the slope of the initial rate plot at low substrate concentrations (48,000 and 13,500 s−1 m−1 for stylopine and THP, respectively), where the effects of substrate and product inhibition are likely less significant.
At a fixed 50 μm concentration of stylopine, GfTNMT showed a typical Michaelis-Menten substrate saturation curve with respect to the cofactor S-adenosylmethionine (SAM), resulting in a calculated Vmax of 16.5 pmol s−1 μg−1, Km of 138 μm, kcat of 0.72 s−1, and kcat/Km of 5200 s−1 m−1. Under optimal conditions, the catalytic rate constants were 0.58 s−1 and 0.27 s−1 for stylopine and THP, respectively.
Overall structure of TNMT
Recombinant GfTNMT was expressed in E. coli and crystallized at pH 7.0. Well-ordered crystals of GfTNMT were grown in the presence of S-adenosylhomocysteine (SAH), SAM, and both SAM + stylopine (which the electron density maps show were converted to the products SAH and SMS), yielding data sets with resolution limits extending to 1.6–1.8 Å (Table 2). Each crystal belongs to space group P3221, with a single protomer lying within the asymmetric unit. The structure of GfTNMT bound to SAH was determined by molecular replacement using the polypeptide chain A of T. flavum pavine N-methyltransferase (PDB 5KOK), which shares 53% sequence identity (11). The structures of the other two complexes were determined starting from the structure of the GfTNMT/SAH complex. The refined models of GfTNMT include 336 out of the 359 residues in the native protein. The three-dimensional structure of 13 residues in the 70s loop (residues 68 to 80) was not defined sufficiently well in electron density maps to be modeled in any of the GfTNMT crystal structures. Additionally, 35 residues at the N terminus of the recombinantly expressed form of GfTNMT were not resolved, of which 25 residues compose the non-natural hexahistidine tag and polypeptide linker added during the construction of the expression vector. The C terminus of GfTNMT and all other regions within the polypeptide chain are clearly defined by electron density maps.
Table 2.
Crystallographic statistics
| Crystal | GfTNMT + SAH | GfTNMT + SAM | GfTNMT + SAH + SMS |
|---|---|---|---|
| PDB code | 6P3M | 6P3N | 6P3O |
| Space group | P3221 | P3221 | P3221 |
| Unit cell dimensions | |||
| a = b, c (Å) | 103.693, 82.812 | 104.023, 82.746 | 103.473, 82.161 |
| α = β, γ (°) | 90, 120 | 90, 120 | 90, 120 |
| Wavelength (Å) | 0.9795 | 0.9795 | 0.9795 |
| Resolution (Å)a | 37.60-1.80 (1.85-1.8) | 39.56-1.60 (1.64-1.60) | 39.34-1.80 (1.85-1.80) |
| Rsymb | 0.055 (1.10) | 0.067 (0.536) | 0.075 (1.02) |
| CC1/2 | 1.00 (0.74) | 1.00 (0.68) | 1.00 (0.84) |
| I/σ | 26.93 (2.18) | 11.59 (1.65) | 17.84 (2.42) |
| Completeness (%) | 99.9 (99.9) | 98.9 (94.1) | 99.9 (100.0) |
| Redundancy | 9.8 (9.4) | 5.4 (5.0) | 13.0 (13.4) |
| Refinement | |||
| Resolution (Å) | 1.80 | 1.60 | 1.80 |
| Unique reflections | 45,474 | 64,394 | 44,981 |
| Rworkc/Rfreed | 0.162/0.197 | 0.178/0.196 | 0.184/0.206 |
| Total no. of atoms | 3106 | 3249 | 3118 |
| Protein atoms | 2753 | 2794 | 2746 |
| Ligand atoms | 31 | 27 | 51 |
| Water atoms | 322 | 428 | 321 |
| Average B-factors (protein) | 35.28 | 34.07 | 39.21 |
| Average B-factors (ligand) | 34.90 | 29.89 | 44.45 |
| Average B-factors (water) | 46.51 | 47.38 | 48.23 |
| r.m.s.d. from ideal geometry | |||
| Bond lengths (Å) | 0.005 | 0.007 | 0.007 |
| Bond angles (°) | 1.18 | 1.15 | 1.34 |
a Values from the outermost resolution shell are given in parentheses.
bRsym = Σi|Ii − 〈I〉|/Σi Ii, where Ii is the ith integrated intensity of a given reflection and 〈I〉 is the weighted mean of all measurements of I.
c Rwork = Σ‖Fo| − |Fc‖/Σ|Fo| for 95% of reflection data used in refinement.
d Rfree = Σ‖Fo| − |Fc‖/Σ|Fo| for 5% of reflection data excluded from refinement.
The overall structure of GfTNMT shows the canonical SAM-binding domain and α/β Rossmann fold conserved in class I methyltransferases (Fig. 2) (26). The remainder of the structure comprises an α-helical domain that is very similar to the corresponding domain primarily responsible for defining substrate specificity in PavNMT and CNMT. Superimposition of the entire α-carbon trace of GfTNMT with PavNMT (PDB 5KOK, chain A) and CNMT (PDB 6GKV) yields RMSD values of 0.686 Å (n = 287) and 0.716 Å (n = 299), respectively. As expected, superimposition of each protein by the SAM-binding domain yields much lower RMSD values of 0.445 Å (n = 120) and 0.462 Å (n = 129), whereas superimposition of each protein by the BIA-binding domain yields RMSD values of 0.758 Å (n = 149) and 0.908 Å (n = 166), indicating that the majority of structural variation occurs within the BIA-binding domain.
Figure 2.

Overall crystal structure of GfTNMT in complex with SAH and SMS. A, single subunit of GfTNMT is drawn in ribbon representation colored along a gradient from the N terminus (blue) to the C terminus (red), with the portion of the structure that is poorly defined by electron density from residues 68 to 81 being represented by a dashed line. SAH (carbon atoms, magenta) and SMS (carbon atoms, gray) are shown in all-atom, space-filling representation. Solvent-accessible surface in semi-transparent light gray is shown superimposed onto the ribbon diagram. B, homodimer of GfTNMT was constructed by applying a crystallographic 2-fold rotational symmetry operator on the asymmetric unit composed of a single protomer.
A crystallographic 2-fold rotational symmetry operator generates a homodimeric assembly that is very similar to PavNMT and CNMT. The dimer interface in TNMT (1019 Å2 of buried surface area) includes six salt bridges and eight hydrogen bonds and is derived from the α3, α12, β6, and β7 secondary structural elements on the opposite side of the SAM-binding domain from the substrate-binding pocket (Fig. 3). The dimer complex is predicted to be favorable, with a predicted ΔGsolvation = −5.7 kcal/mol/monomer upon dimerization (27). The elution volume of GfTNMT observed during size-exclusion chromatography is consistent with the hydrodynamic radius expected for a homodimer (Fig. S5).
Figure 3.

Multiple sequence alignment of representatives from the three main branches of NMT enzymes in BIA biosynthesis. Arrows indicate residues targeted for mutation based on sequence conservation and proximity to the active site. Residues in the dimer interface forming salt bridges, hydrogen bonds, or other interactions are colored green, yellow, and gray, respectively. Residues that interact with the SAM/SAH are colored purple. Residues that interact with SMS are colored red. The names of secondary structural elements are consistent with that of PavNMT (11). GenBankTM accession numbers are as follows: GfTNMT, G. flavum TNMT (20); PsTNMT, P. somniferum TNMT, AAY79177; PsRNMT, P. somniferum RNMT, KX369612; TfPavNMT, T. flavum PavNMT, EU883010; CjCNMT, C. japonica CNMT, BAB71802; GfCNMT, putative G. flavum CNMT (20); PsCNMT, P. somniferum CNMT, AAP45316.
Structural determinants of substrate recognition
In GfTNMT, the substrate-binding pockets lie at the junction of the BIA-binding and SAM-binding domains. The methyl donor SAM forms most of its interactions with the Rossman fold domain, including the highly-conserved GXGXG motif seen in all class Ia NMTs (28). All of the interactions between TNMT and SAM are similar to those seen in PavNMT and CNMT, but are somewhat surprisingly distinct from the interactions seen in other NMTs. This sub-class specificity for SAM binding indicates that the NMTs involved with BIA biosynthesis likely diverged from a common ancestor and perhaps indicate unique aspects of SAM recognition that may also be functionally important in BIA NMTs.
The L-shaped binding pocket for SMS appears to be quite distinct from the BIA-binding pockets seen in the previously determined structures of CNMT and PavNMT (Fig. 4; supporting Movie S1). At the bottom of the “L,” the mostly hydrophobic side chains of Phe-92, Glu-98, Trp-297, Ile-329 and Phe-332 form a 'benzyl pocket' (BP) that surrounds the portion of stylopine derived from the benzyl group of the branch point BIA precursor reticuline. At the top of the L, the mostly hydrophobic side chains of Ile-234, Phe-243, Phe-257, Val-262, Met-290, and Phe-340 form an “isoquinoline pocket” (IP) that is complementary to the portion of stylopine derived from the isoquinoline group of reticuline. At the bend in the L, the mostly hydrophilic side chains of Tyr-81, Glu-204, Glu-207, and His-208 form a small “catalytic pocket” (CP) that surrounds the amino group and methyl donor of SAM. Notably, there is clear electron density for the side chain of Tyr-81, which is located adjacent to a poorly ordered part of the 70s loop. Even though 13 residues from the 70s loop are not ordered well-enough to model in any of the TNMT structures determined, the clear electron density for the side chain of Tyr-81 and the proximity of the phenol group to the amino group of the substrate and product strongly suggest a role in substrate binding and/or catalysis.
