

Abstract
Growth of the cholera bacterium Vibrio cholerae in a biofilm community contributes to both its pathogenicity and survival in aquatic environmental niches. The major components of V. cholerae biofilms include Vibrio polysaccharide (VPS) and the extracellular matrix proteins RbmA, RbmC, and Bap1. To further elucidate the previously observed overlapping roles of Bap1 and RbmC in biofilm architecture and surface attachment, here we investigated the structural and functional properties of Bap1. Soluble expression of Bap1 was possible only after the removal of an internal 57-amino-acid-long hydrophobic insertion sequence. The crystal structure of Bap1 at 1.9 Å resolution revealed a two-domain assembly made up of an eight-bladed β-propeller interrupted by a β-prism domain. The structure also revealed metal-binding sites within canonical calcium blade motifs, which appear to have structural rather than functional roles. Contrary to results previously observed with RbmC, the Bap1 β-prism domain did not exhibit affinity for complex N-glycans, suggesting an altered role of this domain in biofilm-surface adhesion. Native polyacrylamide gel shift analysis did suggest that Bap1 exhibits lectin activity with a preference for anionic or linear polysaccharides. Our results suggest a model for V. cholerae biofilms in which Bap1 and RbmC play dominant but differing adhesive roles in biofilms, allowing bacterial attachment to diverse environmental or host surfaces.
Keywords: microbial pathogenesis, biofilm, amyloid, lectin, glycobiology, Vibrio cholerae, X-ray crystallography, virulence factor, extracellular matrix protein, cholera
Introduction
Vibrio cholerae, the bacterium responsible for pandemic cholera, forms three-dimensional biofilms that aid in V. cholerae transmission, pathogenicity, and environmental persistence (1–3). The biofilm structure is composed of a specialized bacterial community with distinct growth stage properties, held together by an extracellular matrix of polysaccharides, proteins, and nucleic acids (3–5). V. cholerae biofilms have been implicated in mammalian pathogenicity by increasing the infectious dose and by providing resistance to the acidic environment of the stomach (1, 4). In the natural aquatic niche, biofilms aid in the persistence of V. cholerae by providing protection from environmental threats such as predation and nutrient limitation (2, 5–8).
The major components of the V. cholerae biofilm matrix are Vibrio polysaccharide (VPS),3 nucleic acids, and the matrix proteins RbmA, RbmC, and Bap1 (3, 8, 9). Vibrio polysaccharide is formed by repeating units of an acetylated tetrasaccharide unique to V. cholerae, whose synthesis and export are carried out by the products of the vps I and vps II gene clusters (10–12). RbmA and RbmC are two of six proteins encoded by the Rugosity and Biofilm Modulators (rbm) gene cluster (13, 14). Bap1 (Biofilm-Associated Protein 1), which shares substantial sequence identity with a large fragment of RbmC, is encoded by a single gene downstream of the vps and rbm gene clusters. Synchronized up-regulation of vps I, vps II, and rbm gene clusters, as well as bap1, has been shown to occur during the biofilm production life stage of V. cholerae (15). The high-resolution structure of RbmA uncovered a composition of tandem fibronectin type III domains and provided substantial insight into its contribution to the V. cholerae biofilm matrix (16, 17). Less is understood about the structure and molecular mechanisms underlying the scaffolding roles played by Bap1 and RbmC (18).
Insights into the function of the biofilm matrix components have come from knockout mutagenesis studies of the biofilm matrix proteins (RbmA, RbmC, and Bap1) and microscopy utilizing fluorescently-labeled components of the biofilm. Investigation into V. cholerae biofilm formation and architecture often utilizes so-called rugose strains, which exhibit increased biofilm production, wrinkled colony morphologies, and the formation of a floating structure called a pellicle (19). Experiments utilizing deletion mutants of bap1, rbmC, or both genes in the rugose background suggest that Bap1 and RbmC have similar, additive, and essential roles in biofilm formation (14). Further analysis of the V. cholerae biofilm architecture using microscopy techniques provides additional insight implicating RbmA in V. cholerae cell–cell adhesion, Bap1 in surface attachment, and both Bap1 and RbmC in formation of dynamic envelopes that encase clusters of cells in the mature biofilm (9). Deletion of either bap1 or rbmC results in similar biofilm development profiles supporting their partially redundant function, whereas the double deletion results in cell clusters that are unable to attach to surfaces (10, 21). In the context of in vitro growth assays on glass coverslips, the two proteins appear to be functionally redundant, as deletion of either single protein results in biofilms with apparently normal development and surface attachment (20). However, Bap1 and RbmC are not totally redundant as RbmC does not localize to the biofilm surface upon bap1 deletion (9).
Although the complete three-dimensional structures of RbmC and Bap1 are unknown, previous studies utilizing sequence-based prediction methods propose multidomain architectures for Bap1 and RbmC (14). The prediction for Bap1 includes four Vibrio, Colwellia, Bradyrhizobium, and Shewanella (VCBS) domains and four FG-GAP domains (both of which fall under the umbrella of the β-propeller structural motif), a calcium-binding EF-hand domain, and a β-prism–like lectin domain. Predictions for the structural organization of RbmC are similar to that of Bap1, but with one additional C-terminal β-prism domain and two N-terminal repeats resembling the C-terminal βγ-crystallin domain of StcE, a mucinase expressed by enterohemorrhagic Escherichia coli O157:H7 (EHEC O157:H7) (14, 21, 22). Whereas the core-predicted β-propeller domains of Bap1 and RbmC share ∼60% sequence identity, the β-prism domains are more dissimilar with ∼36% sequence identity (excluding gaps). One additional β-prism is also found in the V. cholerae genome attached to the pore-forming toxin V. cholerae cytolysin (VCC) with ∼33% sequence identity to the Bap1 β-prism domain. The three β-prism domains from RbmC and VCC all exhibit low-nanomolar affinity for the central core of complex N-glycans found abundantly on eukaryotic cell surfaces (23). Sequence alignments indicate that Bap1 has a 57-amino acid insertion within the β-prism domain, not present in the RbmC or VCC β-prism domains (23).
Here, we describe the crystal structure of Bap1Δ57 (missing the 57-amino acid β-prism domain insertion) at 1.9 Å resolution. The structure of Bap1Δ57 reveals a two-domain arrangement consisting of an eight-bladed β-propeller with a β-prism domain inserted within blade 6 via a flexible linker. In addition, we show evidence of Bap1Δ57 binding to anionic polysaccharides in a manner that differs significantly from the lectin-binding activity displayed by the β-prism domains of RbmC (23). Our studies support a model of V. cholerae biofilm architecture that allows for varied roles of biofilm scaffolding and surface attachment in aquatic versus host environments. Our results provide a starting point for understanding how Bap1 and RbmC may participate in structural and adhesive roles in building the V. cholerae biofilm matrix.
Results
1.9 Å crystal structure of Bap1Δ57 reveals a two-domain architecture
Initial attempts to express full-length Bap1 (predicted molecular mass of 72.6 kDa) or the isolated Bap1 β-prism domain in E. coli resulted in insoluble material. However, we were successful in expressing a Bap1 construct in which the β-prism domain was genetically excised (Bap1β-propeller), suggesting that this domain contributes to the insolubility of the full-length Bap1 protein. Given that isolated RbmC and VCC β-prism domains express in a soluble form (23) and that the distinguishing feature of the Bap1 β-prism domain is the 57-amino acid insertion, we made Bap1Δ57 and β-prismΔ57 domain constructs with this insertion removed. To aid in expression and screening, we additionally fused a protease-cleavable GFPUV domain to the N terminus of the Bap1Δ57 and β-prismΔ57 constructs. This two-pronged approach drastically improved soluble expression yielding material that exhibited monodisperse behavior by size-exclusion chromatography. Although the Bap1β-propeller construct yielded crystals, these exhibited fiber-like properties and did not suitably diffract X-rays. The Bap1Δ57 construct also yielded three-dimensional crystals that diffracted to better than 2 Å resolution. X-ray data analysis indicated that crystals belonged to space group P41212 with unit cell dimensions of a = b = 71 Å, c = 304 Å, α = β = γ = 90° (Table 1).
Table 1.
X-ray data and refinement statistics
| Bap1Δ57 native | Bap1Δ57 K2PtCl4 | |
|---|---|---|
| Data collection | ||
| Space group | P41212 | P41212 |
| Cell dimensions a, b, c (Å) | 71.0, 71.0, 304.0 | 71.1, 71.1, 302.3 |
| Cell angles α, β, γ (0) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Wavelength (Å) | 1.25414 | 0.91738 |
| Resolution limits (Å) | 47.6–1.9 (1.94–1.90)a | 49.6–2.0 (2.05–2.00) |
| Total reflections | 693,582 (14,468) | 1,383,672 (102,802) |
| Unique reflections | 62,568 (3,772) | 53,632 (3,880) |
| Mosaicity (°) | 0.08 | 0.08 |
| Rsym | 0.11 (0.64) | 0.17 (2.24) |
| Rpim | 0.03 (0.36) | 0.03 (0.44) |
| I/σI | 13.8 (2.0) | 17.6 (2.7) |
| Completeness (%) | 99.7 (96.2) | 99.8 (99.5) |
| Anomalous completeness (%) | 99.9 (99.6) | |
| Multiplicity | 11.1 (3.8) | 25.8 (26.5) |
| Anomalous multiplicity | 13.9 (13.9) | |
| CC½ | 0.998 (0.665) | 0.999 (0.797) |
| Wilson B-factor (Å2) | 17.8 | 29.3 |
| Average phase FOM (after RESOLVE) | 0.8 | |
| Refinement (4,522 protein atoms, 32 ligand atoms, 532 water molecules) | ||
| Resolution (Å) | 47.6–1.9 | |
| No. of reflections (Rfree) | 62,421 | |
| Final Rwork/Rfree (%) | 15.8/17.3 | |
| Average B-factor (Å2) | 27.1 | |
| RMSD | ||
| Bond lengths (Å) | 0.004 | |
| Bond angles (°) | 0.7 | |
| Ramachandran statistics | ||
| Favored | 96.3% | |
| Allowed | 3.5% | |
| Outliers | 0.2% | |
| MolProbity Scores | ||
| Overall score | 1.0 (100%) | |
| All-atom clashscore | 0.7 (100%) |
a Numbers in parentheses denote the highest-resolution shell.
