

Abstract
The glucose-responsive transcription factor carbohydrate response element–binding protein (ChREBP) critically promotes aerobic glycolysis and cell proliferation in colorectal cancer cells. It has been reported that ubiquitination may be important in the regulation of ChREBP protein levels and activities. However, the ChREBP-specific E3 ligase and molecular mechanism of ChREBP ubiquitination remains unclear. Using database exploration and expression analysis, we found here that levels of the E3 ligase SMURF2 (Smad-ubiquitination regulatory factor 2) negatively correlate with those of ChREBP in cancer tissues and cell lines. We observed that SMURF2 interacts with ChREBP and promotes ChREBP ubiquitination and degradation via the proteasome pathway. Interestingly, ectopic SMURF2 expression not only decreased ChREBP levels but also reduced aerobic glycolysis, increased oxygen consumption, and decreased cell proliferation in colorectal cancer cells. Moreover, SMURF2 knockdown increased aerobic glycolysis, decreased oxygen consumption, and enhanced cell proliferation in these cells, mostly because of increased ChREBP accumulation. Furthermore, we identified Ser/Thr kinase AKT as an upstream suppressor of SMURF2 that protects ChREBP from ubiquitin-mediated degradation. Taken together, our results indicate that SMURF2 reduces aerobic glycolysis and cell proliferation by promoting ChREBP ubiquitination and degradation via the proteasome pathway in colorectal cancer cells. We conclude that the SMURF2–ChREBP interaction might represent a potential target for managing colorectal cancer.
Keywords: cell metabolism, cell proliferation, E3 ubiquitin ligase, ubiquitylation (ubiquitination), colorectal cancer, protein degradation, Carbohydrate responsive element binding protein (ChREBP), colorectal cancer, metabolism, Smad ubiquitination regulatory factor 2 (Smurf2), ubiquitin proteasome pathway
Introduction
The transcription factor carbohydrate response element–binding protein (ChREBP)4 is one of the members of the basic helix–loop–helix leucine zipper transcription factor family. It has emerged as a major player in regulating the expression of critical genes involved in glycolysis, gluconeogenesis, and lipogenesis in metabolic tissues (1–6). Our previous study showed that ChREBP may serve as a metabolic switch between aerobic glycolysis and oxidative phosphorylation, as well as influence proliferative and tumorigenic potential of colorectal cancer cells (7). Thus regulating the level and activity of ChREBP can reprogram glucose metabolism. Recent study has suggested that post-translational modifications play important roles in regulating ChREBP level and activity in different types of cells. Glucose-activated p300 acetylates ChREBP on Lys-672 and increases its transcriptional activity in hepatocytes (8). Glucose regulates ChREBP phosphorylation and its subcellular localization (9). Both O-glycosylation and ubiquitination can regulate ChREBP protein level and activity in hepatocytes (10–12). However, it remains unclear how the ChREBP protein level is regulated by ubiquitination in cancer cells.
SMURF2 (Smad ubiquitination regulatory factor 2) is a member of the HECT E3 ubiquitin ligase family and regulates ubiquitination-mediated protein degradation (13). SMURF2 exerts negative functions in transforming growth factor-β (TGF-β) and Bone Morphogenetic Proteins (BMP) signaling pathways (14). SMURF2-deficient mice are prone to a variety of cancers because histone modification and the extent of chromatin compaction is altered in these mice, suggesting that SMURF2 might function as a tumor suppressor (15). So far studies have broadened the repertoire of SMURF2 substrates and extended its function to controlling the cell cycle, proliferation, differentiation, metastasis, and senescence (16–21). However, there is limited evidence to indicate any mechanistic link between SMURF2 and cell metabolism.
In our study, we found that SMURF2 played an important role in the ubiquitination and proteasomal degradation of ChREBP. Thus SMURF2 functioned as an E3 ligase of ChREBP. Moreover, altering SMURF2 expression could regulate aerobic glycolysis, oxidative phosphorylation, and cell proliferation of colorectal cancer cells by altering ChREBP protein levels. AKT decreased SMURF2 levels and protected ChREBP from degradation. Our results indicate that SMURF2 exerts a critical role in the Warburg effect by regulating ChREBP stability in colorectal cancer cells, and the SMURF2–ChREBP axis might be a novel target in the treatment of colorectal cancer.
Results
The E3 ligase SMURF2 decreased ChREBP protein level in colorectal cancer cells
ChREBP plays a critical role in promoting glycolysis and cell proliferation in colorectal cancer cells (7). It has been reported that ubiquitination may be important for regulating ChREBP protein level and activity (10–12). To search for the E3 ligase of ChREBP, analysis of negative correlation of gene expression was performed using the Genevestigator database. Genevestigator showed that SMURF2 ranked first among several E3 ubiquitin ligases that displayed negative expression correlation with ChREBP in cancer. Utilizing a mouse model of colitis-related colorectal carcinoma induced by the carcinogen azoxymethane, followed by the inflammatory agent dextran sodium sulfate, we found that the SMURF2 protein level decreased gradually, whereas ChREBP expression increased gradually in the progression of mouse colorectal cancer (Fig. 1A). By examining ChREBP and SMURF2 expression in different human colorectal cancer cell lines, including CaCO-2, HCT116, SW1116, and SW620, we found a negative correlation between ChREBP and SMURF2 levels (Fig. 1B). These results indicated that the ChREBP protein level was negatively correlated with that of SMURF2 in the progression of mouse colorectal cancer and in human colorectal cancer cells.
Figure 1.

SMURF2 decreased ChREBP protein level without affecting its transcription. A, SMURF2 protein level was negatively correlated with ChREBP expression in progression of mice colorectal cancer. B, SMURF2 and ChREBP protein levels were negatively correlated in human colorectal cancer cell lines. C, SMURF2 decreased ChREBP protein level, whereas the E3 ligase-inactive mutant SMURF2C716A failed to decrease ChREBP protein level in 293T cells. SMURF2 decreased ChREBP protein level in a dose-dependent manner. D, SMURF2 did not change the ChREBP mRNA level. E, luciferase activity assay showed that SMURF2 but not SMURF2C716A decreased the transcriptional activity of ChREBP. The data in A and C are quantified as the mean ± S.D., and the bar indicates the mean. Statistical significance was calculated using Prism 5 (GraphPad Software). *, p < 0.05. AOM, azoxymethane; Ctrl, control; ACC, Acetyl CoA carboxylase.