Figure 4.
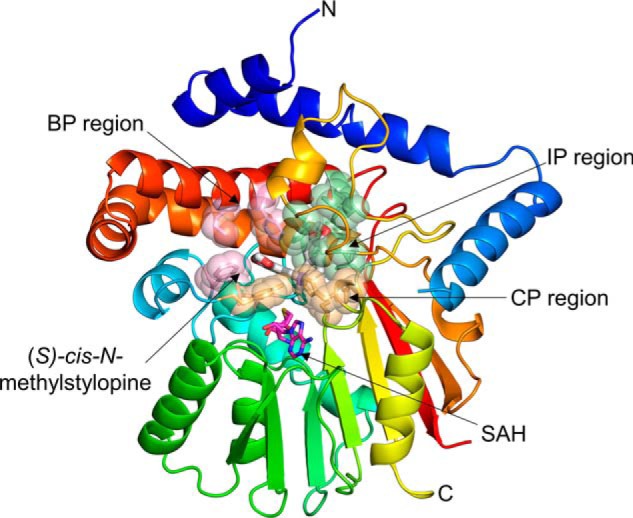
Regions of GfTNMT defining the substrate-binding site. The substrate-binding site is defined by the residues contained within the BP, IP, and CP highlighted in light pink, light green, and light yellow, respectively. SMS is drawn as white sticks, and SAM is drawn as magenta sticks. Residues defining the BP, IP, and CP are shown as translucent spheres.
In addition to the large number of hydrophobic contacts seen in the BP and IP, a limited number of hydrogen bonds appear to be formed with the oxygen atoms in the ether linkages at the ends of the BIA substrate/product. At the end of the BP, the side-chain phenol group of Tyr-81 and carboxyamide group of Glu-98 are close enough to form hydrogen bonds with the oxygen atoms bonded to C-9 and C-10.
Water molecules likely play important roles in binding specificity and affinity. First, in the absence of the BIA substrate, ordered water molecules appear to occupy most of the BP and IP. When SMS is present, most of these water molecules are displaced, but the positions of residues within 5 Å of the BIA substrate or product do not seem to be affected. Displacing these well-ordered water molecules from this somewhat rigid binding site likely contributes a favorable entropic effect upon binding. Second, two water molecules trapped within the interior of the binding pocket by the BIA product facilitate hydrogen bonding between the oxygen at the C-2 position and the side chain of Gln-339 and the main chain carbonyl oxygen of Phe-332 (Fig. 5). These water molecules appear to aid in correctly positioning the substrate within the binding pocket and likely determine in part the enzyme's specificity through recognition of the C-2/C-3 methylenedioxy bridge commonly found in TNMT substrates.
Figure 5.

Interactions between GfTNMT and the N-methylated product SMS. Residues within 4 Å of SMS are shown as lines and presented in all-atom representation. Hydrogen bonds are represented as yellow-dashed lines. A, SMS is held in a bent conformation within the binding-site pocket through hydrophobic interactions between the protein and the protoberberine backbone of SMS. B, presence of a hydrogen bond network between two water molecules and O-16 helps to recognize the terminal methylenedioxy bridge adjacent to ring A of the isoquinoline portion of the BIA substrate.
The size and shape of the NMT-binding pocket appear to be complementary to structures of BIA compounds in which the two central six-membered rings are arranged in an (S)-cis configuration (Fig. 6, A and B). The cis configuration (i.e. hydrogen atoms on the carbon and nitrogen atoms bridging the two central rings both lying on the same side of the C–N bond) causes the best-fit planes of the two six-membered rings to be arranged in an L-shaped conformation in which the planes are roughly 90° relative to each other (Fig. 5C). In contrast, arrangement of these atoms in a trans conformation leads to a dramatically different coplanar arrangement of the best-fit planes for the two adjacent six-membered rings. The refined omit map of GfTNMT crystallized in the presence of stylopine and SAM clearly supports the (S)-cis conformation of the SMS product and also shows the presence of SAH, the other product expected after SAM has donated its methyl group to stylopine (Fig. 7).
Figure 6.

Internal cavities surrounding SMS (white) and SAH (blue) in GfTNMT. A, in GfTNMT, the enzyme product is found in an L-shaped conformation with rings A and B lying within the deepest part of the cavity formed primarily by the IP. B, right view is rotated ∼90° from the view in A. C, structures of (R)-cis, (S)-cis, and (S)-trans, stereoisomers of stylopine, are shown in the low-energy conformations seen in the crystallographic structures of closely-related BIA compounds from the Cambridge Crystallographic Structure Database: UHUYEB (18), YILHUW (60), and LURXOL (58). Structures are shown in an oblique view showing all four rings on the left, followed by a rotated view that shows the flat, planar arrangement of all rings when the hydrogen atoms bonded to the central bridging atoms between rings B and C adopt a trans configuration. Note that for compounds with the central bridging atoms in the cis configuration, rings A and B clearly lie out-of-plane with rings C and D.
Figure 7.
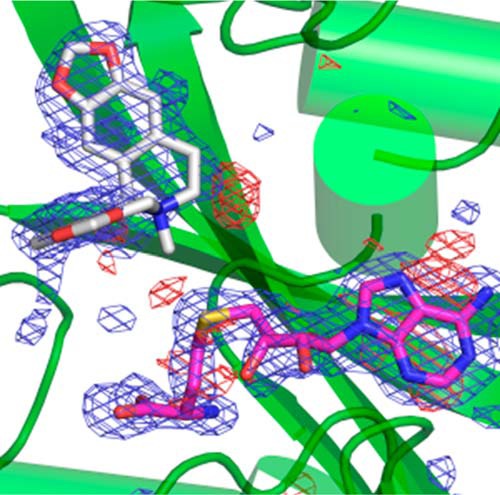
Omit electron density map of SAH (magenta) and SMS (light gray) bound to GfTNMT. The refined |Fo| − |Fc| electron density map was calculated after removing the coordinates for the two ligands and refining the structure for 20 cycles. The map is shown at a contour level three standard deviations above the mean. Note the absence of electron density for a methyl group attached to the sulfur atom of SAH, confirming the transfer of a methyl group from the substrate SAM initially present in the crystallization mixture. Also note the weaker electron density around the N-methyl group and ring C of the isoquinoline ring of SMS. The electron density for this part of the isoquinoline ring is better defined at lower contour levels. Positive electron density is shown as a blue mesh, and negative electron density is shown as a red mesh.
Mutational studies of TNMT
As mentioned previously, the L-shaped binding pocket for SMS can be roughly divided into three main regions that primarily determine substrate specificity and catalysis (Fig. 4). In TNMT, three residues in the α14-helix (His-328, Ile-329, and Phe-332) form one side of the binding pocket defining the BP region. Mutating the equivalent residues in TNMT to alanine also results in substrate-dependent changes in enzyme activity (Fig. 8A). The H328A mutation produces a 5- and 2-fold decrease in activity with stylopine and scoulerine, respectively, but the activity with THP increases 2-fold. Even though the nearest atom of His-328 is located more than 5.9 Å from the BIA substrate, the imidazole side chain positions the side chain of Phe-332 within the active site to interact directly with ring D and the C-9/C-10 methylenedioxy bridge in SMS. Consistent with its more direct role in substrate binding, mutation of Phe-332 to alanine results in a >10-fold reduction in activity for all three BIA substrates. A >5-fold decrease in activity is also seen for all three substrates in the I329A mutant. Previous mutational analysis of residues 328–332 in the α16-helix of CNMT by Bennett et al. (8) confirms the importance of some of these residues for substrate binding and specificity in a related enzyme.
Figure 8.

Relative activity of GfTNMT mutants. A and B, relative activity of putative substrate binding or recognition mutants on TNMT substrates is shown with black bars for stylopine, gray bars for tetrahydropalmatine, and white bars for scoulerine. C, relative activity of putative substrate binding or recognition mutants with respect to PavNMT substrate pavine is shown with black bars for formation of monomethylated product (+14 m/z) and gray bars for di-methylated product (+28 m/z). D, relative activity of putative catalytic mutants on TNMT substrates is shown with black bars for stylopine, gray bars for tetrahydropalmatine, and white bars for scoulerine. E, relative activity of putative catalytic mutants under conditions outside the linear range with respect to the WT GfTNMT is shown for tetrahydropalmatine. Relative product formation was quantified by LC-MS, with the product ion count for WT GfTNMT and each substrate set to 100%, and all other corresponding values were scaled accordingly. E, mutant E207A was set to 100%. Error bars represent standard deviation. nd, no product detected.
Patterns of primary sequence conservation in TNMTs suggest that two residues in the IP region (Met-290 and Phe-340) and one residue in the CP region (Glu-204) may be particularly important for defining substrate specificity. To explore this, we prepared single, double, and triple alanine mutations at these three positions, as well as replacements of these residues to the equivalent residues found in PavNMT. As expected, the single substitutions with alanine or the residues found in PavNMT indicate more modest decreases in binding activity than the double and triple alanine mutations (Fig. 8, B and C). The triple mutant E204A/M290P/F340Y showed the greatest decrease in activity with the native TNMT substrates stylopine, THP, and scoulerine, as well as a significant decrease in the activity with PavNMT-specific substrates pavine. This result shows that replacement of these residues is not sufficient to switch the specificity of TNMT to PavNMT.
