

Abstract
The free radical nitric oxide (NO•) exerts biological effects through the direct and reversible interaction with specific targets (e.g. soluble guanylate cyclase) or through the generation of secondary species, many of which can oxidize, nitrosate or nitrate biomolecules. The NO•-derived reactive species are typically short-lived, and their preferential fates depend on kinetic and compartmentalization aspects. Their detection and quantification are technically challenging. In general, the strategies employed are based either on the detection of relatively stable end products or on the use of synthetic probes, and they are not always selective for a particular species. In this study, we describe the biologically relevant characteristics of the reactive species formed downstream from NO•, and we discuss the approaches currently available for the analysis of NO•, nitrogen dioxide (NO2•), dinitrogen trioxide (N2O3), nitroxyl (HNO), and peroxynitrite (ONOO−/ONOOH), as well as peroxynitrite-derived hydroxyl (HO•) and carbonate anion (CO3•−) radicals. We also discuss the biological origins of and analytical tools for detecting nitrite (NO2−), nitrate (NO3−), nitrosyl–metal complexes, S-nitrosothiols, and 3-nitrotyrosine. Moreover, we highlight state–of–the–art methods, alert readers to caveats of widely used techniques, and encourage retirement of approaches that have been supplanted by more reliable and selective tools for detecting and measuring NO•-derived oxidants. We emphasize that the use of appropriate analytical methods needs to be strongly grounded in a chemical and biochemical understanding of the species and mechanistic pathways involved.
Keywords: nitric oxide, nitrosative stress, oxidation–reduction (redox), oxidative stress, oxygen radicals, nitrosylation, reactive nitrogen species, reactive oxygen species, 3-nitrotyrosine, nitration, nitrogen dioxide, nitrosation, peroxynitrite
Introduction
Soon after the discovery of nitric oxide (NO•) as a physiological mediator in the vascular, nervous, and immune systems, it became evident that this moderately-reactive free radical can give rise to secondary species, many of which are oxidizing, nitrosating, or nitrating agents toward biomolecules (1–4).
The species formed downstream from NO• (i.e. NO•-derived oxidants) include nitrogen dioxide (NO2•), dinitrogen trioxide (N2O3), nitroxyl (HNO), and peroxynitrite (ONOO−/ONOOH), as well as hydroxyl (HO•) and carbonate anion (CO3•−) radicals formed from peroxynitrite (Fig. 1). These species are short-lived (half-lives are typically in the millisecond to microsecond range) and are frequently referred to in general as “reactive nitrogen species.” However, this term should be used with caution because in a similar way as “reactive oxygen species,” with which NO•-derived species are usually grouped, this term may give the inaccurate idea that there exists only one ill-defined species that has a particular type of reactivity and targets all biomolecules (5, 6). In contrast, the different species have unique reactivities and, depending on the particular properties of each, they may lead to oxidation, nitrosation, or nitration. As a further argument against the use of the term “reactive nitrogen species,” some of the species formed downstream from NO• (e.g. CO3•−) do not contain nitrogen. Finally, researchers in the nitrogen fixation field might argue that the reactive nitrogen species are those formed in the activation of nitrogen in the nitrogenase-catalyzed process of ammonia formation. Thus, in line with proposals in the free radical research field (5, 6), we suggest that the name of the identified species should be used whenever possible. When the species that are being referred to are unknown, we suggest using the term “NO•-derived oxidants.”
Figure 1.
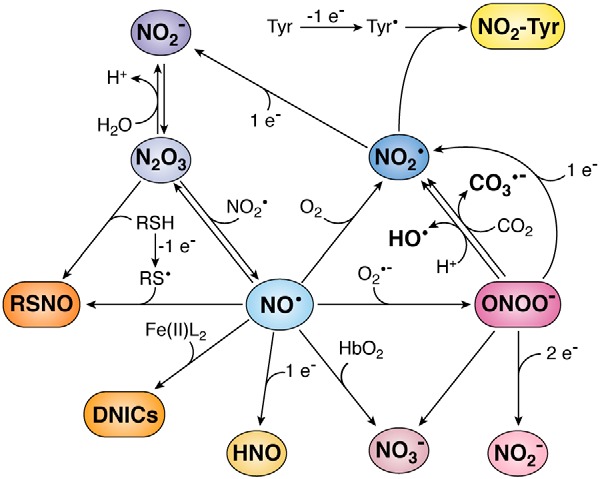
Nitric oxide and its biologically relevant derivatives. Nitric oxide can give rise to several species. Reaction with superoxide (O2•−) generates peroxynitrite (ONOO−); with oxyhemoglobin (HbO2), nitrate (NO3−); with oxygen (O2), nitrogen dioxide (NO2•); with strong one-electron reductants, nitroxyl (HNO); with liganded iron(II) (Fe(II)L2), dinitrosyl iron complexes (DNICs); with thiyl radical (RS•), S-nitrosothiol (RSNO); and with NO2•, dinitrogen trioxide (N2O3). Many of these products are reactive and yield further products. Peroxynitrite at neutral pH will protonate and generate NO3−, as well as NO2• and hydroxyl radicals (HO•) in 30% yield. In the presence of carbon dioxide (CO2), peroxynitrite will generate NO3−, as well as NO2• and carbonate anion radical (CO3•−) in 33% yield. In the presence of reductants, peroxynitrite will be reduced to nitrite (NO2−) or NO2•. Nitrogen dioxide can react with tyrosyl radicals (Tyr•) to generate 3-nitrotyrosine (NO2–Tyr) or with a reductant to form NO2−. Dinitrogen trioxide can be rapidly hydrolyzed to NO2−, it can be formed by NO2− in acidic pH, and it can react with thiols (RSH) to generate RSNO. In this figure, stoichiometries are not always strict, and protons are sometimes omitted for simplicity.
The preferential targets of NO•-derived oxidants in biological systems are typically located in close proximity (in the micrometer distance range) and determined by a combination of factors, including kinetic aspects of rate constants multiplied by target concentration, compartmentalization, and membrane permeability. Some of the NO•-derived oxidants are good one-electron oxidants that start oxygen-dependent chain reactions in both aqueous and lipidic compartments, which may amplify the effects (1, 7).
In many cases, the formation of NO•-derived oxidants is linked to the presence of partially-reduced oxygen species, as exemplified by peroxynitrite, which is formed from the reaction of NO• with the superoxide radical (O2•−). Thus, the formation of NO•-derived oxidants is frequently related to inflammation, in which increased formation of NO• through the inducible nitric-oxide synthase converges with increased formation of O2•− and other oxidants. In fact, the high reactivity of some of the species derived from NO• make them part of the weaponry that immune cells use in their battles against microorganisms (8). In addition to their cell-damaging activity, they can have signaling roles. The recognition that hydrogen peroxide (H2O2) can act as second messenger (9) and that signaling actions can be extended to species derived from NO• (10) have expanded the traditional view of oxidative stress as a misbalance between oxidant formation and antioxidant action to include the view of a disruption in regulatory pathways.
Because of their high reactivity, the species derived from NO• have relatively short half-lives that impede their detection in biological systems through direct spectroscopic techniques. Thus, the analytical strategies used to demonstrate the formation of a certain species in a particular biological context are based either on the measurement of downstream stable products or on the use of probes that react with the species. Because these strategies are not always specific for a certain species, the modulation of the formation or decay pathways of precursors and products provides complementary evidence. For example, the modulation of NO• and O2•− formation, which are the precursors of peroxynitrite, should accompany the results obtained through the detection of the stable product 3-nitrotyrosine or through the use of peroxynitrite probes. The modulation of NO• formation can be carried out using nitric oxide synthase inhibitors, among other strategies.
In the following sections, we briefly describe the reactive species derived from NO• in a biological milieu and NO• itself, as well as some of the stable end products (Fig. 1). We examine methodologies used for their detection and quantification, focusing on strategies aimed at assessing their involvement in biological processes.
Nitric oxide
The discovery of nitric oxide, a free radical, as an endogenously generated effector molecule, was a paradigm shift in biological signaling. Nitric oxide (NO•, IUPAC names nitrogen monoxide, oxidonitrogen(•), or oxoazanyl) is a diatomic free radical produced in animals mainly by the enzymes nitric oxide synthases (NOS)3. Nitric oxide has a low dipole moment (0.159 D (11)), so it has weak intermolecular interactions and it is a gas at 1 atm and 25 °C. It is only sparingly soluble in water (1.94 ± 0.03 mm (12)) but is about 10 times more soluble in organic solvents (13). The partition coefficient in membrane models and human low-density lipoprotein is 4.4–3.4 at 25 °C (14). The diffusion through cell membranes is very rapid (14–17). The permeability coefficients of lipid membranes to NO• range from 18 to 73 cm s−1 (15, 17), similar to that of an equally thick layer of water.
Unlike several other free radical species, NO• is not a one-electron oxidant (E0′ (NO•, H+/HNO) ∼−0.55 V at pH 7) (18, 19). It does not abstract hydrogen atoms, and it does not add to unsaturated bonds. Importantly, NO• does not react directly with thiols (RSH). Among the main targets of NO• in biological systems are metal centers. Coordination to the ferrous heme in soluble guanylate cyclase is responsible for many physiological effects of NO• (20–23). Reaction with oxyhemoglobin to form NO3− is an important sink of NO• (24, 25). Other relevant targets of NO• are other free radical species, in particular O2•− (1). Nitric oxide can also react with oxygen, and this is analyzed in the next section on autoxidation.
The effects of NO• are exerted either via direct reactions with biological targets or indirectly via NO•-derived oxidants. Dysregulation of NO• homeostasis has been linked to neurodegeneration, cardiovascular disease, cancer, and inflammation. Therefore, the detection and quantification of NO• and its derived oxidants in vitro and in vivo are relevant to understanding the molecular bases of physiological as well as pathological processes.
Nitric oxide autoxidation
The reaction of NO• with oxygen (O2), termed autoxidation, is a complex process that gives different products and intermediates relevant to the detection of NO• and several of its derived species. This process is considered to be too slow to be of relevance under most physiological conditions. In the first step, two molecules of NO• and one molecule of O2 give two molecules of NO2• (Equation 1). Next, NO2• reacts with NO• reversibly to give N2O3 (Equation 2) (26, 27). In water, in the absence of other targets, N2O3 is subsequently hydrolyzed to two molecules of NO2− (Equation 3).
| (Eq. 1) |
| (Eq. 2) |
| (Eq. 3) |
The rate of decomposition of NO• is second order in NO• and first order in O2 concentrations, and in water the final stoichiometry is four molecules of NO• per O2, so that the rate equation is expressed as in Equation 4.
| (Eq. 4) |
The limiting reaction in the autoxidation of NO• is the reaction with O2 (Equation 1); trapping subsequent products has no effect on the overall rate (27). The rate constant of the process is third order (k ∼2 × 106 m−2 s−1 (27, 28)). In aqueous solutions, the forward reaction of N2O3 formation (Equation 2) is very fast (k = 1.1 × 109 m−1 s−1), and the dissociation has a rate constant k = 8 × 104 s−1 (29). The hydrolysis of N2O3 is also rapid in water and can be further accelerated by salts such as phosphate and bicarbonate (27, 30, 31).
The autoxidation of NO• is slow in vivo because the rate of NO• decay is second order in NO• concentration, which is expected to be in the nanomolar range under normal conditions. This reaction can be accelerated in hydrophobic environments such as lipid membranes, lipoproteins, and proteins (32–34) due to the increase in concentration of both NO• and O2 because of their hydrophobicity (14, 35). This so-called “lens effect” may be of relevance where NO• concentrations are significantly increased, especially in sites of inflammation.
Detection of nitric oxide
Nitric oxide is difficult to measure in vivo because of its short half-life (typically in the range of 0.1–10 s), reactivity, and low steady-state concentration (i.e. nanomolar to micromolar). Nonetheless, several strategies have been developed to measure NO• or its derived species in vitro or in vivo that involve the use of absorbance, fluorescence, electron paramagnetic resonance (EPR), and electrochemistry (Fig. 2). Furthermore, NO• can be measured by chemiluminescence, a methodology that can be adapted to also measure other species. These methods are described in the following sections.
Figure 2.

Detection of nitric oxide. A, oxidation of oxyhemoglobin. Methemoglobin can be detected spectrophotometrically. B, electrochemical sensor. NO• is oxidized to nitrosonium cation (NO+), which is converted to nitrite (NO2−). The current is directly proportional to NO• concentration. C, reaction of NO• with 2-phenyl-4,4,5,5-tetramethylimidazoline-1-yloxyl-3-oxide (PTIO) to yield NO2• and PTI, the conversion can be followed by EPR. D, fluorogenic probes for NO•-derived species. The 4,5-diaminofluorescein diacetate (DAF-2–DA) is cell-membrane–permeable, and the acetyl groups are removed by intracellular esterases to yield the nonfluorescent DAF-2 that then reacts with NO•-derived species to yield the fluorescent triazole derivative DAF-2 T. At right are shown related fluorescent probes diaminonaphthalene (DAN) and 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM). The mechanisms of triazole formation from diamino-fluorogenic probes involve two possible routes: one is the direct nitrosation by N2O3 to give an intermediary N-nitrosamine that then diazotizes and reacts with the second amine to yield the triazole; the other mechanism requires the oxidation of the diamino probe by NO•-derived and other one-electron oxidants, followed by the reaction of the radical with NO• to form the N-nitrosamine. Either of these pathways implicate NO• but with different stoichiometries. E, ozone-based chemiluminescence. F, reaction catalyzed by nitric oxide synthase to generate NO•. l-Arginine is first hydroxylated to Nω-hydroxy-l-arginine with O2 and NADPH as cosubstrates. In the second step, this intermediate is oxidized by a second O2 and 0.5 eq of NADPH to give l-citrulline and NO•. G, bioassays to measure downstream physiological actions of NO•.
Oxyhemoglobin oxidation
The identification of the endothelial-derived relaxing factor as NO• back in 1987 (36) was in part made by the change in the UV-visible spectrum of deoxyhemoglobin to form nitrosyl hemoglobin, with a corresponding shift of the Soret band from 433 to 406 nm. Nonetheless, the reaction mostly used to quantify NO• in vitro is with oxyhemoglobin, which is stable in air.
Oxyhemoglobin (Fe(II)(Hb)O2) is oxidized by NO• to yield NO3− and methemoglobin (Fe(III)(Hb)), which can be measured spectrophotometrically (Fig. 2A). The absorption of the Soret band is strong; thus the sensitivity of this method is relatively high (submicromolar). The maximum absorbance changes are observed at 401 (increased by reaction with NO•) and 421 nm (decreased), with an isosbestic point at 411 nm. If multiple wavelengths can be measured, the reaction can be followed by the absorbance difference of 401–421 nm (Δϵ401–421 = 77 mm−1 cm−1) (37). If there is interference at these wavelengths, the absorbance at 577 nm can be used (Δϵ577 = 10 mm−1 cm−1) or even both absorbances at 577 and 630 nm to calculate oxy- and methemoglobin concentrations before and after addition of NO• (38). Due to the fast reaction with NO• (k = 3.7 × 107 m−1 s−1) (39), oxyhemoglobin is also often used as NO• scavenger. Nitrite can also oxidize oxy- to methemoglobin but at much lower rates (although autocatalytically), so when high concentrations of NO2− are expected, like with the use of NO• donors for long-time periods, the contribution of NO2− to hemoglobin oxidation should be considered. Peroxynitrite can also oxidize oxyhemoglobin; thus, addition of superoxide dismutase is recommended as a control to prevent peroxynitrite formation from NO• and O2•− so that the methemoglobin formed can be associated with NO•. In addition, a potential O2•−-dependent redox cycling of hemoglobin can be avoided.
Electrochemical sensor
Electrodes specific for NO• are commercially available, but many research laboratories make their own. Typically, they consist of a filament made of carbon or platinum and a coating to provide specificity that either attracts NO• (40) or excludes other oxidizable species (41). At the anode, NO• is oxidized by one-electron to nitrosonium cation (NO+), which is converted to NO2− (Fig. 2B). The current generated from NO• oxidation is directly proportional to NO• concentration with a 10 nm detection limit (a calibration curve should be run with each experiment). The electrode is covered with a gas-permeable membrane that allows diffusion of NO• but not NO2− or other charged species. Temperature should be kept constant considering that solubility of NO• gas is very temperature-sensitive. Microelectrodes have been designed (<1 mm) that allow direct detection in cells in real time (42).
Electron paramagnetic resonance (EPR)
Although NO• is a free radical, i.e. it has an unpaired electron, it is difficult to detect directly by EPR, and spin-trapping techniques have to be used. Nitronyl nitroxides (with nitrone and nitroxide functional groups) are used as NO• probes (43). They react with NO• to give an iminonitroxide (Fig. 2C) with a dramatic change in the EPR spectrum that can be followed in a continuous and quantitative way. For example, 2-phenyl-4,4,5,5-tetramethylimidazoline-1-yloxyl-3-oxide (PTIO) or its water-soluble analogue carboxy-PTIO react with NO• with second-order rates constants of 104 m−1 s−1 and a change in the EPR spectrum from five to seven lines (44, 45).
Due to its fast reaction with NO•, carboxy-PTIO is often used as a scavenger of NO•; however, care should be taken because NO2• is a product of the reaction and has its own reactivities. In addition, biological reductants like thiols, ascorbate, or O2•− can nonspecifically reduce the nitroxides. Encapsulation of PTIO in liposomes has been used to avoid reduction (46).
Hydrophobic and hydrophilic nitroxides are available that allow detection of NO• at different depths of a biological membrane. Collisions of NO• with spin labels located in water or in membranes alter both the linewidth and the spin-lattice relaxation time that can be used to qualitatively and quantitatively measure NO• (17, 47).
Colloid iron diethyldithiocarbamate (Fe(DETC)2) or N-methyl-d-glucamine dithiocarbamate are reliable spin traps for NO• detection. They form iron nitrosyl complexes with characteristic three-line EPR spectra (gav = 2.04; aN = 1.27 mT) at room temperature that are stable in the presence of oxygen (48). Dinitrosyl iron complexes (DNIC) with thiol-containing ligands have been detected in animal and bacterial cells by EPR. These complexes are formed in vivo in the paramagnetic (EPR-active) mononuclear as well as diamagnetic (EPR-silent) binuclear forms. The amount of NO• can calculated from the EPR amplitude signal because the linewidth of the NO-Fe(DETC)2 EPR spectrum may vary considerably with variations in the amount of Fe(DETC)2 in membrane lipids and the amount of Fe(III) present (48, 49).
In addition, deoxyhemoglobin or other hemeproteins in the reduced Fe(II) form react with NO• to form nitrosyl–heme complexes that besides having characteristic UV-visible spectra are paramagnetic and can be followed by EPR. However, because of the instability of the complexes, the EPR spectra should be run at low temperatures (77 K) (50–53).
Fluorogenic probes
Fluorogenic probes have been developed that specifically react with NO•-derived species (i.e. N2O3) to yield fluorescent products, such as diaminonaphthalene (DAN) and diaminofluorescein (DAF) derivatives (Fig. 3D). The most popular of this kind is 4,5-diaminofluorescein (DAF-2), where nitrosation results in the highly-fluorescent triazole DAF-2 T (λexc = 495 nm, λem = 515 nm). The esterified diacetate derivative (DAF-2-DA) is also commercially available. It is highly membrane-permeable and detects intracellular nitrosation of the probe.
Figure 3.
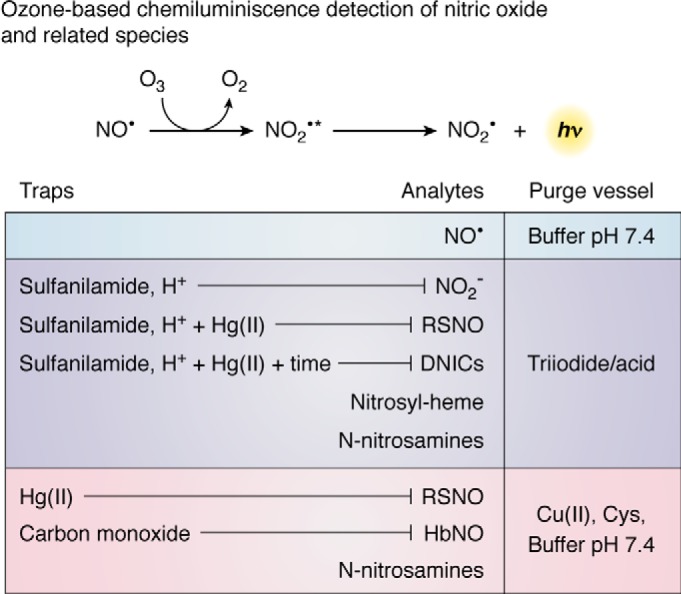
Chemiluminescence detection of nitric oxide and derived species. This sensitive method allows the determination of NO• and several related species in gas or liquid phase. The system includes a purge vessel where the sample is injected. An inert gas transports NO• from the purge vessel to a detector where ozone (O3) reacts with NO• yielding NO2• in the excited state, which decays to the basal state emitting light. Different reagents can be used in the purge vessel to selectively convert certain analytes into NO•. A neutral buffer is used if NO• as such is to be measured. A triiodide/acid solution reduces most derivatives to NO•, and thus it is useful for total quantitation. The sample can be pretreated with reagents that will trap specific species. Thus, the amount of NO2− in a complex sample is obtained by the difference between untreated sample and sample pretreated with sulfanilamide and acid to trap NO2−. RSNO can be removed by pretreatment with Hg(II), sulfanilamide, and acid. DNICs are sensitive to the same treatment but differently from RSNO; they decay over time (10–12 h). The remaining signal after treatment with Hg(II), sulfanilamide, and acid derives mostly from N-nitrosamines, and to a lesser degree from nitrosyl–heme. Alternatively, the purge vessel can contain Cu(II) and cysteine, which does not reduce NO2− to NO• but effectively reduces RSNO to NO•, and it also releases NO• from nitrosyl-hemoglobin and N-nitrosamines. When analyzing nitrosation of hemoglobin or other heme-proteins, carbon monoxide (CO) is added to avoid reaction of NO• with heme-proteins.
The pH should be carefully controlled because DAF-2 T fluorescence is pH-sensitive (54). Other potential artifacts are divalent cations, in particular calcium, which was reported to significantly increase the fluorescent signal from DAF-2, as well as the incident light (55). Multiple and long exposures to excitation light, instead of causing photobleaching of the dye, potentiate the fluorescence response. Thus, minimum periods of light exposure are recommended. The use of 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) is favored because of its increased photostability, stability to pH, and reactivity toward NO•-derived species (54). The sensitivity of DAF-FM is 1.4 times higher than that of DAF-2. This increase of sensitivity is thought to result from the higher rate of the reaction with nitrosating NO+ equivalents due to the electron-donating effect of the methyl group (43).