According to the negative correlation between ChREBP and SMURF2 expression, we hypothesized that the E3 ubiquitin ligase SMURF2 might decrease ChREBP protein levels by promoting its ubiquitination and degradation. To test the hypothesis, we examined whether ectopic expression of Myc-tagged SMURF2 (Myc-SMURF2) decreased the HA-tagged ChREBP (HA-ChREBP) level in 293T cells. As shown in Fig. 1C, WT SMURF2, but not the E3 ligase-inactive mutant SMURF2C716A, decreased ChREBP levels in 293T cells. Moreover, SMURF2 decreased ChREBP levels in a dose-dependent manner (Fig. 1C). We ectopically expressed Myc-SMURF2 in 293T cells and found that the mRNA level of ChREBP had no significant change in the SMURF2-overexpressed cells, suggesting that SMURF2-dependent down-regulation of ChREBP occurred at post-transcriptional levels (Fig. 1D). The 4× ACC ChoRE-Luciferase reporter construct containing four ChoRE elements in the promoter of ACC driving luciferase gene has been applied to detect the activity of ChREBP-mediated transcription as described previously (22). The luciferase assay showed that WT SMURF2, but not SMURF2C716A, decreased the activity of ChREBP (Fig. 1E). These results suggest that SMURF2 decreases ChREBP protein level and activity.
SMURF2 co-immunoprecipitated and co-localized with ChREBP
To explore whether SMURF2 interacted with ChREBP, we performed co-immunoprecipitation experiments. After co-expressing FLAG-ChREBP and Myc-SMURF2 in 293T cells, we found that Myc-SMURF2 could readily co-immunoprecipitated with FLAG-ChREBP (Fig. 2A). We then found that endogenous ChREBP and SMURF2 protein co-immunoprecipitated in HCT116 cells (Fig. 2B). To determine whether SMURF2 and ChREBP co-localized, we ectopically expressed Myc-SMURF2 and HA-ChREBP in HeLa cells and analyzed subcellular localization using immunofluorescent staining. We found that Myc-SMURF2 and HA-ChREBP co-localized in both cytosol and nucleus (Fig. 2C). We also found endogenous SMURF2 co-localized with exogenous HA-ChREBP in both cytosol and nucleus in HCT116 and HeLa cells (Fig. S1, A and B).
Figure 2.

SMURF2 co-immunoprecipitated with ChREBP. A, co-immunoprecipitation analysis for Myc-SMURF2 and FLAG-ChREBP when being co-expressed in 293T cells. B, endogenous SMURF2 co-immunoprecipitated with ChREBP in HCT116 cells. C, ectopically expressed Myc-SMURF2 and HA-ChREBP co-localized in HeLa cells. The scale bar is 20 μm. Red, Myc-SMURF2; green, HA-ChREBP; blue, 4′,6′-diamino-2-phenylindole (DAPI). D, the schematic diagram shows the domain structure of SMURF2. Myc-SMURF2 1–148, and Myc-SMURF2 149–348 but not Myc-SMURF2 349–748 co-immunoprecipitated with FLAG-ChREBP when they are co-expressed in 293T cells. E, the schematic diagram shows the domain structure of ChREBP. FLAG-ChREBP 1–251, FLAG-ChREBP 252–625, and FLAG-ChREBP 626–852 co-immunoprecipitated with Myc-SMURF2 when they are co-expressed in 293T cells. bHLH, basic helix–loop–helix; IB, immunoblotting; IP, immunoprecipitation.
To further analyze which domains of SMURF2 were responsible for interacting with ChREBP, we generated Myc-tagged N-terminal C2 domain of SMURF2 (Myc-SMURF2 1–148), Myc-tagged WW domain of SMURF2 (Myc-SMURF2 149–348), and Myc-tagged C-terminal HECT domain of SMURF2 (Myc-SMURF2 349–748), according to the functional domains of human SMURF2 protein. We found that both Myc-SMURF2 1–148 and Myc-SMURF2 149–348 co-immunoprecipitated with FLAG-ChREBP (Fig. 2D). This finding suggested that both C2 and WW domains of SMURF2 interacted with ChREBP. We also constructed the three ChREBP truncates FLAG-ChREBP 1–251, FLAG-ChREBP 252–625, and FLAG-ChREBP 626–852 according to the functional domains of human ChREBP protein. We found that all three ChREBP truncates co-immunoprecipitated with Myc-SMURF2, suggesting that multiple regions of ChREBP interacted with SMURF2 (Fig. 2E).
SMURF2 promoted ChREBP degradation through ubiquitin proteasome pathway
We performed the cycloheximide (CHX) chase experiment and found that endogenous ChREBP protein in HepG2 cells was unstable (Fig. 3A). Furthermore, we performed the CHX chase experiment and found that the half-life of exogenous HA-ChREBP protein expressed in 293T cells was significantly reduced in SMURF2-overexpressing 293T cells (Fig. 3C). We then explored whether SMURF2 functioned as an E3 ligase to promote ChREBP ubiquitination for degradation. We co-expressed HA-ChREBP and Myc-SMURF2 in 293T cells and found that Myc-SMURF2 decreased HA-ChREBP protein level as expected. Addition of the lysosome inhibitor chloroquine had no effect on the protein level of HA-ChREBP (Fig. 3B), whereas the proteasome inhibitor MG132 prevented HA-ChREBP degradation from Myc-SMURF2 (Fig. 3D), suggesting that Myc-SMURF2 promoted ChREBP degradation via the ubiquitination proteasome pathway. We next examined the effect of SMURF2 on ChREBP ubiquitination. HA-ChREBP was co-transfected with FLAG-ubiquitin and Myc-SMURF2 into 293T cells in the presence of MG132, and the ubiquitinated proteins were immunoprecipitated with the anti-HA antibody and immunoblotted with the anti-FLAG antibody to detect ubiquitinated ChREBP. Increased accumulation of ubiquitinated HA-ChREBP was detected, and Myc-SMURF2 promoted ubiquitination of HA-ChREBP (Fig. 3E). Moreover, we transfected HA-ChREBP, His-ubiquitin, and Myc-SMURF2 or Myc-SMURF2C716A into HCT116 cells in the presence of MG132, followed by His-tag pulldown of ubiquitinated proteins. The HA blot showed that SMURF2 greatly increased ubiquitination of HA-ChREBP, whereas SMURF2C716A had a mild effect (Fig. 3F). Our results indicate that SMURF2 functions as an E3 ligase of ChREBP for ubiquitination and proteasomal degradation.