Arg-41, which lies near the tip of the IP region, was also targeted for mutagenesis studies based on its potential for indirect effects on substrate binding as well as its high level of sequence conservation in BIA NMTs (Figs. 3 and 7). Arg-41 interacts with either three well-defined water molecules in the crystal structures of two different complexes (TNMT + SAM; TNMT + SAH + SMS) or with a negatively-charged sulfate ion in the TNMT + SAH complex, all near the side chains of Gln-339 and Phe-38. Even though the guanidinium group of Arg-41 is quite distant (10.9 Å) from the BIA substrate, it interacts with the side chains of Phe-38, Phe-243, and Gln-339 through positioning of the trapped water molecules or sulfate ion. Phe-38, Phe-243, and Gln-339 all interact directly with the substrate. Somewhat surprisingly, the R41A mutant exhibited increased relative activity for the substrates THP and scoulerine but showed no significant change of activity for stylopine (Fig. 8A).
Residues in the CP region occupy the region of the active site near the nitrogen atom and the methyl donor, thus likely playing roles in the formation and breakdown of the transition state. Sequence conservation and the structures of a diverse range of NMTs suggest that His-208 most likely plays an important role in catalysis, possibly as a general base to accept a proton from the donor amino group to create a stronger nucleophile (Fig. 3) (8, 11, 29). Residues Glu-204 and Glu-207 are also nearby and may also play important roles in catalysis. Previously reported mutagenesis studies suggest roles in stabilizing specific steps of the reaction coordinate (8, 11). In TNMT, single mutations of residues His-208, Glu-204, and Glu-207 to alanine result in <10% of WT activity for three different substrates, with scoulerine showing slightly higher activity (∼10 and ∼20% activity, respectively) in the E204A and E207A mutants (Fig. 8D). Combining these mutations to form three different double mutants and the triple mutant all generate inactive enzymes with trace levels of substrate turnover under standard assay conditions. Assaying the single, double, and triple mutants at high protein concentrations to lower the detection limit showed no substantial difference between each of the E204A and E207A single mutants (Fig. 8E). Similarly, there were no substantial differences between the H208A single mutant and E204A/E207A double mutant although both showed less activity than the previous single mutants. The E204A/H208A and E207A/H208A double mutants showed even lower activity but were not notably different from each other, and the triple mutant showed substantially less activity than all others.
Other potentially interesting residues in the CP region lie in the 70s loop that is generally poorly ordered in the crystal structures of BIA NMTs and other NMTs. Residue Tyr-81 lies at the end of the 70s loop in GfTNMT and is highly-conserved in all BIA NMTs. The crystal structure of the ternary complex places the hydroxyl group of the side chain within 2.9, 3.7, and 4.0 Å of the sulfur of SAH, Nγ of His-208, and nitrogen of SMS, respectively. Based on its position and the potential for this group to play multiple roles during catalysis, a series of point mutants were generated and assayed. Mutations of Tyr-81 to phenylalanine, alanine, or arginine all yield similar results for all three substrates, with 2–5-fold decreases in activity relative to the WT enzyme (Fig. 8A). Torres et al. (11) previously showed that the Y79A mutant in PavNMT showed a significant decrease in activity with preferred substrates, while marginally increasing activity with the minor substrate tetrahydropapaverine.
Discussion
Functional characterization of GfTNMT
Our functional characterization of GfTNMT expands on similar enzymes previously cloned from P. somniferum, Papaver bracteatum, and E. californica (12, 21). Sequence identity and phylogenetic analysis place it firmly as a member of the TNMT family, which appears to be monophyletic, as reported previously (Fig. S1) (21). In contrast to CNMTs and the distantly related BIA NMT-like enzyme recently discovered in Ephedra, GfTNMT did not accept any of the non-BIAs, including tetrahydroisoquinolines, and is evidently more specialized in function (Table 1) (9, 12, 30). This observation is consistent with hypotheses that TNMTs diverged from the CNMTs and are more evolutionarily advanced (21). GfTNMT exhibits a substrate range largely comparable with other TNMTs and also showed an apparent aversion for free hydroxyl groups in its substrates (12, 16, 21). From a plant biosynthetic perspective, this feature is significant because scoulerine, the first protoberberine pathway intermediate, is synthesized in a membrane-bound subcellular compartment (ER or ER-derived vesicles) and any quaternary alkaloids produced therein (e.g. N-methylscoulerine) would be unable to diffuse or be transported out into the cytosol where downstream enzymes reside (25, 31). From a structural perspective, this selectivity for fully O-substituted protoberberines can be partially attributed to indirect effects from Arg-41, which is conserved in all NMTs, as well as to the binding of the tetrahydroprotoberberine substrates within the active site with minimal room within the binding pocket to accommodate the hydrophilic side chains.
The presence of high levels of apparent substrate inhibition and product inhibition makes it challenging to determine the exact values for kinetic parameters. However, the apparent affinity (Km) of GfTNMT for stylopine (∼15 μm) appears to be worse than reported for PsTNMT and PbTNMT (0.6 and 2.0 μm, respectively) but better than for EcTNMT (40 μm; Fig. S4) (12). Nevertheless, the GfTNMT turnover number (kcat) for stylopine (∼0.8 s−1) was substantially better than PbTNMT (0.001 s−1) and similar to EcTNMT (0.3 s−1). The apparent affinity for the cofactor SAM was somewhat worse than expected, with an apparent Km value at 50 μm stylopine (138 μm) nearly 10- and 3-fold greater than reported for PsTNMT (11.5 μm) and EcTNMT (32 μm), respectively, but better than those reported for CjCNMT (390 μm) and PsRNMT (168 μm). The in vitro activity of GfTNMT is consistent with a primary role in biosynthesis of several quaternary alkaloids present in Glaucium spp., including protoberberines SMS and N-methyl-THP, some of which show antimicrobial activity that may confer a selective advantage (2). Prior to this report, next-generation sequencing and integrated metabolomics were used to show that GfTNMT transcripts as well as corresponding BIA substrates and products are abundant in G. flavum root tissues (20, 32). In addition, many of these compounds likely also function as crucial intermediates in the biosynthesis of protopine alkaloids known to accumulate in certain Glaucium species (1, 33) and in the biosynthesis of benzo[c]phenanthridine (e.g. sanguinarine) and phthalideisoquinoline (e.g. noscapine) alkaloids that accumulate in opium poppy and other species.
The apparent substrate inhibition of GfTNMT in the presence of high concentrations of stylopine and THP substrates is comparable with that reported for EcTNMT and PbTNMT (Fig. S4) (12). Although inhibition has not been reported for various CNMTs, both PsRNMT and TfPavNMT displayed modest inhibition with certain substrates (11, 13). For TfPavNMT, a crystal structure showing both (S)- and (R)-tetrahydropapaverine simultaneously bound in the active site leads to a suggestion that the use of racemic (R,S)-tetrahydropapaverine in enzyme assays was responsible for the observed inhibition (11). The physiological significance of substrate inhibition has not been studied in BIA-biosynthesizing plants. However, substrate inhibition has been proposed to help to maintain a steady concentration of products despite varying substrate availability and thus stabilize pathway flux (34). Product inhibition has not been studied for other BIA NMTs, but substantial inhibition by the pathway end-product sanguinarine was demonstrated for an O-methyltransferase from T. flavum (35). For GfTNMT, inhibition by SMS may reflect the enzyme's role in producing a pathway intermediate rather than end-product alkaloid. In this case, product inhibition may help to prevent the accumulation of an N-methylated product that may have undesirable effects on downstream enzymes or impact plant viability.
In contrast to previously reported TNMTs, GfTNMT exhibits a broader substrate range, which includes aporphine and morphinan alkaloids. Given the existence of five additional BIA NMTs in G. flavum, the physiological significance of the apparently broad range of substrates accepted by GfTNMT remains unclear (20). Although quaternary aporphine alkaloids such as those produced by GfTNMT in vitro (e.g. magnoflorine) are widespread in plants, including some Glaucium spp., quaternary morphinans such as N-methylthebaine have only rarely been reported in select Papaver species (33, 36–38). This work is the first to describe an NMT with activity toward morphinans (39), but previously characterized TNMTs do not appear to have been assayed with morphinans. The fairly broad substrate specificity seen in GfTNMT suggests practical applications. For example, the enzyme could be used to add non-natural substituents into semisynthetic opiates such as naloxone (8).
Insights into substrate recognition
When combined with the results of site-directed mutagenesis experiments, the crystal structures of GfTNMT provide a molecular structural explanation for substrate recognition in the TNMT enzymes that also clarifies the strategies used by other BIA NMTs for recognizing distinct substrates. The most deeply buried and perhaps most distinctly formed part of the binding site for protoberberine substrates is the IP. The isoquinoline group of SMS binds in a manner that is quite similar to that which is seen for the methylheliamine product analog co-crystallized with CjCNMT, but this binding mode is quite different from that which is seen for tetrahydropapaverine bound to TfPavNMT, where the benzyl ring occupies the lower part of the IP region (Fig. 9). Met-290 and Phe-340 are highly-conserved positions in the TNMTs and occupy central positions in the IP region adjacent to the isoquinoline ring of SMS bound to GfTNMT. Replacing Met-290 with the TfPavNMT consensus residue proline results in large decreases in activity for the canonical protoberberine substrates but does not affect the enzyme's mono-methylation activity for the canonical PavNMT substrate pavine (Fig. 8). Replacing Phe-340 with the TfPavNMT consensus residue tyrosine also results in large decreases in activity for the canonical protoberberine substrates stylopine and scoulerine, although there is a small increase in activity for tetrahydropalmatine. For this mutant, the mono-methylation of pavine is also not affected, although the di-methylation reaction seems less efficient, as also seen for the M290P mutant. These single-site mutations indicate that relatively minor changes to the closely-packed IP region lead to a change in specificity that makes the enzyme lose most of its ability to use protoberberine substrates, while mostly retaining the ability to use pavine as a substrate, thus becoming slightly more PavNMT-like. Mutating these positions to alanine leads to larger decreases in activity for both the protoberberine and pavine substrates, indicating a beneficial effect for pavine when PavNMT consensus residues are present.