The proposed mechanism of triazole formation involves N2O3 reacting with an amine to form an intermediate N-nitrosamine that at neutral pH can diazotize and then react with the second amine to yield the triazole (Fig. 3D) (56, 57). Alternatively, a radical intermediate of the diamino-probe is formed by NO2• or other strong oxidants (e.g. radicals derived from peroxynitrite or peroxidases/H2O2) that then react with NO• (Fig. 3D) (58). These probes show some specificity issues, in that the triazole is not an exclusive product of NO•, and fluorescent products can be derived from peroxynitrite, nitroxyl (HNO), and ascorbic acid, complicating the interpretation of results (59, 60). Nonetheless, the role of NO• can be confirmed in cells using NOS inhibitors, NO• scavengers, and also by HPLC to isolate DAF-triazole (60–62).
Novel genetically-encoded fluorescent NO• biosensors have been developed (63). For example, one sensor has a fusion between a fluorescent protein and a bacteria-derived NO• domain that selectively binds NO• via a nonheme Fe(II) center. Once NO• binds, the domain gets closer to the fluorescent protein and quenches its emission (64).
Ozone-based chemiluminescence detection of nitric oxide and related species
The chemiluminescence detection of NO• and several other related species presents very good sensitivity and reproducibility and has become the gold standard method for quantification (62, 65–68). The sensitivity is in the nanomolar range. The method can be used with any type of gas or liquid sample, including cell lysates and tissue homogenates (68, 69).
The sample is injected into a purge vessel containing a given reactant such as triiodide. This vessel has fritted glass at the base and is purged at a constant flow rate with nitrogen or helium gas. The NO• that was injected (or generated) in the vessel is carried by this inert gas to the detector (65, 68). The NO• in the carrier gas passes first through a reaction cell where ozone is constantly introduced. The reaction with ozone (O3) generates NO2• in the excited state (NO2•*) that is then carried by the inert gas flow to the detection cell where red and near IR light emission from NO2•* decay to the basal state is measured (68, 70). The intensity of emission is directly proportional to the amount of NO• (Fig. 2E).
This method is not only useful to the study of NO• but also of other oxidation products that can be converted to NO• through different methods, such as NO2•, NO2−, S-nitrosothiols, nitrosyl–metal complexes, and N-nitrosamines (62, 65, 66, 68). The reactant used in the purge vessel determines what species can be quantified (Fig. 3). If buffer at neutral pH is used, only NO• as such will give a signal (69). One of the most popular reactants for chemiluminescent detection of NO• and its derivatives is an acidic triiodide solution (65). It consists of iodine plus iodide and acetic acid (65, 68, 71). The triiodide that forms in this solution can convert NO2−, S-nitrosothiols, N-nitrosamines, and nitrosyl–metal complexes to NO• (65, 66). In the case of a biological sample that contains a mixture of these species, several tubes are prepared that include the parent sample, then one with acidic sulfanilamide to trap NO2−, and another that also includes HgCl2 to decompose S-nitrosothiols (65, 66). The difference in the measured NO• with the different treatments indicate how much NO2− and S-nitrosothiol were in the sample.
Additional methods for more selective chemical reduction of S-nitrosothiols include copper-based assays where the reactant in the purge vessel consists of a buffer at neutral pH and Cu(II) plus an excess of cysteine (66, 71). In this case the copper is reduced to Cu(I) by cysteine, which then reduces S-nitrosothiols to NO• and thiol. The use of Hg(II) is recommended to discriminate the signal from N-nitrosamines (66). For applications in blood, a modification of the method that includes carbon monoxide has been developed that prevents capture of NO• by hemoglobin (72).
Nitric oxide synthase activity
The NOS enzymes catalyze the oxidation of arginine to NO• and stoichiometric amounts of citrulline (Fig. 2F) (73). Therefore, the rate of NO• formation can be estimated from the rate of citrulline formation from arginine and saturating concentrations of NOS cofactors (NADPH, FAD, FMN, tetrahydrobiopterin, and calcium/calmodulin). Radiolabeled arginine is used, and the reaction is stopped with EDTA, which binds calcium and inactivates the enzyme. The radiolabeled citrulline product is separated from arginine by cation-exchange chromatography (cationic arginine is retarded and zwitterionic citrulline is eluted) and measured in a liquid scintillation counter (74). Because citrulline in the cell could be derived from non-NOS pathways, controls should be performed with addition of a NOS inhibitor as well as omission of NADPH.
There are commercially available kits to follow NOS activity indirectly, by measuring the time course of NO2− formation spectrophotometrically using the Griess reaction described below.
Bioassays for nitric oxide
The production of NO• in mammalian cells can be detected indirectly by measuring its biological activities like vasodilation, platelet aggregation, and guanylate cyclase activation (Fig. 2G).
Cyclic GMP
In the cellular context, cyclic GMP (cGMP) is formed not only by guanylate cyclases stimulated by NO• (NO-GC or soluble GC) but also by the membrane natriuretic peptide receptor-coupled guanylate cyclases (GC-A and GC-B). Therefore, to measure levels of cGMP as an indirect measurement of NO•, controls with inhibitors of NOS should be included. The different methods used to determine cGMP have been recently reviewed in Ref. 75 and include radiolabeled [α-32P]GTP (76), a cGMP antibody in a commercially available enzyme-linked immunosorbent assay (ELISA), and fluorescence-based cGMP indicators (77).
Vessel relaxation
The seminal studies that introduced NO• to the biological scenario as a critical regulator of blood flow were related to its identification as an endothelium-derived relaxing factor (36, 67). This function is explained by the formation of NO• from endothelial NOS, subsequent diffusion to the underlying smooth muscle, and activation of soluble guanylate cyclase, which initiates a signaling cascade that ultimately leads to vasodilation and increased blood flow. Thus, a method amply used by physiologists and pharmacologists to detect production of NO• consists of measuring tension in isolated vascular preparations treated with agonist and antagonists of NO•-dependent signaling (67, 78).
Inhibition of platelet aggregation
A way to test NO• production is to follow inhibition of platelet aggregation after activation. It is a very simple, inexpensive method first described in 1962 (79). Washed human platelets are equilibrated at 37 °C in a turbidometric platelet aggregometer in the absence and presence of a system that produces NO•. An activator like thrombin is added to induce aggregation, and turbidimetry is followed with time (80).
Nitrogen dioxide
Nitrogen dioxide (NO2•) is a reddish-brown free radical gas that forms part of air pollution. In biological systems, there are different endogenous sources of NO2•. These include: (a) NO• autoxidation (see section above); (b) NO2− oxidation, a reaction that is catalyzed by different heme-dependent peroxidases (Equations 5–7) and Cu,Zn-superoxide dismutase (81–83) or performed nonenzymatically by strong one-electron oxidants such as CO3•−, HO• or peroxyl radicals (ROO•) (84–86); and (c) homolysis of the peroxo bond of peroxynitrous acid (ONOOH) or of short-lived adducts formed from the reaction of peroxynitrite (ONOO−) with carbon dioxide (CO2), with carbonyl-containing compounds, or with metal centers (87–89).
| (Eq. 5) |
| (Eq. 6) |
| (Eq. 7) |
The solubility of NO2• in water is low. The reported Henry coefficient (∼1.4 × 10−2 m atm−1 at 20 °C) (90, 91) is quite uncertain due to its rapid (k = 4.5 × 108 m−1 s−1 (92)) dimerization to dinitrogen tetroxide (N2O4). The latter has ∼100-fold increased solubility in water (90) and rapidly hydrolyzes to NO2− and NO3− (k = 1000 s−1) (90, 93). However, under most physiological conditions where NO2• concentrations are low (<1 μm), dimerization, which is a reversible process with a Keq = 7 × 104 m−1, is outcompeted by bimolecular reactions of NO2• with different targets, some of which are far more concentrated (93–95). The partition coefficients in organic solvents indicate that NO2• is slightly hydrophobic, although less than NO•, which suggests a minor “lens effect” for NO2• reaction kinetics in membranes or other hydrophobic biological systems (94, 96).
The reduction potential of the NO2•/NO2− pair is 0.99 V at pH 7 (97). Thus, it is a good one-electron oxidant. According to kinetic considerations, NO2• is predicted to react mostly with thiol-containing molecules (kRS− ∼108 m−1 s−1) (98) and ascorbate (k = 1.8–3.5 × 107 m−1 s−1 at pH 7.4) (99), whereas urate (k = 2 × 107 m−1 s−1 at pH 7.4) is a main target in plasma (98). In hydrophobic media, NO2• can initiate lipid peroxidation (100). Furthermore, it can add to alkene double bonds in a fast and reversible process to form nitroalkyl radicals, which eventually undergo cis-trans–isomerization (101) or form nitro-derivatives. The biological formation, characteristics, and relevance of nitrated fatty acids has been reviewed (93, 102–105). Finally, NO2• reacts at diffusion-controlled rates with other radical species, such as tyrosyl radicals in proteins, to form 3-nitrotyrosine (see section on 3-nitrotyrosine below). The reversible reaction with NO• leads to the formation of the nitrosating species N2O3 (Equation 2).
Detection of nitrogen dioxide
The UV-visible absorption spectrum of NO2• shows a broad band peak at ∼400 nm with an absorption coefficient of 200 m−1 cm−1 in aqueous solution (26). The low absorption coefficient, the low stability of the radical, and the need to make corrections for N2O4 and NO2− absorption limit the technique. Nitrogen dioxide is frequently detected and quantified by chemiluminescence methods, in which the intensity of the emitted light is proportional to the concentration of NO2•. Some of these methods rely on the reaction of NO• with ozone (see section above on ozone-based chemiluminescence detection of nitric oxide and related species) and therefore require that NO2• be reduced to NO• using particular catalytic converters. The latter are usually nonspecific due to reduction of other nitrogen-containing compounds (106, 107). Nitrogen dioxide can also be converted to NO• photolytically using UV-LED irradiation (108). In addition, luminol (5-amino-2,3-dihydro-1,4-phthalazinedione) in alkaline solution reacts with NO2• giving rise to intense chemiluminescence, although other one-electron oxidants can also lead to light emission (109).
Detection of NO2• in cells and tissues requires different methodologies. Because NO2• is a strong one-electron oxidant, it can react with typical redox probes such as 2′,7′-dichlorodihydrofluorescein (DCFH2). In experimental designs, addition of NO2− may be useful to convert other one-electron oxidants to NO2•.
One strategy depends on the ability of NO2• to rapidly combine with free or protein tyrosyl radicals to form 3-nitrotyrosine (110). This is analyzed in the section below on 3-nitrotyrosine. Furthermore, nitration of green fluorescent protein (GFP) leads to a decrease in its intrinsic fluorescence and was used to evaluate NO2• formation. Although the decrease in fluorescence intensity is not specific for nitration, it can be utilized in combination with pharmacological modulation of NO• levels to indicate NO2• formation (59).
Finally, because of the radical nature of NO2•, EPR has been utilized either by direct detection of NO2• in salt matrices and low temperatures or by using spin traps such as nitromethane at alkaline pH or nitrone compounds (93, 111–113). Nitroso spin traps do not trap NO2• (113).
Due to the short half-life of NO2• in aqueous solutions even in the absence of other targets, as a result of dimerization and hydrolysis of N2O4, the study of the kinetics of NO2• reactions requires the use of very fast methodologies that allow measurements to be made in the microsecond time scale, such as pulse radiolysis. Furthermore, the low absorption coefficient of NO2• limits its direct detection so that product monitoring or competition kinetics need to be used. Competition with 2,2′-azino-bis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) oxidation to ABTS+ is frequently employed due to the high extinction coefficient of the latter radical at 414 nm (3.6 × 104 m−1 cm−1) (98, 114).
Dinitrogen trioxide and its detection
Dinitrogen trioxide (N2O3) can be formed from NO2• reaction with NO• (26, 27) and is considered an important intermediate in the autoxidation of NO• (27) (see section on autoxidation above). It can also be formed from NO2− at acidic pH, with an equilibrium constant of 3 × 10−3 m−1 (Equation 8) (115).
| (Eq. 8) |
As discussed in the section on autoxidation, N2O3 is rapidly hydrolyzed to NO2− (Equation 3), and this reaction is accelerated by certain salts, such as phosphate and bicarbonate (27, 30, 31). Therefore, the first approach to measure the production of N2O3 is by monitoring NO2− (see section below on nitrite and nitrate).
N2O3 is considered an important nitrosating species in vitro because it reacts rapidly with thiolates and amines to give the corresponding nitrosated species (116, 117), but its role in biological nitrosation is uncertain because of the slow kinetics of NO• autoxidation (116, 118). Because both NO2• and NO• are slightly hydrophobic (94, 96), it was suggested that N2O3 formation should be accelerated in hydrophobic regions. However, the nitrosation of thiols buried in the hydrophobic regions actually decreases because the dissociation to the more reactive thiolate is disfavored (119). Anyway, the formation of S-nitrosothiols is not specific to N2O3, and other mechanisms may be more relevant (116, 118), so that S-nitrosothiols are not necessarily good indicators of N2O3 formation (see section below on S-nitrosothiols).
Besides measuring NO2− and S-nitrosothiols, another method to detect N2O3 is to use fluorogenic probes such as diaminonaphthalene (DAN) and diaminofluorescein (DAF), which were discussed in the section on detection of nitric oxide and suffer from the same issues as S-nitrosothiols.
Nitrosyl–metal complexes and their detection
Intracellular dinitrosyl iron complexes (DNICs) are formed from NO•, a ligand such as GSH, and loosely bound iron, also called labile or chelatable iron pool (120). Recent studies have shown that the exposure of cells to NO•, either exogenous or endogenous, leads to the formation of more DNICs than S-nitrosothiols, in a 4:1 ratio (69). DNICs have been proposed to be relevant precursors in the nitrosation of thiols (121).
The most selective technique to measure DNICs is through EPR (120, 122). Mononuclear DNICs show a characteristic EPR spectrum (48, 122). EPR has several advantages such as the ability to measure signals in optically opaque samples, a good sensitivity (200 nm), and the capacity to distinguish enzymatically generated NO• by the change in the spectrum using [15N]arginine (122).
DNICs made with GSH can also be analyzed by UV-visible spectrophotometry, provided there is a separation step such as HPLC. They show a spectrum with maximum absorbance below 200 nm and characteristic peaks at 310, 360, and 680 nm (ϵ = 9200, 7400, and 200 m−1 cm−1, respectively) for the diamagnetic binuclear form, or 390 nm (ϵ = 3900 m−1 cm−1) for the paramagnetic mononuclear form (48).
Cellular DNICs can also be quantified through ozone-based chemiluminescence, using the triiodide method. Care should be taken in quantification because the signal is time-sensitive and decays within hours, and also because DNICs are sensitive to HgCl2, analogously to S-nitrosothiols (69). To distinguish between signals from S-nitrosothiols and DNICs, it was proposed to stabilize S-nitrosothiols in the cell lysate using a buffer containing diethylenetriaminepentaacetic acid and N-ethylmaleimide and to analyze the sample immediately after extraction and 20 h later to ensure the full decay of DNICs (69).
Formation of protein nitrosyl–metal complexes is particularly relevant in red blood cells, because NO• can react with deoxyhemoglobin to yield nitrosyl-hemoglobin. The detection of this product predominates at low oxygen tensions (123) because deoxyhemoglobin will be more abundant and because oxyhemoglobin will rapidly decompose NO• to NO3− (24). Nitrosyl-hemoglobin can be quantified in vitro through UV-visible spectrophotometry and shows absorption maxima at 403 and 575 nm (36). In red blood cells, nitrosyl-hemoglobin is difficult to quantify by spectrophotometry where there is a mixture of different forms of hemoglobin absorbing at the same wavelength. EPR, in contrast, is specific for nitrosyl-hemoglobin and allows its quantification in packed red blood cells. The limit of quantification was calculated to be 200 nm. Under normal conditions, the amount of nitrosyl-hemoglobin in human blood is below the detection limit. However, patients exposed to 80 ppm NO• inhalation treatment increased its nitrosyl-hemoglobin levels up to 2 μm (124).
S-Nitrosothiols
The formation of S-nitrosothiols is undoubtedly linked to the formation of NO• in biological systems. Nevertheless, the exact chemistry is still under debate. In fact, thiols or rather thiolates can be nitrosated by the products of NO• autoxidation (see section above on autoxidation) in a direct mechanism by N2O3 or stepwise by NO2• and NO• (Equation 9-11) (116, 125).
| (Eq. 9) |
| (Eq. 10) |
| (Eq. 11) |
Although the formation of NO2• and N2O3 can be accelerated by hydrophobic regions in lipid membranes and even proteins (32–34), autoxidation is still too slow to be biologically significant (118). Furthermore, in cells, oxygen inhibits rather than increases thiol nitrosation, arguing against a significant role for NO• autoxidation in biological thiol nitrosation (118).
Regarding the mechanisms of biological thiol nitrosation, there is evidence supporting the intermediacy of nitrosyl–iron complexes (DNICs) (69, 121), as well as the intermediacy of cytochrome c (126).
S-Nitrosothiols undergo further reactions with other thiols, such as trans-nitrosation, where the nitroso moiety is transferred regenerating the original thiol (127). For instance, the trans-nitrosation from S-nitrosoglutathione to cysteine occurs with k = 140 m−1 s−1 (127). Thioredoxin catalyzes both trans-nitrosation and denitrosation (128). Alcohol dehydrogenase class III catalyzes the reduction of S-nitrosoglutathione efficiently and has therefore been called S-nitrosoglutathione reductase (129).
Detection of S-nitrosothiols
Several methods have been developed to quantify total S-nitrosothiols and also to identify proteins that are nitrosated. S-Nitrosothiols show a UV spectrum with a maximum at 335 nm. The absorptivity for S-nitrosoglutathione at 335 nm is 922 m−1 cm−1; therefore, the sensitivity of the spectrophotometric analysis is low (above 50 μm) and depends on having a purified sample or on chromatographic or capillary electrophoresis separation (130).
Another historically important method for S-nitrosothiols is by Saville (131). In this method the S-nitrosothiol is treated with Hg(II). Tight binding to the thiolate releases NO+ that is rapidly hydrolyzed to NO2− (131). The released NO2− is then measured by the Griess method. This method has micromolar sensitivity (see section below on detection of nitrite).
Antibodies against S-nitrosocysteine have been used in immunohistochemical assays, Western blotting, and immunoprecipitation. However, specificity issues and the advent of biotin switch techniques that also allow mapping the modified cysteine within a protein have discouraged their use (66).
The gold standard method to quantify S-nitrosothiols is ozone-based chemiluminescence that provides nanomolar sensitivity and is appropriate for most biological applications (Fig. 3). As discussed in the section on chemiluminescence, S-nitrosothiols can be quantified using the triiodide reaction and also using copper ions and reductants (66).
It was early observed that several proteins could be S-nitrosated (132) but unbiased approaches to the S-nitrosoproteome were only possible after the introduction of the “biotin switch” method in 2001 (133). The original method involved blocking free thiols in S-nitrosated proteins with methyl methanethiosulfonate, then specifically reducing the nitrosated thiols with ascorbate, followed by reaction of these thiols with N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)-propionamide. The biotinylated proteins could then be selectively captured by using the specific binding to immobilized streptavidin (133). Some issues with this method have been pointed out, namely that it is very difficult to ensure that all free thiols are effectively blocked in the first step, that ascorbate does not reduce S-nitrosothiols directly but through the intermediacy of metals in solution (134), and that no chemical trace is left to indicate that the thiol was effectively nitrosated.
Relative quantification of protein S-nitrosation can be achieved through different means, including isotope-coded affinity tags (ICAT) and stable isotope labeling by amino acids in cell culture (SILAC). Both methods are based on using a light and a heavy isotope-containing tag. In ICAT, samples to compare are processed in parallel and tagged with biotin derivatives that include either light or heavy isotope linkers, and then mixed and further processed (135). SILAC involves adding either light or heavy isotope-containing arginine and lysine to control or stimulated cell cultures (136). The cell lysates from both cell cultures can then be mixed and processed as in the biotin switch method. If the same peptide is enriched from both control and treated samples, it will elute at the same time in the LC-MS analysis, but the mass spectra will differ by a known number of Da, and the relative amounts can be calculated from the intensities in the MS peaks (136).
Other approaches to identify S-nitrosated proteins and the location of the modification include the use of organomercurial compounds to either trap or tag S-nitrosothiols, after blocking free thiols (137) or the selective reaction of S-nitrosothiols with derivatized phosphines to tag S-nitrosated peptides in one step through reductive ligation (138).
Nitrite and nitrate
Nitrite (NO2−, IUPAC name dioxidonitrate(1−)) and nitrate (NO3−, IUPAC name trioxidonitrate(1−)) were considered for a long time to be rather inert products of NO• oxidation. The concentration of NO3− in plasma of fasting individuals is 20–40 μm, and it is considered to derive mostly from the reaction between NO• and oxyhemoglobin, but also from the diet (139). The concentration of NO2− in plasma is significantly lower (50–300 nm) because there are several processes by which it can be converted to NO• or further oxidized to NO3− (140). Nitrate is concentrated in saliva and can be converted to NO2− by bacteria in the oral cavity (139). Xanthine oxidase has also been shown to reduce NO3− to NO2−, but it seems to be a minor contribution compared with the oral microbiome (140).
Nitrite can be reduced to NO• by different proteins, including deoxyhemoglobin, deoxymyoglobin, xanthine oxidase, and aldehyde oxidase (141). The reduction by deoxyhemoglobin is thought to be quantitatively the most important pathway for the generation of NO• from NO2− and responsible for the NO•-like effects of NO2− infusion in the presence of red blood cells (141). The NO2− reductase activity of deoxyhemoglobin leads to the formation of NO• and methemoglobin (Equation 12).
| (Eq. 12) |
Detection of nitrite and nitrate
There are several methods to detect NO2− in biological samples. The simplest method to measure NO2− is the Griess method, which sensitivity is in the micromolar range. The method is based on the diazotization of sulfanilamide by NO2− in acidic pH and the subsequent reaction with N-(1-naphthyl)ethylenediamine to yield an intensely pink-colored product with absorption maximum at 540 nm (Fig. 4) (142). The measurement of NO3− is usually done by first converting it to NO2−, either by using vanadium chloride or the enzyme NO3− reductase (142). The analysis can readily be automated to measure NO2− and NO3− (143). Lower concentrations of NO2− (down to 20 nm) can be quantified by the formation of the fluorescent triazole derivative of DAN (see section above on fluorescent detection). In this case, the reaction of NO2− with DAN is done at acidic pH (Fig. 3), and then the fluorescence of the product is measured at alkaline pH (144).