Figure 3.

SMURF2 promoted ChREBP ubiquitination and proteasomal degradation. A, levels of endogenous ChREBP after CHX treatment for indicated time in HepG2 cells. B, addition of lysosome inhibitor chloroquine has no effect on Myc-SMURF2–stimulated HA-ChREBP degradation. C, effect of SMURF2 on the half-life of ChREBP. Myc-SMURF2 was transfected with HA-ChREBP, and cells were treated with CHX at 3 μg/ml for the indicated times. D, addition of proteasome inhibitor MG132 prevents Myc-SMURF2–induced HA-ChREBP degradation. E, SMURF2 promoted ubiquitination (Ub) of ChREBP. F, SMURF2 promoted ubiquitination of ChREBP, whereas SMURF2C716A showed a mild effect. The data in A–E are quantified as the mean ± S.D., and the bar indicates the mean. Statistical significance was calculated using Prism 5 (GraphPad Software). *, p < 0.05. IB, immunoblotting; IP, immunoprecipitation.
SMURF2 promoted ChREBP degradation, reduced aerobic glycolysis and cell proliferation, and increased oxygen consumption in colorectal cancer cells
ChREBP has been shown to enhance aerobic glycolysis, decrease oxygen consumption, and increase proliferation in colorectal cancer cells (7). Considering our findings that SMURF2 promoted ChREBP protein degradation, we next investigated whether SMURF2 regulated metabolic and proliferative activity of colorectal cancer cells. We established Caco-2 cells stably transfected with Myc-tagged GFP cDNA (Myc-GFP), Myc-tagged SMURF2 cDNA (Myc-SMURF2), or Myc-tagged SMURF2C716A cDNA (Myc-SMURF2C716A) and compared their metabolic and proliferative activity. To investigate the effect of SMURF2 on colorectal cancer cell proliferation, cell numbers were counted at days 1–4 after plating. The result showed that overexpression of SMURF2 promoted ChREBP degradation (Fig. 4A) and decreased cell growth rate compared with the control (Fig. 4B). We next compared the glycolytic activity of Caco-2 cells stably transfected with Myc-GFP or Myc-SMURF2 by analyzing glucose uptake and lactate production at 48 h after plating. We found that overexpression of SMURF2 reduced glucose uptake and lactate production (Fig. 4, C and D).
Figure 4.

Ectopic expression of SMURF2 in colorectal cancer cells promoted ChREBP degradation, decreased aerobic glycolysis and cell proliferation, and increased oxygen consumption in colorectal cancer cells. A, ectopic expression of SMURF2 in Caco-2 cells promoted endogenous ChREBP degradation. B, cell proliferation of Caco-2 cells stably expressing either Myc-GFP, Myc-SMURF2 or Myc-SMURF2C716A. C–E, glucose uptake (C), lactate production (D), and oxygen consumption (E) of Caco-2 cells stably expressing Myc-GFP, Myc-SMURF2, or Myc-SMURF2C716A. The data in A are quantified as the mean ± S.D., and the bar indicates the mean. Statistical significance was calculated using Prism 5 (GraphPad Software). *, p < 0.05.
We also compared oxygen consumption rates between Myc-GFP and Myc-SMURF2 and found that Caco-2 cells stably expressing Myc-SMURF2 displayed higher oxygen consumption rates (Fig. 4E). Moreover, Myc-SMURF2C716A overexpression had minimal effect on metabolic and proliferation activity. Consistently, overexpression of SMURF2 in SW1116, another human colorectal cancer cell line, had similar effect on ChREBP expression, aerobic glycolysis, oxygen consumption, and cell proliferation (Fig. S2, A–E). Taken together, our findings suggest that ectopic expression of SMURF2 reduce aerobic glycolysis and cell proliferation as well as increase oxygen consumption rates in colorectal cancer cells.
SMURF2 knockdown promoted ChREBP accumulation, increased aerobic glycolysis and cell proliferation, and decreased oxygen consumption in colorectal cancer cells
To further investigate the effect of SMURF2 suppression on metabolic and proliferative activity, we established HCT116 cells stably transfected with control shRNA or SMURF2 shRNAs (SMURF2 shRNA-1 and SMURF2 shRNA-2). Stable expression of SMURF2 shRNAs decreased SMURF2 levels and promoted ChREBP accumulation (Fig. 5A). In contrast to what was observed in SMURF2 overexpressing colorectal cancer cells, cell proliferation increased in HCT116 cells stably expressing SMURF2 shRNAs compared with control cells (Fig. 5B). ShRNA-mediated suppression of SMURF2 increased glucose uptake and lactate production, accompanied by decreased oxygen consumption (Fig. 5, C–E).
Figure 5.