Figure 9.

Substrate binding in the active sites of related BIA NMTs. A, structures of NMT substrates or substrate analogs (white) are shown relative to the product SAH (space-filling representation) in the binding sites of CjCNMT (left, orange; PDB code 6GKZ), GfTNMT (center, green; PDB code 6P3O), and TfPavNMT (right, cyan; PDB code 5KOK). Each structure is drawn from the same viewpoint relative to the SAH-binding domain. Residues and water molecules within the proximity of the ligand in GfTNMT, and their equivalent position by sequence alignment in CNMT and PavNMT, are shown in all-atom representation within the active site. B, chemical structures of the ligands found within the crystal structures are shown for reference.
Although it was initially hypothesized that mutating both positions 290 and 340 to the PavNMT consensus residues might lead to a more definitive switch in specificity, combining these mutations lead instead to the loss of measurable activity for all substrates tested. This result may indicate the presence of disruptive effects on the binding pocket and protein structure in general due to the presence of both mutations within the TNMT background. Alternatively, the large difference in binding modes seen for GfTNMT (where Met-290 and Phe-340 form direct hydrophobic interactions with the central rings characteristic of the protoberberine scaffold) versus TfPavNMT (where Pro-288 and Tyr-338, interact with the terminal O-methyl groups of the benzyl ring of tetrahydropapaverine) may require a more extensive series of mutations to effect a switch in specificity. In other words, the double mutant may be too disrupted to work with either protoberberine or pavine substrates unless the rest of the IP pocket is converted to a more PavNMT-like state. The modest recovery of activity with pavine observed for the E204A/M290P/F340Y triple mutant is consistent with this interpretation.
The replacement of Arg-41 with alanine was also explored, because Arg-41 is highly-conserved in all BIA NMTs and is located just slightly beyond the tip of the IP region in TNMT. Surprisingly, the R41A mutant showed a >2-fold increase in activity with THP and scoulerine, but no difference in activity compared with the WT enzyme with stylopine (Fig. 8A). Crystal structures show that the side chain of Arg-41 is too far to interact directly with the substrate, but the side chain of Arg-41 can directly interact with residues Phe-38, Phe-243, and Gln-339 in the IP through electrostatic and hydrogen bond interactions, including water-mediated interactions with the guanidinium group that involve three well-ordered water molecules near the IP region. Phe-38, Phe-243, and Gln-339 help to shape the IP-binding pocket around the C-2 and C-3 positions of the isoquinoline ring of the BIA substrate. The loss of the guanidinium group in R41A is expected to expand the size of a small cavity adjacent to the isoquinoline ring that is occupied by water molecules even when the WT enzyme is bound to SMS. As a result, the enzyme's preference for the methylenedioxy bridge of stylopine is reduced in the R41A mutant. The increased activity of the R41A mutant for tetrahydropalmatine and scoulerine may also reflect the increased conformational flexibility at the back of the IP region. This flexibility may allow for the rearrangement of the hydrogen bond network around the C-2 and C-3 positions, possibly allowing the mutant enzyme to form better interactions with THP, scoulerine, and perhaps other BIA compounds.
In contrast to the IP region, the BP region of TNMT appears to be less well-packed and likely more tolerant of structural diversity in substrates. The 70s loop appears to form a lid that can dynamically close and open over the front of the BP region, whereas residues 328–332 from helix α14 form the central part of the binding pocket which accommodates the portion of BIA compounds derived from the benzyl ring. As expected from the structure, alanine replacements lead to much stronger effects for the most deeply buried Phe-332 residue, intermediate effects for Ile-329, and the smallest and least direct effects for His-328 (Figs. 4 and 8). In addition to its role in forming the BP region, Phe-332 forms multiple ring stacking and hydrophobic interactions with the central rings of protoberberine substrates, and it is the most centrally located and deeply buried residue in the BIA-binding site. The 2-fold increase in activity of the H328A mutant with THP and the 5-fold decrease in activity with stylopine suggest indirect effects on binding and catalysis, most likely with the methylenedioxy bridge found at the C-9/C-10 position of stylopine (Fig. 8A). Because His-328 is 5.8 Å away from the nearest atom of SMS, His-328 likely exerts indirect effects on substrate binding and positioning, perhaps through ring-stacking effects on Phe-332 or by interactions with the side chain of Glu-79, a residue in the 70s loop that also interacts with one of the methylenedioxy ether oxygens of SMS. In both cases, the imidazole side chain could be stabilizing side chains that directly interact with the bound substrate. The I329A and F332A mutants both show stronger effects, with >10-fold losses in activity with stylopine, THP, and scoulerine. Ile-329 forms a van der Waals contact with the terminal bridging methylene group. In addition, loss of the branched aliphatic side chain would be expected to open a cavity that would allow the side chain of Phe-332 to move away from the substrate and destabilize binding interactions (Fig. 4). Previous structural and mutagenesis studies on CjCNMT also suggest interactions between the benzyl-derived ring of the BIA substrate and residues 328–332 of helix α16 (8).
Ile-329 also interacts with a hydrophobic cluster involving Leu-83, Phe-87, Leu-301, and Val-325, which may help to stabilize more closed conformations of the 70s loop. The dynamic nature of the 70s loop is suggested by the weak electron density of major portions of this loop in multiple crystal structures of BIA NMTs (8, 11). Several roles for assisting with substrate binding and catalysis have been suggested for specific residues in this loop based on mutagenesis studies and crystal structures (8, 11, 29, 40–42). Some of these roles will be further discussed below.
Insights into stereoselectivity
TNMTs have previously been shown to specifically produce the (S)-cis-stereoisomers of N-methyltetrahydroprotoberberines (16, 17). However, the structural mechanism by which TNMTs stereoselectively produce the (S)-cis-N-methylated product has not been previously explored. The crystal structures reported here reveal for the first time how an L-shaped BIA-binding pocket in GfTNMT selectively binds to a bent conformation that is possible for (S)-cis-protoberberine compounds but not (S)-trans compounds. (S)-trans-Protoberberine compounds are expected to be less flexible and much more likely to adopt a flat and nearly planar structure. Attempts to model a planar stereoisomer of (S)-trans-stylopine show a much poorer fit to the L-shaped binding pocket seen in the crystal structures of the SAH, SAM, and SAH/SMS complexes. Our extensive but unsuccessful efforts to soak and cocrystallize GfTNMT with (S)-trans stereoisomers of stylopine and canadine support the notion that (S)-cis stereoisomers may be preferred by the enzyme based on steric considerations.
Because the pKa value of stylopine has been measured to be 6.9, a nearly equimolar mixture of protonated and deprotonated forms is expected under physiological conditions, as well as the slightly acidic conditions used for crystallization, at pH 6.75 (43). Previous studies of stylopine indicate the presence of a 3:1 ratio of trans-cis conformers of (S)-stylopine under neutral pH conditions at room temperature (18, 44). The modest energetic barrier expected for interconverting these conformers via inversion of the stereochemistry of the nitrogen atom of the deprotonated species suggests that these two conformers are likely in dynamic equilibrium under physiological conditions. Consistent with the presence of a dynamic equilibrium and the stereoselectivity of GfTNMT for a specific stereoisomer, overnight incubations of GfTNMT with the sample of stylopine used for both activity assays and crystallization trials indicate that all of the stylopine can be converted to SMS (Fig. S6).
Chiral chromatography and the conversion of 100% of the stylopine substrate after long reaction times are also consistent with the absence of the (R)-isomer and the stereospecificity of GfTNMT for the (S)-isomer of stylopine (16, 17). Total conversion would not be expected if the (R)-isomer is present and is not used as a substrate. Using the (R)-isomer as a substrate, we would also expect the presence of a distinct peak following chiral chromatography. The only other explanation that is consistent with the data are that chiral chromatography fails to resolve both substrate and product enantiomers, and GfTNMT is able to utilize both (R)- and (S)-stereoisomers. (R)-Stylopine also has not been observed bound to the protein in electron density maps calculated from several high-resolution data sets of GfTNMT co-crystallized with stylopine at millimolar concentrations, which is consistent with unsuccessful attempts to model a binding mode for (R)-cis-stylopine or (R)-trans-stylopine in which the nitrogen is positioned near the catalytic center without generating steric clashes between the C-9/C-10 methylenedioxy bridge and residues Phe-257 and Phe-243. These results agree with previous observations found in planta, in which only the (S)-stereoisomers of the products are observed (14, 19, 33).
Insights into mechanisms of catalysis
The high level of sequence conservation and the proximity of the side-chain imidazole group of His-208 to the amino group of SMS bound to GfTNMT both suggest an important role in catalysis. Replacement of His-208 with alanine leads to <5% of WT activity for three different protoberberine substrates (Fig. 8D), which is consistent with mutagenesis studies of the same residue in TfPavNMT and CjCNMT homologs (8, 11). This residue has been proposed to act as a general base catalyst to deprotonate the amino group of the alkaloid substrate and increase the nucleophilicity of the nitrogen atom, but other roles consistent with the structures and mutagenesis data are possible.