Figure 4.

Detection of nitrite and nitrate with the Griess method. Sulfanilamide reacts with NO2− in acidic pH to yield a diazonium intermediate that subsequently reacts with N-(1-naphthyl)ethylenediamine to yield the intensely colored Griess product with absorption maximum at 540 nm. NO3− can be converted to NO2− with vanadium chloride or NO3− reductase to allow both NO3− and NO2− to be measured.
Nitrite and nitrate can also be quantified by HPLC, capillary electrophoresis, and GC-MS (145, 146). For low concentrations such as those often encountered in biological samples, the ozone-based chemiluminescence method (see section on chemiluminescence and Fig. 3) offers the required nanomolar sensitivity. In this case, the purge vessel needs to be filled with the triiodide acidic solution that converts NO2− to NO• that is then carried to the detection cell by the carrier gas. Nitrate is measured by first reducing it chemically or enzymatically to NO2−. The contribution of other species such as S-nitrosothiols to the signal is controlled by running samples treated with acidic sulfanilamide to trap all free NO2−.
Nitroxyl
The product of the one-electron reduction of NO• is HNO (nitroxyl, azanone, nitrosyl hydride, and hydrogen oxonitrate). The reduction potential of this process, E0′ (NO•, H+/HNO) ∼−0.55 V at pH 7 (18, 19), is quite low, but high enough to make endogenous HNO formation a possibility. Biological studies are usually performed using nitroxyl donors (e.g. Angeli's salt). The ground state of HNO is a singlet in which all the electrons are spin-paired, whereas that of NO− (nitroxyl anion, oxonitrate (1−)) is a triplet with two unpaired electrons (18, 19). Thus, deprotonation is spin-forbidden and slow, and the pKa of HNO is 11.4 (147).
Nitroxyl can react with soft electrophiles (18). In vivo, the preferential reactions of HNO are with thiols and metal centers. For example, the reaction between HNO and GSH, which is present in millimolar concentrations inside cells, has a rate constant of 3.1 × 106 m−1 s−1 (148). In addition, HNO can dimerize yielding nitrous oxide (N2O) and water (k = 8 × 106 m−1 s−1) (147). Nitroxyl can also react with oxygen to form peroxynitrite with a rate constant of 1.8–2 × 104 m−1 s−1 at pH 7.4 (148), but this process is relatively slow under biological conditions and has low relevance. More information on the biochemistry of HNO can be found in Refs. 149–152.
Detection of nitroxyl
Nitroxyl can be detected by observing the dimerization product, N2O, by gas chromatography (GC) (152). It can also be detected by membrane inlet MS, in which HNO traverses a membrane before reaching the mass spectrometer (153) (Fig. 5, A and B).
Figure 5.

Detection of nitroxyl. A, detection based on the HNO dimerization product N2O by GC. B, membrane inlet MS for the detection of HNO and its decay products. C, reaction with metalloporphyrins for spectrophotometric detection. D, electrochemical sensor based on the reaction of HNO with a cobalt(III) porphyrin. E, fluorogenic probe based on the reaction of copper(II) to copper(I). F, reaction between HNO and a nitroxide TEMPO derivative to form NO• and a hydroxylamine which, appropriately derivatized, is fluorescent. G, reaction between HNO and a 2-mercapto-2-methylpropionic acid fluorogenic derivative. H, reaction of HNO with an ester of 2-(diphenylphosphino)benzoic acid to give a benzamide phosphine oxide and an alcohol that, appropriately derivatized, is fluorescent. Some protons are omitted for simplicity.
Iron(III) porphyrins react with HNO forming nitrosyl–iron(II) porphyrins (154). The products can be detected both spectrophotometrically and by the typical three-line EPR signal of a ferrous nitrosyl complex under anaerobic conditions (155). Manganese(III) porphyrins also react with HNO leading to a large shift in the UV-visible Soret band, which can be used for colorimetric detection of HNO (Fig. 5C) (156).
Cobalt(III) porphyrins react with HNO and constitute the basis of an amperometric electrochemical sensor for HNO (Fig. 5D). In the resting state, the polarized electrode (0.8 V) contains Co(III) porphyrin. When the porphyrin reacts with HNO it forms a Co(III)–NO− complex that is oxidized releasing NO• and the Co(III) porphyrin, ready for another cycle. The current intensity is proportional to HNO, and the sensitivity is in the nanomolar range. The success of the electrode is based on the fact that HNO reacts with Co(III) and not with Co(II) porphyrins, whereas NO• reacts with Co(II) and not with Co(III). This is an advantage of Co(III) over Fe(III) porphyrins, which react both with NO• and HNO (149, 157, 158).
Nitroxyl can reduce Cu(II) to Cu(I) and NO•. This is the basis of a group of fluorogenic probes in which the reduction of the metal ion is concomitant with the release of fluorescence quenching (Fig. 5E). The probes should be used with caution for the potential reduction by other reductants, as well as interference from hydrogen sulfide (H2S), S-nitrosothiols and oxygen (159–161).
Nitroxyl reacts with stable nitroxide free radicals such as (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO) with rate constants of 104–105 m−1 s−1 forming the hydroxylamine and NO• (Fig. 5F) (162, 163). Fluorogenic TEMPO derivatives have been prepared in which the nitroxide group quenches the fluorescence, which is released when the nitroxide is converted to the hydroxylamine (160, 164). Due to the complex chemistry and to the potential to react with other reductants and oxidants, the use of the nitroxide probes in biological systems is limited.
Nitroxyl reacts fast with thiols. The formation of GSH sulfinamide (GS(O)NH2) from the reaction of GSH with HNO can be used as footprint for HNO. An N-hydroxysulfenamide is formed as an intermediate, and the final sulfinamide can be separated and detected by HPLC or MS (Equation 13) (165, 166).
| (Eq. 13) |
A probe has been developed that consists of an ester of 2-mercapto-2-methylpropionic acid and a fluorescent compound. The reaction of HNO with the thiol forms an N-hydroxysulfenamide intermediate that cyclizes releasing the fluorophore (Fig. 5G) (160, 167).
Nitroxyl reacts fast with arylphosphines to yield phosphine oxides and azaylides (Equation 14). The rate constants are in the order of 106 m−1 s−1 (168, 169). The azaylides are indicative of the formation of HNO and can be detected by NMR and MS, although, depending on the phosphine used, they may hydrolyze to the corresponding phosphine oxide. Although arylphosphines are resistant to reductants, possible interference by S-nitrosothiols is a potential concern (170).
| (Eq. 14) |
The azaylides are nucleophilic and can react with an adjacent electrophilic group such as an ester or a carbamate. When the azaylide attacks the carbonyl, alcohol is released, and a unique amide phosphine oxide product is formed (Fig. 5H). This product, as well as the alcohol, can serve as reporters for HNO. The hydrolysis of the probe should be controlled as well as possible interference from S-nitrosothiols (160, 169–171).
Despite the progress in the development of methods to measure HNO, the potential limitations should be carefully addressed. More than one method should be used, preferentially in combination with HPLC or MS detection of HNO-specific products (160).
Peroxynitrite
Peroxynitrite (ONOO−) and peroxynitrous acid (ONOOH) are formed through the diffusion-controlled reaction between O2•− and NO• (Equation 15). IUPAC names for ONOO− and ONOOH are (dioxido)oxidonitrate(1−) and (hydridodioxido)oxidonitrogen, respectively. In this text, the term peroxynitrite is used for the mixture of ONOO− and ONOOH, unless specified.
| (Eq. 15) |
Peroxynitrite is a powerful one- and two-electron oxidant; the reduction potentials are E0′ (ONOOH, H+/NO2•, H2O) = 1.6 V and E0′ (ONOOH, H+/NO2−, H2O) = 1.3 V (172). Peroxynitrous acid can traverse membranes through simple diffusion, whereas ONOO− can use anion channels. The anion is a good nucleophile, and ONOOH can act as an electrophile. In buffer, ONOOH (pKa 6.8, Equation 16) can decay to nitric acid (HNO3) plus a 30% fraction of HO• and NO2• radicals (Equation 17), but this process (k = 0.9 s−1 at pH 7.4 and 37 °C) is relatively slow and has limited physiological significance. The most relevant biological targets for peroxynitrite are peroxiredoxins, GSH peroxidases, CO2, and metal centers (1, 7, 86). The peroxiredoxins are thiol-dependent peroxidases that constitute the most efficient peroxynitrite scavengers known to date, with rate constants of ∼0.1–10 × 107 m−1 s−1 and high concentrations in different cellular compartments (173). In addition, peroxynitrite can react fast (k ∼104 m−1 s−1 at pH 7.4) (174) with CO2, which is abundant in tissues (1.2 mm), leading to the formation of secondary radicals, CO3•− and NO2•, in 33% yield (Equation 18) (89).
| (Eq. 16) |
| (Eq. 17) |
| (Eq. 18) |
The reactions with metal centers are diverse. Peroxynitrite can be reduced by one electron yielding NO2• as the metal center is oxidized, or by two electrons yielding NO2−. In addition, some hemeproteins (e.g. methemoglobin) catalyze peroxynitrite isomerization to NO3−, whereas others (e.g. Fe(III) cytochrome c) do not react at all (1, 7).
Peroxynitrite detection
Because of its short life in cells and tissues (∼1 ms) (7), peroxynitrite cannot be detected in biological samples through direct spectroscopic techniques. Nevertheless, the UV absorbance of ONOO− (ϵ302 = 1700 m−1 cm−1) (175) has proven to be very useful for the quantification of stock solutions in the laboratory at alkaline pH, as well as for following ONOO− decay in stopped-flow kinetic experiments.
One analytical approach for the detection of peroxynitrite is the use of probes that react with peroxynitrite itself or with its downstream radicals (NO2•, CO3•−, and HO•). Because the specificity of the probes is not always straightforward, particularly for the latter, the modulation of O2•− and NO• formations, which are the precursors of peroxynitrite, should accompany the results obtained with probes. Another analytical approach to evidence the involvement of peroxynitrite in a certain biological process is the detection of nitrotyrosine, a stable product formed from the reaction of radicals derived from peroxynitrite with tyrosine residues. As in the case of the probes, confirmatory evidence is required. These approaches are described in the next sections.
A growing number of small fluorogenic organic molecules designed and synthesized to detect peroxynitrite are reported constantly, having different selectivity and sensitivity toward this oxidant. The basic common characteristic is to have weak basal fluorescence, which is largely increased upon oxidation (176–178). In terms of the reaction mechanism, fluorogenic probes can be divided in two main groups: 1) probes that react with the radicals derived from peroxynitrite and yield a fluorescent end product by a radical mechanism; and 2) probes that react directly through a nucleophilic attack by peroxynitrite anion (ONOO−) to a particular functional group of the electrophilic probe, releasing masked fluorescence. The probes that react directly with ONOO− are potentially more straightforward, specific, and quantitative. They must react fast (>105–106 m−1 s−1) and outcompete other routes of decay. Besides, genetically-encoded fluorescent protein sensors for peroxynitrite have been described recently (179, 180). They use similar principles as some of the chemical probes that lead to direct detection of this oxidant (i.e. boronate-based compounds, see below).
Importantly, detection methods based on probes reveal only a minor fraction of total peroxynitrite, because a large proportion reacts with other targets in the cell. Moreover, the fraction trapped by the probe may vary with cell type or metabolic state according to the abundance of alternative targets (181). For a full review of chemical probes for peroxynitrite detection, see Refs. 176, 181.
Probes that react with the radicals derived from peroxynitrite
Probes frequently used for oxidant detection in biological systems are reduced dyes like 2′,7′-dichlorodihydrofluorescein (DCFH2) and dihydrorhodamine (DHR-123). Although extensively used, they present a series of limitations and caveats (5). The general reaction mechanism is a one-electron oxidation by potent one-electron oxidants such as those derived from peroxynitrite (NO2•, CO3•−, and HO•) (182) yielding a radical intermediate (DCF•−), which is afterward oxidized to highly resonant moieties responsible for the increase in fluorescence emission (DCF) (Fig. 6A). These probes do not react directly with peroxynitrite (178, 183). Neither NO• nor O2•− are able to oxidize either probe at significant yields; however, these radicals may react with the radical intermediate (DCF•−) in termination reactions giving nonfluorescent products (184). In addition to peroxynitrite-derived radicals, other potent one-electron oxidants such as those produced from heme peroxidases and other metalloproteins in the presence of H2O2 can generate fluorescent DCF (177). Thiyl radicals (RS•) derived from the oxidation of GSH can also oxidize DCFH2 with a significantly high rate constant of ∼107 m−1 s−1 at pH 7.4 (185). Moreover, DCF-dependent fluorescence can be self-amplified by redox-cycling of the one-electron oxidized dye (5).
Figure 6.

Detection of peroxynitrite. A, mechanism of 2′,7′-dichlorodihydrofluorescein (DCFH2) oxidation. The reduced probe (DCFH2) is oxidized by peroxynitrite-derived radicals and other one-electron oxidants yielding a radical intermediate (DCF•−) that is oxidized by oxygen to yield fluorescent DCF. Thin arrows show alternative reactions and redox cycles. AH− stands for ascorbate. B, boronate oxidation by peroxynitrite. The major pathway (>85%, above) consists of heterolytic cleavage of the peroxyl bond leading to phenol which, appropriately derivatized, is fluorescent. The minor radical pathway (below) involves homolytic cleavage of the peroxyl bond giving NO2• and a phenyl-type radical that yields the nitro-derivative (201). C, structures of boronate-derived probes. CBA and CBE indicate a boronic acid or a boronic pinacolate ester attached to a coumarin scaffold, respectively (193). Fl-B (194), FlAmBE (192), and FBBE (205) are boronic pinacolate ester derivatives linked to a fluorescein scaffold with structural modifications as shown. D, genetically-encoded boronate-based GFP for the detection of peroxynitrite. Nucleophilic attack by the peroxynitrite anion to the phenylalanine boron moiety results in the formation of tyrosine and fluorescence. Modified from Ref. 176.
Luminol chemiluminescence can also be used to detect the radicals derived from peroxynitrite (NO2•, CO3•−, and HO•) as well as other strong one-electron oxidants. The mechanism involves the one-electron oxidation of luminol to initiate the reactions that lead to light emission. Chemiluminescence is increased in the presence of CO2 and inhibited by thiols, urate, and NO• (176, 184, 186).
The lack of specificity of these probes constitutes an important limitation. DCFH2, DHR, and luminol are not specific for peroxynitrite and cannot be used as unique tools. These probes may be useful to detect an overall increase in oxidant generation in cells, but they do not allow us to identify the particular species involved. Their use for the detection of peroxynitrite or any other particular oxidant is ambiguous and inconclusive and should be avoided unless complemented with other more specific methods.
Probes that react directly with peroxynitrite anion
Another mechanistic possibility for the detection of peroxynitrite involves a nucleophilic attack by the oxidant to an electrophilic functional group supported on the probe, leading to fluorescence. Different functional groups have been proposed as electrophilic centers, like activated ketones (187–189), α-ketoamides (190, 191), and boronates (boronic acids or boronic esters) (192–194).
A group of probes named HKGreen (HK is Hong Kong) numbered 1–4, in which the electrophilic center is an activated ketone, has been reported for peroxynitrite detection. Structurally, HKGreen-1 holds a trifluoro-ketone linked to a dichlorofluorescein scaffold through an aryl ether linkage (187); HKGreen-2 holds a trifluoro-ketone linked to BODIPY through phenol (188); and HKGreen-3 holds a trifluoro-ketone linked to a rhodol scaffold (189). All of them can react with peroxynitrite by a mechanism where the ketone generates a dioxirane intermediate upon reaction with the oxidant, which oxidizes the phenyl ring and releases the fluorescent molecule. The poor yields of the reactions of HKGreen-1–3 with peroxynitrite (187–189) limit their utility in biological systems.
Another possible electrophilic functional group is α-ketoamide. DCM-KA, an α-ketoamine moiety attached to dicyanomethylene-4H-pyran, was recently proposed (191). Although it selectively responds to peroxynitrite with fluorescence emission in the near-IR region (λem >600 nm), the probe has a slow response (90 s), although complete kinetic analyses are lacking (191).
The arrival of organoborane compounds to the redox biology field over the last decade represented a major breakthrough. Boronate-based probes are successfully applied to the assessment of peroxynitrite in biological systems (192, 194). Even though they were originally conceived for hydrogen peroxide detection (195, 196), it was later demonstrated that boronate derivatives react considerably faster with peroxynitrite than with hydrogen peroxide (192–194) (see below). These compounds are particularly promising due to the simplicity of their reaction with peroxynitrite and are arising as the most suitable probes for detecting and even quantifying this oxidant. They can also be directed to compartments (e.g. mitochondria) (197).
Structurally, boronic acids are trivalent boron-containing organic compounds that hold one alkyl or aryl substituent and two hydroxyl groups to fill the remaining valences on the boron atom (RB(OH)2). Replacement of the hydroxyl groups of boronic acids by alkoxy or aryloxy groups gives boronic esters (RB(OR′)2). The sp2-hybridized boron atom possesses a vacant p orbital due to the deficiency of two electrons and contains only six valence electrons, thus making boronic acids and boronic esters electrophilic centers and mild Lewis acids (198). Therefore, several nucleophiles like peroxynitrite (ONOO−), ionized hydrogen peroxide (HOO−), peroxymonocarbonate (HCO4−), amino acid hydroperoxides (ROO−), or hypochlorite (ClO−) are prone to react with the electrophilic center of boronates (Equation 19). The rate constant of the reaction with ONOO− is several orders of magnitude higher (kONOO− ∼106, kClO− 102, kHCO4− 170, kROO− ∼20, and kH2O2 ∼1–2 m−1 s−1) at physiological pH (193, 194, 199, 200).
| (Eq. 19) |
The reaction mechanism with peroxynitrite, similar to that with H2O2, involves a nucleophilic addition of ONOO− (or HOO−) to the electrophilic boron atom generating an anionic quaternary intermediate that suffers a subsequent heterolytic cleavage at the peroxyl bond and hydrolysis giving phenol as the major nonradical end product (∼85–99% yield) (194, 201). The phenol, appropriately derivatized, is fluorescent. In addition, the reaction with ONOO− typically involves a minor pathway where homolytic cleavage of the peroxyl bond gives radical intermediates, NO2• and a phenyl-type radical, that combine to form a nitrobenzene-type product. This product is a peroxynitrite footprint, because the major phenol product is common for all ROO− or ClO− (Fig. 6B) (201). The mechanism of oxidation of boronates by ONOO− has been studied, both experimentally and theoretically, using density functional theory calculations (193, 201). Moreover, work using isotope-labeling studies is ongoing to track the origin of oxygen atoms in the oxidation products of the redox probes.
Although the high rate constant predicts preferential reaction of boronate probes with peroxynitrite versus other oxidants, confirmation of peroxynitrite involvement in a certain process should be provided by the detection of the products of the minor radicalar pathway of reaction, as well as by modulation of the formation and decay of peroxynitrite precursors.
Several boronic acids or boronic esters attached to different fluorescent scaffolds (coumarin-derivatives (202–204), fluorescein-derivatives (192, 194, 205), or BODIPY-derivatives (206, 207)) are described as fluorogenic probes, where the reaction with the oxidant releases the oxidized fluorescent product (Fig. 6C).
Fluorescein-dimethylamide boronate (FlAmBE) (192), fluorescein-boronate (Fl-B) (194), and 4-(pinacol boronate)benzyl-derivative fluorescein methyl ester (FBBE) (205) were recently reported as fluorogenic probes derived from modified fluorescein attached to a pinacol boronic ester for monitoring peroxynitrite in biological systems. Unlike Fl-B or FlAmBE, where there is a straight reaction mechanism with peroxynitrite as described above, the oxidative conversion of FBBE to the fluorescent product (fluorescein methyl ester) is a two-step reaction (205). The reaction of an oxidant (ROO− or ClO−) toward the boronate group leading to the corresponding phenol is the first step of this process. The second step is a slow p-methide quinone elimination leading to the formation of fluorescein methyl ester. Although the rate constant for the reaction of FBBE with ONOO− is high (>105 m−1 s−1), the buildup of the fluorescent product is slow (k = 0.09 s−1), due to a gradual p-quinone methide elimination (205).
An improvement of boronate probes derived from fluorescein scaffolds with respect to coumarin scaffolds is that the spectroscopic properties of the former (FlAmBE: λex = 485 nm and λem = 535 nm (192); Fl-B: λex = 492 nm and λem = 515 nm (194); and FBBE: λex = 494 nm and λem = 518 nm (205)) allow their use in common laboratory equipment for cellular assays, such as flow cytometry and epi-fluorescence microscopy. In contrast, other reported probes such as coumarin-7-boronic acid (CBA) (202) and/or coumarin-7-boronic acid pinacolate ester (CBE) (λex = 332 nm and λem = 470 nm) present limitations for those techniques. Moreover, although coumarin-derived and fluorescein-derived boronic probes react with comparable kinetic rate constants toward peroxynitrite (k ∼1 × 106 m−1 s−1), Fl is a more efficient fluorophore than 7-hydroxycoumarin (COH), and it has a higher molar absorption coefficient (ϵFl, 490 nm = 76,900 m−1 cm−1; ϵCOH, 323 nm = 11,200 m−1 cm−1) and higher quantum yield (ΦF, Fl = 0.93; ΦF, COH = 0.15) (208, 209). Therefore, Fl-B is more sensitive than CBA. Recently, Fl-B enabled the detection of basal rates of peroxynitrite generation in vascular endothelial cells dependent on endothelial NOS activation, secondary to ionomycin-induced calcium ion influx (194).
Curiously, a boronate-derived probe named GSH-ABAH, supported on 4-amino-2-(benzo[d]thiazol-2-yl)phenol (ABAH) and with a thiol-reactive maleimide substituting the amino group, is proposed to simultaneously detect ONOO− and biological thiols by a combination of excited state intramolecular proton transfer and photoinduced electron transfer, giving an increase in fluorescence emission when both analytes are present (210). This unusual strategy may need an extensive characterization of the reaction and independent validation under biologically-relevant conditions.