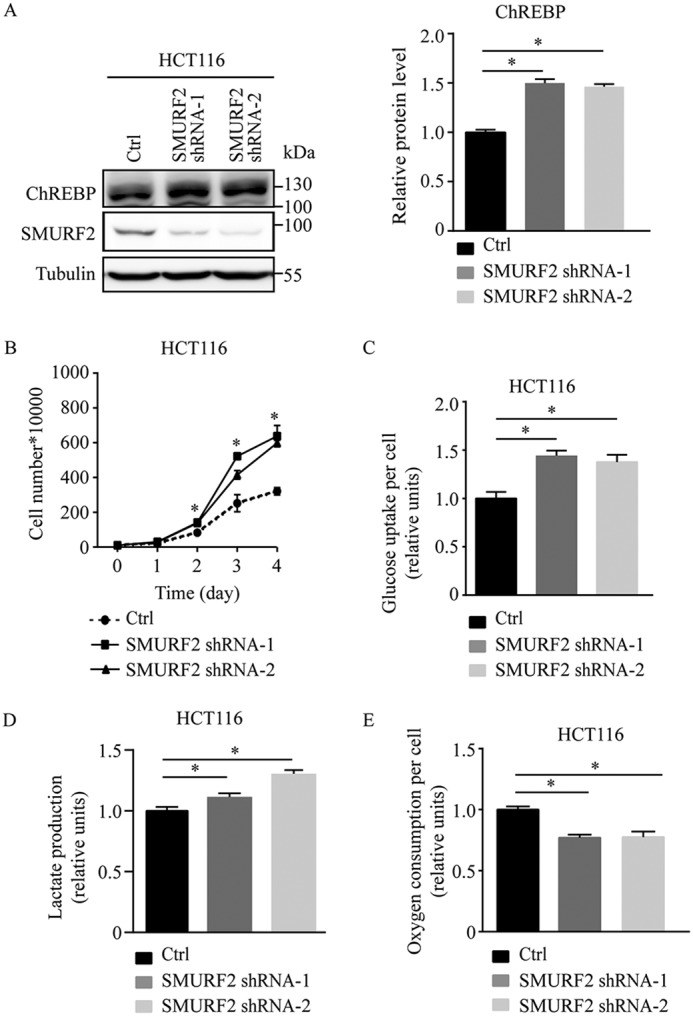
SMURF2 knockdown promoted ChREBP accumulation, increased aerobic glycolysis and cell proliferation, and decreased oxygen consumption in HCT116 cells. A, SMURF2 knockdown promoted endogenous ChREBP accumulation in HCT116 cells. B, cell proliferation of HCT116 cells stably expressing either control (Ctrl) or SMURF2 shRNAs. C–E, glucose uptake (C), lactate production (D), and oxygen consumption (E) of HCT116 stably expressing either control or SMURF2 shRNAs. The data in A are summarized as the means ± S.D., and the bar indicates the mean. Statistical significance was calculated using Prism 5 (GraphPad Software). *, p < 0.05.
To assess whether these changes in metabolism and proliferation were indeed due to suppression of SMURF2, we constructed HCT116 cells stably transfected with either the control or nonsuppressible SMURF2 mutant cDNA in the presence of the control shRNA or SMURF2 shRNA-2. The nonsuppressible SMURF2 mutant cDNA contained several mutations that did not alter amino acid sequence in the region targeted by SMURF2 shRNA-2. The nonsuppressible SMURF2 cDNA could rescue the observed phenotypes in metabolism and proliferation of SMURF2 shRNA-transfected HCT116 cells (Fig. S3, A–E). Therefore, our results suggested that the observed phenotypes of SMURF2 knockdown HCT116 cells were due to suppression of SMURF2 instead of a nonspecific effect of RNAi.
Furthermore, knockdown of SMURF2 in LoVo, another colorectal cancer cell line, also increased ChREBP expression, aerobic glycolysis and cell proliferation while decreasing oxygen consumption (Fig. S4, A–E). These results showed that suppression of SMURF2 led to ChREBP accumulation, accompanied by increased aerobic glycolysis and cell proliferation, as well as decreased oxygen consumption in colorectal cancer cells.
ChREBP played an important role in SMURF2-dependent alterations in metabolism and proliferation in colorectal cancer cells
To investigate whether ChREBP was responsible for SMURF2-dependent alterations in metabolism and proliferation, we used ChREBP siRNAs to suppress the expression of ChREBP in HCT116 cells stably expressing either control shRNAs or SMURF2 shRNAs. ChREBP protein level was higher in SMURF2 shRNAs-transfected HCT116 cells compared with control shRNAs-transfected HCT116 cells (Fig. 6A). Transient transfection of ChREBP siRNAs resulted in reduced ChREBP protein level in both control and SMURF2 shRNAs-transfected HCT116 cells (Fig. 6A). Cell proliferation, glycolytic activity, and oxygen consumption analyses were performed to examine whether ChREBP contributed to the SMURF2-dependent metabolic and growth phenotype. Cell proliferation increased in HCT116 cells stably expressing SMURF2 shRNAs compared with control cells. However, the growth rate increase was blocked in ChREBP siRNAs-transfected HCT116 cells stably expressing SMURF2 shRNA1 or shRNA2 (Fig. 6, B and B′). Glucose uptake decreased in ChREBP siRNAs-transfected cells compared with control cells (Fig. 6C), which was consistent with our previous study (7). Glucose uptake increased in SMURF2 shRNAs-transfected cells compared with control shRNAs-transfected cells (Fig. 6C). Furthermore, glucose uptake induction was abolished in ChREBP siRNAs-transfected HCT116 cells stably expressing SMURF2 shRNAs (Fig. 6C). Similarly, lactate production induction and oxygen consumption reduction were blocked in ChREBP siRNAs-transfected HCT116 stably expressing SMURF2 shRNAs (Fig. 6, D and E). These observations indicate that ChREBP play an important role in SMURF2-dependent alterations in metabolism and proliferation in colorectal cancer cells.
Figure 6.

ChREBP played an important role in mediating SMURF2-dependent growth and metabolic alterations. A, Western blotting analysis of HCT116 cells stably expressing either control (Ctrl) or SMURF2 shRNAs transfected with either control or ChREBP siRNA using the indicated antibodies. B–E, cell proliferations (B and B′), glucose uptake (C), lactate production (D), and oxygen consumption (E) of HCT116 cells stably expressing either control or SMURF2 shRNAs transfected with siRNA for control and ChREBP.
AKT enhanced ChREBP protein stability by decreasing endogenous SMURF2 in colorectal cancer cells and during progression of mouse colorectal cancer
TGF-β and AKT have been reported to regulate the expression of SMURF2 (23, 24). We have tested the possibility of TGF-β or AKT regulating SMURF2-dependent degradation of ChREBP. The TGF-β signal decreased ChREBP mRNA level in colorectal cancer cells (data not shown), which made it difficult to determine whether TGF-β altered SMURF2-dependent ubiquitination and degradation of ChREBP. On the other hand, the phosphatidylinositol 3-kinase signaling pathway is often activated in human cancers (25, 26). Phosphorylation of AKT, induced in the progress of colorectal carcinogenesis, has been found to promote cell proliferation and inhibit apoptosis in human colorectal cancer cells (27, 28). Utilizing a mouse model as described in Fig. 1A, we found that total AKT expression was similar in progression of mouse colorectal cancer, but the level of Ser-473 phosphorylated AKT increased in the progression of mouse colorectal cancer (Fig. 7A). Interestingly, levels of SMURF2 gradually reduced in the progression of mouse colorectal cancer (Fig. 1A). Recent study suggested that AKT interacted with and phosphorylated SMURF2 and then induced SMURF2 degradation by increasing its ubiquitination (29). We found that transient transfection of AKT reduced endogenous SMURF2 protein level and increased endogenous ChREBP protein level in HCT116 cells (Fig. 7B). We also found that AKT did not change the mRNA levels of ChREBP and SMURF2 (Fig. 7C). Similarly, in another human colorectal cancer LoVo cell line, AKT overexpression reduced and increased endogenous SMURF2 and ChREBP protein levels, respectively (Fig. 7D). To further investigate whether AKT was an upstream regulator of SMURF2-dependent ChREBP degradation, we treated HCT116 and SW1116 cells with the AKT phosphorylation inhibitor MK2206 (35). We observed that MK2206 decreased AKT phosphorylation, accompanied by SMURF2 accumulation and ChREBP reduction (Fig. 7, E and F). Our findings suggest that AKT might regulate SMURF2-dependent ChREBP degradation in colorectal cancer cells (Fig. 8). However, the upstream signal regulating the activity of the AKT–SMURF2–ChREBP axis requires further study.