The two side-chain carboxylate groups of two other highly-conserved residues, Glu-204 and Glu-207, are also nearby and may play roles in catalysis. The E204A and E207A mutants show <5% of WT activity for stylopine and THP, but show a bit more activity for scoulerine. Although the arrangement of Glu-204 and Glu-207 in GfTNMT is quite similar to the same residues in CjCNMT, the precise positions of the nitrogen atom nucleophile and methyl acceptor differ. In addition, the E207A mutation has very little effect on CjCNMT activity (60–70% of WT), whereas the E204A mutation shows a much stronger effect, having only 4–6% of WT activity. The more dramatic effect of the E207A mutation in GfTNMT may suggest a role to electrostatically stabilize the buildup of positive charge during the methylation of the amino group and formation of the quaternary nitrogen in the SMS product. The similarly large effects of the E204A mutation in both GfTNMT and CjCNMT are consistent with the previous proposal by Bennett et al. (8) that the negatively-charged carboxylate group may provide a stabilizing electrostatic effect on the positively-charged amino group. In contrast, in TfPavNMT Ala-202 is found in the same position as Glu-204 in the TNMT and CNMT enzymes, but the short hydrophobic side chain clearly fails to provide any stabilizing electrostatic effects. In TfPavNMT, Glu-205 is located in the equivalent position in the primary structure as Glu-207 in CjCNMT and GfTNMT, but the side chain occupies a very different location. The negatively-charged carboxylate group of Glu-205 in TfPavNMT accepts a hydrogen bond from the amino group of tetrahydropapaverine in a way that is quite similar to Glu-204 in CjCNMT and GfTNMT. Replacing the side-chain carboxylate group of Glu-205 in TfPavNMT with the nonpolar methyl group of alanine has different effects depending upon the substrate, ranging from almost no activity for reticuline, to 40% of WT activity for tetrahydropapaverine, and to 75% of WT activity for pavine (11). The somewhat variable effects that are seen in GfTNMT, CjCNMT, and TfPavNMT for mutations to the glutamate residues at positions 204 and 207 in GfTNMT suggest that the specific interactions of these residues can differ depending on the specific substrates and transition states that are formed, thus providing some flexibility or plasticity in the stabilization of different complexes in each enzyme.
In addition to the roles of the putative catalytic residues mentioned above, Tyr-81 near the C-terminal end of the 70s loop may also play a role in catalysis. Mutations replacing Tyr-81 with phenylalanine, arginine, and alanine all lead to large decreases in activity with stylopine, significant but smaller decreases with scoulerine, and more modest decreases with THP (Fig. 8A). Similarly large decreases in activity were seen for the equivalent Y79A mutant of TfPavNMT using pavine and reticuline as substrates, but activity actually improved with this mutation when tetrahydropapaverine was used as the methyl acceptor. The location at the C-terminal end of the dynamic 70s loop and the high level of sequence conservation indicate that the specific interactions seen in the crystal structures of the ground-state abortive complex may hint at the stabilizing interactions possible at the transition state. Although the structure of Rattus norvegicus glycine NMT (PDB code 1NBH) differs significantly from GfTNMT and the other BIA NMTs, Tyr-21 occupies nearly the same location in the active site relative to the methyl donor and nitrogen acceptor as Tyr-81 in GfTNMT (45). In glycine NMT, Tyr-21 has been proposed to promote catalysis by excluding water from the catalytic center, thereby increasing the effect of charge–charge interactions, while also contributing a hydrogen bond to potentially stabilize a reaction intermediate. If Tyr-81 is performing a similar function in GfTNMT, the equivalent hydrogen bond would be expected to help restrict the conformational flexibility of residues in the 70s loop over the entrance to the substrate-binding pocket and also help to exclude water from the catalytic center. Stabilization of the transition state, as proposed by Swiderek et al. (46), may also occur. Although no equivalent residues to Glu-204 or Glu-207 are apparent in glycine NMT, the related malarial parasite Plasmodium falciparum phosphoethanolamine NMT contains a tyrosine/histidine dyad and a more distant aspartate residue that have been implicated in catalysis by mutagenesis (40). Some of the details relating to the mechanism of catalysis in P. falciparum's phosphoethanolamine NMT also remain uncertain. The equivalent tyrosine residue has also been proposed to function as a general base catalyst or for transition-state stabilization (29, 40).
This work provides the first opportunity to compare the structure–function relationships of all three major classes of BIA NMTs. Our results highlight several key differences in substrate binding and recognition, as well as differences in the catalytic mechanism itself. The shape of the GfTNMT active site and use of N-methylation to lock the chirality of the nitrogen center provide a striking mechanism for enforcing the stereoselective production of N-methylated BIA compounds in the (S)-cis conformation. Understanding the basis of stereospecific substrate and product selection in GfTNMT provides a landmark case for understanding or engineering higher levels of stereospecificity in other enzymes in the BIA biosynthetic pathway and other pathways, especially for metabolic engineering purposes. Several important questions require additional investigation, especially relating to finer details of the catalytic mechanism and mechanisms underlying substrate specificity compared with PavNMT and CNMT. Additional studies on mutant or engineered enzymes, as well as other examples of natural enzymes with distinct substrate specificities are expected to provide further insight into the specific mechanisms involved (32).
Experimental procedures
Chemicals
Cryptopine and allocryptopine were purchased from MP Biomedicals. (S)-Bulbocapnine, glaucine, isocorydine hydrochloride, and isothebaine were purchased from Sequoia Research Products. S-(5′-Adenosyl)-l-methionine chloride dihydrochloride, boldine, dextromethorphan hydrobromide, (±)-ephedrine hydrochloride, mescaline hydrochloride, 1-methyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline hydrobromide (heliamine), 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride (salsolinol), noscapine hydrochloride, reserpine, and tryptamine were purchased from Sigma. (S)-Coclaurine hydrochloride, (R,S)-norcoclaurine hydrobromide, and (R,S)-norlaudanosoline hydrobromide were purchased from Toronto Research Chemicals. (R,S)-Canadine, codeine, hydrastine, morphine, papaverine, (R,S)-pavine, (R)-reticuline, (S)-reticuline, (S)-scoulerine, stylopine, tetrahydrocolumbamine, tetrahydropalmatine, and thebaine were obtained as reported previously (13, 21). Stylopine used in crystallization was further separated from nonsoluble trace contaminants by washing 1 mg of stylopine with 3× 300 μl of 50% methanol, followed by centrifugation and separation by pipette. Propranolol was isolated from a Teva-Propranolol tablet by acid/base organic extraction. Harmine and harmaline were isolated from seeds of Peganum harmala (47). Mitragynine was isolated from dried leaves of Mitragyna speciosa (48). N-Methylcoclaurine was prepared enzymatically using GfNMT1 and subsequently purified by acid/base organic extraction and preparative TLC. SMS and THP were prepared enzymatically from stylopine and tetrahydropalmatine using GfTNMT, purified by solid-phase extraction and preparative TLC, and authenticated by LC-MS/MS analysis (Fig. S2). Pentaerythritol ethoxylate (15:4 EO/OH) was purchased from Hampton Research. All other reagents were obtained from Sigma or BioShop Canada.
The stereochemical identity and purity of the stylopine used in this work was assessed by chiral LC-MS under conditions previously reported to separate tetrahydroprotoberberine enantiomers (Fig. S6) (49). The detection of a single peak for both stylopine and SMS is consistent with the presence of a single enantiomer. According to literature reports indicating that TNMT enzymes do not accept (R)-protoberberine substrates, our observation of the complete conversion of stylopine by GfTNMT is consistent with an assignment of (S)-stereochemistry (16, 17). Based on these analyses, and taking into consideration the unequivocal observation of only the (S)-stereoisomer of N-methylstylopine in the reported GfTNMT crystal structure, it is likely that the stylopine used in this work was (S)-stylopine. However, we take a conservative approach and avoid making interpretations that depend on this fact.
Mutagenesis
GfTNMT (GFLNMT2) was cloned from G. flavum cDNA as described previously (20). The ORF was ligated into E. coli expression vector pET47b via NdeI and XhoI restriction sites such that the encoded polypeptide included an N-terminal hexahistidine tag and linker. Codons were targeted for site-directed mutagenesis using a PCR-based method (Table S2) (50). All constructs were verified by Sanger sequencing at the University of Calgary DNA Sequencing and Genetic Analysis Lab (Calgary, Alberta, Canada). All oligonucleotide primers were obtained from Integrated DNA Technologies.
Recombinant gene expression and protein purification
Single colonies of E. coli ArcticExpress transformed with pET47b-GfTNMT (WT or mutant derivatives) expression plasmid were used for the expression and purification of proteins for enzyme assays. Starter growth cultures were prepared in 50 ml of lysogeny broth supplemented with 50 μg/ml kanamycin sulfate and 20 μg/ml gentamicin sulfate. Cultures were grown at 30 °C for 14 h with shaking at 200 rpm, after which they were used as seed cultures to inoculate 1 liter of fresh media to an initial OD600 reading of ∼0.2. Cultures were grown at 30 °C to an OD600 reading of ∼0.5–0.6 and cooled to 16 °C prior to initiating recombinant gene expression by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside. After a 16-h period, cells were harvested by centrifugation at 14,000 × g and 4 °C for 20 min and flash-cooled in liquid nitrogen prior to storage at −80 °C until purification.