Recently, a novel genetically-encoded boronate-based GFP was reported for the detection of peroxynitrite in mammalian cells (179, 180). GFP fluorescence requires the presence of a key tyrosine residue (Tyr-66) that together with Ser-65 and Gly-67 participate in a cyclized structure inside a protein barrel that constitutes the actual fluorophore (211). Modern synthetic biology techniques allow the incorporation of unnatural amino acids during protein translation, by which Tyr-66 is substituted by phenylalanine boronic acid (p-B(OH)2Phe), resulting in a nonfluorescent version of GFP. Upon exposure to peroxynitrite, the boronate-modified Phe is converted to Tyr generating the actual GFP fluorophore (Fig. 6D). It is interesting to point out that GFP had been previously proposed as a way to follow the formation of peroxynitrite and other nitrating species (59). However, in this early report, the critical Tyr-66 became nitrated to 3-nitrotyrosine, which resulted in a decrease of fluorescence intensity.
Because the available methodologies for peroxynitrite detection have a series of limitations, there are several practical aspects that must be taken into consideration. When studying peroxynitrite generation in biological systems, the pharmacological modulation is mandatory to confirm the identity of the radical precursors and oxidants that are being generated. For instance, the use of enzyme inhibitors is useful for unequivocally identifying peroxynitrite formation (i.e. modulation by NOS inhibitors or by suppression of O2•− formation). For a full discussion on practical considerations when assessing peroxynitrite in cells using fluorogenic probes, see Refs. 176, 181.
3-Nitrotyrosine
Peroxynitrite can lead to the nitration of free and protein-bound tyrosines. The mechanism of tyrosine nitration is free radical-dependent and does not directly involve peroxynitrite. Instead, it involves a strong one-electron oxidant and NO2•. The initial one-electron oxidant can be CO3•−, NO2•, oxometal complexes such as Compounds I and II of hemeperoxidases, HO•, and lipid peroxyl radicals (LOO•), among others (Fig. 7). Tryptophan, nucleic acid bases, and polyunsaturated fatty acids can also become nitrated, but the better studied is tyrosine (1).
Figure 7.

Free radical-mediated formation of nitrotyrosine. Strong one-electron oxidants oxidize tyrosine to tyrosyl radical, which combines with NO2• forming 3-nitrotyrosine. Some protons are omitted for simplicity.
Due to the radical nature of the mechanism of tyrosine nitration, other sources of one-electron oxidants and NO2• that do not involve peroxynitrite can give rise to 3-nitrotyrosine. This is the case of hemeperoxidases in the presence of hydrogen peroxide and NO2− (82). In addition, tyrosine can also become nitrated by the reaction of tyrosyl radicals with NO• followed by two oxidation steps (212). Nitrating species can also be generated by NO2− under acidic conditions (213). Thus, the detection of 3-nitrotyrosine alone cannot be considered a specific footprint of peroxynitrite unless in the light of additional confirmatory evidence.
Nitration modifies the properties of the tyrosine. First, the acidity of the phenolic group is increased (pKa of 3-nitrotyrosine 7.5), generating an extra negative charge in the protein at neutral pH. This is useful for separative strategies based on isoelectric focusing of nitrated proteins (214, 215). Second, the 274 nm absorbance maximum shifts to 360 nm at acidic pH (ϵ360 = 2790 m−1 cm−1) or 440 nm at alkaline pH (ϵ440 = 4400 m−1 cm−1) (184). Although this red shift is useful for chromatographic separations with UV-visible detection and for the quantification of 3-nitrotyrosine standards, it can lead to photochemical decomposition in MALDI-based MS approaches with 337 nm lasers. Finally, the hydrophobicity of a 3-nitrotyrosine-containing peptide increases with respect to the unmodified one. 3-Nitrotyrosine is considered a relatively stable end product. Putative 3-nitrotyrosine denitrase and reductase activities that catalyze the removal of the nitro group or its reduction to amino in 3-nitrotyrosine-containing proteins, respectively, have been reported in mammalian tissues and in bacteria. The enzymes responsible have not yet been identified (216–218).
Tyrosine nitration is a low-yield process. For instance, peroxynitrite-dependent tyrosine nitration in phosphate buffer ranges from ∼6 to ∼18% of the peroxynitrite added in the absence or presence of bicarbonate, respectively, and much less in the presence of competing biomolecules (174, 219). In cells and tissues, a relatively small number of proteins is found significantly nitrated and in only a few tyrosine residues. This introduces challenges in the analytical strategies, because the detection of about one 3-nitrotyrosine within one million tyrosines is required (1, 4, 220–222).
Another challenge is the artifactual formation of 3-nitrotyrosine during sample processing, for example, from residual NO2− under acidic conditions. This is a major methodological concern that has contributed to the thousand-fold scattering of 3-nitrotyrosine values in biological samples (222–225). Separation of 3-nitrotyrosine from its precursors (226) or reduction to 3-aminotyrosine by sodium dithionite (227), early in sample preparation protocols, is useful in preventing artifactual nitration. In MS-based procedures, tyrosine labeled with stable isotopes can be added to the sample to control for artifactual nitration, in addition to serving as internal standard for tyrosine measurement (224, 228).
The analytical challenges increase even further if the aim is to detect a nitrated peptide or protein rather than free 3-nitrotyrosine, because there is the need to extract, separate, and enrich modified proteins and to hydrolyze them into peptides or amino acids.
Several approaches have been developed for the qualitative and quantitative assessment of free or protein-bound 3-nitrotyrosine. These methods include immunoassays, HPLC with various detection methods, and gas or liquid chromatography coupled to MS. Due to the potential for sensitivity and selectivity, strategies based on MS have become the gold standard of 3-nitrotyrosine determination and have started to yield reference values. For example, the concentration of 3-nitrotyrosine in plasma is subnanomolar (222). A thorough discussion of the method of validation applied to 3-nitrotyrosine analysis can be found in Refs. 221, 222. In the following sections, a brief description of available methods is provided.
Antibody-based methods for 3-nitrotyrosine detection
Nitration changes the immunogenicity of a protein through the generation of new epitopes. This has biomedical consequences in the triggering of immune reactions and in the generation of autoimmune responses (229, 230). In addition, this is the basis of procedures for the detection of 3-nitrotyrosine rooted on the use of antibodies. Poly- and monoclonal antibodies have been raised and are now commercially available. Immunohistochemical methods, immunoblotting techniques, and ELISAs have been developed and applied to the study of biological samples (184, 231–234). The typical readout of antibody-based methods is the difference in immunoreactivity in samples versus controls. However, procedures are subject to wide variations, particularly regarding the selectivity of the antibody, and unless validated, they lead to qualitative or at best semi-quantitative results (223). Recently, a highly-sensitive electrochemiluminescence-based ELISA was introduced for the assessment of biological samples and validated with MS (235).
Analysis of free 3-nitrotyrosine by HPLC-based methods
HPLC with UV-visible detection (236) or fluorescence detection (following derivatization with fluorogenic compounds) (237) has been employed to measure 3-nitrotyrosine. However, these approaches are not sensitive or selective enough for their application to biological samples. Reduction to 3-aminotyrosine followed by HPLC with electrochemical detection presents improved sensitivity (225), but it cannot match the selectivity provided by MS-based approaches (222, 223).
Analysis of free 3-nitrotyrosine by GC-MS, GC-MS/MS, and LC-MS/MS
Strategies based on GC for the separation of the analytes must follow the conversion of 3-nitrotyrosine to a volatile form, either by reduction to 3-aminotyrosine and/or derivatization. Particularly useful is the incorporation of fluorine atoms in the derivative through the use of fluorinated reagents in the case of MS procedures based on negative ion electron capture detection (226, 238–240). The perfluorinated derivatives undergo electron capture and dissociation to yield a single intense anion that can be monitored by selected ion monitoring (GC-MS analysis). Subsequent analysis by selected reaction monitoring after collision-induced dissociation (GC-MS/MS) increases selectivity.
The use of LC instead of GC presents the advantage that no derivatization steps are needed. Positive electrospray ionization mode is usually used. LC-MS is not selective enough, but collision-induced dissociation of 3-nitrotyrosine in LC-MS/MS analysis produces a few characteristic ions that can be used for quantification. Specifically, protonated unlabeled 3-nitrotyrosine (m/z 227) yields species with m/z 181 and m/z 90.
In both GC and LC-based MS methods, quantitative data can be obtained by comparison with the signals produced by an internal standard, which allows corrections for variability, sample losses, and matrix effects. The internal standard for MS consists of 3-nitrotyrosine labeled with the stable isotopes 13C or 15N. These isotopic homologues behave identically to nonlabeled 3-nitrotyrosine in extraction, chromatography and ionization steps but provide ions with increased m/z. Some of these standards are commercially available. In a typical procedure, labeled 3-nitrotyrosine is added to the samples and the calibration standards in known amounts. The area or height of the desired peak is related to that of the internal standard in the samples and is compared with the calibration data. The inclusion of labeled tyrosine in addition to labeled 3-nitrotyrosine allows the quantification of total tyrosine and the control of artifactual nitration (224, 228). For example, in a typical procedure 3-[13C6]nitrotyrosine is added in a known amount to a sample as internal standard for 3-nitrotyrosine quantification; [13C915N1]tyrosine is added as internal standard for total tyrosine quantification, and [13C6]tyrosine is added to control for potential artifactual generation of 3-nitrotyrosine during sample processing (241).
Analysis of 3-nitrotyrosine in proteins
Quantitative data on the amount of 3-nitrotyrosine in a protein-containing sample can be obtained by extraction of total proteins followed by hydrolysis by chemical or enzymatic methods and, finally, analysis of free 3-nitrotyrosine by the procedures described above. The extraction step, typically with an organic solvent, is useful for NO2− elimination as well as protein precipitation. Total hydrolysis can be achieved with 6 m hydrochloric acid at 116 °C in vacuo, following NO2− removal (225, 228). Alkaline hydrolysis has also been tried (239). Alternatively, a mixture of proteolytic enzymes (Pronase) can be used, but the former is preferred, because enzymatic hydrolysis is not always complete, and the enzymes themselves can contribute to the values of tyrosine and 3-nitrotyrosine measured.
If information on a particular protein instead of a whole mixture is required, separation or enrichment steps can be included. These steps include mono- or two-dimensional electrophoresis and chromatographic or immunoaffinity procedures. A useful aspect to take into account in the design of separation steps is the increase in acidity of tyrosine residues upon nitration, which leads to a decrease in the pI of the protein (214, 215).
To reveal the sites of nitration in the proteins, peptide mapping can be performed by hydrolysis with trypsin to obtain the tryptic fragments followed by LC-MS/MS to separate the peptides and analyze them. The modified peptides are then identified by search algorithms. Importantly, the results should be confirmed through targeted MS/MS approaches, in which prior knowledge of the modification site allows for selected/multiple reaction monitoring and parallel reaction monitoring (221, 241, 242).
In MALDI-based procedures for peptide and protein analysis, 3-nitrotyrosine residues absorb the light of typical lasers (337 nm) and produce a characteristic pattern of ions in which the m/z is decreased by 16 and 30. The unique triplet signal enables to further identify the nitrated peptides (243, 244). Nevertheless, methods based on electrospray ionization present the advantages that photochemical decomposition is avoided and that the typical ion with m/z 181 can be detected and serve as a footprint for 3-nitrotyrosine (221).
A typical proteomics approach for the detection of nitrated proteins in complex samples would involve two-dimensional separation, immunoblotting, manual removal of selected spots, in-gel digestion, and MS/MS identification, with confirmation by targeted approaches (220–222, 245–247). A critical discussion of caveats in nitroproteomics can be found in Refs. 220–222.
Carbonate anion radical
Carbonate anion radical (CO3•−, IUPAC name trioxocarbonate(−1)) is a strong one-electron oxidant with a standard reduction potential (E0′ (CO3•−, H+/HCO3−)) of +1.77 V (248). In biological systems, it can be generated by the homolysis of the initial adduct formed by the reaction of ONOO− and CO2, ONOOCO2−. Although some debate exists in relation to the lifetime of the adduct and the yields of the CO3•− and NO2• radicals (249), it is now accepted that the adduct has a very short half-life (<1 μs) that precludes its participation in bimolecular reactions and that it homolyzes to CO3•− and NO2• in ∼33% yields. The experimental and theoretical evidence in support of the radical model has been recently reviewed (89).
Carbonate anion radical can also be formed in biological systems by the one-electron oxidation of bicarbonate or its conjugate base, carbonate anion (CO32−), by strong enough one-electron oxidants such as HO• (E0′ = +2.31 V at pH 7) (250, 251). This route is expected to be of relevance inside the phagosomes of activated immune cells, where bicarbonate is considered to be a main target for HO• (252). Compounds I and II of heme peroxidases cannot oxidize HCO3− (93). In contrast, CO2/HCO3− enhances the peroxidatic activity of Cu,Zn-superoxide dismutase (253, 254), and although the mechanism is controversial, it leads to CO3•− formation through an enzyme-bound intermediate (199, 255, 256). Xanthine oxidase can also lead to CO3•− (257–259).
The pKa of HCO3· is <0, indicating that the radical only exists in its anionic form in biological systems (260, 261). Accordingly, it cannot diffuse through membranes (262), and it is confined to water-soluble compartments. It can react both by electron transfer or hydrogen abstraction mechanisms (259). It can also participate in oxygen transfer reactions, particularly when reacting with itself or with other free radicals. Furthermore, although some examples of CO3•− addition reactions have been proposed, the products formed are usually unstable, which makes their detection difficult.
The main intracellular targets of CO3•− are thiol-containing moieties, particularly GSH due to its high cellular abundance (k = 5.3 × 106 m−1 s−1 at pH 7) (93, 263), solvent-exposed sulfur-containing residues (k = 4.5 × 107 and 3.6 × 107 m−1 s−1 at pH 7 for Cys and Met, respectively), as well as aromatic residues in proteins (k = 4.6 × 107 and 7 × 108 m−1 s−1 at pH 7 for Tyr and Trp, respectively), ascorbate (k ∼109 m−1 s−1 at pH 11.4), guanine bases in nucleic acids (k ∼108 m−1 s−1), and particularly in plasma, urate (kurate estimated to be ∼108 m−1 s−1) (86, 264). Carbonate anion radical promotes protein–protein (particularly ditryptophan) or DNA–DNA cross-links (265, 266). It also rapidly reacts with transition metal centers or organic cofactors (267–269). Interestingly, bicarbonate enhanced the susceptibility of bacterial suspensions to ionizing radiation, which implies extracellular HO• (252). The higher toxicity caused by extracellular CO3•− compared with HO• is a consequence of the higher diffusion distances of the former, derived from its lower reactivity and longer lifetime.
Detection of carbonate anion radical
The lack of specific biomarkers for CO3•− complicates its detection, both in vitro or in vivo. Carbonate anion radical has a characteristic light absorption spectrum with a peak at 600 nm (ϵ600 = 1930 m−1 cm−1) (260, 270). This has been utilized for measuring the kinetics of the reaction of the radical with different compounds by pulse radiolysis techniques (263, 264, 268). Time-resolved resonance Raman spectroscopy has also been used to characterize CO3•−, which shows a strong polarized and intense band at 1062 cm−1 (261). Furthermore, CO3•− can be directly detected by fast-flow EPR where it shows a singlet band at g = 2.0113 (89, 271). Due to the short half-life of the radical, which is in the microsecond range, direct detection requires rapid or fast flow methodologies, which are not always available or require high reactant amounts (259). Alternatively, spin-trapping EPR methodologies have also been utilized. Indeed, the reaction of CO3•− with DMPO yields an initial unstable adduct, which has been proposed as the O-centered radical DMPO-OCO2·, that then decays to DMPO-OH• (272, 273). Carbonate anion radical can also be detected by means of chemiluminescence. Spontaneous chemiluminescence has been proposed to rely on radical dimerization to an excited species (CO2)2* that decays to the ground state with light emission at ∼440 nm (Equations 20 and 21) (274–277).
| (Eq. 20) |
| (Eq. 21) |
Additionally, the presence of bicarbonate enhanced the yield of luminol chemiluminescence caused either by hydrogen peroxide plus catalyzers or by peroxynitrite, which can be ascribed to CO3•− formation (186, 257). This is in accordance with CO3•− oxidation of aromatic and heterocyclic molecules with second-order rate constants ranging from 5 × 105 to 5 × I07 m−1 s−1 at pH 7 (263, 278).
Hydroxyl radical and its detection
The extremely reactive hydroxyl radical (HO•) can be formed from the homolysis of peroxynitrous acid, which gives NO2• and HO• in 30% yield, while the rest decays to nitric acid (Equation 16-18). This spontaneous decay has a relatively slow rate constant (0.9 s−1 at pH 7.4 and 37 °C) and will be outcompeted by direct reactions with targets such as thiol or selenoproteins, metal centers, or CO2, minimizing the relevance of peroxynitrous acid homolysis in biological HO• generation (1). Nevertheless, it is possible that a very small amount of peroxynitrite-derived HO• initiates oxygen-dependent chain reactions in which oxidation events are amplified, particularly in lipid-rich compartments (279). Undoubtedly, the formation of HO• is relevant in in vitro systems in the absence of alternative targets that react directly with peroxynitrite. In fact, the detection of HO• formation from peroxynitrite contributed to build the concept that NO• can be the precursor of strong oxidants (280). In biological systems, HO• is mainly generated through the Fenton reaction, i.e. through the reduction of hydrogen peroxide to HO• and H2O by reduced metal ions, Fe(II) or Cu(I). It can also be formed from photochemical processes (e.g. photolysis of hydrogen peroxide) and through the radiolysis of water. With a reduction potential of +2.31 V at pH 7 (250), HO• is so oxidizing that it can react with most biomolecules. In addition, the rate constants are close to the diffusion limit, with values of 109–1010 m−1 s−1. The mechanisms of HO• reactions include one-electron abstraction, hydrogen atom abstraction, and addition to double bonds or aromatic rings.
The detection of HO• can be carried out by different approaches that include laser-induced fluorescence and EPR associated with spin trapping (281). The kinetics of HO• reactions have been studied by pulse radiolysis combined with competition kinetics. Hydroxyl radical can also be detected with the use of probes. Due to the very high reactivity of HO• with biomolecules, very high probe loading is required, which can raise concerns. Because HO• adds to aromatic rings, classical methods involve the hydroxylation of salicylic acid or phenylalanine (to m-tyrosine) (282). Often, the hydroxylation reaction can be followed by fluorescence. Various coumarins and many other probes have been developed recently and are reviewed in Refs. 177, 281, 283.
Some reflections on the detection of nitric oxide–derived oxidants
The fate of a certain species is determined by kinetics (i.e. rate constant multiplied by target concentration) as well as by compartmentalization. Detection methods based on probes reveal only a fraction of the total species, because a large fraction reacts with targets and remains undetected. Knowledge of the rate constants and concentrations of probe and targets can help estimate the actual amount of oxidant produced from the amount detected. For example, the rate of peroxynitrite formation that was detected in endothelial cells with a boronate probe was ∼0.01 μm s−1, but it translated into a total of ∼0.1 μm s−1 when the scavenging by other targets was taken into account (194).
Given the loose specificity of some of the methodologies for the detection of NO•-derived oxidants, modulation of NO• formation is mandatory. This can be pharmacologically achieved through the use of NOS inhibitors such as N-nitro-l-arginine methyl ester or NG-monomethyl-l-arginine. Nitric oxide donors may be used to recapitulate the actions of NO•-derived oxidants following NOS inhibition. Alternatively, genetically engineered cell lines or animals in which NOS are knocked out or overexpressed can be used (284). Complementary evidence can be achieved with inhibitors of the formation of other precursors. For example, the levels of the peroxynitrite precursor, O2•−, can be turned down with NADPH oxidase inhibition or superoxide dismutase overexpression. The role of NO• in a certain process can also be probed using a scavenger such as oxyhemoglobin, which reacts fast with NO• forming NO3−. In this regard, the heterologous expression in mammalian cells of bacterial enzymes with strong NO• dioxygenase activity has been proposed as a tool for NO• depletion (285).
Finally, when a certain NO•-derived oxidant or downstream product is detected in the context of a pathophysiological process, it is tempting to conclude that the relation is causative and that the NO•-derived oxidant or downstream product participates in the development of the pathology. However, the relation may be only associative; the NO•-derived oxidant or downstream product may constitute, at best, a biomarker. Further evidence involving pharmacological or genetic modulation of NO•-dependent pathways and their impact on biological outcome is needed to reveal causality.
Concluding remarks
Three decades of research have contributed to building the concept that NO• can exert biological effects both through the direct and usually reversible interaction with specific targets or through the generation of secondary species, many of which can oxidize, nitrosate, or nitrate biomolecules. The reactive species derived from NO• are typically short-lived, and their preferential targets depend on kinetic and compartmentalization aspects. Their elusive character imposes technical challenges in their detection and quantification. In general terms, the strategies are based either on the detection of stable end products or on the use of synthetic probes. These methods are summarized in Table 1. Rigorous use of these strategies requires understanding of the mechanistic pathways involved. In many cases, probes are able to react with several species, and efforts are under way to expand the toolset with probes of suitable selectivity. The evidence about the role of a particular species in a certain pathophysiological process should combine data obtained through more than one analytical or immunochemical procedure as well as complementary evidence coming from the modulation of the formation or decay pathways of the species. A better identification and quantification of NO•-derived oxidants and of their relevant molecular targets, strongly grounded in chemical and biochemical knowledge, will assist to expand rational applications of the NO• field to biology and medicine.
Table 1.