Figure 7.

AKT modulated SMURF2 and ChREBP protein levels. A, Western blotting analysis of p-AKT, AKT, and actin protein levels during progression of mouse colorectal cancer. B, Western blotting analysis of HCT116 cells transfected with either HA-GFP or HA-AKT using the indicated antibodies. C, real-time PCR analysis for mRNA levels of ChREBP and SMURF2 after HA-GFP or HA-AKT was transfected in HCT116 cells. D, Western blotting analysis of LoVo cells transfected with either HA-GFP or HA-AKT. E, Western blotting analysis of HCT116 cells treated with 1 μm MK2206 for 48 h. F, Western blotting analysis of SW1116 cells treated with 1 μm MK2206 for 48 h. The data in A, B, and D–F are quantified as the mean ± S.D., and the bar indicates the mean. Statistical significance was calculated using Prism 5 (GraphPad Software). *, p < 0.05.
Figure 8.

A mechanistic model showing that SMURF2 regulates aerobic glycolysis and cell proliferation in colorectal cancer cell by enhancing ChREBP ubiquitination.
Discussion
Previously, we found that ChREBP played pivotal roles in regulating metabolism and proliferation in colorectal cancer cells (7). This study identified SMURF2, a HECT type ubiquitin ligase, as an E3 ligase of ChREBP which reduced ChREBP protein levels through proteasomal degradation. ChREBP mediated SMURF2-dependent alterations in metabolism and proliferation in colorectal cancer cells. Therefore, we have revealed a novel metabolic role of SMURF2 by acting as an E3 ligase for ChREBP in colorectal cancer cells.
Post-translational modifications play an important role in regulating ChREBP function. Glucose regulates ChREBP phosphorylation, subcellular localization, and DNA-binding activity (9). Glucose-induced ChREBP acetylation increases its transcriptional activity (8). ChREBP O-glycosylation enhances its DNA-binding activity and protein stability (10–12). Interestingly, O-glycosylation could decrease ubiquitin-mediated degradation of ChREBP (11, 12). Although much study has been carried out in investigating post-translational modifications of ChREBP, the mechanism by which the protein stability of ChREBP is regulated is still unknown.
SMURF2 has been reported to play a dual role in cancer by functioning as both tumor promoter and suppressor by regulating the stability of proteins in tumorigenesis (15, 19, 30–33). However, the role of SMURF2 in regulating proliferation in colorectal cancer cells is unclear. SMURF2 gene is not frequently mutated in colorectal cancer (http://www.cbioportal.org/) (36, 37).5 Interestingly, changes in SMURF2 expression correlated with poor prognosis in colon cancer (https://www.proteinatlas.org/) (38).5 Our findings suggest that SMURF2 decreases aerobic glycolysis and cell proliferation in colorectal cancer cells.
SMURF2 was originally identified as a regulator for the TGF-β signaling pathway (14). TGF-β is reported to regulate the expression of SMURF2 (23, 24). However, we did not observe TGF-β–induced SMURF2-mediated ChREBP degradation in colorectal cancer cells. Here we found that transient transfection of AKT reduced SMURF2 protein level and increased ChREBP protein level in HCT116 cells. AKT has been reported to promote aerobic glycolysis in human colorectal cancer cells (34). Suppression of ChREBP decreased aerobic glycolysis in colorectal cancer cells (7). However, it remains unclear whether AKT regulates ChREBP level or activity in cancer cells. Our findings suggest a possible mechanistic link between AKT and ChREBP by showing that AKT may promote ChREBP stabilization through decreasing SMURF2 levels. The upstream signal regulating the activity of the AKT–SMURF2–ChREBP axis requires further study.
In summary, we have uncovered a novel role of SMURF2 in promoting ChREBP degradation. SMURF2 regulates the metabolic and proliferative activity in colorectal cancer cells by promoting ChREBP ubiquitination and degradation. AKT might promote the function of ChREBP by decreasing the level of SMURF2. The newly identified AKT–SMURF2–ChREBP axis plays an important role in regulating the metabolic and proliferative activity of colorectal cancer cells.
Materials and methods
Cell culture and treatment
HCT116, CaCO-2, SW1116, LoVo human colorectal cancer cells, 293T human embryonic kidney cells, and HeLa human cervical cancer cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 1 mmol/liter sodium pyruvate, 2 mmol/liter l-glutamine, 100-unit/ml penicillin, and 100 μg/ml streptomycin under the standard culture condition (at 37 °C in a 5% CO2 humidified atmosphere). For the MK2206 treatment, HCT116 cells were treated with 1 μm MK2206, an AKT phosphorylation inhibitor (Cell Signaling Technology) for 48 h before further analysis.
Mice
Female C57BL/6 mice were obtained from Shanghai Jiao Tong University School of Medicine Animal Centre and housed in controlled temperature rooms and had free access to food and water, with a 12-h light/dark cycle. Our animal research has been approved by the Shanghai Jiao Tong University School of Medicine Animal Care and Use Committee.
Western blotting analysis
Total protein extraction was disintegrated using Triton X-100 lysis buffer (1 mm EDTA, 40 mm Tris-HCl, pH 8, 100 mm NaCl, and 0.5% Nonidet P-40, 1% Triton X-100). The protein content was quantified using the BCA protein assay kit (Pierce).