Stored cell pellets were thawed on ice and resuspended in protein extraction buffer (100 mm Tris-Cl, pH 8.0, 300 mm NaCl and 10% (v/v) glycerol) supplemented with 10 μg/ml lysozyme, followed by sonication on ice at 50% duty cycle for 5 min. Crude lysates were centrifuged at 14,000 × g and 4 °C for 20 min, and the supernatant was transferred to a fresh tube containing 1 ml of TALON cobalt-affinity purification resin. Following a 1-h incubation period on ice with gentle shaking, the resin was washed with two 20-ml volumes of protein extraction buffer, followed by 20 ml of protein extraction buffer containing 20 mm imidazole. Bound protein was eluted in fractions using five 1-ml portions of protein extraction buffer containing 200 mm imidazole. The eluate fractions most abundant in protein were identified using a Bradford assay and pooled. Subsequent spin dialysis and concentration in a Millipore 30-kDa molecular mass cutoff spin filter yielded a protein solution in protein extraction buffer containing less than 0.5 mm imidazole. The resulting protein preparations were analyzed by SDS-PAGE and spectrophotometry at 280 nm to confirm acceptable purity and concentration, respectively. Extinction coefficients for GfTNMT proteins were calculated using ExPASy ProtParam tool (51).
Purification of WT GfTNMT for crystallization was performed as noted above, with some modifications. The pET47b-GfTNMT expression plasmid was expressed in E. coli Rosetta 2 cells. The clarified lysate (40 ml) from 1.67 liters of induced cell culture was loaded to a 1-ml HisTrap column (GE Healthcare) and eluted over a linear 0–0.5 m imidazole gradient in protein extraction buffer. The protein was dialyzed overnight against dialysis buffer A (20 mm Tris-Cl, 30 mm NaCl, 0.25 mm EDTA, 1 mm DTT, pH 8.0) in 30-kDa dialysis tubing (Millipore) followed by purification by anion-exchange chromatography (HiTrap Q HP 1 ml pre-equilibrated in dialysis buffer A; GE Healthcare). The protein was loaded and eluted at 1 ml/min in a linear gradient from 20 mm to 1 m sodium chloride in buffer A. Peak fractions were pooled and dialyzed overnight into dialysis buffer B buffer (20 mm Tris-Cl, 30 mm KCl, 10% w/v glycerol, 1 mm DTT, 0.25 mm EDTA, pH 7.5). Final protein solutions were concentrated to above 6 mg/ml and flash-cooled by liquid nitrogen in 100-μl aliquots in PCR tubes for storage at −80 °C.
Enzyme assays
Initial substrate range assays were carried out in 100 mm Tris-HCl, pH 8.0, using 100 μm alkaloid, 500 μm SAM, and 10 μg of purified recombinant protein in a 50-μl volume at 37 °C for 16 h. Assays were quenched with the addition of 200 μl of methanol, centrifuged to pellet-precipitated protein, and stored at −20 °C until LC-MS analysis. Substrates with which GfTNMT produced a reaction product consistent with N-methylation (+14 or +28 m/z with respect to the substrate) were investigated in more detail. Relative turnover was measured in triplicate assays under the same conditions as the initial assays above, except that 2 μg of protein and 10 min incubation times were used to allow linearity of product formation. The substrate showing the highest turnover, as measured by the decrease in the substrate peak area relative to the boiled protein negative control, was set to 100%. All other substrates were scaled proportionally.
For determination of pH optimum, assays were performed as described above for relative turnover, except that the buffer system was 100 mm Bistris propane, pH 6.0–9.5, and the alkaloid substrate was (S)-scoulerine. For determination of temperature optimum, assays were performed as described above for relative turnover, except that the alkaloid substrate was THP. Reaction tubes were incubated in water baths at various temperatures (4–60 °C) for 10 min prior to and during the assay period, and 0.5 μg of protein was used per reaction. Kinetic parameters were measured as described above for relative turnover, except that 0.5 μg of protein and 5-min incubation times were used to ensure linearity of product formation at all substrate concentrations. Stylopine concentration was varied from 2.5 to 200 μm, and THP concentration was varied from 5 to 500 μm, whereas SAM remained fixed at 500 μm. Subsequently, SAM concentration was varied from 5 to 1000 μm with stylopine concentration fixed at 50 μm. Product formation was quantified by reference to a 5-point standard curve of the SMS or N-methyl-THP. Saturation curves and kinetic constants were calculated according to the Michaelis-Menten P model implemented in Prism5 (GraphPad). Product inhibition assays were carried out as described above for relative turnover, except that the substrate was THP, 0.5 μg of protein was used per reaction, and N-methylstylopine was added to the reaction mixture at concentrations ranging from 1 to 40 μm. Product formation in the absence of SMS was set to 100%, and inhibited activities were scaled proportionally. For determination of GfTNMT mutant proteins' relative activities, assays were performed as described above for relative turnover except that 0.5 μg of protein was used per reaction. For further comparison of low activities remaining for putative catalytic residue mutants, assays were also performed with 10 μg of protein. Product formation for the WT GfTNMT was set to 100%, and the mutant activities were scaled proportionally.
LC-MS analysis
LC-MS was performed using a 1200 HPLC coupled to a 6410 triple-quadrupole MS (Agilent). 5-μl samples were injected onto a 2.1 × 100-mm Poroshell 120 SB-C18 2.7-μm column, and analytes were eluted in a gradient of solvent A (10 mm ammonium acetate, pH 5.5, 5% acetonitrile) to solvent B (100% acetonitrile) at a flow rate of 0.4 ml/min. The gradient began at 0% solvent B, reached 80% solvent B by 8 min, and increased to 100% solvent B at 9 min. After a 1-min wash, the solvent mix returned to 0% solvent B for a 3-min re-equilibration period prior to the next injection. Analytes were applied to the MS using an electrospray ionization probe operating in positive mode with the following conditions: capillary voltage, 4000 V; fragmentor voltage, 100 V; source temperature, 350 °C; nebulizer pressure, 50 p.s.i.; and gas flow, 10 liters/min. For full-scan analysis (substrate, temperature, and pH range assays), quadrupoles 1 and 2 were set to radio frequency only, whereas the third quadrupole scanned from 200 to 700 m/z. For all other analyses, multiple-reaction monitoring mode was used. Precursor ions were selected in quadrupole 1, and collision energies of 10–20 eV were applied in quadrupole 2 with an argon gas pressure of 1.8 × 10−3 torr. The resulting fragment ions were resolved in quadrupole 3, and the most abundant was used as a quantifier (SMS, 338.2 > 190.2; N-methyltetrahydropalmatine, 370.2 > 206.1).
Crystallization
For initial crystallization screening, GfTNMT in dialysis buffer B was diluted to 5.3 mg/ml with 1 mm SAH and screened using sitting-drop vapor diffusion using the commercially available IndexHT and CrystalHT screens (Hampton Research). Screens were set up using a Hydra-II–Plus-One pipetting robot placing 0.4 μl of protein mixture and 0.4 μl of well-solution in a 96-well sitting-drop plate (Corning Inc.). An initial crystallization condition identified from the IndexHT screen was subsequently optimized by hanging-drop vapor diffusion to yield well-diffracting crystals of GfTNMT with SAH (PDB code 6P3M) in drops containing 1.5 μl of protein solution to 1.5 μl of well-solution (40 mm ammonium sulfate, 30% v/v pentaerythritol, 15:4 EO/OH, 12.5% v/v glycerol, 100 mm Tris-Cl, pH 6.98) equilibrated over 0.8 ml of well-solution at 22 °C. Crystals were observed overnight and continued to grow to full size over 2 weeks, yielding rectangular crystals with average dimensions of ∼200 × 100 × 100 μm. Crystallization of GfTNMT with SAM and GfTNMT with SAM and stylopine was performed from the optimized conditions obtained from GfTNMT with SAH crystals. However, initial protein solutions were prepared containing 5.3 mg/ml GfTNMT with 1 mm SAM. Where appropriate, 0.5 mm stylopine was added to the protein solution immediately prior to the crystallization setup. Final optimized conditions for GfTNMT with SAM (PDB code 6P3N) contained 10 mm ammonium sulfate, 25% v/v pentaerythritol (15:4 EO/OH), 100 mm Tris-Cl, pH 6.98, and yielded square crystals ∼300 × 200 × 200 μm in dimension after 2 weeks. Final optimized conditions for GfTNMT with SAM and stylopine (PDB code 6P3O) contained 20 mm ammonium sulfate, 22.5% v/v pentaerythritol (15:4 EO/OH), 100 mm Tris-Cl, pH 6.75, and yielded square crystals ∼300 × 200 × 200 μm in dimension after 3 weeks. Although SAM and stylopine were used in crystallization, the crystal was determined to contain the products SAH and SMS based on electron density maps.
Crystal structure determination
Single crystals were mounted on a polymer film MicroLoop (Mitegen) and flash-cooled by plunging into liquid nitrogen. The crystals were stored under liquid nitrogen and transferred to the Canadian Light Source beamline 08B1-1 or Stanford Synchrotron Radiation Laboratory beamlines 12-2 or 14-1 for data collection. Complete datasets were measured from individual crystals; diffraction images were indexed, integrated, and scaled using XDS and XSCALE (52). Initial phases were calculated by molecular replacement using PHASER (53), with TfPavNMT (PDB 5KOK) as the search model (52% sequence identity to GfTNMT). Reciprocal space refinement calculations were performed using REFMAC (54) from the CCP4 program suite (55), and COOT (56) was used to visualize electron density maps and model building. Model quality was assessed using MolProbity (57). The geometric parameters for SMS were obtained from the crystal structure of (S)-trans-stylopine (58). Restraints were generated by REFMAC (54). The refined omit electron density map shown in Fig. 7 was calculated after the SAH and SMS ligands were removed, and the remaining model was subjected to 20 rounds of refinement to reduce the effects of model bias.