Summary of methods for the detection of nitric oxide–derived oxidants
| Method | NO• | NO2• | N2O3 | NO2− | NO3− | DNICs | RSNO | HNO | ONOO−/ONOOH | CO3•− | HO• |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Oxyhemoglobin oxidation, UV-visible, <1 μm | Yes, fast | Yes, but very slow | Yes, slower than NO• | ||||||||
| NO•-selective electrode | Yes, in neutral buffer | No, but NO2− forms NO• in acid | No, only with Cu(II) and cysteine | ||||||||
| EPR with spin traps | Yes, cPTIO (forms NO2•) or Fe(MGD)2 | Yes, nitromethane and nitrone traps | Yes, DMPO and other traps | ||||||||
| Direct EPR, 200 nm sensitivity | Nitrosyl heme with Fe(II) heme, 77 K | Yes | Nitrosyl heme with Fe(III) heme, 77 K | Fast flow | |||||||
| Fluorogenic diamino probes (DAN and DAF) | Not direct, but through N2O3 or oxidants and NO• | Yes, fast and direct reaction | Yes, in acidic pH | ||||||||
| NOS activity, radiolabeled arginine | Yes, controls with NOS inhibitors and without NADPH | ||||||||||
| Ozone-based chemiluminescence, high sensitivity, 10 nm | Yes, neutral buffer in purge vessel | Reductant in purge vessel | Acidic triiodide in purge vessel | Acidic triiodide in purge vessel | Acidic triiodide in purge vessel | ||||||
| Luminol chemiluminescence | Yes | Not direct but through derived free radicals | Yes | Yes | |||||||
| DCFH2 and DHR fluorescence | Yes | Not direct but through derived free radicals | Yes | Yes | |||||||
| HPLC | Yes | Yes | Yes | Low Mr RSNO | Glutathione sulfinamide | m-Tyrosine or hydroxylated probes | |||||
| UV spectrophotometry | Pulse radiolysis | Pulse radiolysis | Must be purified | Low sensitivity (50 μm) | Stocks and stopped-flow | Pulse radiolysis | |||||
| Griess for nitrite detection (micromolar sensitivity) | No, but oxidation gives NO2− | No, but hydrolysis product is NO2− | Yes | Not direct, but after reduction to NO2− | Not direct, but with Hg(II) | ||||||
| Biotin-switch proteomics | Yes, provides protein identity | ||||||||||
| N2O detection | Yes | ||||||||||
| Oxidized metalloporphyrins, UV-visible or EPR | Yes, interference by reductants and NO• | ||||||||||
| Co(III)-porphyrin electrode | Yes | ||||||||||
| Cu(II)-fluorogenic probes | Yes, interference by reductants | ||||||||||
| Nitroxide probes | Yes, interference by reductants | ||||||||||
| 2-Mercapto-2-methylpropionic acid fluorogenic probe | Yes, confirm specific product | ||||||||||
| Arylphosphine probes | Yes, RSNO interference, confirm specific product | ||||||||||
| Detection of 3-nitrotyrosine, several methods, MS confirmation desirable | Not direct, only in the presence of strong oxidants | Yes, product of NO2• with tyrosyl radical | Yes, in acid | Not direct but through derived free radicals | Forms tyrosyl radical | Forms tyrosyl radical | |||||
| Boronic acid probes | Yes, direct and fast, H2O2 reaction very slow |
This work was supported by Comisión Sectorial de Investigación Científica Grupos_2018 (to R. R. and A. D.), Espacio Interdisciplinario 2015 (to R. R.), Universidad de la República, Uruguay, and Agencia Nacional de Investigación e Innovación Grant FCE_1_2017_1_136043 (to M. N. M). The authors declare that they have no conflicts of interest with the contents of this article.
- NOS
- nitric oxide synthase
- EPR
- electron paramagnetic resonance
- GC
- gas chromatography
- LC
- liquid chromatography
- MS
- mass spectrometry
- MALDI
- matrix-assisted laser desorption/ionization
- DCF
- 2′,7′-dichlorofluorescein
- DCFH2
- 2′,7′-dichlorodihydrofluorescein
- CBA
- coumarin boronic acid
- CBE
- coumarin boronic ester
- Fl-B
- fluorescein-boronate
- FlAmBE
- fluorescein-dimethylamide boronate
- FBBE
- 4-(pinacol boronate)benzyl-derivative of fluorescein methyl ester
- DNIC
- dinitrosyl iron complex
- DAN
- diaminonaphthalene
- DAF
- diaminofluorescein
- DAF-FM
- 4-amino-5-methylamino-2′,7′-difluorofluorescein
- DAF-2–DA
- 4,5-diaminofluorescein diacetate
- PTIO
- 2-phenyl-4,4,5,5-tetramethylimidazoline-1-yloxyl-3-oxide
- Fe(DETC)2
- iron diethyldithiocarbamate
- ICAT
- isotope-coded affinity tag
- SILAC
- stable isotope labeling by amino acids in cell culture
- TEMPO
- (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl
- Fl
- fluorescein
- ABTS
- 2,2′-azino-bis-(3-ethylbenzothiazoline-6-sulfonic acid)
- COH
- 7-hydroxycoumarin
- DMPO
- 5,5-dimethyl-1-pyrroline-N-oxide.
References
- 1. Ferrer-Sueta G., Campolo N., Trujillo M., Bartesaghi S., Carballal S., Romero N., Alvarez B., and Radi R. (2018) Biochemistry of peroxynitrite and protein tyrosine nitration. Chem. Rev. 118, 1338–1408 10.1021/acs.chemrev.7b00568 [DOI] [PubMed] [Google Scholar]
- 2. Heinrich T. A., da Silva R. S., Miranda K. M., Switzer C. H., Wink D. A., and Fukuto J. M. (2013) Biological nitric oxide signalling: chemistry and terminology. Br. J. Pharmacol. 169, 1417–1429 10.1111/bph.12217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ignarro L. J. (1990) Biosynthesis and metabolism of endothelium-derived nitric oxide. Annu. Rev. Pharmacol. Toxicol. 30, 535–560 10.1146/annurev.pa.30.040190.002535 [DOI] [PubMed] [Google Scholar]
- 4. Radi R. (2004) Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. U.S.A. 101, 4003–4008 10.1073/pnas.0307446101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Forman H. J., Augusto O., Brigelius-Flohe R., Dennery P. A., Kalyanaraman B., Ischiropoulos H., Mann G. E., Radi R., Roberts L. J. 2nd., Vina J., and Davis K. J. (2015) Even free radicals should follow some rules: a guide to free radical research terminology and methodology. Free Radic. Biol. Med. 78, 233–235 10.1016/j.freeradbiomed.2014.10.504 [DOI] [PubMed] [Google Scholar]
- 6. Winterbourn C. C. (2008) Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 4, 278–286 10.1038/nchembio.85 [DOI] [PubMed] [Google Scholar]
- 7. Ferrer-Sueta G., and Radi R. (2009) Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem. Biol. 4, 161–177 10.1021/cb800279q [DOI] [PubMed] [Google Scholar]
- 8. Piacenza L., Trujillo M., and Radi R. (2019) Reactive species and pathogen antioxidant networks during phagocytosis. J. Exp. Med. 216, 501–516 10.1084/jem.20181886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sundaresan M., Yu Z. X., Ferrans V. J., Irani K., and Finkel T. (1995) Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 270, 296–299 10.1126/science.270.5234.296 [DOI] [PubMed] [Google Scholar]
- 10. Marine A., Krager K. J., Aykin-Burns N., and Macmillan-Crow L. A. (2014) Peroxynitrite induced mitochondrial biogenesis following MnSOD knockdown in normal rat kidney (NRK) cells. Redox Biol. 2, 348–357 10.1016/j.redox.2014.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lide D. R. (2004) Permittivity (Dielectric Constant) of Gases. 85th Ed., pp. 174, CRC Press, Boca Raton, FL [Google Scholar]
- 12. Zacharia I. G., and Deen W. M. (2005) Diffusivity and solubility of nitric oxide in water and saline. Ann. Biomed. Eng. 33, 214–222 10.1007/s10439-005-8980-9 [DOI] [PubMed] [Google Scholar]
- 13. Shaw A. W., and Vosper A. J. (1976) Solubility of nitric oxide in aqueous and nonaqueous solvents. J. Chem. Soc. Faraday Trans. 1: Physical Chemistry in Condensed Phases 8, 1239–1244 10.1039/F19777301239 [DOI] [Google Scholar]
- 14. Möller M., Botti H., Batthyany C., Rubbo H., Radi R., and Denicola A. (2005) Direct measurement of nitric oxide and oxygen partitioning into liposomes and low density lipoprotein. J. Biol. Chem. 280, 8850–8854 10.1074/jbc.M413699200 [DOI] [PubMed] [Google Scholar]
- 15. Denicola A., Souza J. M., Radi R., and Lissi E. (1996) Nitric oxide diffusion in membranes determined by fluorescence quenching. Arch. Biochem. Biophys. 328, 208–212 10.1006/abbi.1996.0162 [DOI] [PubMed] [Google Scholar]
- 16. Möller M. N., and Denicola A. (2018) Diffusion of nitric oxide and oxygen in lipoproteins and membranes studied by pyrene fluorescence quenching. Free Radic. Biol. Med. 128, 137–143 10.1016/j.freeradbiomed.2018.04.553 [DOI] [PubMed] [Google Scholar]
- 17. Subczynski W. K., Lomnicka M., and Hyde J. S. (1996) Permeability of nitric oxide through lipid bilayer membranes. Free Radic. Res. 24, 343–349 10.3109/10715769609088032 [DOI] [PubMed] [Google Scholar]
- 18. Bartberger M. D., Liu W., Ford E., Miranda K. M., Switzer C., Fukuto J. M., Farmer P. J., Wink D. A., and Houk K. N. (2002) The reduction potential of nitric oxide (NO) and its importance to NO biochemistry. Proc. Natl. Acad. Sci. U.S.A. 99, 10958–10963 10.1073/pnas.162095599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shafirovich V., and Lymar S. V. (2002) Nitroxyl and its anion in aqueous solutions: spin states, protic equilibria, and reactivities toward oxygen and nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 99, 7340–7345 10.1073/pnas.112202099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Enemark J. H., and Feltham R. D. (1974) Principles of structure, bonding, and reactivity for metal nitrosyl complexes. Coord. Chem. Rev. 13, 339–406 10.1016/S0010-8545(00)80259-3 [DOI] [Google Scholar]
- 21. Goodrich L. E., Paulat F., Praneeth V. K., and Lehnert N. (2010) Electronic structure of heme-nitrosyls and its significance for nitric oxide reactivity, sensing, transport, and toxicity in biological systems. Inorg. Chem. 49, 6293–6316 10.1021/ic902304a [DOI] [PubMed] [Google Scholar]
- 22. Toledo J. C. Jr., and Augusto O. (2012) Connecting the chemical and biological properties of nitric oxide. Chem. Res. Toxicol. 25, 975–989 10.1021/tx300042g [DOI] [PubMed] [Google Scholar]
- 23. Radi R. (1996) Reactions of nitric oxide with metalloproteins. Chem. Res. Toxicol. 9, 828–835 10.1021/tx950176s [DOI] [PubMed] [Google Scholar]
- 24. Doyle M. P., and Hoekstra J. W. (1981) Oxidation of nitrogen oxides by bound dioxygen in hemoproteins. J. Inorg. Biochem. 14, 351–358 10.1016/S0162-0134(00)80291-3 [DOI] [PubMed] [Google Scholar]
- 25. Gardner P. R. (2005) Nitric oxide dioxygenase function and mechanism of flavohemoglobin, hemoglobin, myoglobin and their associated reductases. J. Inorg. Biochem. 99, 247–266 10.1016/j.jinorgbio.2004.10.003 [DOI] [PubMed] [Google Scholar]
- 26. Treinin A., and Hayon E. (1970) Absorption spectra and reaction kinetics of NO2, N2O3, and N2O4 in aqueous solution. J. Am. Chem. Soc. 92, 5821–5828 10.1021/ja00723a001 [DOI] [PubMed] [Google Scholar]
- 27. Goldstein S., and Czapski G. (1995) Kinetics of nitric oxide autoxidation in aqueous solution in the absence and presence of various reductants. The nature of the oxidizing intermediates. J. Am. Chem. Soc. 117, 12078–12084 10.1021/ja00154a007 [DOI] [Google Scholar]
- 28. Ford P. C., Wink D. A., and Stanbury D. M. (1993) Autoxidation kinetics of aqueous nitric oxide. FEBS Lett. 326, 1–3 10.1016/0014-5793(93)81748-O [DOI] [PubMed] [Google Scholar]
- 29. Graetzel M., Taniguchi S., and Henglein A. (1970) Pulsradiolytische untersuchung der NO-oxydation und des gleichgewichts N2O3 NO + NO2 in waessriger loesung. Ber. Bunsenges. Phys. Chem. 74, Issue 5 10.1002/bbpc.19700740513 [DOI] [Google Scholar]
- 30. Lewis R. S., Tannenbaum S. R., and Deen W. M. (1995) Kinetics of N-nitrosation in oxygenated nitric oxide solutions at physiological pH: role of nitrous anhydride and effects of phosphate and chloride. J. Am. Chem. Soc. 117, 3933–3939 10.1021/ja00119a006 [DOI] [Google Scholar]
- 31. Caulfield J. L., Singh S. P., Wishnok J. S., Deen W. M., and Tannenbaum S. R. (1996) Bicarbonate inhibits N-nitrosation in oxygenated nitric oxide solutions. J. Biol. Chem. 271, 25859–25863 10.1074/jbc.271.42.25859 [DOI] [PubMed] [Google Scholar]
- 32. Liu X., Miller M. J., Joshi M. S., Thomas D. D., and Lancaster J. R. (1998) Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc. Natl. Acad. Sci. U.S.A. 95, 2175–2179 10.1073/pnas.95.5.2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Möller M. N., Li Q., Vitturi D. A., Robinson J. M., Lancaster J. R. Jr., and Denicola A. (2007) Membrane “Lens” effect: focusing the formation of reactive nitrogen oxides from the •NO/O2 reaction. Chem. Res. Toxicol. 20, 709–714 10.1021/tx700010h [DOI] [PubMed] [Google Scholar]
- 34. Möller M. N., and Denicola A. (2019) Acceleration of the autoxidation of nitric oxide by proteins. Nitric Oxide 85, 28–34 10.1016/j.niox.2019.01.014 [DOI] [PubMed] [Google Scholar]
- 35. Möller M. N., Li Q., Chinnaraj M., Cheung H. C., Lancaster J. R. Jr., and Denicola A. (2016) Solubility and diffusion of oxygen in phospholipid membranes. Biochim. Biophys. Acta 1858, 2923–2930 10.1016/j.bbamem.2016.09.003 [DOI] [PubMed] [Google Scholar]
- 36. Ignarro L. J., Buga G. M., Wood K. S., Byrns R. E., and Chaudhuri G. (1987) Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 84, 9265–9269 10.1073/pnas.84.24.9265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Murphy M. E., and Noack E. (1994) Nitric oxide assay using hemoglobin method. Methods Enzymol. 233, 240–250 10.1016/S0076-6879(94)33027-1 [DOI] [PubMed] [Google Scholar]
- 38. Winterbourn C. C. (1990) Oxidative reactions of hemoglobin. Methods Enzymol. 186, 265–272 10.1016/0076-6879(90)86118-F [DOI] [PubMed] [Google Scholar]
- 39. Eich R. F., Li T., Lemon D. D., Doherty D. H., Curry S. R., Aitken J. F., Mathews A. J., Johnson K. A., Smith R. D., Phillips G. N. Jr., and Olson J. S. (1996) Mechanism of NO-induced oxidation of myoglobin and hemoglobin. Biochemistry 35, 6976–6983 10.1021/bi960442g [DOI] [PubMed] [Google Scholar]
- 40. Malinski T., and Taha Z. (1992) Nitric oxide release from a single cell measured in situ by a porphyrinic-based microsensor. Nature 358, 676–678 10.1038/358676a0 [DOI] [PubMed] [Google Scholar]
- 41. Friedemann M. N., Robinson S. W., and Gerhardt G. A. (1996) o-Phenylenediamine-modified carbon fiber electrodes for the detection of nitric oxide. Anal. Chem. 68, 2621–2628 10.1021/ac960093w [DOI] [PubMed] [Google Scholar]
- 42. Cha W., Tung Y. C., Meyerhoff M. E., and Takayama S. (2010) Patterned electrode-based amperometric gas sensor for direct nitric oxide detection within microfluidic devices. Anal. Chem. 82, 3300–3305 10.1021/ac100085w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nagano T., and Yoshimura T. (2002) Bioimaging of nitric oxide. Chem. Rev. 102, 1235–1270 10.1021/cr010152s [DOI] [PubMed] [Google Scholar]
- 44. Akaike T., Yoshida M., Miyamoto Y., Sato K., Kohno M., Sasamoto K., Miyazaki K., Ueda S., and Maeda H. (1993) Antagonistic action of imidazolineoxyl N-oxides against endothelium-derived relaxing factor/•NO through a radical reaction. Biochemistry 32, 827–832 10.1021/bi00054a013 [DOI] [PubMed] [Google Scholar]
- 45. Goldstein S., Russo A., and Samuni A. (2003) Reactions of PTIO and carboxy-PTIO with •NO, •NO2, and O2•−. J. Biol. Chem. 278, 50949–50955 10.1074/jbc.M308317200 [DOI] [PubMed] [Google Scholar]
- 46. Akaike T., and Maeda H. (1996) Quantitation of nitric oxide using 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (PTIO). Methods Enzymol. 268, 211–221 10.1016/S0076-6879(96)68023-9 [DOI] [PubMed] [Google Scholar]
- 47. Singh R. J., Hogg N., Mchaourab H. S., and Kalyanaraman B. (1994) Physical and chemical interactions between nitric oxide and nitroxides. Biochim. Biophys. Acta 1201, 437–441 10.1016/0304-4165(94)90073-6 [DOI] [PubMed] [Google Scholar]
- 48. Vanin A. F. (2018) EPR characterization of dinitrosyl iron complexes with thiol-containing ligands as an approach to their identification in biological objects: an overview. Cell Biochem. Biophys. 76, 3–17 10.1007/s12013-017-0811-8 [DOI] [PubMed] [Google Scholar]
- 49. Lancaster J. R. Jr., and Hibbs J. B. Jr. (1990) EPR demonstration of iron-nitrosyl complex formation by cytotoxic activated macrophages. Proc. Natl. Acad. Sci. U.S.A. 87, 1223–1227 10.1073/pnas.87.3.1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dikalov S., and Fink B. (2005) ESR techniques for the detection of nitric oxide in vivo and in tissues. Methods Enzymol. 396, 597–610 10.1016/S0076-6879(05)96052-7 [DOI] [PubMed] [Google Scholar]
- 51. Giorgio S., Linares E., Capurro Mde L., de Bianchi A. G., and Augusto O. (1996) Formation of nitrosyl hemoglobin and nitrotyrosine during murine leishmaniasis. Photochem. Photobiol. 63, 750–754 10.1111/j.1751-1097.1996.tb09626.x [DOI] [PubMed] [Google Scholar]
- 52. Hall D. M., and Buettner G. R. (1996) In vivo spin trapping of nitric oxide by heme: electron paramagnetic resonance detection ex vivo. Methods Enzymol. 268, 188–192 10.1016/S0076-6879(96)68020-3 [DOI] [PubMed] [Google Scholar]
- 53. Wang Q. Z., Jacobs J., DeLeo J., Kruszyna H., Kruszyna R., Smith R., and Wilcox D. (1991) Nitric oxide hemoglobin in mice and rats in endotoxic shock. Life Sci. 49, PL55–PL60 10.1016/0024-3205(91)90251-6 [DOI] [PubMed] [Google Scholar]
- 54. Itoh Y., Ma F. H., Hoshi H., Oka M., Noda K., Ukai Y., Kojima H., Nagano T., and Toda N. (2000) Determination and bioimaging method for nitric oxide in biological specimens by diaminofluorescein fluorometry. Anal. Biochem. 287, 203–209 10.1006/abio.2000.4859 [DOI] [PubMed] [Google Scholar]
- 55. Broillet M., Randin O., and Chatton J. (2001) Photoactivation and calcium sensitivity of the fluorescent NO indicator 4,5-diaminofluorescein (DAF-2): implications for cellular NO imaging. FEBS Lett. 491, 227–232 10.1016/S0014-5793(01)02206-2 [DOI] [PubMed] [Google Scholar]
- 56. Ralt D., Wishnok J. S., Fitts R., and Tannenbaum S. R. (1988) Bacterial catalysis of nitrosation: involvement of the nar operon of Escherichia coli. J. Bacteriol. 170, 359–364 10.1128/jb.170.1.359-364.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kojima H., Urano Y., Kikuchi K., Higuchi T., Hirata Y., and Nagano T. (1999) Fluorescent indicators for imaging nitric oxide production. Angew. Chem. Int. Ed. Engl. 38, 3209–3212 10.1002/(SICI)1521-3773(19991102)38:21<3209::AID-ANIE3209>3.0.CO;2-6 [DOI] [PubMed] [Google Scholar]
- 58. Jourd'heuil D. (2002) Increased nitric oxide-dependent nitrosylation of 4,5-diaminofluorescein by oxidants: implications for the measurement of intracellular nitric oxide. Free Radic. Biol. Med. 33, 676–684 10.1016/S0891-5849(02)00955-3 [DOI] [PubMed] [Google Scholar]
- 59. Espey M. G., Xavier S., Thomas D. D., Miranda K. M., and Wink D. A. (2002) Direct real-time evaluation of nitration with green fluorescent protein in solution and within human cells reveals the impact of nitrogen dioxide vs. peroxynitrite mechanisms. Proc. Natl. Acad. Sci. U.S.A. 99, 3481–3486 10.1073/pnas.062604199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang X., Kim W.-S., Hatcher N., Potgieter K., Moroz L. L., Gillette R., and Sweedler J. V. (2002) Interfering with nitric oxide measurements 4,5-diaminofluorescein reacts with dehydroascorbic acid and ascorbic acid. J. Biol. Chem. 277, 48472–48478 10.1074/jbc.M209130200 [DOI] [PubMed] [Google Scholar]
- 61. Cortese-Krott M. M., Rodriguez-Mateos A., Kuhnle G. G., Brown G., Feelisch M., and Kelm M. (2012) A multilevel analytical approach for detection and visualization of intracellular NO production and nitrosation events using diaminofluoresceins. Free Radic. Biol. Med. 53, 2146–2158 10.1016/j.freeradbiomed.2012.09.008 [DOI] [PubMed] [Google Scholar]
- 62. Hogg N., Zielonka J., and Kalyanaraman B. (2017) in Nitric Oxide (Ignarro L. J., and Freeman B. A., eds) 3rd Ed., pp. 23–44, Academic Press, New York [Google Scholar]