The structure of Bap1Δ57 was solved by single isomorphous replacement with anomalous scattering (SIRAS) utilizing a crystal derivative soaked in K2Cl4Pt. Two platinum sites contributed to phasing, yielding maps with a clear molecular outline, and subsequent density modification led to interpretable electron density maps. Refinement against a 1.9 Å native data set resulted in a final Rwork of 15.8 and Rfree of 17.3. The overall structure reveals a β-propeller structural core tethered to an accessory β-prism domain by linkers at both the N- and C-terminal ends of the β-prism (Fig. 1A). These extended linkers contained higher B-factors in general than the two structural domains and did not participate in crystal-packing interactions. This arrangement suggests that the linkers are likely flexible in solution and that the relative orientation between the two domains results from the crystal lattice, although this cannot be confirmed as only one copy of the molecule was present per asymmetric unit.
Figure 1.
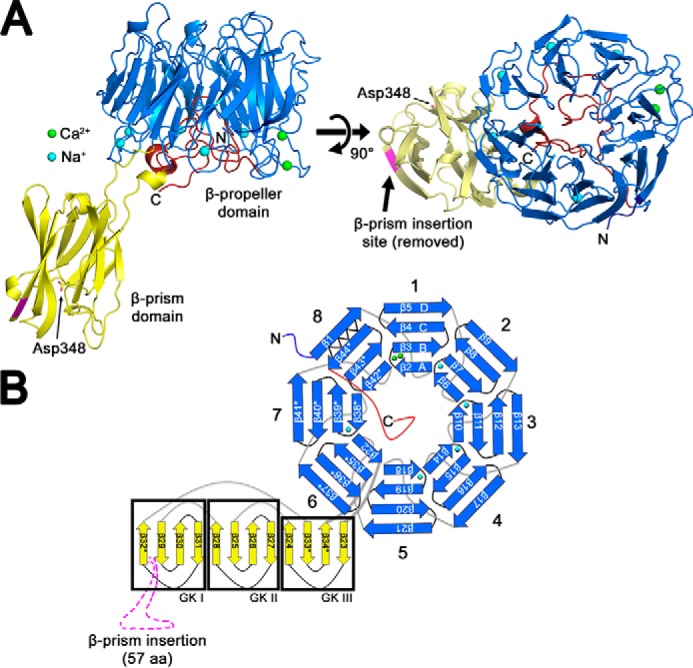
Crystal structure of Bap1Δ57 reveals a two-domain architecture. A, Bap1Δ57 consists of two structural domains: an eight-bladed β-propeller with a β-prism domain inserted within a loop of blade 6. The protein structure is shown in cartoon representation with the β-propeller colored in marine and the β-prism colored in yellow. Two modeled Ca2+ ions and five Na+ ions are shown as green and blue spheres, respectively. The C-terminal region that forms a plug within the central β-propeller cavity is colored red. A key aspartic acid residue (Asp348) that forms essential contacts with bound carbohydrates in homologous β-prism lectin domains from Vibrio cholerae (in RbmC and VCC) is shown in stick representation. B, schematic diagram illustrating Bap1Δ57 topology. The so-called “Velcro closure” of the β-propeller domain is represented by a zig-zag line, and the location of the 57-amino acid insert that has been removed genetically is represented by an arrow in A and a dashed magenta line in B. All structural representations generated using the PyMOL Molecular Graphics System, Version 2.2.0 Schrödinger, LLC.
Bap1 β-propeller domain adopts a canonical β-propeller fold with distinct features
Analysis of the topology of the Bap1 fold reveals an eight-bladed β-propeller core with a β-prism accessory domain interrupting the β-propeller motif at a loop within blade 6 (Fig. 1B). The fact that the Bap1 β-propeller domain can be expressed as a soluble protein (by deleting the β-prism domain from the loop in blade 6) suggests that the β-prism domain is not necessary for folding or stability of the β-propeller structure. Similarly, removal of the 57-amino acid insertion within the β-prism does not appear to affect folding of this domain, which exhibits a fold similar to β-prism domains from RbmC and VCC, with RMSDs of 1.0 and 1.1 Å, respectively. We observed no evidence for higher-ordered oligomeric structures in solution or in the crystal packing lattice, suggesting that Bap1Δ57 exists as a monomer.
Each of the eight propeller blades consists of a four-stranded antiparallel β-sheet, with β-strands radiating out from their N-terminal strands (β-strand A, βA) at the center of the toroid to their C-terminal strands (β-strand D, βD) forming the outer edge of the propeller (shown in blade 1 of the β-propeller topology diagram in Fig. 1B). As is commonly found in β-propeller proteins, the Bap1 β-propeller domain contains a Velcro closure with the most N-terminal residues (Asp-31–Ser-41) acting as βD of blade 8, zipping the disc together via β-sheet hydrogen bonding (Fig. 1B). A relatively unstructured C-terminal loop extends from β-strand C (βC) of blade 8 to the center of the cation-binding face (described below) and inserts into the core of the β-propeller, acting as a cone-shaped plug in what exists as a central cavity in other β-propellers.
A phased anomalous difference density map calculated from X-ray data collected at a wavelength of 1.25 Å showed two strong peaks (>5.0 σ) in blade 1 of the β-propeller, which were interpreted as Ca2+ ions (Figs. 1A and 2A). The presence of calcium blade motifs ((D/N)X(D/N)GDGXX(D/E)) (24) between βA and βB in blades 2–5 and 7 of the Bap1 sequence, as well as the orientation of these residues in the structural model, suggested the presence of additional ions; however, no peaks were observed for these locations in the anomalous difference maps (Fig. 2B). Analysis of the Bap1Δ57 model using the CheckMyMetal server (https://csgid.org/metal_sites),4 which analyzes the coordination geometry of ions in refined structures (25), suggested the presence of Na+ coordinated by five of the calcium blade motifs, and thus five Na+ ions were modeled into in the βA–βB loop of blades 2–5 and 7 (one Na+ per blade; Fig. 1, A and B). At this wavelength (1.25 Å), we would expect an observable anomalous signal from the Ca2+ K-edge (f″ = 0.89 e) but a much weaker signal for Na+ (f″ = 0.08 e), supporting our interpretation. Based on the similarity to other calcium blade motifs, as well as the observation that exposure of Bap1Δ57–GFPUV to EDTA causes precipitation of the protein, it is possible that the ions present in the Bap1Δ57 model play a role in structural stability, rather than a functional or enzymatic role (24).
Figure 2.
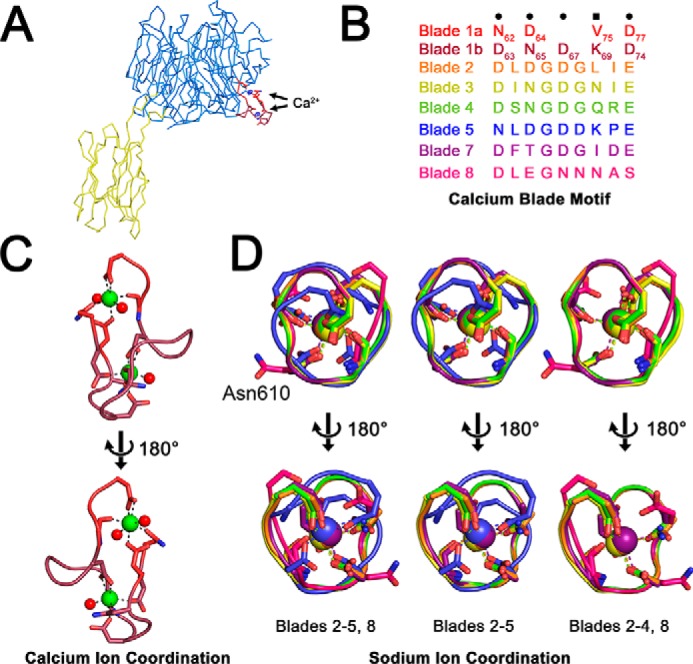
Metal-binding sites. A, phased anomalous difference map calculated at 5.0 σ shows electron density peaks for two putative calcium sites in the βA–βB loop of blade 1. No anomalous density was observed for additional five coordination sites, which were modeled as sodium atoms. Bap1 is displayed in Cα ribbon representation with β-propeller domain in marine and β-prism domain in yellow. B, amino acid sequences for individual coordination loops in the β-propeller domain. Blade 1 coordinates two Ca2+ ions via two intertwined calcium blade motifs, and the Na+ ions are coordinated by individual calcium blade motifs in propeller blades 2–5 and 7. The loop 6 site is interrupted by the β-prism domain, and blade 8 exhibits no density for a bound ion. C, close-up of putative Ca2+ coordination sites shows side chains that coordinate ions. Ca2+ ions are depicted as green spheres and water molecules as red spheres. D, superposition of remaining ion-coordination sites colored as depicted in B. The unoccupied loop in blade 8 is shown for comparison showing the flipped-out orientation of Asn-610 in magenta.