Comparative structural analysis
Superimpositions of related NMT structures were performed using the “align” command for manual selections of the β-sheet components of the SAH-binding domain in PyMOL (59). Surface area predictions and analysis of the dimer interface were performed using ePISA (27).
Author contributions
D. E. L., J. S. M., P. J. F., and K. K. N. conceptualization; D. E. L., P. J. F., and K. K. N. resources; D. E. L., J. S. M., M. R., P. J. F., and K. K. N. data curation; D. E. L., J. S. M., M. R., M. A. T., V. A. M., P. J. F., and K. K. N. formal analysis; D. E. L., J. S. M., M. A. T., P. J. F., and K. K. N. supervision; D. E. L., J. S. M., P. J. F., and K. K. N. validation; D. E. L., J. S. M., M. R., M. A. T., V. A. M., P. J. F., and K. K. N. investigation; D. E. L., J. S. M., M. R., M. A. T., P. J. F., and K. K. N. visualization; D. E. L., J. S. M., M. R., M. A. T., V. A. M., P. J. F., and K. K. N. methodology; D. E. L., J. S. M., P. J. F., and K. K. N. writing-original draft; D. E. L., J. S. M., P. J. F., and K. K. N. project administration; D. E. L., J. S. M., P. J. F., and K. K. N. writing-review and editing; P. J. F. and K. K. N. funding acquisition.
Supplementary Material
Acknowledgments
Some of the X-ray diffraction data were measured at beamlines 08B-1 of the Canadian Light Source (CLS), which is supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), the National Research Council of Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan. Some of the diffraction data were also measured at beamlines 12-2 and 14-1 at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the United States Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by National Institutes of Health NIGMS Grant P41GM103393 and NCRR Grant P41RR001209.
This work was supported in part by Discovery Grants from the Natural Sciences and Engineering Research Council of Canada (NSERC) Grant 05728 (to K. K. S. N.) and Grant 183573 (to P. J. F.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6, Tables S1 and S2, supporting Movie S1, and supporting Refs. 1–4.
The atomic coordinates and structure factors (codes 6P3M, 6P3N, and 6P3O) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- BIA
- benzylisoquinoline alkaloid
- NMT
- N-methyltransferase
- TNMT
- tetrahydroprotoberberine N-methyltransferase
- PavNMT
- pavine N-methyltransferase
- CNMT
- coclaurine N-methyltransferase
- RNMT
- reticuline N-methyltransferase
- GfTNMT
- G. flavum TNMT
- CjCNMT
- C. japonica CNMT
- TfPavNMT
- T. flavum PavNMT
- SMS
- (S)-cis-N-methylstylopine
- THP
- tetrahydropalmatine
- BP
- benzyl pocket
- IP
- isoquinoline pocket
- CP
- catalytic pocket
- SAM
- S-adenosyl-l-methionine
- SAH
- S-adenosyl-l-homocysteine
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- PDB
- Protein Data Bank
- RMSD
- root mean square deviation
- ER
- endoplasmic reticulum.
References
- 1. Hagel J. M., and Facchini P. J. (2013) Benzylisoquinoline alkaloid metabolism: a century of discovery and a brave new world. Plant Cell Physiol 54, 647–672 10.1093/pcp/pct020 [DOI] [PubMed] [Google Scholar]
- 2. Iwasa K., Kamigauchi M., Ueki M., and Taniguchi M. (1996) Antibacterial activity and structure-activity relationships of berberine analogs. Eur. J. Med. Chem. 31, 469–478 10.1016/0223-5234(96)85167-1 [DOI] [Google Scholar]
- 3. Dastmalchi M., Park M. R., Morris J. S., and Facchini P. (2018) Family portraits: the enzymes behind benzylisoquinoline alkaloid diversity. Phytochemistry Rev. 17, 249–277 10.1007/s11101-017-9519-z [DOI] [Google Scholar]
- 4. Li Y., Li S., Thodey K., Trenchard I., Cravens A., and Smolke C. D. (2018) Complete biosynthesis of noscapine and halogenated alkaloids in yeast. Proc. Natl. Acad. Sci. U.S.A. 115, E3922–E3931 10.1073/pnas.1721469115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Narcross L., Fossati E., Bourgeois L., Dueber J. E., and Martin V. J. J. (2016) Microbial factories for the production of benzylisoquinoline alkaloids. Trends Biotechnol. 34, 228–241 10.1016/j.tibtech.2015.12.005 [DOI] [PubMed] [Google Scholar]
- 6. Schläger S., and Dräger B. (2016) Exploiting plant alkaloids. Curr. Opin. Biotechnol. 37, 155–164 10.1016/j.copbio.2015.12.003 [DOI] [PubMed] [Google Scholar]
- 7. Beaudoin G. A., and Facchini P. J. (2014) Benzylisoquinoline alkaloid biosynthesis in opium poppy. Planta 240, 19–32 10.1007/s00425-014-2056-8 [DOI] [PubMed] [Google Scholar]
- 8. Bennett M. R., Thompson M. L., Shepherd S. A., Dunstan M. S., Herbert A. J., Smith D. R. M., Cronin V. A., Menon B. R. K., Levy C., and Micklefield J. (2018) Structure and biocatalytic scope of coclaurine N-methyltransferase. Angew. Chem. Int. Ed. Engl. 57, 10600–10604 10.1002/anie.201805060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Choi K. B., Morishige T., Shitan N., Yazaki K., and Sato F. (2002) Molecular cloning and characterization of coclaurine N-methyltransferase from cultured cells of Coptis japonica. J. Biol. Chem. 277, 830–835 10.1074/jbc.M106405200 [DOI] [PubMed] [Google Scholar]
- 10. Desgagné-Penix I., and Facchini P. J. (2012) Systematic silencing of benzylisoquinoline alkaloid biosynthetic genes reveals the major route to papaverine in opium poppy. Plant J. 72, 331–344 10.1111/j.1365-313X.2012.05084.x [DOI] [PubMed] [Google Scholar]
- 11. Torres M. A., Hoffarth E., Eugenio L., Savtchouk J., Chen X., Morris J. S., Facchini P. J., and Ng K. K. (2016) Structural and functional studies of pavine N-methyltransferase from Thalictrum flavum reveal novel insights into substrate recognition and catalytic mechanism. J. Biol. Chem. 291, 23403–23415 10.1074/jbc.M116.747261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liscombe D. K., Ziegler J., Schmidt J., Ammer C., and Facchini P. J. (2009) Targeted metabolite and transcript profiling for elucidating enzyme function: isolation of novel N-methyltransferases from three benzylisoquinoline alkaloid-producing species. Plant J. 60, 729–743 10.1111/j.1365-313X.2009.03980.x [DOI] [PubMed] [Google Scholar]
- 13. Morris J. S., and Facchini P. J. (2016) Isolation and characterization of reticuline N-methyltransferase involved in biosynthesis of the aporphine alkaloid magnoflorine in opium poppy. J. Biol. Chem. 291, 23416–23427 10.1074/jbc.M116.750893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takao N., Kamigauchi M., and Okada M. (1983) Biosynthesis of benzo[c]phenanthridine alkaloids sanguinarine, chelirubine and macarpine. Helv. Chim. Acta 66, 473–484 10.1002/hlca.19830660208 [DOI] [Google Scholar]
- 15. Winzer T., Gazda V., He Z., Kaminski F., Kern M., Larson T. R., Li Y., Meade F., Teodor R., Vaistij F. E., Walker C., Bowser T. A., and Graham I. A. (2012) A Papaver somniferum 10-gene cluster for synthesis of the anticancer alkaloid noscapine. Science 336, 1704–1708 10.1126/science.1220757 [DOI] [PubMed] [Google Scholar]
- 16. Rueffer M., Zumstein G., and Zenk M. H. (1990) Partial purification and properties of S-adenosyl-l-methionine: (S)-tetrahydroprotoberberinecis-N-methyltransferase from suspension-cultured cells of Eschscholtzia and Corydalis. Phytochemistry 29, 3727–3733 10.1016/0031-9422(90)85321-6 [DOI] [Google Scholar]
- 17. Rüeffer M., and Zenk M. H. (1986) S-Adenosyl-l-methionine:(S)-7,8,13,14-tetrahydroberberine N-methyltransferase, a branch point enzyme in the biosynthesis of benzophenanthridine and protopine alkaloids. Tetrahedron Lett. 5603–5604 [Google Scholar]
- 18. Kamigauchi M., Yoshida M., Noda Y., Nishijo J., In Y., Tomoo K., Ohishi H., and Ishida T. (2003) Difference between enzymatic and chemical N-methylations of protoberberine-type alkaloid, dependent on the stereoisomer of (−)-N-methyl-7,8,13,13α-tetrahydroberberinium salt. Bull. Chemical Soc. Japan 76, 587–593 10.1246/bcsj.76.587 [DOI] [Google Scholar]
- 19. Iwasa K. (1995) The biotransformation of protoberberine alkaloids by plant tissue cultures. In The Alkaloids Vol. 46 (Cordell G.A. ed.) pp. 273–346, Academic Press, New York, New York [Google Scholar]
- 20. Hagel J. M., Morris J. S., Lee E. J., Desgagné-Penix I., Bross C. D., Chang L., Chen X., Farrow S. C., Zhang Y., Soh J., Sensen C. W., and Facchini P. J. (2015) Transcriptome analysis of 20 taxonomically related benzylisoquinoline alkaloid-producing plants. BMC Plant Biol. 15, 227 10.1186/s12870-015-0596-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liscombe D. K., and Facchini P. J. (2007) Molecular cloning and characterization of tetrahydroprotoberberine cis-N-methyltransferase, an enzyme involved in alkaloid biosynthesis in opium poppy. J. Biol. Chem. 282, 14741–14751 10.1074/jbc.M611908200 [DOI] [PubMed] [Google Scholar]