Equal samples were electrophoresed on 7.5% SDS-PAGE gel. Proteins were transferred to Immuno-Blot polyvinylidene difluoride membrane (Millipore) and then blocked in 5% fat-free milk in Tris-buffered saline with 0.1% Tween 20, followed by incubation with primary antibodies overnight at 4 °C. Secondary peroxidase-labeled anti-rabbit or anti-mouse IgG antibodies (Santa Cruz) was applied, respectively. The signals were detected with an enhanced chemiluminescent solution (Millipore). Protein was visualized using Amersham Biosciences Imager 600 (GE Healthcare). The following primary antibodies were used: ChREBP (Novus), SMURF2, AKT, and p-AKT (Ser-473) (Cell Signaling Technology); FLAG and tubulin (Sigma); and HA and Myc (Santa Cruz).
Cell proliferation
A total of 3000, 5000, 8000, or 10,000 cells were plated in 6-well plates in triplicate. The plates were incubated at 37 °C with 5% CO2. The cell numbers were counted at days 1–4 after plating.
Co-immunoprecipitation
The cells were lysed in Triton X-100 buffer, followed by incubation with appropriate primary antibodies overnight at 4 °C and an additional 2-h incubation with protein G Plus/protein A-agarose beads (Santa Cruz) at 4 °C. The beads were then washed three times with lysis buffer and boiled in 2× sample loading buffer for 10 min. Cell lysates and immunoprecipitates were performed by Western blotting analysis.
His tag pulldown assay
HA-ChREBP, His-ubiquitin, and Myc-SMURF2 or Myc-SMURF2C716A constructs were co-transfected into HCT116 cells. After 48 h, the cells were washed with PBS and lysed with buffer 1 (8 m urea, 100 mm Na2HPO4, 10 mm Tris-HCl, pH 8.0, 0.2% Triton X-100, and 10 mm imidazole) at room temperature for 30 min. The lysates were incubated with nickel–nitrilotriacetic acid beads (Qiagen) for 3 h at room temperature. After incubation, the beads were washed twice with buffer 1, twice with buffer 2 (8 m urea, 100 mm Na2HPO4, 10 mm Tris-HCl, pH 6.3, 0.2% Triton X-100, and 10 mm imidazole), and finally once with buffer 3 (100 mm NaCl, 20% glycerol, 20 mm Tris-HCl, pH 8.0, 1 mm DTT, and 10 mm imidazole). The proteins were eluted with 2× sample loading buffer for 10 min and subjected to Western blotting analysis.
shRNA and cDNA transfection
The SMURF2 shRNAs were purchased from GIPZ human ubiquitin conjugation subset 1 library with the following shRNA-1 sequence (sense): 5′-CAGTTAATCCGGAACATTT-3′ and shRNA-2 sequence (sense): 5′-AGCGAGACCTGGTTCAGAA-3′. The cDNA encoding human SMURF2 was gifted from Professor Bin Li at Shanghai Jiao Tong University School of Medicine and verified by sequencing. The SMURF2 cDNA was then subcloned into the lentiviral vector pLVX-IRES-puro (Clontech). The nonsuppressible SMURF2 cDNA to SMURF2 shRNA-2 contained several mutations identified by the letters in bold, 5′-AAG GGA TCT CGT CCA GAA ACT AA-3′. These mutations did not alter the amino acid sequence. The nonsuppressible SMURF2 cDNA was then subcloned into the lentiviral vector pLenti6.3-IRES-mCherry-Blasticidin, which was provided by Dr. Jiong Deng at Shanghai Jiao Tong University School of Medicine. The SMURF2 shRNAs and SMURF2 cDNA were transfected into 293T cells with the packaging plasmids PMD2G and psPAX2 using Lipofectamine 2000 (Invitrogen Life Technologies) according to the manufacturer's instructions. Viral supernatant was collected after 48 h. 5 ml of viral supernatant and 5 ml of fresh medium were then added to HCT116 and Caco-2 cells/10-cm dish, and 24 h later, the medium was replaced by fresh medium. Then HCT116 cells stably transfected with different shRNAs and Caco-2 cells stably transfected with SMURF2 cDNA were selected using 2 and 5 μg/ml puromycin, respectively.
siRNA transfection
Predesigned siRNAs against human ChREBP and control scrambled siRNA were synthesized by Jima pharmaceutical company (Shanghai, China). The ChREBP siRNA sequences are 5′-GCACCCUUGGCAAACCUUUUU-3′ and 5′-AAAGGUUUGCCAAGGGUGCUU-3′. siRNA transfections were performed using Lipofectamine RNAiMAX (Invitrogen Life Technologies) according to the manufacturer's instructions.
RNA extraction and real-time PCR analysis
Total RNA was extracted from treated cells using TRIzol (Invitrogen Life Technologies) according to the manufacturer's instructions. Total RNA was reverse transcribed into cDNA using the PrimeScript RT reagent kit (Takara Bio Inc., Japan). Real-time PCR analysis was performed by using a StepOnePlus real-time PCR System (Applied Biosystems). Absolute quantification of mRNA abundance was performed according to user's guide of Applied Biosystems. The forward and reverse primers set for ChREBP were 5′-AACTGGAAGTTCTGGGTGTTC-3′ and 5′-AGGGAGTTCAGGACAGTTGG-3′, respectively. The forward and reverse primers set for SMURF2 were 5′-AGGCTCAATTCTTGGCTCTG-3′ and 5′-AATCTTGCTCGTCGCTCTTC-3′, respectively. The forward and reverse primers set for 18S rRNA were 5′-GTAACCCGTTGAACCCCATT-3′ and 5′-CCATCCAATCGGTAGTAGCG-3′. 18S rRNA was used as the endogenous control.
Luciferase reporter assay
293T cells were transiently transfected with HA-ChREBP, Myc-SMURF2, Myc-SMURF2C716A, FLAG-MLX, 4× ACC ChoRE-Luc reporter, and β-galactosidase using Lipofectamine 2000 (Invitrogen). The total amount of transfected DNA was kept constant by using pcDNA3. β-Galactosidase was served as internal control to normalize transfection efficiency. The cells were lysed 48 h post-transfection with luciferase lysis buffer and assayed using the luciferase reporter assay system (Promega) and β-galactosidase assay kit (Clontech) according to the manufacturer's instructions.