Although our structure contains both Ca2+ (Fig. 2C) and Na+ (Fig. 2D) ions, it is possible that all sites are typically occupied by calcium (24) and that physiological cations were replaced by Na+ during the protein purification process. Blade 6 (which contains the β-prism insertion) and blade 8 do not appear to bind ions. Comparing the sequence of the βA–βB loop in blade 8 with the sequences of the βA–βB loops of the five Na+-coordinating blades reveals a sequence divergence from the calcium-blade motif that could plausibly abolish cation binding (Fig. 2B). All ions in the Bap1Δ57 model are present on the same face of the β-propeller, which we therefore refer to as the cation-binding face, situated on the side of the propeller where the β-prism domain is attached (Fig. 1A). The face opposite the cation-binding face of the β-propeller contains a central pocket surrounded by several aromatic residues, with ∼800 Å2 of exposed solvent-accessible surface area (Fig. 3, A and B, area including yellow and marine surfaces). A surface electrostatic potential map does not indicate any overwhelming charge density near this pocket, whereas the cation-binding face is acidic in nature (Fig. 3C).
Figure 3.

Bap1Δ57 β-propeller domain. A, side-cutaway view of the Bap1Δ57 β-propeller domain. The C-terminal ∼45-amino acid tail folds up and fills the central pocket of the β-propeller. The surface representation was generated without the plug region. Four β-strands from a single β-propeller blade are highlighted and labeled. B, top surface representation view of the β-propeller domain shows broad cavity with side and floor residues colored marine and yellow, respectively. Aromatic residues lining the cavity are shown in stick representation. C, electrostatic potential surface generated by the APBS method (58) as implemented in PyMOL. The putative carbohydrate-binding pocket of the β-prism domain is basic as compared with the rest of the protein and is highlighted by an arrow. D, structure-based sequence alignment of the Bap1Δ57 blades generated using Swiss-PdbViewer (59). The gray-shaded area denotes the calcium/sodium-binding motifs. The secondary structure is shown above with the location of the β-prism domain noted in blade 6. Sequence alignment figure generated by ESPript 3 (60) using the % Multalin coloring scheme and a 0.5 similarity score.
A DALI search of the Bap1Δ57 structure identified β-propeller homologs of Bap1, with the lowest RMSD at 2.4 Å (26) (top 10 are shown in Fig. S1A). Proteins identified by the DALI algorithm can be organized into three major functional categories, including scaffolding proteins, enzymes, and chaperones. In addition, two homologs identified by the DALI search are lectins. The protein homologs identified by the DALI search appear to represent mainly structural similarity, rather than functional homology due to a lack of conservation in residues critical to activity. For example, YesW and YesX are bacterial lyases identified by the DALI search with RMSDs of 3.7 and 3.8 Å, respectively (Fig. S1B). These enzymes are the only bacterial proteins identified in our search that share the calcium blade motif with Bap1 and also contain eight blades. However, it is not likely that these bacterial lyases are functional homologs of Bap1 because the lyase activity of YesW and YesX relies on a deep pocket and a calcium ion located in the central channel of the β-propeller (27), neither of which are present in the Bap1Δ57 crystal structure. In addition, several seven-bladed integrin family proteins were identified via the DALI search (RMSD 4.0 Å or higher), likely due to their coordination of calcium ions via calcium blade motifs (24).
β-propeller domains are common in prokaryotes with diverse functions, including enzymatic activities. Several families of glycoside hydrolases (GH) exist with β-propeller folds, although mostly in five-bladed (GH families 43 and 62) and six-bladed (GH families 33, 34, 83, and 93) forms. These families typically contain active sites near the central axis of the β-propeller and utilize Tyr/Glu or Asp/Glu pairs/triads to catalyze hydrolysis (28, 29) (although some exceptions exist, including a seven-bladed rhamnosidase that utilizes a single histidine in the active site (30)). Inspection of the central cavity of Bap1Δ57 did not reveal Tyr/Glu or Asp/Glu pairs consistent with hydrolase or sialidase activities, and a structure-based sequence alignment (Fig. 3D) of the eight Bap1Δ57 blades did not indicate the presence of Asp-box motifs found in some glycoside hydrolase β-propeller families (31). Although the absence of these motifs does not preclude Bap1 exhibiting glycoside hydrolase activity, it suggests that if this were the case a noncanonical mechanism might be at play or that the active site may be located somewhere outside of the central cavity.
β-Propeller lectins, also called PropLecs, are another common utilization of the β-propeller fold that typically contain carbohydrate-binding sites at the interfaces between blades. An algorithm for detecting PropLec proteins was recently described resulting in a database of predicted sequences based on conserved families of PropLec domains (32). Although the method did predict a number of Vibrio PropLec proteins, Bap1 was not identified by the screen. The Bap1 structural alignment (Fig. 3D) indicates that the sequence identity between blades is quite low (5–35%), and we do not observe any conserved sequences (aside from the calcium/sodium-binding sites) or pockets between the interfaces of multiple blades. Although Bap1 may not fit into the profile for a typical PropLec β-propeller protein, this does not preclude that additional carbohydrate-binding sites may be present.
Bap1Δ57 β-prism domain
The Bap1Δ57 β-prism domain falls into the jacalin-related lectin (JRL) protein family, which consists of a pseudo-3-fold arrangement of Greek key motifs. Most JRLs bind carbohydrates in only one sugar site, located at the top of Greek key I, but three previously characterized β-prism I domains expressed by V. cholerae (two from RbmC and one from VCC) bind their carbohydrate ligands at a single site at the top of Greek key II (23, 33).
The β-prism domain of Bap1Δ57 is located sequentially between βA and βB (β22 and β35* in context of the full structure) of blade 6 within the β-propeller domain. The Bap1Δ57 β-prism domain adopts a canonical β-prism I architecture, made up of 12 β-strands arranged into three antiparallel β-sheets with Greek key folds. The surface of the Bap1Δ57 β-prism domain opposite its attachment to the β-propeller makes up the region that constitutes a carbohydrate-binding site in the other three β-prisms expressed by V. cholerae: RbmCβ-prism 1, RbmCβ-prism 2, and VCCβ-prism (23, 33). In the Bap1Δ57 structure, this end of the β-prism features a positively-charged, lysine-rich surface with a central cavity not found in the other V. cholerae β-prism I lectin domains (Fig. 4A). A bound citrate molecule is situated on one side of this lysine-rich groove (Fig. 4A). Because citrate was required to obtain the optimal crystal form and because this molecule is located in an area with extensive crystal contacts, it is possible that its interaction with the Bap1Δ57 β-prism is an artifact of crystallization. However, it is curious that the citrate molecule is found occupying the same site as the primary mannose in the RbmCβ-prisms 1, 2 and VCCβ-prism structures (Fig. 4, B and C) (23, 33). The citrate molecule is coordinated by hydrogen bonding through backbone amines of Gly-344, Ala-345, and Val-346, and the terminal amine of the Lys-501 side chain, and by van der Waals interactions with Asp-348 and His-500 (Fig. 4D). The citrate coordination pattern is of interest because all of these residues, except His-500 (which is not present in the other β-prisms, as described below), are structurally homologous to those identified in carbohydrate binding by the crystal structures of RbmCβ-prism 2 and VCCβ-prism (Fig. S2) (23, 33).
Figure 4.
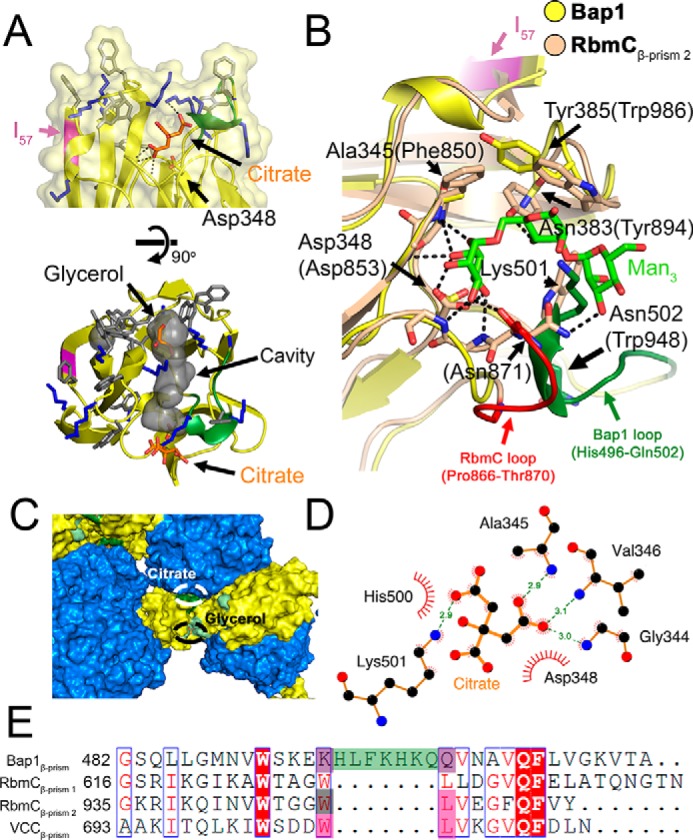
Structural details of the Bap1Δ57 β-prism domain. A, side and top close-up view of the putative carbohydrate-binding pocket of the Bap1Δ57 β-prism domain. Aromatic and lysine residues are shown in gray and blue stick representation, respectively. A citrate molecule from the crystallization buffer was found in close proximity to Asp-348 with putative hydrogen bonding interactions highlighted by dotted lines. A short seven-residue loop unique to Bap1 is shown in green, and the location of the 57-amino acid insertion is shown in magenta. An extended linear cavity is outlined in gray space-surface representation (determined using the cavities and pockets feature of PyMOL with a 7-Å cavity detection radius and a three-solvent radius cavity detection cutoff). B, superposition of the Bap1Δ57 (yellow) β-prism domain and RbmC2 (wheat) β-prism domain with bound mannotriose (green stick representation). Key residues involved in RbmC2/mannotriose interactions are shown as dotted lines (23). Short insertion loops unique to RbmC2 (red) and Bap1 (green) occupy similar locations. Key Bap1 residues are indicated by arrows with RbmC2 residue numbers in parentheses. C, a citrate molecule is located in a region with several crystal contacts (β-propeller domain is colored marine; β-prism domain is colored yellow; and residues involved in citrate coordination are colored dark green). D, schematic shows polar and nonpolar interactions between citrate molecule and the Bap1 β-prism domain. At the crystallization pH (5.5), the citrate molecule is expected to be at least partially protonated, allowing Asp-348 to make additional hydrogen-bonding interactions with a citrate carboxylate group. Putative hydrogen-bonding interactions are shown as green dotted lines and van der Waals interactions as red arcs. Data were generated using LigPlot+ version 2.1 (61). E, sequence alignment shows a key region of the Bap1, RbmCβ-prism 1, RbmCβ-prism 2, and VCCβ-prism domains. The Bap1 7-amino acid insertion is highlighted in green. Sequence alignment was generated using the MUSCLE algorithm implemented in MEGA version 7.0 (62) and ESPript 3 (60).