- 22. Narcross L., Bourgeois L., Fossati E., Burton E., and Martin V. J. (2016) Mining enzyme diversity of transcriptome libraries through DNA synthesis for benzylisoquinoline alkaloid pathway optimization in yeast. ACS Synth. Biol. 5, 1505–1518 10.1021/acssynbio.6b00119 [DOI] [PubMed] [Google Scholar]
- 23. Emanuelsson O., Brunak S., von Heijne G., and Nielsen H. (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2, 953–971 10.1038/nprot.2007.131 [DOI] [PubMed] [Google Scholar]
- 24. Horton P., Park K. J., Obayashi T., Fujita N., Harada H., Adams-Collier C. J., and Nakai K. (2007) WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587 10.1093/nar/gkm259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hagel J. M., and Facchini P. J. (2012) Subcellular localization of sanguinarine biosynthetic enzymes in cultured opium poppy cells. In Vitro Cell Dev. Biol. 48, 233–240 10.1007/s11627-012-9426-3 [DOI] [Google Scholar]
- 26. Liscombe D. K., Louie G. V., and Noel J. P. (2012) Architectures, mechanisms and molecular evolution of natural product methyltransferases. Nat. Prod. Rep. 29, 1238–1250 10.1039/c2np20029e [DOI] [PubMed] [Google Scholar]
- 27. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 28. Gana R., Rao S., Huang H., Wu C., and Vasudevan S. (2013) Structural and functional studies of S-adenosyl-l-methionine binding proteins: a ligand-centric approach. BMC Struct. Biol. 13, 6 10.1186/1472-6807-13-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee S. G., Kim Y., Alpert T. D., Nagata A., and Jez J. M. (2012) Structure and reaction mechanism of phosphoethanolamine methyltransferase from the malaria parasite Plasmodium falciparum: an antiparasitic drug target. J. Biol. Chem. 287, 1426–1434 10.1074/jbc.M111.315267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morris J. S., Groves R. A., Hagel J. M., and Facchini P. J. (2018) An N-methyltransferase from Ephedra sinica catalyzing the formation of ephedrine and pseudoephedrine enables microbial phenylalkylamine production. J. Biol. Chem. 293, 13364–13376 10.1074/jbc.RA118.004067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Amann M., Wanner G., and Zenk M. H. (1986) Intracellular compartmentation of two enzymes of berberine biosynthesis in plant cell cultures. Planta 167, 310–320 10.1007/BF00391333 [DOI] [PubMed] [Google Scholar]
- 32. Hagel J. M., Mandal R., Han B., Han J., Dinsmore D. R., Borchers C. H., Wishart D. S., and Facchini P. J. (2015) Metabolome analysis of 20 taxonomically related benzylisoquinoline alkaloid-producing plants. BMC Plant Biol. 15, 220 10.1186/s12870-015-0594-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shulgin A. T., and Perry W. E. (2002) The Simple Plant Isoquinolines, pp. 1–624, Transform Press, Berkeley, California [Google Scholar]
- 34. Reed M. C., Lieb A., and Nijhout H. F. (2010) The biological significance of substrate inhibition: a mechanism with diverse functions. Bioessays 32, 422–429 10.1002/bies.200900167 [DOI] [PubMed] [Google Scholar]
- 35. Robin A. Y., Giustini C., Graindorge M., Matringe M., and Dumas R. (2016) Crystal structure of norcoclaurine-6-O-methyltransferase, a key rate-limiting step in the synthesis of benzylisoquinoline alkaloids. Plant J. 87, 641–653 10.1111/tpj.13225 [DOI] [PubMed] [Google Scholar]
- 36. Slavík J., and Slavíková L. (1985) Alkaloids from Papaver bracteatum LINDL. Coll. Czechoslovk Chem. Commun. 50, 1216–1226 10.1135/cccc19851216 [DOI] [Google Scholar]
- 37. Slavík J., and Slavíková L. (1990) Alkaloids from Papaver albiflorum PACZ. subsp. albiflorum and P. cf. stevenianum AD Mikheev. Coll. Czechoslovk Chem. Commun. 55, 1812–1816 10.1135/cccc19901812 [DOI] [Google Scholar]
- 38. Slavík J., and Slavíková L. (1996) Alkaloids from Papaver setigerum DC. Coll. Czechoslovk Chem. Commun. 61, 1047–1052 10.1135/cccc19961047 [DOI] [Google Scholar]
- 39. Grobe N., Ren X., Kutchan T. M., and Zenk M. H. (2011) An (R)-specific N-methyltransferase involved in human morphine biosynthesis. Arch. Biochem. Biophys. 506, 42–47 10.1016/j.abb.2010.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saen-Oon S., Lee S. G., Jez J. M., and Guallar V. (2014) An alternative mechanism for the methylation of phosphoethanolamine catalyzed by Plasmodium falciparum phosphoethanolamine methyltransferase. J. Biol. Chem. 289, 33815–33825 10.1074/jbc.M114.611319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stratton C. F., Poulin M. B., Du Q., and Schramm V. L. (2017) Kinetic isotope effects and transition state structure for human phenylethanolamine N-methyltransferase. ACS Chem. Biol. 12, 342–346 10.1021/acschembio.6b00922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang J., and Klinman J. P. (2016) Convergent mechanistic features between the structurally diverse N- and O-methyltransferases: glycine N-methyltransferase and catechol O-methyltransferase. J. Am. Chem. Soc. 138, 9158–9165 10.1021/jacs.6b03462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kulp M., Bragina O., Kogerman P., and Kaljurand M. (2011) Capillary electrophoresis with LED-induced native fluorescence detection for determination of isoquinoline alkaloids and their cytotoxicity in extracts of Chelidonium majus L. J. Chromatogr. A. 1218, 5298–5304 10.1016/j.chroma.2011.06.016 [DOI] [PubMed] [Google Scholar]
- 44. Lehn J.M. (1970) Nitrogen inversion. In Dynamic Stereochemistry. Fortschritte der Chemischen Forschung, vol 15/3 (Baldwin J.E., Fleming R.H., Lehn J.M., Tochtermann W. ed.) pp. 311–377, Springer, Berlin, Heidelberg [Google Scholar]
- 45. Takata Y., Huang Y., Komoto J., Yamada T., Konishi K., Ogawa H., Gomi T., Fujioka M., and Takusagawa F. (2003) Catalytic mechanism of glycine N-methyltransferase. Biochemistry 42, 8394–8402 10.1021/bi034245a [DOI] [PubMed] [Google Scholar]
- 46. Świderek K., Tuñón I., Williams I. H., and Moliner V. (2018) Insights on the origin of catalysis on glycine N-methyltransferase from computational modeling. J. Am. Chem. Soc. 140, 4327–4334 10.1021/jacs.7b13655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brobst A., Lewis J., Klett B., Haustein C., and Shriver J. (2009) The free base extraction of harmaline from Peganum harmala. Am. J. Undergrad Res. 8, 2–3 [Google Scholar]
- 48. Beng G. T., Hamdan M. R., Siddiqui M. J., Mordi M. N., and Mansor S. M. (2011) A simple and cost effective isolation and purification protocol of mitragynine from Mitragyna speciosa Korth (Ketum) leaves. Malaysian J. Anal. Sci. 15, 54–60 [Google Scholar]
- 49. Ma Z.-Z., Xu W., Jensen N. H., Roth B. L., Liu-Chen L.-Y., and Lee D. Y. (2008) Isoquinoline alkaloids isolated from Corydalis yanhusuo and their binding affinities at the dopamine D1 receptor. Molecules 13, 2303–2312 10.3390/molecules13092303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zheng L., Baumann U., and Reymond J. L. (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115 10.1093/nar/gnh110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wilkins M. R., Gasteiger E., Bairoch A., Sanchez J. C., Williams K. L., Appel R. D., and Hochstrasser D. F. (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552 [DOI] [PubMed] [Google Scholar]
- 52. Kabsch W. (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26, 795–800 10.1107/S0021889893005588 [DOI] [Google Scholar]
- 53. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 55. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., and et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 57. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd, Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 10.1093/nar/gkm216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Marek R., Marek J., Dostál J., Táborská E., Slavík J., and Dommisse R. (2002) Isoquinoline alkaloids: a 15N NMR and x-ray study. Magn. Reson. Chem. 40, 687–692 10.1002/mrc.1084 [DOI] [Google Scholar]
- 59. Schrödinger, LLC (2015) The PyMOL Molecular Graphics System, Version 1.8, Schrödinger, LLC, New York [Google Scholar]
- 60. Kamigauchi M., Noda Y., Iwasa K., Nishijo Z., Ishida T., and In Y. (1995) Conformational features of 7,8,13β,14α-tetrahydro-N7-methylcorysaminium, a biosynthetic intermediate to the protopine-type alkaloid corycavine. Helv. Chim. Acta 78, 80–86 10.1002/hlca.19950780108 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.