

Riboswitches are typically RNA elements located in the 5′ untranslated region of mRNAs. They are highly structured and specifically recognize and respond to a given chemical cue to alter transcription termination or translation initiation. In this work, we report a novel mechanism of riboswitch-mediated gene regulation in Vibrio cholerae whereby a 3′ riboswitch, named Vc2, controls the stability of upstream untranslated RNA upon binding to its cognate ligand, the second messenger cyclic di-GMP, leading to the accumulation of previously undescribed putative sRNAs. We further demonstrate that binding of the ligand to the riboswitch prevents RNA degradation. As binding of riboswitches to their ligands often produces compactly structured RNA, we hypothesize this mechanism of gene regulation is widespread.
KEYWORDS: RNA degradation, Vc2, Vibrio cholerae, cyclic nucleotides, riboswitch, sRNA
ABSTRACT
Cyclic di-GMP (c-di-GMP) is a bacterial second messenger molecule that is important in the biology of Vibrio cholerae, but the molecular mechanisms by which this molecule regulates downstream phenotypes have not been fully characterized. We have previously shown that the Vc2 c-di-GMP-binding riboswitch, encoded upstream of the gene tfoY, functions as an off switch in response to c-di-GMP. However, the mechanism by which c-di-GMP controls expression of tfoY has not been fully elucidated. During our studies of this mechanism, we determined that c-di-GMP binding to Vc2 also controls the abundance and stability of upstream noncoding RNAs with 3′ ends located immediately downstream of the Vc2 riboswitch. Our results suggest these putative small RNAs (sRNAs) are not generated by transcriptional termination but rather by preventing degradation of the upstream untranslated RNA when c-di-GMP is bound to Vc2.
IMPORTANCE Riboswitches are typically RNA elements located in the 5′ untranslated region of mRNAs. They are highly structured and specifically recognize and respond to a given chemical cue to alter transcription termination or translation initiation. In this work, we report a novel mechanism of riboswitch-mediated gene regulation in Vibrio cholerae whereby a 3′ riboswitch, named Vc2, controls the stability of upstream untranslated RNA upon binding to its cognate ligand, the second messenger cyclic di-GMP, leading to the accumulation of previously undescribed putative sRNAs. We further demonstrate that binding of the ligand to the riboswitch prevents RNA degradation. As binding of riboswitches to their ligands often produces compactly structured RNA, we hypothesize this mechanism of gene regulation is widespread.
INTRODUCTION
Vibrio cholerae, the causative agent of cholera, lives in diverse environments, from reservoirs to microcompartments within the human host gastrointestinal tract (1). The bacterial second messenger cyclic di-GMP (c-di-GMP) is a central signaling molecule in this bacterium that regulates diverse traits to enable specific adaptation to these changing conditions (2–6). V. cholerae encodes three known transcription factors, VpsR, VpsT, and FlrA, that bind and respond to c-di-GMP to control gene expression by regulating the transcription of genes that mediate biofilm formation, motility, type II secretion, and DNA repair (4, 5, 7–9). In addition, V. cholerae also encodes two riboswitches that directly bind and respond to changes in the intracellular concentration of c-di-GMP, although the function of these riboswitches in the adaptation of V. cholerae is not well understood (10, 11).
c-di-GMP-binding riboswitches can be found as two distinct structural classes (12). The riboswitches encoded by V. cholerae, named Vc1 and Vc2, are both designated class I c-di-GMP-binding riboswitches (10, 11). Vc1 is encoded by the 5′ untranslated region (5′-UTR) of the putative adhesin gbpA, and binding of c-di-GMP to Vc1 enhances production of GbpA, making this riboswitch an on switch (11). Vc2, which has extensive sequence identity with Vc1, is encoded by the 5′-UTR of the tfoY gene (13). We recently demonstrated that Vc2 functions as an off switch, as it represses TfoY production when bound to c-di-GMP, leading to a decrease in TfoY-mediated dispersive motility (10). Other bacterial species also encode cyclic dinucleotide binding riboswitches. For example, Clostridium difficile encodes 11 class I and 4 class II c-di-GMP-binding riboswitches (14). A recent analysis in C. difficile suggested that the class I riboswitches are primarily off switches while the class II riboswitches are on switches (14). Geobacter sulfurreducens encodes riboswitches that can discriminate between c-di-GMP and bacterial cyclic GMP-AMP, and these riboswitches appear to regulate genes involved in exoelectrogenesis (15, 16).