A conserved aspartic acid residue (Asp-348 in Bap1β-prism, Asp-539 in RbmCβ-prism 1, Asp-853 in RbmCβ-prism 2, and Asp-617 in VCCβ-prism) plays a crucial role in the sugar-binding activity of RbmCβ-prism 1, RbmCβ-prism 2, and VCCβ-prism (23, 33) through hydrogen-bonding interactions. Mutation of this aspartate residue position to alanine results in a >50-fold loss of hemolytic activity in VCC and a loss of measurable binding affinity for mannotriose and asialofetuin (which contains target N-glycans) in RbmCβ-prism 2 (23). The citrate forms putative hydrogen-bonding interactions with Asp-348, the key residue involved in carbohydrate binding by other Vibrio β-prism domains.
The citrate molecule present in the Bap1Δ57 structure binds in the carbohydrate-binding site found in the other V. cholerae β-prism domains, and further comparison of the four V. cholerae β-prism domains presents a perplexing conundrum as it appears that the Bap1Δ57 β-prism should be capable of binding carbohydrates in a manner similar to its homologs, yet experimental evidence suggests it does not. The most significant structural divergence of Bap1β-prism from the other V. cholerae β-prism lectins lies in the β11–β12 loop, which contains a seven-amino acid loop insertion between β11 and β12 (β33* and β34* in context of Bap1Δ57) (Fig. 4, B and E). Whereas sequence alignments suggest that Trp-629/948/706 (RbmCβ-prism 1/RbmCβ-prism 2/VCCβ-prism) and Leu-630/949/707 are substituted in Bap1 by Lys-495 and Gln-503 (respectively) with the seven-amino acid loop between them (Fig. 4E), structural alignment shows that Lys-501 and Gln-503 of Bap1 are present in the spatial location of the WL motif and should, in theory, be able to accommodate binding to mannose or the N-glycan pentasaccharide core, especially considering that Lys-501 forms a hydrogen bond with the citrate molecule in the Bap1Δ57 structure (Fig. 4D). An additional major difference between the Bap1Δ57 β-prism and the other V. cholerae β-prism domains is with regard to surface electrostatics. The surface of the Bap1Δ57 β-prism near the homologous carbohydrate-binding site has a lysine-rich and positively charged groove (Figs. 3C and 4A). Although this region could reasonably facilitate binding of Bap1 to anionic polysaccharides or surfaces, it cannot explain why Bap1Δ57 does not bind carbohydrates in a fashion similar to its homologs or why it does not colocalize with VPS at the surface of the biofilm (20).
57-Amino acid insertion in the Bap1 β-prism domain
Bap1 has been shown to contribute to the hydrophobicity of V. cholerae biofilms, specifically within pellicles formed at the air–water interface (34). In the absence of Bap1, V. cholerae biofilm pellicles have decreased elasticity and are more hydrophilic than WT rugose biofilms (34). Whereas Bap1 and RbmC have been shown to play somewhat redundant roles in the V. cholerae biofilm matrix (9, 20, 35), the hydrophobic nature of WT V. cholerae pellicles represents an instance where RbmC cannot rescue the absence of Bap1. The failure of RbmC to rescue the hydrophobic nature of the pellicle formed by a bap1 deletion mutant suggests that some component of Bap1 that is absent in RbmC contributes significantly to the hydrophobicity of the V. cholerae biofilm pellicle. The 57-amino acid insertion is the lone region of Bap1 absent in RbmC and exhibits traits that could aid in maintaining the hydrophobicity of the pellicle (Fig. S3A). Secondary structure prediction using the JPred server suggests the 57-amino acid insertion adopts mainly β-sheet structure, and multiple amyloid prediction algorithms suggest that it may also exhibit amyloidogenic propensities (Fig. 5A) (36–42). The 57-amino acid insertion occurs at the N-terminal end of the 10th β-strand in the β-prism domain (β32* in context of the full structure, Fig. 1B). Residues of this β-strand as well as the loop connecting the 9th and 10th β-strands are not involved in carbohydrate binding in any known β-prism I homologs. Therefore, the removal of this insertion from Bap1 is not expected to alter any potential lectin activity of the Bap1Δ57 β-prism domain. This is not to say, however, that removal of the insertion does not impede other functional roles played by Bap1 in the V. cholerae biofilm matrix.
Figure 5.
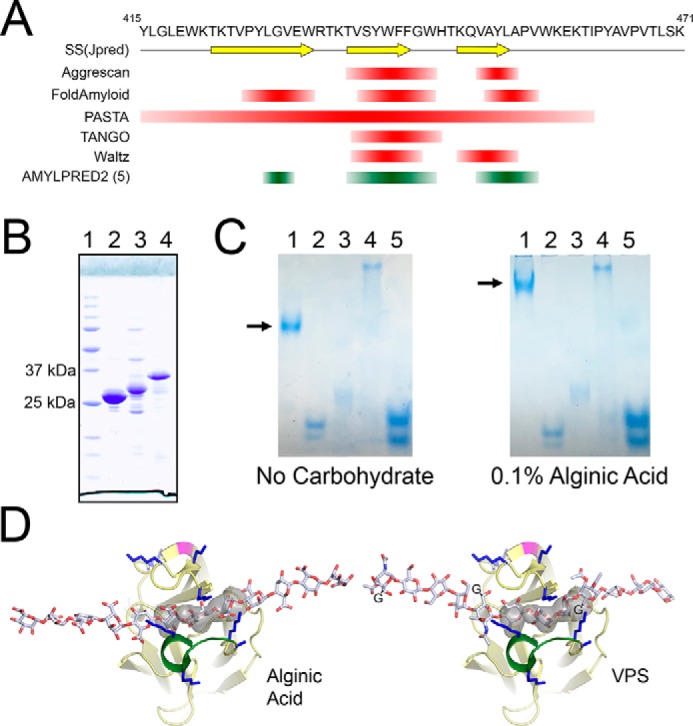
Bap1 β-prism domain exhibits properties from RbmC and VCC β-prism domains. A, 57-amino acid insertion within the Bap1 β-prism is predicted to exhibit amyloid-like properties. The single-letter amino acid sequence is shown with the secondary structure prediction using Jpred4 (42) below (arrows denote β-strands). Amyloid prediction was performed on the Bap1 57-amino acid insertion sequence using a number of individual amyloid-prediction algorithms. Results are shown from Aggrescan (37), FoldAmyloid (38), PASTA (41), TANGO (36), and Waltz (39). The five out of 10 method consensus sequence generated by AMYLPRED2 (40) (http://aias.biol.uoa.gr/AMYLPRED2/)4 is shown in green. B, expression of GFPUV fused to the 57-aa insertion results in insoluble material. (Lanes are as follows: 1, protein standards; 2, Ni-purified GFPUV alone; 3, Ni-purified Insertion57–GFPUV fusion from cleared cell supernatant; 4, Ni-purified cell pellets solubilized and purified in 8 m urea.) The Ni-purified soluble fraction resulted in a truncated protein species presumably missing the 57-aa insertion. C, gel-shift assay suggests Bap1Δ57 interacts with anionic polysaccharides. Native PAGE gel-shift assay provides evidence that Bap1Δ57 β-propeller domain, but not β-prism domain, interacts with alginic acid (Lanes are as follows: 1, Bap1Δ57; 2, Bap1β-propeller; 3, BSA; 4, concanavalin A; 5, GFPUV.) Arrow illustrates the band shift of Bap1Δ57. D, figure shows modeled alginic acid and VPS polysaccharides manually docked into the β-prism cavity. The glycine adducts on the VPS guluronic acid residues were not included in the all-atom model but are represented by the letter G.