- 63. Newman R. H., Fosbrink M. D., and Zhang J. (2011) Genetically encodable fluorescent biosensors for tracking signaling dynamics in living cells. Chem. Rev. 111, 3614–3666 10.1021/cr100002u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Eroglu E., Gottschalk B., Charoensin S., Blass S., Bischof H., Rost R., Madreiter-Sokolowski C. T., Pelzmann B., Bernhart E., Sattler W., Hallström S., Malinski T., Waldeck-Weiermair M., Graier W. F., and Malli R. (2016) Development of novel FP-based probes for live-cell imaging of nitric oxide dynamics. Nat. Commun. 7, 10623 10.1038/ncomms10623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Samouilov A., and Zweier J. L. (1998) Development of chemiluminescence-based methods for specific quantitation of nitrosylated thiols. Anal. Biochem. 258, 322–330 10.1006/abio.1998.2609 [DOI] [PubMed] [Google Scholar]
- 66. Diers A. R., Keszler A., and Hogg N. (2014) Detection of S-nitrosothiols. Biochim. Biophys. Acta 1840, 892–900 10.1016/j.bbagen.2013.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Palmer R. M., Ferrige A. G., and Moncada S. (1987) Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327, 524–526 10.1038/327524a0 [DOI] [PubMed] [Google Scholar]
- 68. MacArthur P. H., Shiva S., and Gladwin M. T. (2007) Measurement of circulating nitrite and S-nitrosothiols by reductive chemiluminescence. J. Chromatogr. Analyt Technol Biomed Life Sci. 851, 93–105 10.1016/j.jchromb.2006.12.012 [DOI] [PubMed] [Google Scholar]
- 69. Keszler A., Diers A. R., Ding Z., and Hogg N. (2017) Thiolate-based dinitrosyl iron complexes: decomposition and detection and differentiation from S-nitrosothiols. Nitric Oxide 65, 1–9 10.1016/j.niox.2017.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Clough P. N., and Thrush B. A. (1967) Mechanism of chemiluminescent reaction between nitric oxide and ozone. Trans. Faraday Soc. 63, 915–925 10.1039/tf9676300915 [DOI] [Google Scholar]
- 71. Basu S., Wang X., Gladwin M. T., and Kim-Shapiro D. B. (2008) Chemiluminescent detection of S-nitrosated proteins: comparison of tri-iodide, copper/CO/cysteine, and modified copper/cysteine methods. Methods Enzymol. 440, 137–156 10.1016/S0076-6879(07)00808-7 [DOI] [PubMed] [Google Scholar]
- 72. Doctor A., Platt R., Sheram M. L., Eischeid A., McMahon T., Maxey T., Doherty J., Axelrod M., Kline J., Gurka M., Gow A., and Gaston B. (2005) Hemoglobin conformation couples erythrocyte S-nitrosothiol content to O2 gradients. Proc. Natl. Acad. Sci. U.S.A. 102, 5709–5714 10.1073/pnas.0407490102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Stuehr D. J., Santolini J., Wang Z.-Q., Wei C.-C., and Adak S. (2004) Update on mechanism and catalytic regulation in the NO synthases. J. Biol. Chem. 279, 36167–36170 10.1074/jbc.R400017200 [DOI] [PubMed] [Google Scholar]
- 74. Bredt D. S., and Snyder S. H. (1990) Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. U.S.A. 87, 682–685 10.1073/pnas.87.2.682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Russwurm M., and Koesling D. (2018) Measurement of cGMP-generating and -degrading activities and cGMP levels in cells and tissues: focus on FRET-based cGMP indicators. Nitric Oxide 77, 44–52 10.1016/j.niox.2018.04.006 [DOI] [PubMed] [Google Scholar]
- 76. Russwurm M., and Koesling D. (2005) Purification and characterization of NO-sensitive guanylyl cyclase. Methods Enzymol. 396, 492–501 10.1016/S0076-6879(05)96041-2 [DOI] [PubMed] [Google Scholar]
- 77. Honda A., Adams S. R., Sawyer C. L., Lev-Ram V., Tsien R. Y., and Dostmann W. R. (2001) Spatiotemporal dynamics of guanosine 3′,5′-cyclic monophosphate revealed by a genetically encoded, fluorescent indicator. Proc. Natl. Acad. Sci. U.S.A. 98, 2437–2442 10.1073/pnas.051631298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Isbell T. S., Koenitzer J. R., Crawford J. H., White C. R., Kraus D. W., and Patel R. P. (2005) Assessing NO-dependent vasodilatation using vessel bioassays at defined oxygen tensions. Methods Enzymol. 396, 553–568 10.1016/S0076-6879(05)96047-3 [DOI] [PubMed] [Google Scholar]
- 79. Born G. V. (1962) Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 194, 927–929 10.1038/194927b0 [DOI] [PubMed] [Google Scholar]
- 80. Zucker M. B. (1989) Platelet aggregation measured by the photometric method. Methods Enzymol. 169, 117–133 10.1016/0076-6879(89)69054-4 [DOI] [PubMed] [Google Scholar]
- 81. Brennan M. L., Wu W., Fu X., Shen Z., Song W., Frost H., Vadseth C., Narine L., Lenkiewicz E., Borchers M. T., Lusis A. J., Lee J. J., Lee N. A., Abu-Soud H. M., Ischiropoulos H., and Hazen S. L. (2002) A tale of two controversies: defining both the role of peroxidases in nitrotyrosine formation in vivo using eosinophil peroxidase and myeloperoxidase-deficient mice, and the nature of peroxidase-generated reactive nitrogen species. J. Biol. Chem. 277, 17415–17427 10.1074/jbc.M112400200 [DOI] [PubMed] [Google Scholar]
- 82. van der Vliet A., Eiserich J. P., Halliwell B., and Cross C. E. (1997) Formation of reactive nitrogen species during peroxidase-catalyzed oxidation of nitrite. A potential additional mechanism of nitric oxide-dependent toxicity. J. Biol. Chem. 272, 7617–7625 10.1074/jbc.272.12.7617 [DOI] [PubMed] [Google Scholar]
- 83. Singh R. J., Goss S. P., Joseph J., and Kalyanaraman B. (1998) Nitration of γ-tocopherol and oxidation of α-tocopherol by copper-zinc superoxide dismutase/H2O2/NO2−: role of nitrogen dioxide free radical. Proc. Natl. Acad. Sci. U.S.A. 95, 12912–12917 10.1073/pnas.95.22.12912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. El-Agamey A. (2009) Laser flash photolysis of new water-soluble peroxyl radical precursor. J. Photochem. Photobiol. A 203, 13–17 10.1016/j.jphotochem.2008.12.015 [DOI] [Google Scholar]
- 85. Adams G. E., Boag J. W., and Michael B. D. (1965) Reactions of the hydroxyl radical. Part 2. Determination of absolute rate constants. Trans. Faraday Soc. 61, 1417–1424 10.1039/TF9656101417 [DOI] [Google Scholar]
- 86. Huie R. E., Shoute L. C. T., and Neta P. (1991) Temperature dependence of the rate constants for reactions of the carbonate radical with organic and inorganic reductants. Int. J. Chem. Kinet. 23, 541–552 10.1002/kin.550230606 [DOI] [Google Scholar]
- 87. Merényi G., Lind J., and Goldstein S. (2002) The rate of homolysis of adducts of peroxynitrite to the C=O double bond. J. Am. Chem. Soc. 124, 40–48 10.1021/ja011799x [DOI] [PubMed] [Google Scholar]
- 88. Lymar S. V., Khairutdinov R. F., and Hurst J. K. (2003) Hydroxyl radical formation by O–O bond homolysis in peroxynitrous acid. Inorg. Chem. 42, 5259–5266 10.1021/ic030104l [DOI] [PubMed] [Google Scholar]
- 89. Augusto O., Goldstein S., Hurst J. K., Lind J., Lymar S. V., Merenyi G., and Radi R. (2019) Carbon dioxide-catalyzed peroxynitrite reactivity- the resilience of the radical mechanism after two decades of research. Free Radic. Biol. Med. 135, 210–215 10.1016/j.freeradbiomed.2019.02.026 [DOI] [PubMed] [Google Scholar]
- 90. Schwartz S. E., and White W. H. (1983) in Advances in Environmental Science and Technology (Schwartz S. E., ed) pp. 1–116, John Wiley & Sons, Inc., New York [Google Scholar]
- 91. Cheung J. L., Li Y. Q., Boniface J., Shi Q., Dacidovits P., Worsnop D. R., Jayne J. T., and, Kolb C. E. (2000) Heterogeneous interactions of NO2 with aqueous surfaces. J. Phys. Chem. A 104, 2655–2662 10.1021/jp992929f [DOI] [Google Scholar]
- 92. Grätzel M., Henglein A., Lilie Ja., Beck G. (1969) Pulse radiolytic study of some elementary processes of nitrite ion oxidation and reduction. Ber Bunsenges Phys. Chem. 73, 646–653 [Google Scholar]
- 93. Augusto O., Bonini M. G., Amanso A. M., Linares E., Santos C. C., and De Menezes S. L. (2002) Nitrogen dioxide and carbonate radical anion: two emerging radicals in biology. Free Radic. Biol. Med. 32, 841–859 10.1016/S0891-5849(02)00786-4 [DOI] [PubMed] [Google Scholar]
- 94. Squadrito G. L., and Postlethwait E. M. (2009) On the hydrophobicity of nitrogen dioxide: could there be a “lens” effect for NO2 reaction kinetics? Nitric Oxide 21, 104–109 10.1016/j.niox.2009.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wardman P. (1998) in N-Centered Radicals (Alfassi Z. B., ed) pp. 155–179, John Wiley & Sons, Chichester, UK [Google Scholar]
- 96. Signorelli S., Möller M. N., Coitiño E. L., and Denicola A. (2011) Nitrogen dioxide solubility and permeation in lipid membranes. Arch. Biochem. Biophys. 512, 190–196 10.1016/j.abb.2011.06.003 [DOI] [PubMed] [Google Scholar]
- 97. Koppenol W. H., Moreno J. J., Pryor W. A., Ischiropoulos H., and Beckman J. S. (1992) Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide. Chem. Res. Toxicol. 5, 834–842 10.1021/tx00030a017 [DOI] [PubMed] [Google Scholar]
- 98. Ford E., Hughes M. N., and Wardman P. (2002) Kinetics of the reactions of nitrogen dioxide with glutathione, cysteine, and uric acid at physiological pH. Free Radic. Biol. Med. 32, 1314–1323 10.1016/S0891-5849(02)00850-X [DOI] [PubMed] [Google Scholar]
- 99. May J. M. (2012) Vitamin C transport and its role in the central nervous system. Subcell. Biochem. 56, 85–103 10.1007/978-94-007-2199-9_6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Pryor W. A., and Lightsey J. W. (1981) Mechanisms of nitrogen dioxide reactions: initiation of lipid peroxidation and the production of nitrous acid. Science 214, 435–437 10.1126/science.214.4519.435 [DOI] [PubMed] [Google Scholar]
- 101. Jiang H., Kruger N., Lahiri D. R., Wang D., Vatèle J. M., and Balazy M. (1999) Nitrogen dioxide induces cis-trans-isomerization of arachidonic acid within cellular phospholipids. Detection of trans-arachidonic acids in vivo. J. Biol. Chem. 274, 16235–16241 10.1074/jbc.274.23.16235 [DOI] [PubMed] [Google Scholar]
- 102. Freeman B. A., Baker P. R., Schopfer F. J., Woodcock S. R., Napolitano A., and d'Ischia M. (2008) Nitro-fatty acid formation and signaling. J. Biol. Chem. 283, 15515–15519 10.1074/jbc.R800004200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Trostchansky A., Bonilla L., González-Perilli L., and Rubbo H. (2013) Nitro-fatty acids: formation, redox signaling, and therapeutic potential. Antioxid. Redox Signal. 19, 1257–1265 10.1089/ars.2012.5023 [DOI] [PubMed] [Google Scholar]
- 104. Schopfer F. J., Cipollina C., and Freeman B. A. (2011) Formation and signaling actions of electrophilic lipids. Chem. Rev. 111, 5997–6021 10.1021/cr200131e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Turell L., Steglich M., and Alvarez B. (2018) The chemical foundations of nitroalkene fatty acid signaling through addition reactions with thiols. Nitric Oxide 78, 161–169 10.1016/j.niox.2018.03.014 [DOI] [PubMed] [Google Scholar]
- 106. Clyne M., Thrush B., and Wayne R. (1964) Kinetics of the chemiluminescent reaction between nitric oxide and ozone. Trans. Faraday Soc. 60, 359–370 10.1039/tf9646000359 [DOI] [Google Scholar]
- 107. Hampl V., Waters C. L., and Archer S. L. (1996) in Methods in Nitric Oxide Research (Feelisch M., and Stamler J., eds) pp. 309–318, John Wiley and Sons, New York [Google Scholar]
- 108. Reed C., Evans M. J., Di Carlo P., Lee J. D., and Carpenter L. J. (2016) Interferences in photolytic NO2 measurements: explanation for an apparent missing oxidant? Atmos. Chem. Phys. 16, 4707–4724 10.5194/acp-16-4707-2016 [DOI] [Google Scholar]
- 109. Kelly T. J., Spicer C. W., and Ward G. F. (1990) An assessment of the luminol chemiluminescence technique for measurement of NO2 in ambient air. Atmos. Environ. 24A, 2397–2403 10.1016/0960-1686(90)90332-H [DOI] [Google Scholar]
- 110. Prütz W. A., Mönig H., Butler J., and Land E. J. (1985) Reactions of nitrogen dioxide in aqueous model systems: oxidation of tyrosine units in peptides and proteins. Arch. Biochem. Biophys. 243, 125–134 10.1016/0003-9861(85)90780-5 [DOI] [PubMed] [Google Scholar]
- 111. Rowlands J. R., and Gause E. M. (1971) Reaction of nitrogen dioxide with blood and lung components. Electron spin resonance studies. Arch. Intern. Med. 128, 94–100 10.1001/archinte.1971.00310190098012 [DOI] [PubMed] [Google Scholar]
- 112. Pace M. D., and Kalyanaraman B. (1993) Spin trapping of nitrogen dioxide radical from photolytic decomposition of nitramines. Free Radic. Biol. Med. 15, 337–342 10.1016/0891-5849(93)90080-E [DOI] [PubMed] [Google Scholar]
- 113. Astolfi P., Greci L., and Panagiotaki M. (2005) Spin trapping of nitrogen dioxide and of radicals generated from nitrous acid. Free Radic. Res. 39, 137–144 10.1080/10715760500045582 [DOI] [PubMed] [Google Scholar]
- 114. Carballal S., Trujillo M., Cuevasanta E., Bartesaghi S., Möller M. N., Folkes L. K., García-Bereguiain M. A., Gutiérrez-Merino C., Wardman P., Denicola A., Radi R., and Alvarez B. (2011) Reactivity of hydrogen sulfide with peroxynitrite and other oxidants of biological interest. Free Radic. Biol. Med. 50, 196–205 10.1016/j.freeradbiomed.2010.10.705 [DOI] [PubMed] [Google Scholar]
- 115. Markovits G. Y., Schwartz S. E., and Newman L. (1981) Hydrolysis equilibrium of dinitrogen trioxide in dilute acid solution. Inorg. Chem. 20, 445–450 10.1021/ic50216a025 [DOI] [Google Scholar]
- 116. Goldstein S., and Czapski G. (1996) Mechanism of the nitrosation of thiols and amines by oxygenated •NO solutions: the nature of the nitrosating intermediates. J. Am. Chem. Soc. 118, 3419–3425 10.1021/ja9536680 [DOI] [Google Scholar]
- 117. Vitturi D. A., Minarrieta L., Salvatore S. R., Postlethwait E. M., Fazzari M., Ferrer-Sueta G., Lancaster J. R. Jr., Freeman B. A., and Schopfer F. J. (2015) Convergence of biological nitration and nitrosation via symmetrical nitrous anhydride. Nat. Chem. Biol. 11, 504–510 10.1038/nchembio.1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lancaster J. R., Jr. (2017) How are nitrosothiols formed de novo in vivo? Arch. Biochem. Biophys. 617, 137–144 10.1016/j.abb.2016.10.015 [DOI] [PubMed] [Google Scholar]
- 119. Zhang H., Andrekopoulos C., Xu Y., Joseph J., Hogg N., Feix J., and Kalyanaraman B. (2009) Decreased S-nitrosation of peptide thiols in the membrane interior. Free Radic. Biol. Med. 47, 962–968 10.1016/j.freeradbiomed.2009.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Toledo J. C. Jr., Bosworth C. A., Hennon S. W., Mahtani H. A., Bergonia H. A., and Lancaster J. R. Jr. (2008) Nitric oxide-induced conversion of cellular chelatable iron into macromolecule-bound paramagnetic dinitrosyliron complexes. J. Biol. Chem. 283, 28926–28933 10.1074/jbc.M707862200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Bosworth C. A., Toledo J. C. Jr., Zmijewski J. W., Li Q., and Lancaster J. R. Jr. (2009) Dinitrosyliron complexes and the mechanism(s) of cellular protein nitrosothiol formation from nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 106, 4671–4676 10.1073/pnas.0710416106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Hogg N. (2010) Detection of nitric oxide by electron paramagnetic resonance spectroscopy. Free Radic. Biol. Med. 49, 122–129 10.1016/j.freeradbiomed.2010.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Dei Zotti F., Lobysheva I. I., and Balligand J.-L. (2018) Nitrosyl-hemoglobin formation in rodent and human venous erythrocytes reflects NO formation from the vasculature in vivo. PLoS ONE 13, e0200352 10.1371/journal.pone.0200352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Piknova B., Gladwin M. T., Schechter A. N., and Hogg N. (2005) Electron paramagnetic resonance analysis of nitrosylhemoglobin in humans during NO inhalation. J. Biol. Chem. 280, 40583–40588 10.1074/jbc.M506292200 [DOI] [PubMed] [Google Scholar]
- 125. Broniowska K. A., and Hogg N. (2012) The chemical biology of S-nitrosothiols. Antiox. Redox Signal. 17, 969–980 10.1089/ars.2012.4590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Basu S., Keszler A., Azarova N. A., Nwanze N., Perlegas A., Shiva S., Broniowska K. A., Hogg N., and Kim-Shapiro D. B. (2010) A novel role for cytochrome c: efficient catalysis of S-nitrosothiol formation. Free Radic. Biol. Med. 48, 255–263 10.1016/j.freeradbiomed.2009.10.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hogg N. (1999) The kinetics of S-trans-nitrosation—a reversible second-order reaction. Anal. Biochem. 272, 257–262 10.1006/abio.1999.4199 [DOI] [PubMed] [Google Scholar]
- 128. Sengupta R., and Holmgren A. (2012) Thioredoxin and thioredoxin reductase in relation to reversible S-nitrosylation. Antiox. Redox Signal. 18, 259–269 10.1089/ars.2012.4716 [DOI] [PubMed] [Google Scholar]
- 129. Jensen D. E., Belka G. K., and Du Bois G. C. (1998) S-Nitrosoglutathione is a substrate for rat alcohol dehydrogenase class III isoenzyme. Biochem. J. 331, 659–668 10.1042/bj3310659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Broniowska K. A., Diers A. R., and Hogg N. (2013) S-Nitrosoglutathione. Biochim. Biophys. Acta 1830, 3173–3181 10.1016/j.bbagen.2013.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Saville B. (1958) A scheme for the colorimetric determination of microgram amounts of thiols. Analyst 83, 670–672 10.1039/an9588300670 [DOI] [Google Scholar]
- 132. Stamler J. S., Jaraki O., Osborne J., Simon D. I., Keaney J., Vita J., Singel D., Valeri C. R., and Loscalzo J. (1992) Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc. Natl. Acad. Sci. U.S.A. 89, 7674–7677 10.1073/pnas.89.16.7674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Jaffrey S. R., and Snyder S. H. (2001) The biotin switch method for the detection of S-nitrosylated proteins. Sci. STKE 2001, pl1 [DOI] [PubMed] [Google Scholar]
- 134. Wang X., Kettenhofen N. J., Shiva S., Hogg N., and Gladwin M. T. (2008) Copper dependence of the biotin switch assay: modified assay for measuring cellular and blood nitrosated proteins. Free Radic. Biol. Med. 44, 1362–1372 10.1016/j.freeradbiomed.2007.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Chouchani E. T., James A. M., Methner C., Pell V. R., Prime T. A., Erickson B. K., Forkink M., Lau G. Y., Bright T. P., Menger K. E., Fearnley I. M., Krieg T., and Murphy M. P. (2017) Identification and quantification of protein S-nitrosation by nitrite in the mouse heart during ischemia. J. Biol. Chem. 292, 14486–14495 10.1074/jbc.M117.798744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Zhou X., Han P., Li J., Zhang X., Huang B., Ruan H.-Q., and Chen C. (2010) ESNOQ, proteomic quantification of endogenous S-nitrosation. PLoS ONE 5, e10015 10.1371/journal.pone.0010015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Raju K., Doulias P.-T., Tenopoulou M., Greene J. L., and Ischiropoulos H. (2012) Strategies and tools to explore protein S-nitrosylation. Biochim. Biophys. Acta 1820, 684–688 10.1016/j.bbagen.2011.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Zhang J., Li S., Zhang D., Wang H., Whorton A. R., and Xian M. (2010) Reductive ligation mediated one-step disulfide formation of S-nitrosothiols. Org. Lett. 12, 4208–4211 10.1021/ol101863s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Lundberg J. O., and Weitzberg E. (2017) in Nitric Oxide (Ignarro L. J., and Freeman B. A., eds) 3rd. Ed., pp. 157–171, Academic Press, New York [Google Scholar]
- 140. DeMartino A. W., Kim-Shapiro D. B., Patel R. P., and Gladwin M. T. (2019) Nitrite and nitrate chemical biology and signalling. Br. J. Pharmacol. 176, 228–245 10.1111/bph.14484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kim-Shapiro D. B., and Gladwin M. T. (2014) Mechanisms of nitrite bioactivation. Nitric Oxide 38, 58–68 10.1016/j.niox.2013.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Tsikas D. (2007) Analysis of nitrite and nitrate in biological fluids by assays based on the Griess reaction: appraisal of the Griess reaction in the l-arginine/nitric oxide area of research. J. Chromatog. B Analyt Technol. Biomed. Life Sci. 851, 51–70 10.1016/j.jchromb.2006.07.054 [DOI] [PubMed] [Google Scholar]