Metabolic assays
Oxygen consumption was measured using an Oxytherm system (Hansatech, Kings Lynn, UK). 80% confluent cells in 10-cm dishes were trypsinized and resuspended in 3 ml of medium. Glucose uptake and lactate production were measured using the glucose assay kit (Shanghai Rongsheng Biotech, Shanghai, China) and lactate assay kit (Sigma), respectively, according to manufacturers' instructions at 48 h after cells were plated in 6-well plates.
Immunofluorescent staining
The cells grown on coverslips were cultured in medium. The cells were washed twice with PBS, fixed in 4% paraformaldehyde for 10 min, permeabilized with 0.1% Triton X-100 for 5 min at room temperature, blocked in 1% BSA for 30 min at room temperature, and incubated with primary antibodies overnight at 4 °C. Alexa 488– and 546–conjugated secondary antibodies were added to the cells and incubated for 30 min at room temperature. After two washes in PBS, the coverslips were mounted with ProLong Gold with 4′,6′-diamino-2-phenylindole (Invitrogen) and photographed using an LSM 710 laser scanning confocal microscope (Zeiss).
Statistical analysis
The experiments were performed at least three times independently, and one representative experiment is shown. The data are summarized as the means ± S.D. using Prism 5 (GraphPad Software).
Author contributions
Y. Li data curation; Y. Li, D. Y., N. T., J. M., M. F., and L. H. formal analysis; Y. Li, D. Y., N. T., Q. L., and L. Z. methodology; Y. Li writing-original draft; P. Z., Y. Z., L. W., and X. T. supervision; Y. Lu and L. T. software; J. Y. Y. and X. T. writing-review and editing; X. T. conceptualization; X. T. funding acquisition; X. T. project administration.
Supplementary Material
This work was supported by National Natural Science Foundation of China Grants 81472239, 81672322, and 31601118; National Key Research and Development Program of China Grant 2016YFC1304800; and Grant SYjdyx18007 from the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning and Construction Plan of Laboratory Technical Team in Shanghai Universities. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- ChREBP
- carbohydrate response element–binding protein
- SMURF2
- Smad-ubiquitination regulatory factor 2
- TGF
- transforming growth factor
- CHX
- cycloheximide.
References
- 1. Yamashita H., Takenoshita M., Sakurai M., Bruick R. K., Henzel W. J., Shillinglaw W., Arnot D., and Uyeda K. (2001) A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. U.S.A. 98, 9116–9121 10.1073/pnas.161284298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang H., and Wollheim C. B. (2002) ChREBP rather than USF2 regulates glucose stimulation of endogenous l-pyruvate kinase expression in insulin-secreting cells. J. Biol. Chem. 277, 32746–32752 10.1074/jbc.M201635200 [DOI] [PubMed] [Google Scholar]
- 3. Collier J. J., Zhang P., Pedersen K. B., Burke S. J., Haycock J. W., and Scott D. K. (2007) c-Myc and ChREBP regulate glucose-mediated expression of the L-type pyruvate kinase gene in INS-1-derived 832/13 cells. Am. J. Physiol. Endocrinol Metab. 293, E48–E56 10.1152/ajpendo.00357.2006 [DOI] [PubMed] [Google Scholar]
- 4. da Silva Xavier G., Rutter G. A., Diraison F., Andreolas C., and Leclerc I. (2006) ChREBP binding to fatty acid synthase and L-type pyruvate kinase genes is stimulated by glucose in pancreatic β-cells. J. Lipid Res. 47, 2482–2491 10.1194/jlr.M600289-JLR200 [DOI] [PubMed] [Google Scholar]
- 5. Dentin R., Pégorier J. P., Benhamed F., Foufelle F., Ferré P., Fauveau V., Magnuson M. A., Girard J., and Postic C. (2004) Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J. Biol. Chem. 279, 20314–20326 10.1074/jbc.M312475200 [DOI] [PubMed] [Google Scholar]
- 6. Billin A. N., and Ayer D. E. (2006) The Mlx network: evidence for a parallel Max-like transcriptional network that regulates energy metabolism. Curr. Top. Microbiol. Immunol. 302, 255–278 [DOI] [PubMed] [Google Scholar]
- 7. Tong X., Zhao F., Mancuso A., Gruber J. J., and Thompson C. B. (2009) The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 106, 21660–21665 10.1073/pnas.0911316106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bricambert J., Miranda J., Benhamed F., Girard J., Postic C., and Dentin R. (2010) Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J. Clin. Invest. 120, 4316–4331 10.1172/JCI41624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uyeda K., and Repa J. J. (2006) Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 4, 107–110 10.1016/j.cmet.2006.06.008 [DOI] [PubMed] [Google Scholar]
- 10. Sakiyama H., Fujiwara N., Noguchi T., Eguchi H., Yoshihara D., Uyeda K., and Suzuki K. (2010) The role of O-linked GlcNAc modification on the glucose response of ChREBP. Biochem. Biophys. Res. Commun. 402, 784–789 10.1016/j.bbrc.2010.10.113 [DOI] [PubMed] [Google Scholar]
- 11. Ido-Kitamura Y., Sasaki T., Kobayashi M., Kim H. J., Lee Y. S., Kikuchi O., Yokota-Hashimoto H., Iizuka K., Accili D., and Kitamura T. (2012) Hepatic FoxO1 integrates glucose utilization and lipid synthesis through regulation of Chrebp O-glycosylation. PLoS One 7, e47231 10.1371/journal.pone.0047231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guinez C., Filhoulaud G., Rayah-Benhamed F., Marmier S., Dubuquoy C., Dentin R., Moldes M., Burnol A. F., Yang X., Lefebvre T., Girard J., and Postic C. (2011) O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 60, 1399–1413 10.2337/db10-0452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rotin D., and Kumar S. (2009) Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 10, 398–409 10.1038/nrm2690 [DOI] [PubMed] [Google Scholar]
- 14. Izzi L., and Attisano L. (2004) Regulation of the TGFβ signalling pathway by ubiquitin-mediated degradation. Oncogene 23, 2071–2078 10.1038/sj.onc.1207412 [DOI] [PubMed] [Google Scholar]