Bap1, including the 57-aa insertion, can be expressed in an E. coli system as a GFPUV-fusion; however, all the recombinant protein product is found in the insoluble cell lysate pellet. Conversely, removal of the insertion in the Bap1 β-prism domain in Bap1Δ57 alleviates this solubility issue and yields abundant protein in the cell lysate supernatant fraction. In an attempt to produce the 57-aa insertion peptide for further analysis, we created a cleavable GFPUV fusion (Insertion57–GFPUV) and expressed this construct in E. coli. As we observed with the full-length Bap1 construct, this new construct was completely insoluble, with only truncated GFPUV material missing the fusion insertion present in the cell supernatant (Fig. 5B). The insoluble inclusion body pellet could be solubilized in 8 m urea and purified by nickel-nitrilotriacetic acid chromatography, but attempts at refolding the fusion on the column (by slowly reducing the urea concentration), by dilution, or by dialysis failed to produce soluble material. This suggests two possibilities: the insertion renders the full-length protein (whether Bap1 or Insertion57–GFPUV) insoluble, or an interaction between the insertion and some component of Bap1 or GFPUV results in cross-linked aggregation or misfolding of the protein. Of course, Bap1 is secreted functionally from V. cholerae where the protein is presumably directed through a secretory pathway, which might also involve chaperones to assist with folding or solubility. Evaluation using Kyte-Doolittle hydropathy values indicates that 63.7% of the residues in the insertion display a hydrophobic nature (Fig. S3B) (43), which may support the former hypothesis. However, the possibility of this insertion having amyloidogenic propensities could support the latter hypothesis (Fig. 5A). It is plausible that the insertion acts to help attach the V. cholerae biofilm matrix to hydrophobic surfaces (19), such as the lipid membrane of a cell, or that amyloid formation within the insertion might aid in biofilm formation as seen in biofilms formed by other bacteria (44). A strongly hydrophobic or amyloidogenic subdomain present in Bap1 could explain its localization to the attachment surface observed in fluorescence microscopy studies on V. cholerae biofilm architecture (9, 20, 35).
Functional analysis of Bap1Δ57 suggests putative lectin activity
Based on super-resolution microscopy of developing biofilms, Bap1 plays a role in cell-surface adhesion and formation of dynamic cell-encasing envelopes with VPS and RbmC (9). In our investigation of the putative sugar-binding activity of Bap1, we determined that unlike the β-prism domains present on RbmC and VCC (23), fluorescently-labeled Bap1Δ57 did not appear to bind complex mammalian sugars present on the Mammalian Glycan Screen version 5.3 (Fig. S4). Furthermore, analysis using isothermal titration calorimetry (ITC) did not reveal binding activity of Bap1Δ57 constructs to the complex glycosylated protein asialofetuin or monosaccharides (l-guluronic acid, d-galactose, or d-glucose) present in VPS (Fig. S5A).
To test the possibility of Bap1Δ57 binding to linear polysaccharides like VPS, we employed a native gel-shift experiment using substrates similar to approaches used with extracellular polysaccharide-interacting proteins from Pseudomonas aeruginosa (45). For polysaccharides, we used alginic acid, bacterial alginate, dextran, 2-hydroxyethylcellulose, and xanthan gum. As controls, we included several proteins with compatible electrophoretic mobilities and varying molecular masses, including BSA, concanavalin A, and GFPUV. Our prior experience demonstrates that the RbmC β-prism domain demonstrates some affinity for dextran and dextran-containing purification resins, which contain α1,3- and α1,6-branches like those found in core N-glycans (23).
Native polyacrylamide gel–shift assays reveal association of Bap1Δ57 but not Bap1β-propeller to alginic acid (Fig. 5C). Polysaccharide binding by Bap1β-prism could not be analyzed by native polyacrylamide gel shift as its pI is above that of the gel buffering system. In addition, Bap1Δ57 displayed a gel shift with xanthan gum in the native polyacrylamide gel shift assay, but not with dextran (Fig. S5B). Taken together, these data suggest that unlike RbmC, Bap1Δ57 does not bind complex mammalian N-glycans, but rather displays an affinity for anionic polysaccharides or polysaccharides with a linear backbone, as both alginic acid and xanthan gum are negatively charged. This is particularly interesting as VPS present in V. cholerae biofilms is an acetylated linear polysaccharide with a glycine adduct (with a free carboxyl group) linked to guluronic acid (11, 12, 46).
To test whether a linear polysaccharide could occupy the Bap1Δ57 β-prism cavity, models for alginic acid and VPS were prepared using CarbBuilder version 2.1.30 (47) and manually docked into the cavity density calculated by PyMOL. Although the exact register and orientation of the ligands may vary, the width of the polysaccharides is comparable with the size of the cavity (Fig. 5D). In RbmC, we observe flexibility in this region in the presence and absence of ligands (23), and in Bap1 this region is near a crystal contact that may influence its unliganded conformation. These considerations might provide additional flexibility in the size of the cavity, particularly when side-chain rearrangements are allowed. An attractive feature of this model is that acetyl groups on polysaccharides would be in close proximity to lysine groups surrounding the β-prism cavity potentially forming polar interactions. The VPS glycine carboxyl on the guluronic acid moiety could additionally form a salt bridge with nearby lysine residues, further strengthening this interaction.
RbmC model based on Bap1Δ57 structure
To gain better insight into the comparative structural features of RbmC and Bap1, we constructed a model for RbmC based on available structural and sequence-based information (the comparative domain organization is shown in Fig. 6A). The β-propeller domains of Bap1 and RbmC are 67.5% identical in sequence, suggesting a high degree of structural similarity. For our model, we attached the previously determined RbmCβ-prism 2 lectin domain (23) to the C terminus and tandem βγ-crystallin domains from E. coli StcE (22) to the N terminus of our Bap1Δ57 model (Fig. 6B). The linker lengths between domains were modeled using the database sequence for RbmC and are assumed to be flexible similar to the linkers connecting the β-prism and β-propeller domains in Bap1Δ57. Based on the linker distance constraints, it is likely that both StcE domains and both β-prism domains protrude from the same cation-binding face of the β-propeller domain in RbmC. Furthermore, the carbohydrate-binding pockets of RbmCβ-prism 1 and RbmCβ-prism 2 are likely oriented in the same outward position on the bottom face of the RbmC molecule (Fig. 6B). Because both β-prism domains in RbmC bind similar N-glycan core structures (23) commonly found on cell-surface proteins with low nanomolar affinity, the possibility exists for strong avidity effects from polyvalent binding to cell-surface ligands. The integration of two N-glycan–binding sites, two putative StcE domain–binding targets (22), and a possible β-propeller ligand suggests that RbmC could serve to bridge multiple biofilm matrix components to target cell surfaces. Bap1, however, might form an overlapping but more limited scaffold for a different subset of matrix and surface ligands.
Figure 6.
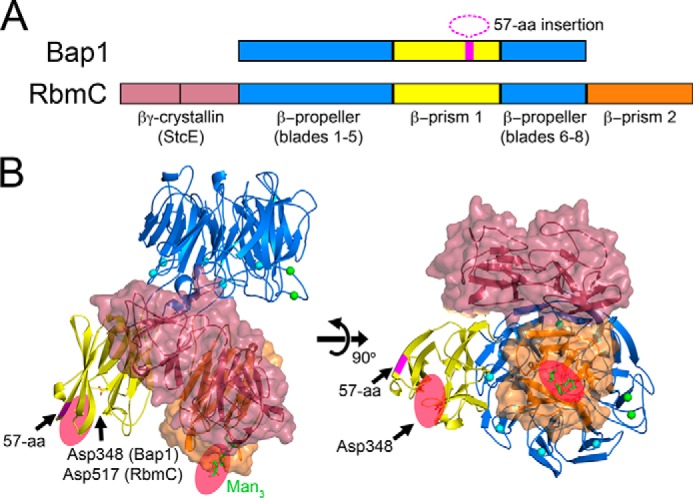
Structural model of RbmC. A, RbmC shares a core β-propeller and β-prism domain with Bap1 and additionally contains a second C-terminal β-prism domain and two N-terminal domains with a predicted βγ-crystallin fold. The 57-amino acid insertion within the Bap1 β-prism domain (magenta) is absent in RbmC. B, proposed structural model for RbmC made by attaching two βγ-crystallin domains from the StcE protein (PDB 3UJZ) to the N terminus, and the C-terminal β-prism domain from RbmC to the C terminus (PDB 5V6F). The model suggests general locations of the accessory RbmC domains in relation to β-propeller, rather than absolute orientation. The sequence-predicted linker lengths between domains were maintained to illustrate that all four accessory domains in RbmC likely occupy the bottom face of the β-propeller domain. Our model also suggests that the two N-glycan–binding pockets of RbmC are also likely on the same face of the molecule (shown as red ovals).
Discussion
We report the 1.9 Å crystal structure of the V. cholerae biofilm matrix protein Bap1Δ57 and demonstrate that the Bap1Δ57 β-prism domain displays a sugar-binding profile that differs from other V. cholerae β-prism domains (RbmC and VCC), with putative specificity for anionic polysaccharides and/or polysaccharides with a linear backbone. Additionally, we suggest that a 57-amino acid insertion that was deleted for E. coli expression might modulate the solubility of Bap1 leading to the surface deposition (9) and hydrophobic properties of biofilms observed in functional studies (34). This might help to explain the observation that surface-attached Bap1 appears to grow radially outward from founder cells (9) as presumably newly secreted Bap1 molecules deposit onto the surface or grow outward through interactions mediated by the 57-aa insertion. Consistent with published microscopy studies, the β-prism domains of Bap1 and RbmC would be primarily involved in adhesive interactions between the biofilm matrix and environmental surfaces, whereas the β-propeller domains might form interactions with protein or other substrates within the biofilm matrix, including other matrix proteins and VPS. The Bap1 β-prism domain may also interact with VPS, or other charged polysaccharides found in the marine environment (like alginates). Whereas RbmC and Bap1 are both sufficient on their own to support biofilm attachment in vitro (20), specific and potentially strong adhesive interactions mediated by β-prism domains might play a more important role on diverse surfaces encountered within the V. cholerae lifecycle.