- 143. Green L. C., Wagner D. A., Glogowski J., Skipper P. L., Wishnok J. S., and Tannenbaum S. R. (1982) Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem. 126, 131–138 10.1016/0003-2697(82)90118-X [DOI] [PubMed] [Google Scholar]
- 144. Misko T. P., Schilling R. J., Salvemini D., Moore W. M., and Currie M. G. (1993) A fluorometric assay for the measurement of nitrite in biological samples. Anal. Biochem. 214, 11–16 10.1006/abio.1993.1449 [DOI] [PubMed] [Google Scholar]
- 145. Tsikas D. (2005) Review methods of quantitative analysis of the nitric oxide metabolites nitrite and nitrate in human biological fluids. Free Radic. Res. 39, 797–815 10.1080/10715760500053651 [DOI] [PubMed] [Google Scholar]
- 146. Tsikas D. (2000) Simultaneous derivatization and quantification of the nitric oxide metabolites nitrite and nitrate in biological fluids by gas chromatography/mass spectrometry. Anal. Chem. 72, 4064–4072 10.1021/ac9913255 [DOI] [PubMed] [Google Scholar]
- 147. Shafirovich V., and Lymar S. V. (2003) Spin-forbidden deprotonation of aqueous nitroxyl (HNO). J. Am. Chem. Soc. 125, 6547–6552 10.1021/ja034378j [DOI] [PubMed] [Google Scholar]
- 148. Smulik R., Dębski D., Zielonka J., Michałowski B., Adamus J., Marcinek A., Kalyanaraman B., and Sikora A. (2014) Nitroxyl (HNO) reacts with molecular oxygen and forms peroxynitrite at physiological pH. Biological implications. J. Biol. Chem. 289, 35570–35581 10.1074/jbc.M114.597740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Doctorovich F., Bikiel D. E., Pellegrino J., Suárez S. A., and Martí M. A. (2014) Reactions of HNO with metal porphyrins: underscoring the biological relevance of HNO. Acc. Chem. Res. 47, 2907–2916 10.1021/ar500153c [DOI] [PubMed] [Google Scholar]
- 150. Bianco C. L., Toscano J. P., Bartberger M. D., and Fukuto J. M. (2017) The chemical biology of HNO signaling. Arch. Biochem. Biophys. 617, 129–136 10.1016/j.abb.2016.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Fukuto J. M., Bartberger M. D., Dutton A. S., Paolocci N., Wink D. A., and Houk K. N. (2005) The physiological chemistry and biological activity of nitroxyl (HNO): the neglected, misunderstood, and enigmatic nitrogen oxide. Chem. Res. Toxicol. 18, 790–801 10.1021/tx0496800 [DOI] [PubMed] [Google Scholar]
- 152. Miranda K. M. (2005) The chemistry of nitroxyl (HNO) and implications in biology. Coord. Chem. Rev. 249, 433–455 10.1016/j.ccr.2004.08.010 [DOI] [Google Scholar]
- 153. Cline M. R., Tu C., Silverman D. N., and Toscano J. P. (2011) Detection of nitroxyl (HNO) by membrane inlet mass spectrometry. Free Radic. Biol. Med. 50, 1274–1279 10.1016/j.freeradbiomed.2011.02.008 [DOI] [PubMed] [Google Scholar]
- 154. Bari S. E., Martí M. A., Amorebieta V. T., Estrin D. A., and Doctorovich F. (2003) Fast nitroxyl trapping by ferric porphyrins. J. Am. Chem. Soc. 125, 15272–15273 10.1021/ja036370f [DOI] [PubMed] [Google Scholar]
- 155. Adachi Y., Nakagawa H., Matsuo K., Suzuki T., and Miyata N. (2008) Photoactivatable HNO-releasing compounds using the retro-Diels–Alder reaction. Chem. Commun., 5149–5151 10.1039/B811985F [DOI] [PubMed] [Google Scholar]
- 156. Martí M. A., Bari S. E., Estrin D. A., and Doctorovich F. (2005) Discrimination of nitroxyl and nitric oxide by water-soluble Mn(III) porphyrins. J. Am. Chem. Soc. 127, 4680–4684 10.1021/ja044632n [DOI] [PubMed] [Google Scholar]
- 157. Suárez S. A., Bikiel D. E., Wetzler D. E., Martí M. A., and Doctorovich F. (2013) Time-resolved electrochemical quantification of azanone (HNO) at low nanomolar level. Anal. Chem. 85, 10262–10269 10.1021/ac402134b [DOI] [PubMed] [Google Scholar]
- 158. Suarez S. A., Munoz M., Bikiel D. E., Marti M. A., and Doctorovich F. (2017) in The Chemistry and Biology of Nitroxyl (HNO) (Doctorovich F., Farmer P. J., and Marti M. A., eds) pp. 239–253, Elsevier, Amsterdam, the Netherlands [Google Scholar]
- 159. Rosenthal J., and Lippard S. J. (2010) Direct detection of nitroxyl in aqueous solution using a tripodal copper(II) BODIPY complex. J. Am. Chem. Soc. 132, 5536–5537 10.1021/ja909148v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Smulik-Izydorczyk R., Dębowska K., Pięta J., Michalski R., Marcinek A., and Sikora A. (2018) Fluorescent probes for the detection of nitroxyl (HNO). Free Radic. Biol. Med. 128, 69–83 10.1016/j.freeradbiomed.2018.04.564 [DOI] [PubMed] [Google Scholar]
- 161. Tennyson A. G., Do L., Smith R. C., and Lippard S. J. (2007) Selective fluorescence detection of nitroxyl over nitric oxide in buffered aqueous solution using a conjugated metallopolymer. Polyhedron 26, 4625–4630 10.1016/j.poly.2007.04.003 [DOI] [Google Scholar]
- 162. Miranda K. M., Paolocci N., Katori T., Thomas D. D., Ford E., Bartberger M. D., Espey M. G., Kass D. A., Feelisch M., Fukuto J. M., and Wink D. A. (2003) A biochemical rationale for the discrete behavior of nitroxyl and nitric oxide in the cardiovascular system. Proc. Natl. Acad. Sci. U.S.A. 100, 9196–9201 10.1073/pnas.1430507100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Samuni Y., Samuni U., and Goldstein S. (2013) The use of cyclic nitroxide radicals as HNO scavengers. J. Inorg. Biochem. 118, 155–161 10.1016/j.jinorgbio.2012.10.002 [DOI] [PubMed] [Google Scholar]
- 164. Cline M. R., and Toscano J. P. (2011) Detection of nitroxyl (HNO) by a prefluorescent probe. J. Phys. Org. Chem. 24, 993–998 10.1002/poc.1871 [DOI] [Google Scholar]
- 165. Donzelli S., Espey M. G., Thomas D. D., Mancardi D., Tocchetti C. G., Ridnour L. A., Paolocci N., King S. B., Miranda K. M., Lazzarino G., Fukuto J. M., and Wink D. A. (2006) Discriminating formation of HNO from other reactive nitrogen oxide species. Free Radic. Biol. Med. 40, 1056–1066 10.1016/j.freeradbiomed.2005.10.058 [DOI] [PubMed] [Google Scholar]
- 166. Wong P. S., Hyun J., Fukuto J. M., Shirota F. N., DeMaster E. G., Shoeman D. W., and Nagasawa H. T. (1998) Reaction between S-nitrosothiols and thiols: generation of nitroxyl (HNO) and subsequent chemistry. Biochemistry 37, 5362–5371 10.1021/bi973153g [DOI] [PubMed] [Google Scholar]
- 167. Pino N. W., Davis J. 3rd., Yu Z., and Chan J. (2017) NitroxylFluor: a thiol-based fluorescent probe for live-cell imaging of nitroxyl. J. Am. Chem. Soc. 139, 18476–18479 10.1021/jacs.7b11471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Reisz J. A., Klorig E. B., Wright M. W., and King S. B. (2009) Reductive phosphine-mediated ligation of nitroxyl (HNO). Org. Lett. 11, 2719–2721 10.1021/ol900914s [DOI] [PubMed] [Google Scholar]
- 169. Reisz J. A., Zink C. N., and King S. B. (2011) Rapid and selective nitroxyl (HNO) trapping by phosphines: kinetics and new aqueous ligations for HNO detection and quantitation. J. Am. Chem. Soc. 133, 11675–11685 10.1021/ja203652z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Miao Z., and King S. B. (2017) in The Chemistry and Biology of Nitroxyl (HNO) (Doctorovich F., Farmer P. J., and Marti M. A., eds) pp. 225–238, Elsevier, Amsterdam, the Netherlands [Google Scholar]
- 171. Kawai K., Ieda N., Aizawa K., Suzuki T., Miyata N., and Nakagawa H. (2013) A reductant-resistant and metal-free fluorescent probe for nitroxyl applicable to living cells. J. Am. Chem. Soc. 135, 12690–12696 10.1021/ja404757s [DOI] [PubMed] [Google Scholar]
- 172. Koppenol W. H., and Kissner R. (1998) Can O=NOOH undergo homolysis? Chem. Res. Toxicol. 11, 87–90 10.1021/tx970200x [DOI] [PubMed] [Google Scholar]
- 173. Perkins A., Poole L. B., and Karplus P. A. (2014) Tuning of peroxiredoxin catalysis for various physiological roles. Biochemistry 53, 7693–7705 10.1021/bi5013222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Denicola A., Freeman B. A., Trujillo M., and Radi R. (1996) Peroxynitrite reaction with carbon dioxide/bicarbonate: kinetics and influence on peroxynitrite-mediated oxidations. Arch. Biochem. Biophys. 333, 49–58 10.1006/abbi.1996.0363 [DOI] [PubMed] [Google Scholar]
- 175. Bohle D. S., Hansert B., Paulson S. C., and Smith B. D. (1994) Biomimetic synthesis of the putative cytotoxin peroxynitrite, ONOO−, and its characterization as a tetramethylammonium salt. J. Am. Chem. Soc. 116, 7423–7424 10.1021/ja00095a062 [DOI] [Google Scholar]
- 176. Prolo C., Rios N., Piacenza L., Álvarez M. N., and Radi R. (2018) Fluorescence and chemiluminescence approaches for peroxynitrite detection. Free Radic. Biol. Med. 128, 59–68 10.1016/j.freeradbiomed.2018.02.017 [DOI] [PubMed] [Google Scholar]
- 177. Wardman P. (2007) Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic. Biol. Med. 43, 995–1022 10.1016/j.freeradbiomed.2007.06.026 [DOI] [PubMed] [Google Scholar]
- 178. Winterbourn C. C. (2014) The challenges of using fluorescent probes to detect and quantify specific reactive oxygen species in living cells. Biochim. Biophys. Acta 1840, 730–738 10.1016/j.bbagen.2013.05.004 [DOI] [PubMed] [Google Scholar]
- 179. Chen Z.-J., Ren W., Wright Q. E., and Ai H.-W. (2013) Genetically encoded fluorescent probe for the selective detection of peroxynitrite. J. Am. Chem. Soc. 135, 14940–14943 10.1021/ja408011q [DOI] [PubMed] [Google Scholar]
- 180. Chen Z.-J., Tian Z., Kallio K., Oleson A. L., Ji A., Borchardt D., Jiang D. E., Remington S. J., and Ai H.-W. (2016) The N–B interaction through a water bridge: understanding the chemoselectivity of a fluorescent protein based probe for peroxynitrite. J. Am. Chem. Soc. 138, 4900–4907 10.1021/jacs.6b01285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Ríos N., Prolo C., Álvarez M. N., Piacenza L., and Radi R. (2017) Nitric Oxide, 3rd Ed., (Ignarro L., and Freeman B. A.) pp. 271–288, Elsevier, New York [Google Scholar]
- 182. Wrona M., Patel K., and Wardman P. (2005) Reactivity of 2′,7′-dichlorodihydrofluorescein and dihydrorhodamine 123 and their oxidized forms toward carbonate, nitrogen dioxide, and hydroxyl radicals. Free Radic. Biol. Med. 38, 262–270 10.1016/j.freeradbiomed.2004.10.022 [DOI] [PubMed] [Google Scholar]
- 183. Wardman P. (2008) Methods to measure the reactivity of peroxynitrite-derived oxidants toward reduced fluoresceins and rhodamines. Methods Enzymol. 441, 261–282 10.1016/S0076-6879(08)01214-7 [DOI] [PubMed] [Google Scholar]
- 184. Radi R., Peluffo G., Alvarez M. N., Naviliat M., and Cayota A. (2001) Unraveling peroxynitrite formation in biological systems. Free Radic. Biol. Med. 30, 463–488 10.1016/S0891-5849(00)00373-7 [DOI] [PubMed] [Google Scholar]
- 185. Wrona M., Patel K. B., and Wardman P. (2008) The roles of thiol-derived radicals in the use of 2′,7′-dichlorodihydrofluorescein as a probe for oxidative stress. Free Radic. Biol. Med. 44, 56–62 10.1016/j.freeradbiomed.2007.09.005 [DOI] [PubMed] [Google Scholar]
- 186. Radi R., Cosgrove T. P., Beckman J. S., and Freeman B. A. (1993) Peroxynitrite-induced luminol chemiluminescence. Biochem. J. 290, 51–57 10.1042/bj2900051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Yang D., Wang H.-L., Sun Z.-N., Chung N.-W., and Shen J.-G. (2006) A highly selective fluorescent probe for the detection and imaging of peroxynitrite in living cells. J. Am. Chem. Soc. 128, 6004–6005 10.1021/ja0603756 [DOI] [PubMed] [Google Scholar]
- 188. Sun Z.-N., Wang H.-L., Liu F.-Q., Chen Y., Tam P. K., and Yang D. (2009) BODIPY-based fluorescent probe for peroxynitrite detection and imaging in living cells. Org. Lett. 11, 1887–1890 10.1021/ol900279z [DOI] [PubMed] [Google Scholar]
- 189. Peng T., and Yang D. (2010) HKGreen-3: a rhodol-based fluorescent probe for peroxynitrite. Org. Lett. 12, 4932–4935 10.1021/ol102182j [DOI] [PubMed] [Google Scholar]
- 190. Li Y., Xie X., Yang X., Li M., Jiao X., Sun Y., Wang X., and Tang B. (2017) Two-photon fluorescent probe for revealing drug-induced hepatotoxicity via mapping fluctuation of peroxynitrite. Chem. Sci. 8, 4006–4011 10.1039/C7SC00303J [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Qu W., Niu C., Zhang X., Chen W., Yu F., Liu H., Zhang X., and Wang S. (2019) Construction of a novel far-red fluorescence light-up probe for visualizing intracellular peroxynitrite. Talanta 197, 431–435 10.1016/j.talanta.2019.01.065 [DOI] [PubMed] [Google Scholar]
- 192. Zielonka J., Zielonka M., Sikora A., Adamus J., Joseph J., Hardy M., Ouari O., Dranka B. P., and Kalyanaraman B. (2012) Global profiling of reactive oxygen and nitrogen species in biological systems high-throughput real-time analyses. J. Biol. Chem. 287, 2984–2995 10.1074/jbc.M111.309062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Sikora A., Zielonka J., Lopez M., Joseph J., and Kalyanaraman B. (2009) Direct oxidation of boronates by peroxynitrite: mechanism and implications in fluorescence imaging of peroxynitrite. Free Radic. Biol. Med. 47, 1401–1407 10.1016/j.freeradbiomed.2009.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Rios N., Piacenza L., Trujillo M., Martínez A., Demicheli V., Prolo C., Álvarez M. N., López M. N., and Radi R. (2016) Sensitive detection and estimation of cell-derived peroxynitrite fluxes using fluorescein-boronate. Free Radic. Biol. Med. 101, 284–295 10.1016/j.freeradbiomed.2016.08.033 [DOI] [PubMed] [Google Scholar]
- 195. Du L., Li M., Zheng S., and Wang B. (2008) Rational design of a fluorescent hydrogen peroxide probe based on the umbelliferone fluorophore. Tetrahedron Lett. 49, 3045–3048 10.1016/j.tetlet.2008.03.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Dickinson B. C., Huynh C., and Chang C. J. (2010) A palette of fluorescent probes with varying emission colors for imaging hydrogen peroxide signaling in living cells. J. Am. Chem. Soc. 132, 5906–5915 10.1021/ja1014103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Zielonka J., Sikora A., Adamus J., and Kalyanaraman B. (2015) in Mitochondrial Medicine: Volume I, Probing Mitochondrial Function (Weissig V., and Edeas M., eds) pp. 171–181, Springer, New York [Google Scholar]
- 198. Hall D. G. (2005) Structure, Properties, and Preparation of Boronic Acid Derivatives. Overview of Their Reactions and Applications. In Boronic Acids, John Wiley & Sons, New York [Google Scholar]
- 199. Ramos Truzzi D., and Augusto O. (2018) in Hydrogen Peroxide Metabolism in Health and Disease (Vissers M. C., Hampton M., and Kettle A. J., eds) pp. 81–99, CRC Press, Los Angeles, CA [Google Scholar]
- 200. Michalski R., Zielonka J., Gapys E., Marcinek A., Joseph J., and Kalyanaraman B. (2014) Real-time measurements of amino acid and protein hydroperoxides using coumarin boronic acid. J. Biol. Chem. 289, 22536–22553 10.1074/jbc.M114.553727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Sikora A., Zielonka J., Lopez M., Dybala-Defratyka A., Joseph J., Marcinek A., and Kalyanaraman B. (2011) Reaction between peroxynitrite and boronates: EPR spin-trapping, HPLC analyses, and quantum mechanical study of the free radical pathway. Chem. Res. Toxicol. 24, 687–697 10.1021/tx100439a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Zielonka J., Sikora A., Joseph J., and Kalyanaraman B. (2010) Peroxynitrite is the major species formed from different flux ratios of co-generated nitric oxide and superoxide direct reaction with boronate-based fluorescent probe. J. Biol. Chem. 285, 14210–14216 10.1074/jbc.M110.110080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Kim J., Park J., Lee H., Choi Y., and Kim Y. (2014) A boronate-based fluorescent probe for the selective detection of cellular peroxynitrite. Chem. Commun. 50, 9353–9356 10.1039/C4CC02943G [DOI] [PubMed] [Google Scholar]
- 204. Palanisamy S., Wu P.-Y., Wu S.-C., Chen Y.-J., Tzou S.-C., Wang C.-H., Chen C.-Y., and Wang Y.-M. (2017) In vitro and in vivo imaging of peroxynitrite by a ratiometric boronate-based fluorescent probe. Biosens. Bioelectron. 91, 849–856 10.1016/j.bios.2017.01.027 [DOI] [PubMed] [Google Scholar]
- 205. Dębowska K., Dębski D., Michałowski B., Dybala-Defratyka A., Wójcik T., Michalski R., Jakubowska M., Selmi A., Smulik R., Piotrowski Ł., Adamus J., Marcinek A., Chlopicki S., and Sikora A. (2016) Characterization of fluorescein-based monoboronate probe and its application to the detection of peroxynitrite in endothelial cells treated with doxorubicin. Chem. Res. Toxicol. 29, 735–746 10.1021/acs.chemrestox.5b00431 [DOI] [PubMed] [Google Scholar]
- 206. Purdey M. S., McLennan H. J., Sutton-McDowall M. L., Drumm D. W., Zhang X., Capon P. K., Heng S., Thompson J. G., and Abell A. D. (2018) Biological hydrogen peroxide detection with aryl boronate and benzil BODIPY-based fluorescent probes. Sensors and Actuators B: Chemical 262, 750–757 10.1016/j.snb.2018.01.198 [DOI] [Google Scholar]
- 207. Xu J., Li Q., Yue Y., Guo Y., and Shao S. (2014) A water-soluble BODIPY derivative as a highly selective “Turn-On” fluorescent sensor for H2O2 sensing in vivo. Biosens. Bioelectron. 56, 58–63 10.1016/j.bios.2013.12.065 [DOI] [PubMed] [Google Scholar]
- 208. Sjöback R., Nygren J., and Kubista M. (1995) Absorption and fluorescence properties of fluorescein. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 51, L7–L21 10.1016/0584-8539(95)01421-P [DOI] [Google Scholar]
- 209. Heldt J. R., Heldt J., Stoń M., and Diehl H. A. (1995) Photophysical properties of 4-alkyl- and 7-alkoxycoumarin derivatives. Absorption and emission spectra, fluorescence quantum yield and decay time. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 51, 1549–1563 10.1016/0584-8539(95)01467-9 30246201 [DOI] [Google Scholar]
- 210. Wu L., Han H.-H., Liu L., Gardiner J. E., Sedgwick A. C., Huang C., Bull S. D., He X.-P., and James T. D. (2018) ESIPT-based fluorescence probe for the rapid detection of peroxynitrite 'AND' biological thiols. Chem. Commun. 54, 11336–11339 10.1039/C8CC06917D [DOI] [PubMed] [Google Scholar]
- 211. Barondeau D. P., Kassmann C. J., Tainer J. A., and Getzoff E. D. (2005) Understanding GFP chromophore biosynthesis: controlling backbone cyclization and modifying post-translational chemistry. Biochemistry 44, 1960–1970 10.1021/bi0479205 [DOI] [PubMed] [Google Scholar]
- 212. Gunther M. R., Hsi L. C., Curtis J. F., Gierse J. K., Marnett L. J., Eling T. E., and Mason R. P. (1997) Nitric oxide trapping of the tyrosyl radical of prostaglandin H synthase-2 leads to tyrosine iminoxyl radical and nitrotyrosine formation. J. Biol. Chem. 272, 17086–17090 10.1074/jbc.272.27.17086 [DOI] [PubMed] [Google Scholar]
- 213. Rocha B. S., Gago B., Barbosa R. M., Lundberg J. O., Radi R., and Laranjinha J. (2012) Intragastric nitration by dietary nitrite: Implications for modulation of protein and lipid signaling. Free Radic. Biol. Med. 52, 693–698 10.1016/j.freeradbiomed.2011.11.011 [DOI] [PubMed] [Google Scholar]
- 214. Cassina A. M., Hodara R., Souza J. M., Thomson L., Castro L., Ischiropoulos H., Freeman B. A., and Radi R. (2000) Cytochrome c nitration by peroxynitrite. J. Biol. Chem. 275, 21409–21415 10.1074/jbc.M909978199 [DOI] [PubMed] [Google Scholar]
- 215. Demicheli V., Moreno D. M., Jara G. E., Lima A., Carballal S., Ríos N., Batthyany C., Ferrer-Sueta G., Quijano C., Estrıìn D. A., Martí M. A., and Radi R. (2016) Mechanism of the reaction of human manganese superoxide dismutase with peroxynitrite: nitration of critical tyrosine 34. Biochemistry 55, 3403–3417 10.1021/acs.biochem.6b00045 [DOI] [PubMed] [Google Scholar]