- 15. Blank M., Tang Y., Yamashita M., Burkett S. S., Cheng S. Y., and Zhang Y. E. (2012) A tumor suppressor function of SMURF2 associated with controlling chromatin landscape and genome stability through RNF20. Nat. Med. 18, 227–234 10.1038/nm.2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Osmundson E. C., Ray D., Moore F. E., Gao Q., Thomsen G. H., and Kiyokawa H. (2008) The HECT E3 ligase SMURF2 is required for Mad2-dependent spindle assembly checkpoint. J. Cell Biol. 183, 267–277 10.1083/jcb.200801049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramkumar C., Kong Y., Cui H., Hao S., Jones S. N., Gerstein R. M., and Zhang H. (2012) SMURF2 regulates the senescence response and suppresses tumorigenesis in mice. Cancer Res. 72, 2714–2719 10.1158/0008-5472.CAN-11-3773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang H., and Cohen S. N. (2004) SMURF2 up-regulation activates telomere-dependent senescence. Genes Dev. 18, 3028–3040 10.1101/gad.1253004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jin C., Yang Y. A., Anver M. R., Morris N., Wang X., and Zhang Y. E. (2009) Smad ubiquitination regulatory factor 2 promotes metastasis of breast cancer cells by enhancingmigration and invasiveness. Cancer Res. 69, 735–740 10.1158/0008-5472.CAN-08-1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fukuchi M., Fukai Y., Masuda N., Miyazaki T., Nakajima M., Sohda M., Manda R., Tsukada K., Kato H., and Kuwano H. (2002) High-level expression of the Smad ubiquitin ligase SMURF2 correlates with poor prognosis inpatients with esophageal squamous cell carcinoma. Cancer Res. 62, 7162–7165 [PubMed] [Google Scholar]
- 21. Du J. X., Hagos E. G., Nandan M. O., Bialkowska A. B., Yu B., and Yang V. W. (2011) The E3 ubiquitin ligase SMAD ubiquitination regulatory factor 2 negatively regulates Krüppel-likefactor 5 protein. J. Biol. Chem. 286, 40354–40364 10.1074/jbc.M111.258707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Callaghan B. L., Koo S. H., Wu Y., Freake H. C., and Towle H. C. (2001) Glucose regulation of the acetyl-CoA carboxylase promoter PI in rat hepatocytes. J. Biol. Chem. 276, 16033–16039 10.1074/jbc.M101557200 [DOI] [PubMed] [Google Scholar]
- 23. Ohashi N., Yamamoto T., Uchida C., Togawa A., Fukasawa H., Fujigaki Y., Suzuki S., Kitagawa K., Hattori T., Oda T., Hayashi H., Hishida A., and Kitagawa M. (2005) Transcriptional induction of SMURF2 ubiquitin ligase by TGF-β. FEBS Lett. 579, 2557–2563 10.1016/j.febslet.2005.03.069 [DOI] [PubMed] [Google Scholar]
- 24. Hsu H. Y., Lin T. Y., Wu Y. C., Tsao S. M., Hwang P. A., Shih Y. W., and Hsu J. (2014) Fucoidan inhibition of lung cancer in vivo and in vitro: role of the SMURF2-dependent ubiquitin proteasome pathway in TGFβ receptor degradation. Oncotarget 5, 7870–7885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Engelman J. A., Luo J., and Cantley L. C. (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7, 606–619 10.1038/nrg1879 [DOI] [PubMed] [Google Scholar]
- 26. Vivanco I., and Sawyers C. L. (2002) The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat. Rev. Cancer. 2, 489–501 10.1038/nrc839 [DOI] [PubMed] [Google Scholar]
- 27. Itoh N., Semba S., Ito M., Takeda H., Kawata S., and Yamakawa M. (2002) Phosphorylation of AKT/PKB is required for suppression of cancer cell apoptosis and tumor progression in human colorectal carcinoma. Cancer 94, 3127–3134 10.1002/cncr.10591 [DOI] [PubMed] [Google Scholar]
- 28. Khaleghpour K., Li Y., Banville D., Yu Z., and Shen S. H. (2004) Involvement of the PI 3-kinase signaling pathway in progression of colon adenocarcinoma. Carcinogenesis 25, 241–248 [DOI] [PubMed] [Google Scholar]
- 29. Choi Y. H., Kim Y. J., Jeong H. M., Jin Y. H., Yeo C. Y., and Lee K. Y. (2014) AKT enhances Runx2 protein stability by regulating SMURF2 function during osteoblast differentiation. Febs j. 281, 3656–3666 10.1111/febs.12887 [DOI] [PubMed] [Google Scholar]
- 30. Ramkumar C., Cui H., Kong Y., Jones S. N., Gerstein R. M., and Zhang H. (2013) SMURF2 suppresses B-cell proliferation and lymphomagenesis by mediating ubiquitination anddegradation of YY1. Nat. Commun. 4, 2598 10.1038/ncomms3598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nie J., Xie P., Liu L., Xing G., Chang Z., Yin Y., Tian C., He F., and Zhang L. (2010) Smad ubiquitylation regulatory factor 1/2 (Smurf1/2) promotes p53 degradation by stabilizing the E3 ligase MDM2. J. Biol. Chem. 285, 22818–22830 10.1074/jbc.M110.126920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li H., and Seth A. (2004) An RNF11: SMURF2 complex mediates ubiquitination of the AMSH protein. Oncogene 23, 1801–1808 10.1038/sj.onc.1207319 [DOI] [PubMed] [Google Scholar]
- 33. Han G., Li A. G., Liang Y. Y., Owens P., He W., Lu S., Yoshimatsu Y., Wang D., Ten Dijke P., Lin X., and Wang X. J. (2006) Smad7-induced β-catenin degradation alters epidermal appendage development. Dev. Cell. 11, 301–312 10.1016/j.devcel.2006.06.014 [DOI] [PubMed] [Google Scholar]
- 34. Franke T. F. (2008) PI3K/AKT: getting it right matters. Oncogene 27, 6473–6488 10.1038/onc.2008.313 [DOI] [PubMed] [Google Scholar]
- 35. Yap T. A., Yan L., Patnaik A., Fearen I., Olmos D., Papadopoulos K., Baird R. D., Delgado L., Taylor A., Lupinacci L., Riisnaes R., Pope L. L., Heaton S. P., Thomas G., Garrett M. D., et al. (2011) First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with adcanced solid tumors. J. Clin. Oncol. 29, 4688–4695 10.1200/JCO.2011.35.5263 [DOI] [PubMed] [Google Scholar]
- 36. Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., and Schultz N. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., and Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., et al. (2015) Proteomics: Tissue-based map of the human proteome. Science 347, 1260419 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.