In the aquatic niche, the ability of Bap1 to bind anionic polysaccharides or abiotic surfaces (via the β-prism insertion) would provide a survival advantage by promoting attachment to a multitude of substrates, including extracellular polysaccharides found on phyto- and zooplankton or macroflora such as macroalgae. In addition, the increased elasticity provided to the biofilm matrix by Bap1 may confer increased tensile strength that aids in survival in environments where dynamic movement (such as ocean currents) is abundant (34). Furthermore, the putative hydrophobic and/or amyloidogenic nature of the 57-amino acid insertion in the Bap1 β-prism may play a role in attachment to hydrophobic or other abiotic surfaces. The putative lectin activity of Bap1Δ57 presented here and the contribution of Bap1 to the hydrophobic nature of V. cholerae biofilms (34) support our hypothesis of a dual role played by Bap1 in attaching V. cholerae biofilms to both biotic and abiotic surfaces in the aquatic niche.
In the host gut environment, we propose that RbmC plays a dominant role by binding complex N-glycans (23). Thus, in effect, ingested biofilm fragments decorated with RbmC could be “captured” by ligands on epithelial cell surfaces. This role may be aided by the N-terminal βγ-crystallin repeat domain, as the homologous domain in EHEC O157:H7 mucinase, StcE, has been shown to play a role in binding to cell surfaces (22). In support of this hypothesis, a study performed by Liu et al. (48) provides experimental evidence of interaction between mucin and V. cholerae biofilms.
Additional investigation of the unique specificity of lectin activity displayed by Bap1 and RbmC is needed. Because of the lack of hits in the mammalian glycan screen and apparent preference for linear and/or anionic polysaccharides, it is not likely that Bap1 plays a role in penetration of the mucosal layer of the mammalian gut. However, it is crucial to further investigate the polysaccharide specificity of Bap1 in the context of its interactions with other matrix components. These studies in conjunction with structures of Bap1 complexed with carbohydrate ligands will provide a framework for understanding the network of complex molecular interactions that underlie biofilm assembly and adhesion in V. cholerae.
Experimental procedures
Cloning, expression, and protein purification
Bap1 (VC_1888, NP_231522.1) constructs were generated by amplification of fragments from V. cholerae (strain N16961) genomic DNA via PCR stitching and insertion into pNGFP-BC (49) using NcoI and XhoI restriction sites to yield N-terminal, His-tagged GFPUV fusions, cleavable by thrombin or trypsin. The Bap1Δ57 construct (residues 25–414 and 473–691) results from removal of the predicted secretion signal (residues 1–24) and deletion of the 57-amino acid insertion in its β-prism domain. Bap1β-propeller–GFPUV contains the β-propeller domain alone (residues 25–316 and 515–691). Bap1β-prism–GFPUV contains the version of the β-prism domain also present in the Bap1Δ57–GFPUV construct (residues 316–414 and 473–514). Insertion57–GFPUV contains the 57-aa insertion (residues 415–472) cloned to the C terminus of GFPUV, separated by a poly-asparagine linker and thrombin site. Because of cloning artifacts, the sequence preceding Tyr-415 is SAMAFT (following the LVPRG thrombin site).
GFPUV fusion constructs were transformed into T7Express E. coli (New England Biolabs) or BL21-CodonPlus (DE3)-RIL (Agilent) E. coli (for Insertion57–GFPUV), and overnight cultures were diluted 20-fold into fresh LB-Miller media supplemented with 50 μg/ml carbenicillin. Expression cultures were subsequently grown with shaking (210 rpm) at 37 °C until reaching an A600 of 0.5 to 0.7, at which point the cells were induced with 1 mm isopropyl β-d-1-thiogalactopyranoside (IPTG). Following induction with IPTG, Bap1Δ57–GFPUV and Bap1β-propeller–GFPUV were grown for an additional 18 h at 18 °C, while Bap1β-prism–GFPUV was grown for an additional 4 h at 30 °C. Following expression, cells were pelleted by centrifugation in a Sorvall LYNX 6000 centrifuge using an F9-6 × 1000 rotor (Thermo Fisher Scientific) at 3,900 × g for 12 min at 4 °C. Cell pellets were resuspended in 1× TBS (20 mm Tris, pH 7.5, 150 mm NaCl) to a final volume of ∼10 ml per pellet from 1 liter of expression growth. Samples were stored at −80 °C until the time of purification. Thawed cell cultures were supplemented with protease inhibitor mixture (Roche Applied Science) and 10 mm imidazole. Cell lysis was performed via passage three times through an EmulsiFlex-C5 high pressure homogenizer (Avestin, Inc.) at ∼18,000 p.s.i. The cell lysate was cleared via centrifugation in a Sorvall LYNX 6000, at 40,000 × g for 25 min using an F20-12×50 rotor (Thermo Fisher Scientific).
Recombinant Bap1Δ57–GFPUV was purified from the cleared supernatant by a combination of nickel-affinity (5-ml HisTrap HF, GE Healthcare) and desalting (50-ml Bio-Scale Mini Bio-Gel, Bio-Rad) chromatography columns on a Profinia Protein Purification System (Bio-Rad). After sample loading, the nickel column was washed with 2 column volumes (CV) of 1× TBS and 5 CV of 1× TBS plus 40 mm imidazole. Bap1Δ57-GFPUV was eluted from the nickel column using 1× TBS supplemented with 250 mm imidazole and desalted into 30 ml of ion-exchange Buffer A (IEX Buffer A: 20 mm Tris, pH 7.5, 50 mm NaCl). GFPUV was cleaved from the fusion protein by incubating with a 1:1000 w/w ratio of human α-thrombin (Hematologic Technologies, Inc.) at room temperature for 1.5 h. The cleavage reaction was stopped using 1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride. Cleaved Bap1Δ57 was separated from its His-tagged GFPUV fusion partner via anionic exchange on a 5-ml Q-column HF (GE Healthcare) equilibrated in IEX Buffer A. GFPUV was eluted from the Q-column in 20 mm Tris, pH 7.5, 200 mm NaCl, and Bap1Δ57 eluted in 60 ml of 20 mm Tris, pH 7.5, 250 mm NaCl. Eluted Bap1Δ57 was concentrated in a 30-kDa cutoff Vivaspin20 centrifugal filter (GE Healthcare) and buffer exchanged by passage over a Superdex 200 Increase 10/300 GL (s200i, GE Healthcare) gel-filtration column in 1× TBS.
Bap1β-propeller–GFPUV was purified from cleared cell lysate in a similar fashion as Bap1Δ57–GFPUV, desalted into 1× TBS, and cleaved using a 1:1000 w/w ratio of trypsin (Sigma) at room temperature for 1.5 h. Cleaved Bap1β-propeller was separated from the His-tagged GFPUV by tandem nickel-affinity and gel-filtration chromatography, with a 1-ml HisTrap HF column (GE Healthcare) attached to the top of the s200i gel-filtration column, and eluted in a buffer appropriate for downstream applications. Bap1β-prism–GFPUV was purified from the cleared cell lysate by nickel-affinity chromatography using Toyopearl 650 M AF-chelate resin (Tosoh Biosciences) charged with nickel sulfate, using the procedure described above for the HisTrap HF column. Bap1β-prism–GFPUV eluted from the Toyopearl chelate resin was cleaved from the fusion protein by a 1:500 w/w trypsin digest carried out at room temperature for 1.5 h. Bap1β-prism was separated from GFPUV by passage over an s200i gel-filtration column. Purity of all constructs was determined by SDS-PAGE.
Crystallization of Bap1 constructs
Bap1Δ57 was purified and separated from its GFPUV fusion partner as described above and concentrated to 7.5 mg/ml for crystallization studies. Crystallization screening and optimization were performed using commercial sparse matrix and grid screening by the hanging-drop method in 24-well VDX plates (Hampton Research). Native crystals were grown by pipetting a 1:1 v/v ratio of protein sample and precipitant solution (0.1 m sodium citrate and 10% PEG 3,350) onto siliconized coverslips and suspending them over 0.5 ml of reservoir solution. Crystals were harvested after approximately 2 weeks. Native crystals were cryoprotected in mother liquor containing 20% glycerol, and flash-cooled in liquid nitrogen. Heavy atom derivatization was achieved by soaking native crystals in a drop of mother liquor with 10 mm K2Cl4Pt for 3 min, followed by back soaking in mother liquor plus 20% glycerol, without the heavy atom for ∼30 s, and flash-cooling in liquid nitrogen.
Bap1β-propeller domain crystals were grown via hanging-drop vapor diffusion with a 1:1 v/v ratio of 8.75 mg/ml protein and precipitant solution (0.2 m BisTris, pH 7.0, 15% PEG 8,000, and 20% glycerol) at either 17 or 25 °C. Crystals were harvested and flash-cooled in liquid nitrogen, with no additional cryoprotectant as the precipitant solution contained 20% glycerol.
X-ray structure determination of Bap1Δ57
X-ray data were collected at the NSLS-II 17-ID-1 (AMX) beamline at Brookhaven National Laboratory equipped with a Dectris Eiger X 9 M pixel-array detector. The structure of Bap1Δ57 was solved via single isomorphous replacement with anomalous signal with native crystals soaked in K2Cl4Pt. Data were indexed using XDS (50) and scaled with Aimless (51). The structure was phased by SIRAS using native and K2Cl4Pt derivative data using the AutoSol module of Phenix (52). Density modification (∼60% solvent) and automatic model building by Phenix using data to 1.9 Å led to the placement of 538 residues (of 608 expected residues in the construct) with a map-model correlation coefficient of 0.84 and Rwork and Rfree values of 0.25 and 0.27, respectively. Automatic model rebuilding was carried out using ARP/wARP version 7.6 (53) resulting in a new model with 600 residues and Rwork and Rfree values of 0.193 and 0.239, respectively. The final model was refined using phenix.refine (54) utilizing automatic water-picking and target weight optimization algorithms as implemented in Phenix (54). Refinement progress was monitored by tracking the Rwork/Rfree ratio (with Rfree representing 5% of total reflections). Iterative model rebuilding (into 2Fo-Fc and Fo-Fc maps) and comprehensive validation was carried out with Coot (55) and the Phenix implementation of MolProbity (56) to final Rwork and Rfree values of 0.158 and 0.173, respectively.