- 216. Deeb R. S., Nuriel T., Cheung C., Summers B., Lamon B. D., Gross S. S., and Hajjar D. P. (2013) Characterization of a cellular denitrase activity that reverses nitration of cyclooxygenase. Am. J. Physiol. Heart Circ. 305, H687–H698 10.1152/ajpheart.00876.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Gerding H. R., Karreman C., Daiber A., Delp J., Hammler D., Mex M., Schildknecht S., and Leist M. (2019) Reductive modification of genetically encoded 3-nitrotyrosine sites in α-synuclein expressed in E. coli. Redox Biol. 26, 101251 10.1016/j.redox.2019.101251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Kuo W.-N., Kanadia R. N., Shanbhag V. P., and Toro R. (1999) Denitration of peroxynitrite-treated proteins by 'protein nitratases' from rat brain and heart. Mol. Cell. Biochem. 201, 11–16 10.1023/A:1007024126947 [DOI] [PubMed] [Google Scholar]
- 219. Beckman J. S., Ischiropoulos H., Zhu L., van der Woerd M., Smith C., Chen J., Harrison J., Martin J. C., and Tsai M. (1992) Kinetics of superoxide dismutase- and iron-catalyzed nitration of phenolics by peroxynitrite. Arch. Biochem. Biophys. 298, 438–445 10.1016/0003-9861(92)90432-V [DOI] [PubMed] [Google Scholar]
- 220. Abello N., Kerstjens H. A., Postma D. S., and Bischoff R. (2009) Protein tyrosine nitration: selectivity, physicochemical and biological consequences, denitration, and proteomics methods for the identification of tyrosine-nitrated proteins. J. Proteome Res. 8, 3222–3238 10.1021/pr900039c [DOI] [PubMed] [Google Scholar]
- 221. Batthyány C., Bartesaghi S., Mastrogiovanni M., Lima A., Demicheli V., and Radi R. (2017) Tyrosine-nitrated proteins: proteomic and bioanalytical aspects. Antioxid. Redox Signal. 26, 313–328 10.1089/ars.2016.6787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Tsikas D., and Duncan M. W. (2014) Mass spectrometry and 3-nitrotyrosine: strategies, controversies, and our current perspective. Mass Spectrom. Rev. 33, 237–276 10.1002/mas.21396 [DOI] [PubMed] [Google Scholar]
- 223. Duncan M. W. (2003) A review of approaches to the analysis of 3-nitrotyrosine. Amino Acids 25, 351–361 10.1007/s00726-003-0022-z [DOI] [PubMed] [Google Scholar]
- 224. Nicholls S. J., Shen Z., Fu X., Levison B. S., and Hazen S. L. (2005) Quantification of 3-nitrotyrosine levels using a benchtop ion trap mass spectrometry method. Methods Enzymol. 396, 245–266 10.1016/S0076-6879(05)96022-9 [DOI] [PubMed] [Google Scholar]
- 225. Shigenaga M. K., Lee H. H., Blount B. C., Christen S., Shigeno E. T., Yip H., and Ames B. N. (1997) Inflammation and NO(X)-induced nitration: assay for 3-nitrotyrosine by HPLC with electrochemical detection. Proc. Natl. Acad. Sci. U.S.A. 94, 3211–3216 10.1073/pnas.94.7.3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Schwedhelm E., Tsikas D., Gutzki F.-M., and Frölich J. C. (1999) Gas chromatographic–tandem mass spectrometric quantification of free 3-nitrotyrosine in human plasma at the basal state. Anal. Biochem. 276, 195–203 10.1006/abio.1999.4361 [DOI] [PubMed] [Google Scholar]
- 227. Söderling A.-S., Ryberg H., Gabrielsson A., Lärstad M., Torén K., Niari S., and Caidahl K. (2003) A derivatization assay using gas chromatography/negative chemical ionization tandem mass spectrometry to quantify 3-nitrotyrosine in human plasma. J. Mass Spectrom. 38, 1187–1196 10.1002/jms.543 [DOI] [PubMed] [Google Scholar]
- 228. Yi D., Ingelse B. A., Duncan M. W., and Smythe G. A. (2000) Quantification of 3-nitrotyrosine in biological tissues and fluids: generating valid results by eliminating artifactual formation. J. Am. Soc. Mass Spectrom. 11, 578–586 10.1016/S1044-0305(00)00113-6 [DOI] [PubMed] [Google Scholar]
- 229. Khan F., and Siddiqui A. A. (2006) Prevalence of anti-3-nitrotyrosine antibodies in the joint synovial fluid of patients with rheumatoid arthritis, osteoarthritis and systemic lupus erythematosus. Clin. Chim. Acta 370, 100–107 10.1016/j.cca.2006.01.020 [DOI] [PubMed] [Google Scholar]
- 230. Thomson L., Tenopoulou M., Lightfoot R., Tsika E., Parastatidis I., Martinez M., Greco Todd M., Doulias P.-T., Wu Y., Tang W. H., Hazen S. L., and Ischiropoulos H. (2012) Immunoglobulins against tyrosine-nitrated epitopes in coronary artery disease. Circulation 126, 2392–2401 10.1161/CIRCULATIONAHA.112.103796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Beckmann J. S., Ye Y. Z., Anderson P. G., Chen J., Accavitti M. A., Tarpey M. M., and White C. R. (1994) Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol. Chem. Hoppe Seyler 375, 81–88 10.1515/bchm3.1994.375.1.81 [DOI] [PubMed] [Google Scholar]
- 232. Brito C., Naviliat M., Tiscornia A. C., Vuillier F., Gualco G., Dighiero G., Radi R., and Cayota A. M. (1999) Peroxynitrite inhibits T lymphocyte activation and proliferation by promoting impairment of tyrosine phosphorylation and peroxynitrite-driven apoptotic death. J. Immunol. 162, 3356–3366 [PubMed] [Google Scholar]
- 233. Viera L., Ye Y. Z., Estévez A. G., and Beckman J. S. (1999) Immunohistochemical methods to detect nitrotyrosine. Methods Enzymol. 301, 373–381 10.1016/S0076-6879(99)01101-5 [DOI] [PubMed] [Google Scholar]
- 234. Ye Y. Z., Strong M., Huang Z.-Q., and Beckman J. S. (1996) Antibodies that recognize nitrotyrosine. Methods Enzymol. 269, 201–209 10.1016/S0076-6879(96)69022-3 [DOI] [PubMed] [Google Scholar]
- 235. Knight A. R., Taylor E. L., Lukaszewski R., Jensen K. T., Jones H. E., Carré J. E., Isupov M. N., Littlechild J. A., Bailey S. J., Brewer E., McDonald T. J., Pitt A. R., Spickett C. M., and Winyard P. G. (2018) A high-sensitivity electrochemiluminescence-based ELISA for the measurement of the oxidative stress biomarker, 3-nitrotyrosine, in human blood serum and cells. Free Radic. Biol. Med. 120, 246–254 10.1016/j.freeradbiomed.2018.03.026 [DOI] [PubMed] [Google Scholar]
- 236. Crow J. P., and Ischiropoulos H. (1996) Detection and quantitation of nitrotyrosine residues in proteins: in vivo marker of peroxynitrite. Methods Enzymol. 269, 185–194 10.1016/S0076-6879(96)69020-X [DOI] [PubMed] [Google Scholar]
- 237. Kamisaki Y., Wada K., Nakamoto K., Kishimoto Y., Kitano M., and Itoh T. (1996) Sensitive determination of nitrotyrosine in human plasma by isocratic high-performance liquid chromatography. J. Chromatog. B Biomed. Appl. 685, 343–347 10.1016/S0378-4347(96)00202-2 [DOI] [PubMed] [Google Scholar]
- 238. Crowley J. R., Yarasheski K., Leeuwenburgh C., Turk J., and Heinecke J. W. (1998) Isotope dilution mass spectrometric quantification of 3-nitrotyrosine in proteins and tissues is facilitated by reduction to 3-aminotyrosine. Anal. Biochem. 259, 127–135 10.1006/abio.1998.2635 [DOI] [PubMed] [Google Scholar]
- 239. Frost M. T., Halliwell B., and Moore K. P. (2000) Analysis of free and protein-bound nitrotyrosine in human plasma by a gas chromatography/mass spectrometry method that avoids nitration artifacts. Biochem. J. 345, 453–458 10.1042/bj3450453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Jiang H., and Balazy M. (1998) Detection of 3-nitrotyrosine in human platelets exposed to peroxynitrite by a new gas chromatography/mass spectrometry assay. Nitric Oxide 2, 350–359 10.1006/niox.1998.0196 [DOI] [PubMed] [Google Scholar]
- 241. Martinez A., Peluffo G., Petruk A. A., Hugo M., Piñeyro D., Demicheli V., Moreno D. M., Lima A., Batthyány C., Durán R., Robello C., Martí M. A., Larrieux N., Buschiazzo A., Trujillo M., et al. (2014) Structural and molecular basis of the peroxynitrite-mediated nitration and inactivation of Trypanosoma cruzi iron-superoxide dismutases (Fe-SODs) A and B: disparate susceptibilities due to the repair of Tyr-35 radical by Cys-83 in Fe-SODB through intramolecular electron transfer. J. Biol. Chem. 289, 12760–12778 10.1074/jbc.M113.545590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. McDonagh B. (2017) Detection of ROS induced proteomic signatures by mass spectrometry. Front. Physiol. 8, 470 10.3389/fphys.2017.00470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Batthyány C., Souza J. M., Durán R., Cassina A., Cerveñansky C., and Radi R. (2005) Time course and site(s) of cytochrome c tyrosine nitration by peroxynitrite. Biochemistry 44, 8038–8046 10.1021/bi0474620 [DOI] [PubMed] [Google Scholar]
- 244. Sarver A., Scheffler N. K., Shetlar M. D., and Gibson B. W. (2001) Analysis of peptides and proteins containing nitrotyrosine by matrix-assisted laser desorption/ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 12, 439–448 10.1016/S1044-0305(01)00213-6 [DOI] [PubMed] [Google Scholar]
- 245. Ghosh S., Janocha A. J., Aronica M. A., Swaidani S., Comhair S. A., Xu W., Zheng L., Kaveti S., Kinter M., Hazen S. L., and Erzurum S. C. (2006) Nitrotyrosine proteome survey in asthma identifies oxidative mechanism of catalase inactivation. J. Immunol. 176, 5587–5597 10.4049/jimmunol.176.9.5587 [DOI] [PubMed] [Google Scholar]
- 246. Kanski J., and Schöneich C. (2005) Protein nitration in biological aging: proteomic and tandem mass spectrometric characterization of nitrated sites. Methods Enzymol. 396, 160–171 10.1016/S0076-6879(05)96016-3 [DOI] [PubMed] [Google Scholar]
- 247. Peng F., Li J., Guo T., Yang H., Li M., Sang S., Li X., Desiderio D. M., and Zhan X. (2015) Nitroproteins in human astrocytomas discovered by gel electrophoresis and tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 26, 2062–2076 10.1007/s13361-015-1270-3 [DOI] [PubMed] [Google Scholar]
- 248. Armstrong D. A., Huie R. E., Koppenol W. H., Lymar S. V., Merényi G., Neta P., Ruscic B., Stanbury D. M., Steenken S., and Wardman P. (2015) Standard electrode potentials involving radicals in aqueous solution: inorganic radicals (IUPAC Technical Report). Pure Appl. Chem. 87, 1139–1150 10.1515/pac-2014-0502 [DOI] [Google Scholar]
- 249. Serrano-Luginbuehl S., Kissner R., and Koppenol W. H. (2018) Reaction of CO2 with ONOO(−): one molecule of CO2 is not enough. Chem. Res. Toxicol. 31, 721–730 10.1021/acs.chemrestox.8b00068 [DOI] [PubMed] [Google Scholar]
- 250. Koppenol W. H., Stanbury D. M., and Bounds P. L. (2010) Electrode potentials of partially reduced oxygen species, from dioxygen to water. Free Radic. Biol. Med. 49, 317–322 10.1016/j.freeradbiomed.2010.04.011 [DOI] [PubMed] [Google Scholar]
- 251. Buxton G. V., and Elliot A. J. (1986) Rate constant for reaction of hydroxyl radicals with bicarbonate ions. Int. J. Radiat. Appl. Instrum. C Radiat. Phys. Chem. 27, 241–243 [Google Scholar]
- 252. Wolcott R. G., Franks B. S., Hannum D. M., and Hurst J. K. (1994) Bactericidal potency of hydroxyl radical in physiological environments. J. Biol. Chem. 269, 9721–9728 [PubMed] [Google Scholar]
- 253. Liochev S. I., and Fridovich I. (2004) Bicarbonate-enhanced peroxidase activity of Cu,Zn SOD: is the distal oxidant bound or diffusible? Arch. Biochem. Biophys. 421, 255–259 10.1016/j.abb.2003.11.015 [DOI] [PubMed] [Google Scholar]
- 254. Liochev S. I., and Fridovich I. (2004) CO2, not HCO3−, facilitates oxidations by Cu,Zn superoxide dismutase plus H2O2. Proc. Natl. Acad. Sci. U.S.A. 101, 743–744 10.1073/pnas.0307635100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Karunakaran C., Zhang H., Joseph J., Antholine W. E., and Kalyanaraman B. (2005) Thiol oxidase activity of copper, zinc superoxide dismutase stimulates bicarbonate-dependent peroxidase activity via formation of a carbonate radical. Chem. Res. Toxicol. 18, 494–500 10.1021/tx049747j [DOI] [PubMed] [Google Scholar]
- 256. Mason R. P., Ganini D., Bonini M. G., and Ranguelova K. (2012) Two hypotheses for the oxidation of SOD1-Cu(I). Free Radic. Biol. Med. 53, 1991–1992 10.1016/j.freeradbiomed.2012.10.524 [DOI] [PubMed] [Google Scholar]
- 257. Michelson A. M., and Maral J. (1983) Carbonate anions; effects on the oxidation of luminol, oxidative hemolysis, γ-irradiation and the reaction of activated oxygen species with enzymes containing various active centres. Biochimie 65, 95–104 10.1016/S0300-9084(83)80179-5 [DOI] [PubMed] [Google Scholar]
- 258. Bonini M. G., Miyamoto S., Di Mascio P., and Augusto O. (2004) Production of the carbonate radical anion during xanthine oxidase turnover in the presence of bicarbonate. J. Biol. Chem. 279, 51836–51843 10.1074/jbc.M406929200 [DOI] [PubMed] [Google Scholar]
- 259. Medinas D. B., Cerchiaro G., Trindade D. F., and Augusto O. (2007) The carbonate radical and related oxidants derived from bicarbonate buffer. IUBMB Life 59, 255–262 10.1080/15216540701230511 [DOI] [PubMed] [Google Scholar]
- 260. Czapski G., Lymar S. V., and Schwarz H. A. (1999) Acidity of the carbonate radical. J. Phys. Chem. A 103, 3447–3450 10.1021/jp984769y [DOI] [Google Scholar]
- 261. Bisby R. H., Johnson S. A., Parker A. W., and Tavender S. M. (1998) Time-resolved resonance Raman spectroscopy of the carbonate radical. Chem. Soc. Faraday Trans. 94, 2069–2072 10.1039/a801239c [DOI] [Google Scholar]
- 262. Bartesaghi S., Valez V., Trujillo M., Peluffo G., Romero N., Zhang H., Kalyanaraman B., and Radi R. (2006) Mechanistic studies of peroxynitrite-mediated tyrosine nitration in membranes using the hydrophobic probe N-t-BOC-l-tyrosine tert-butyl ester. Biochemistry 45, 6813–6825 10.1021/bi060363x [DOI] [PubMed] [Google Scholar]
- 263. Chen S. N., and Hoffman M. Z. (1973) Rate constants for the reaction of the carbonate radical with compounds of biochemical interest in neutral aqueous solution. Radiat. Res. 56, 40–47 10.2307/3573789 [DOI] [PubMed] [Google Scholar]
- 264. Shafirovich V., Dourandin A., Huang W., and Geacintov N. E. (2001) The carbonate radical is a site-selective oxidizing agent of guanine in double-stranded oligonucleotides. J. Biol. Chem. 276, 24621–24626 10.1074/jbc.M101131200 [DOI] [PubMed] [Google Scholar]
- 265. Medinas D. B., Gozzo F. C., Santos L. F., Iglesias A. H., and Augusto O. (2010) A ditryptophan cross-link is responsible for the covalent dimerization of human superoxide dismutase 1 during its bicarbonate-dependent peroxidase activity. Free Radic. Biol. Med. 49, 1046–1053 10.1016/j.freeradbiomed.2010.06.018 [DOI] [PubMed] [Google Scholar]
- 266. Yun B. H., Geacintov N. E., and Shafirovich V. (2011) Generation of guanine–thymidine cross-links in DNA by peroxynitrite/carbon dioxide. Chem. Res. Toxicol. 24, 1144–1152 10.1021/tx200139c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Tórtora V., Quijano C., Freeman B., Radi R., and Castro L. (2007) Mitochondrial aconitase reaction with nitric oxide, S-nitrosoglutathione, and peroxynitrite: mechanisms and relative contributions to aconitase inactivation. Free Radic. Biol. Med. 42, 1075–1088 10.1016/j.freeradbiomed.2007.01.007 [DOI] [PubMed] [Google Scholar]
- 268. Trujillo M., Folkes L., Bartesaghi S., Kalyanaraman B., Wardman P., and Radi R. (2005) Peroxynitrite-derived carbonate and nitrogen dioxide radicals readily react with lipoic and dihydrolipoic acid. Free Radic. Biol. Med. 39, 279–288 10.1016/j.freeradbiomed.2005.03.014 [DOI] [PubMed] [Google Scholar]
- 269. Dassanayake R. S., Shelley J. T., Cabelli D. E., and Brasch N. E. (2015) Pulse radiolysis and ultra-high-performance liquid chromatography/high-resolution mass spectrometry studies on the reactions of the carbonate radical with vitamin B12 derivatives. Chemistry 21, 6409–6419 10.1002/chem.201406269 [DOI] [PubMed] [Google Scholar]
- 270. Weeks J. L., and Rabani J. (1966) The pulse radiolysis of deaerated aqueous carbonate solutions. I. Transient optical spectrum and mechanism. 11. pK for OH radicals. J. Phys. Chem. 70, 2100–2106 10.1021/j100879a005 [DOI] [Google Scholar]
- 271. Bonini M. G., Radi R., Ferrer-Sueta G., Ferreira A. M., and Augusto O. (1999) Direct EPR detection of the carbonate radical anion produced from peroxynitrite and carbon dioxide. J. Biol. Chem. 274, 10802–10806 10.1074/jbc.274.16.10802 [DOI] [PubMed] [Google Scholar]
- 272. Alvarez M. N., Peluffo G., Folkes L., Wardman P., and Radi R. (2007) Reaction of the carbonate radical with the spin-trap 5,5-dimethyl-1-pyrroline-N-oxide in chemical and cellular systems: pulse radiolysis, electron paramagnetic resonance, and kinetic-competition studies. Free Radic. Biol. Med. 43, 1523–1533 10.1016/j.freeradbiomed.2007.08.002 [DOI] [PubMed] [Google Scholar]
- 273. Villamena F. A., Locigno E. J., Rockenbauer A., Hadad C. M., and Zweier J. L. (2007) Theoretical and experimental studies of the spin trapping of inorganic radicals by 5,5-dimethyl-1-pyrroline-N-oxide (DMPO). 2. Carbonate radical anion. J. Phys. Chem. A 111, 384–391 10.1021/jp065692d [DOI] [PubMed] [Google Scholar]
- 274. Epperlein M. M., Nourooz-Zadeh J., Jayasena S. D., Hothersall J. S., Noronha-Dutra A., and Neild G. H. (1998) Nature and biological significance of free radicals generated during bicarbonate hemodialysis. J. Am. Soc. Nephrol. 9, 457–463 [DOI] [PubMed] [Google Scholar]
- 275. Hardeland R., Poeggeler B., Niebergall R., and Zelosko V. (2003) Oxidation of melatonin by carbonate radicals and chemiluminescence emitted during pyrrole ring cleavage. J. Pineal Res. 34, 17–25 10.1034/j.1600-079X.2003.02941.x [DOI] [PubMed] [Google Scholar]
- 276. Bartesaghi S., Trujillo M., Denicola A., Folkes L., Wardman P., and Radi R. (2004) Reactions of desferrioxamine with peroxynitrite-derived carbonate and nitrogen dioxide radicals. Free Radic. Biol. Med. 36, 471–483 10.1016/j.freeradbiomed.2003.10.011 [DOI] [PubMed] [Google Scholar]
- 277. Denicola A., Rubbo H., Rodríguez D., and Radi R. (1993) Peroxynitrite-mediated cytotoxicity to Trypanosoma cruzi. Arch. Biochem. Biophys. 304, 279–286 10.1006/abbi.1993.1350 [DOI] [PubMed] [Google Scholar]
- 278. Yeh H. C., and Lin W. Y. (2003) Stopped-flow study of the enhanced chemiluminescence for the oxidation of luminol with hydrogen peroxide catalyzed by microperoxidase 8. Talanta 59, 1029–1038 10.1016/S0039-9140(03)00003-1 [DOI] [PubMed] [Google Scholar]
- 279. Stadler K., Bonini M. G., Dallas S., Jiang J., Radi R., Mason R. P., and Kadiiska M. B. (2008) Involvement of inducible nitric-oxide synthase in hydroxyl radical-mediated lipid peroxidation in streptozotocin-induced diabetes. Free Radic. Biol. Med. 45, 866–874 10.1016/j.freeradbiomed.2008.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Beckman J. S., Beckman T. W., Chen J., Marshall P. A., and Freeman B. A. (1990) Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 87, 1620–1624 10.1073/pnas.87.4.1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Gligorovski S., Strekowski R., Barbati S., and Vione D. (2015) Environmental implications of hydroxyl radicals (•OH). Chem. Rev. 115, 13051–13092 10.1021/cr500310b [DOI] [PubMed] [Google Scholar]
- 282. Freinbichler W., Bianchi L., Colivicchi M. A., Ballini C., Tipton K. F., Linert W., and Corte L. D. (2008) The detection of hydroxyl radicals in vivo. J. Inorg. Biochem. 102, 1329–1333 10.1016/j.jinorgbio.2007.12.017 [DOI] [PubMed] [Google Scholar]
- 283. Żamojć K., Zdrowowicz M., Jacewicz D., Wyrzykowski D., and Chmurzyński L. (2016) Fluorescent and luminescent probes for monitoring hydroxyl radical under biological conditions. Crit. Rev. Anal. Chem. 46, 160–169 10.1080/10408347.2015.1045118 [DOI] [PubMed] [Google Scholar]
- 284. Huang P. L., Dawson T. M., Bredt D. S., Snyder S. H., and Fishman M. C. (1993) Targeted disruption of the neuronal nitric-oxide synthase gene. Cell 75, 1273–1286 10.1016/0092-8674(93)90615-W [DOI] [PubMed] [Google Scholar]
- 285. Forrester M. T., Eyler C. E., and Rich J. N. (2011) Bacterial flavohemoglobin: a molecular tool to probe mammalian nitric oxide biology. BioTechniques 50, 41–45 10.2144/000113586 [DOI] [PMC free article] [PubMed] [Google Scholar]