Mammalian glycan array
Bap1Δ57-GFPUV was purified as described above and eluted from the s200i column in 0.1 m sodium bicarbonate, pH 8.3. Purified protein was concentrated in a 30-kDa cutoff Vivaspin6 (GE Healthcare) to 2.1 mg/ml and fluorescently labeled using a Molecular Probes AlexaFluor488 labeling kit (Life Technologies, Inc.). The labeled protein sample was sent to the Consortium for Functional Glycomics (www.functionalglycomics.org)4 for analysis, as described previously (23), at concentrations of 5 and 50 μg/ml.
Polysaccharide interaction by native acrylamide gel electrophoresis
Bap1Δ57 and Bap1β-propeller were purified as described, with final elution from gel filtration in 50 mm imidazole, pH 7.0, 50 mm NaCl. Lyophilized BSA and concanavalin A (Thermo Fisher Scientific) were prepared as controls by dissolving in 50 mm imidazole, pH 7.0, 50 mm NaCl. GFPUV was expressed in T7 SHuffleExpress (New England Biolabs) and purified by Ni-affinity and gel-filtration chromatography using conventional methods, with a final elution in 1× TBS. 6 μg per lane of the appropriate protein sample was used in gel-shift analysis. Gels were prepared with final concentrations of 7.5% bisacrylamide (Bio-Rad), 25 mm Tris, pH 8.3 (Thermo Fisher Scientific), 250 mm glycine (Research Products International), 0.05% ammonium persulfate (Thermo Fisher Scientific), and 0.025% TEMED (Bio-Rad). Running buffer consisted of 25 mm Tris, pH 8.5, and 192 mm glycine. Gel electrophoresis was carried out at 200 V for 120 min at room temperature. After electrophoresis, gels were fixed in 50% methanol, 10% acetic acid for 15 min, stained in 10% acetic acid, 0.02% Coomassie Brilliant Blue G-250 for 1 h or overnight, and then destained in 10% acetic acid.
Isothermal titration calorimetry
ITC data were collected using a Nano ITC calorimeter (TA Instruments) at 25 °C. Bap1Δ57 protein in 1× TBS buffer was loaded into the chamber (170 μl) at a concentration of ∼0.1 mm. Fifteen injections of 3 μl each were made for ligands dissolved in 1× TBS buffer (0.1 mm alginic acid sodium salt, 0.03 mm asialofetuin, 10 mm l-guluronic acid, 20 mm d-galactose, 5 mm d-glucose). Raw thermograms were baseline corrected by NITPIC version 1.2.7 (57) and displayed using GUSSI version 1.4.2 (downloaded from http://biophysics.swmed.edu/MBR/software.html).
Author contributions
K. K. and R. O. formal analysis; K. K., A. B., E. C., J. L., C. V., and R. O. investigation; K. K., E. C., and R. O. methodology; K. K. writing-original draft; R. O. conceptualization; R. O. resources; R. O. data curation; R. O. software; R. O. supervision; R. O. funding acquisition; R. O. validation; R. O. visualization; R. O. project administration; R. O. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Dr. Bing Hao at the University of Connecticut Health Center and Dr. Victoria Robinson at the University of Connecticut, Storrs, for allowing use of their home X-ray sources. Additional thanks to Prof. Dr. Markus Wahl and Dr. Ursula Neu for support during manuscript revisions. High-resolution X-ray data were collected at the 17-ID-1 (AMX) beamline of the National Synchrotron Light Source II, a United States Department of Energy Office (DOE) of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. We thank NSLS-II beamline staff personnel Dr. Jean Jakoncic and Dr. Alexei Soares for assistance in X-ray data collection and processing. V. cholerae N16961 DNA was provided by Dr. Erika Taylor. Glycan data were obtained from the Protein-Glycan Interaction Resource of the CFG (National Institutes of Health Supporting Grant R24 GM098791) and the National Center for Functional Glycomics (NCFG) at Beth Israel Deaconess Medical Center, Harvard Medical School (National Institutes of Health Supporting Grant P41 GM103694).
This work was also supported by NIH Training Grant T32 GM008271 in molecular biophysics (to K. K.).
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S5 and supporting Refs. 1–3.
The atomic coordinates and structure factors (code 6MLT) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- VPS
- Vibrio polysaccharide
- RMSD
- root mean square deviation
- aa
- amino acid
- PDB
- Protein Data Bank
- SIRAS
- single isomorphous replacement with anomalous scattering
- VCC
- V. cholerae cytolysin
- GH
- glycoside hydrolase
- JRL
- jacalin-related lectin
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- TEMED
- N,N,N′,N′-tetramethylethylenediamine
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- CV
- column volume
- ITC
- isothermal titration calorimetry
- IEX
- ion-exchange.
References
- 1. Tamayo R., Patimalla B., and Camilli A. (2010) Growth in a biofilm induces a hyperinfectious phenotype in Vibrio cholerae. Infect. Immun. 78, 3560–3569 10.1128/IAI.00048-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huq A., Grim C. J., and Colwell R. R. (2011) in Epidemiological and Molecular Aspects on Cholera (Ramamurthy T. and Bhattacharya S. K. eds) pp. 311–339, Springer, New York [Google Scholar]
- 3. Teschler J. K., Zamorano-Sánchez D., Utada A. S., Warner C. J., Wong G. C., Linington R. G., and Yildiz F. H. (2015) Living in the matrix: assembly and control of Vibrio cholerae biofilms. Nat. Rev. Microbiol. 13, 255–268 10.1038/nrmicro3433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miller C. J., Drasar B. S., and Feachem R. G. (1984) Response of toxigenic Vibrio cholerae 01 to physico-chemical stresses in aquatic environments. J. Hyg. 93, 475–495 10.1017/S0022172400065074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huq A., Small E. B., West P. A., Huq M. I., Rahman R., and Colwell R. R. (1983) Ecological relationships between Vibrio cholerae and planktonic crustacean copepods. Appl. Environ. Microbiol. 45, 275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tamplin M. L., Gauzens A. L., Huq A., Sack D. A., and Colwell R. R. (1990) Attachment of Vibrio cholerae serogroup O1 to zooplankton and phytoplankton of Bangladesh waters. Appl. Environ. Microbiol. 56, 1977–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Islam M. S., Jahid M. I., Rahman M. M., Rahman M. Z., Islam M. S., Kabir M. S., Sack D. A., and Schoolnik G. K. (2007) Biofilm acts as a microenvironment for plankton-associated Vibrio cholerae in the aquatic environment of Bangladesh. Microbiol. Immunol. 51, 369–379 10.1111/j.1348-0421.2007.tb03924.x [DOI] [PubMed] [Google Scholar]
- 8. Conner J. G., Teschler J. K., Jones C. J., and Yildiz F. H. (2016) Staying alive: Vibrio cholerae's cycle of environmental survival, transmission, and dissemination. Microbiol. Spectr. 4, 593–633 10.1128/microbiolspec.VMBF-0015-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berk V., Fong J. C., Dempsey G. T., Develioglu O. N., Zhuang X., Liphardt J., Yildiz F. H., and Chu S. (2012) Molecular architecture and assembly principles of Vibrio cholerae biofilms. Science 337, 236–239 10.1126/science.1222981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fong J. C., Syed K. A., Klose K. E., and Yildiz F. H. (2010) Role of Vibrio polysaccharide (vps) genes in VPS production, biofilm formation and Vibrio cholerae pathogenesis. Microbiology 156, 2757–2769 10.1099/mic.0.040196-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yildiz F., Fong J., Sadovskaya I., Grard T., and Vinogradov E. (2014) Structural characterization of the extracellular polysaccharide from Vibrio cholerae O1 El-Tor. PLoS ONE 9, e86751 10.1371/journal.pone.0086751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reichhardt C., Fong J. C., Yildiz F., and Cegelski L. (2015) Characterization of the Vibrio cholerae extracellular matrix: a top-down solid-state NMR approach. Biochim. Biophys. Acta 1848, 378–383 10.1016/j.bbamem.2014.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fong J. C., Karplus K., Schoolnik G. K., and Yildiz F. H. (2006) Identification and characterization of RbmA, a novel protein required for the development of rugose colony morphology and biofilm structure in Vibrio cholerae. J. Bacteriol. 188, 1049–1059 10.1128/JB.188.3.1049-1059.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fong J. C., and Yildiz F. H. (2007) The rbmBCDEF gene cluster modulates development of rugose colony morphology and biofilm formation in Vibrio cholerae. J. Bacteriol. 189, 2319–2330 10.1128/JB.01569-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moorthy S., and Watnick P. I. (2005) Identification of novel stage-specific genetic requirements through whole genome transcription profiling of Vibrio cholerae biofilm development. Mol. Microbiol. 57, 1623–1635 10.1111/j.1365-2958.2005.04797.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giglio K. M., Fong J. C., Yildiz F. H., and Sondermann H. (2013) Structural basis for biofilm formation via the Vibrio cholerae matrix protein RbmA. J. Bacteriol. 195, 3277–3286 10.1128/JB.00374-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maestre-Reyna M., Wu W.-J., and Wang A. H. (2013) Structural insights into RbmA, a biofilm scaffolding protein of V. cholerae. PLoS ONE 8, e82458 10.1371/journal.pone.0082458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fong J. C., Rogers A., Michael A. K., Parsley N. C., Cornell W.-C., Lin Y.-C., Singh P. K., Hartmann R., Drescher K., Vinogradov E., Dietrich L. E., Partch C. L., and Yildiz F. H. (2017) Structural dynamics of RbmA governs plasticity of Vibrio cholerae biofilms. Elife 6, e26163 10.7554/eLife.26163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yildiz F. H., and Schoolnik G. K. (1999) Vibrio cholerae O1 El Tor: identification of a gene cluster required for the rugose colony type, exopolysaccharide production, chlorine resistance, and biofilm formation. Proc. Natl. Acad. Sci. U.S.A. 96, 4028–4033 10.1073/pnas.96.7.4028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan J., Sharo A. G., Stone H. A., Wingreen N. S., and Bassler B. L. (2016) Vibrio cholerae biofilm growth program and architecture revealed by single-cell live imaging. Proc. Natl. Acad. Sci. U.S.A. 113, E5337–E5343 10.1073/pnas.1611494113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Absalon C., Van Dellen K., and Watnick P. I. (2011) A communal bacterial adhesin anchors biofilm and bystander cells to surfaces. PLoS Pathog. 7, e1002210 10.1371/journal.ppat.1002210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu A. C., Worrall L. J., and Strynadka N. C. (2012) Structural insight into the bacterial mucinase StcE essential to adhesion and immune evasion during enterohemorrhagic E. coli infection. Structure 20, 707–717 10.1016/j.str.2012.02.015 [DOI] [PubMed] [Google Scholar]
- 23. De S., Kaus K., Sinclair S., Case B. C., and Olson R. (2018) Structural basis of mammalian glycan targeting by Vibrio cholerae cytolysin and biofilm proteins. PLoS Pathog. 14, e1006841 10.1371/journal.ppat.1006841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rigden D. J., Woodhead D. D., Wong P. W., and Galperin M. Y. (2011) New structural and functional contexts of the Dx[DN]xDG linear motif: insights into evolution of calcium-binding proteins. PLoS ONE 6, e21507 10.1371/journal.pone.0021507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng H., Chordia M. D., Cooper D. R., Chruszcz M., Müller P., Sheldrick G. M., and Minor W. (2014) Validation of metal-binding sites in macromolecular structures with the CheckMyMetal web server. Nat. Protoc. 9, 156–170 10.1038/nprot.2013.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 10.1093/nar/gkq366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ochiai A., Itoh T., Mikami B., Hashimoto W., and Murata K. (2009) Structural determinants responsible for substrate recognition and mode of action in family 11 polysaccharide lyases. J. Biol. Chem. 284, 10181–10189 10.1074/jbc.M807799200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zechel D. L., and Withers S. G. (2000) Glycosidase mechanisms: anatomy of a finely tuned catalyst. Acc. Chem. Res. 33, 11–18 10.1021/ar970172 [DOI] [PubMed] [Google Scholar]
- 29. Pons T., Naumoff D. G., Martínez-Fleites C., and Hernández L. (2004) Three acidic residues are at the active site of a β-propeller architecture in glycoside hydrolase families 32, 43, 62, and 68. Proteins 54, 424–432 10.1002/prot.10604 [DOI] [PubMed] [Google Scholar]
- 30. Munoz-Munoz J., Cartmell A., Terrapon N., Henrissat B., and Gilbert H. J. (2017) Unusual active site location and catalytic apparatus in a glycoside hydrolase family. Proc. Natl. Acad. Sci. U.S.A. 114, 4936–4941 10.1073/pnas.1701130114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quistgaard E. M., and Thirup S. S. (2009) Sequence and structural analysis of the Asp-box motif and Asp-box β-propellers; a widespread propeller-type characteristic of the Vps10 domain family and several glycoside hydrolase families. BMC Struct. Biol. 9, 46 10.1186/1472-6807-9-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bonnardel F., Kumar A., Wimmerova M., Lahmann M., Perez S., Varrot A., Lisacek F., and Imberty A. (2019) Architecture and evolution of blade assembly in β-propeller lectins. Structure 27, 764–775.e3 10.1016/j.str.2019.02.002 [DOI] [PubMed] [Google Scholar]
- 33. Levan S., De S., and Olson R. (2013) Vibrio cholerae cytolysin recognizes the heptasaccharide core of complex N-glycans with nanomolar affinity. J. Mol. Biol. 425, 944–957 10.1016/j.jmb.2012.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hollenbeck E. C., Fong J. C., Lim J. Y., Yildiz F. H., Fuller G. G., and Cegelski L. (2014) Molecular determinants of mechanical properties of V. cholerae biofilms at the air–liquid interface. Biophys. J. 107, 2245–2252 10.1016/j.bpj.2014.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Drescher K., Dunkel J., Nadell C. D., van Teeffelen S., Grnja I., Wingreen N. S., Stone H. A., and Bassler B. L. (2016) Architectural transitions in Vibrio cholerae biofilms at single-cell resolution. Proc. Natl. Acad. Sci. U.S.A. 113, E2066–E2072 10.1073/pnas.1601702113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fernandez-Escamilla A.-M., Rousseau F., Schymkowitz J., and Serrano L. (2004) Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 22, 1302–1306 10.1038/nbt1012 [DOI] [PubMed] [Google Scholar]
- 37. Conchillo-Solé O., de Groot N. S., Avilés F. X., Vendrell J., Daura X., and Ventura S. (2007) AGGRESCAN: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinformatics 8, 65 10.1186/1471-2105-8-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Garbuzynskiy S. O., Lobanov M. Y., and Galzitskaya O. V. (2010) FoldAmyloid: a method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 26, 326–332 10.1093/bioinformatics/btp691 [DOI] [PubMed] [Google Scholar]
- 39. Maurer-Stroh S., Debulpaep M., Kuemmerer N., Lopez de la Paz M., Martins I. C., Reumers J., Morris K. L., Copland A., Serpell L., Serrano L., Schymkowitz J. W., and Rousseau F. (2010) Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 7, 237–242 10.1038/nmeth.1432 [DOI] [PubMed] [Google Scholar]
- 40. Tsolis A. C., Papandreou N. C., Iconomidou V. A., and Hamodrakas S. J. (2013) A consensus method for the prediction of “aggregation-prone” peptides in globular proteins. PLoS ONE 8, e54175 10.1371/journal.pone.0054175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Walsh I., Seno F., Tosatto S. C., and Trovato A. (2014) PASTA 2.0: an improved server for protein aggregation prediction. Nucleic Acids Res. 42, W301–W307 10.1093/nar/gku399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Drozdetskiy A., Cole C., Procter J., and Barton G. J. (2015) JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 43, W389–W394 10.1093/nar/gkv332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., and Bairoch A. (2005) Protein identification and analysis tools on the ExPASy server. In (Walker J. M. ed) The Proteomics Protocols Handbook, Humana Press, Totowa, NJ [Google Scholar]
- 44. Taglialegna A., Lasa I., and Valle J. (2016) Amyloid structures as biofilm matrix scaffolds. J. Bacteriol. 198, 2579–2588 10.1128/JB.00122-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tielen P., Kuhn H., Rosenau F., Jaeger K.-E., Flemming H.-C., and Wingender J. (2013) Interaction between extracellular lipase LipA and the polysaccharide alginate of Pseudomonas aeruginosa. BMC Microbiol. 13, 159 10.1186/1471-2180-13-159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wai S. N., Mizunoe Y., Takade A., Kawabata S. I., and Yoshida S. I. (1998) Vibrio cholerae O1 strain TSI-4 produces the exopolysaccharide materials that determine colony morphology, stress resistance, and biofilm formation. Appl. Environ. Microbiol. 64, 3648–3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kuttel M. M., Ståhle J., and Widmalm G. (2016) CarbBuilder: software for building molecular models of complex oligo- and polysaccharide structures. J. Comput. Chem. 37, 2098–2105 10.1002/jcc.24428 [DOI] [PubMed] [Google Scholar]
- 48. Liu Z., Wang Y., Liu S., Sheng Y., Rueggeberg K.-G., Wang H., Li J., Gu F. X., Zhong Z., Kan B., and Zhu J. (2015) Vibrio cholerae represses polysaccharide synthesis to promote motility in mucosa. Infect. Immun. 83, 1114–1121 10.1128/IAI.02841-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kawate T., and Gouaux E. (2006) Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14, 673–681 10.1016/j.str.2006.01.013 [DOI] [PubMed] [Google Scholar]
- 50. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 10.1107/S0907444913000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Langer G., Cohen S. X., Lamzin V. S., and Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 10.1038/nprot.2008.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen V. B., Arendall W. B. 3rd., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Scheuermann T. H., and Brautigam C. A. (2015) High-precision, automated integration of multiple isothermal titration calorimetric thermograms: new features of NITPIC. Methods 76, 87–98 10.1016/j.ymeth.2014.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jurrus E., Engel D., Star K., Monson K., Brandi J., Felberg L. E., Brookes D. H., Wilson L., Chen J., Liles K., Chun M., Li P., Gohara D. W., Dolinsky T., Konecny R., et al. (2018) Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 10.1002/pro.3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Guex N., and Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 10.1002/elps.1150181505 [DOI] [PubMed] [Google Scholar]
- 60. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Laskowski R. A., and Swindells M. B. (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 10.1021/ci200227u [DOI] [PubMed] [Google Scholar]
- 62. Tamura K., Peterson D., Peterson N., Stecher G., Nei M., and Kumar S. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.