Our previous studies showed that the regulation of the tfoY gene is complex, consisting of four promoters we have designated P1-tfoY to P4-tfoY and the Vc2 c-di-GMP binding riboswitch (Fig. 1A) (10). This regulatory system discriminates between at least three distinct intracellular c-di-GMP concentrations in V. cholerae when grown in rich medium in the laboratory: low, generated by overexpressing an active EAL phosphodiesterase; intermediate, the unaltered wild-type (WT) levels of c-di-GMP; or high, generated by overexpressing an active diguanylate cyclase (DGC). Importantly, we demonstrated that the concentrations of c-di-GMP observed in these three states are physiologically relevant (10). Transcription from the promoters P3-tfoY and P4-tfoY, which are located within and to the 3′ end of Vc2, respectively, and located closest to the translation start site of tfoY, does not lead to transcription of a complete Vc2 riboswitch; thus, expression of tfoY from these transcripts is Vc2 independent (Fig. 1A). Rather, these promoters are induced in a VpsR-dependent manner at high intracellular c-di-GMP. Transcription from the upstream promoters, P1-tfoY and P2-tfoY, does generate the full-length Vc2 riboswitch, and we demonstrated that binding of c-di-GMP to Vc2 leads to inhibition of TfoY production at intermediate and high c-di-GMP concentrations via an unknown mechanism (10).
FIG 1.

3′-RACE of P1-tfoY transcripts. (A) This map shows the four transcriptional start sites identified for tfoY transcripts and their location on the V. cholerae chromosome. Numbering is relative to the start of the tfoY coding sequence. The locations of the sRNAs identified in this study are indicated below the map. The box with “term” indicates the location of the factor-independent termination discussed in the text. (B) Numbering of the x axis corresponds to the genomic map in panel A, except for position zero, which is included here for reference but does not represent a nucleotide position on the genome. The y axis indicates the number of P1-tfoY transcript sequences recovered whose 3′ end mapped to the position indicated. Asterisks indicate sites where sequences contained 3′-end tails with extragenomic nucleotides. Sixty-six sequences are depicted in total.
These studies suggested a model whereby one function of Vc2 was to inhibit production of TfoY from the upstream P1-tfoY and P2-tfoY at intermediate and high concentrations of c-di-GMP, while transcription initiation of tfoY from P3-tfoY and P4-tfoY at high c-di-GMP concentrations was induced by c-di-GMP binding to the transcription factor VpsR. At low concentrations of c-di-GMP, TfoY stimulates a rapid, flagellum-based swimming motility that we termed dispersive motility (10), but the function of increased TfoY at high c-di-GMP concentrations is not known, although a recent paper did not see such c-di-GMP induction of TfoY production in another V. cholerae strain (17). Both in vitro and in vivo evidence suggested that Vc2 inhibits translation of tfoY when bound to c-di-GMP with the ribosome binding site (RBS) of tfoY sequestered by an anti-RBS element located downstream of the Vc2 riboswitch (18, 19), but the mechanism of this inhibition has not yet been fully established in the native sequence of the tfoY 5′-UTR.
While further exploring the mechanism by which Vc2 functions as an off switch, we observed that most of the 3′ ends of transcripts derived from the P1-tfoY promoter were located at the 3′ end of the Vc2 riboswitch, leading us to elucidate that c-di-GMP binding to the Vc2 riboswitch directs accumulation of a novel noncoding RNA. This putative sRNA, which we named P1-Vc2, initiates at the upstream P1-tfoY promoter and has a 3′ end located at the end of the Vc2 riboswitch. P1-Vc2 is not generated by transcription termination; rather, it accumulates at intermediate and high intracellular c-di-GMP concentrations due to increased stability when Vc2 is bound to c-di-GMP. Our results describe a novel form of gene regulation whereby a 3′-riboswitch controls the accumulation of an untranslated RNA by preventing its degradation.
RESULTS
Identification of putative sRNAs that contain Vc2 on their 3′ end.
In our previous study, primer extension analyses determined that the majority of transcripts containing the tfoY open reading frame (ORF) originate from P3-tfoY and P4-tfoY, even though P1-tfoY and P2-tfoY appeared to be stronger promoters based on expression analysis of gfp transcriptional fusions (10). Therefore, the fate of transcripts initiated at P1-tfoY and P2-tfoY was unclear. To further explore this finding, we conducted a 3′ rapid amplification of cDNA ends (3′-RACE) assay to identify the location of the 3′ ends of transcripts originating from P1-tfoY in cells grown to mid-log phase. These studies were performed with wild-type V. cholerae and thus were at intermediate concentrations of c-di-GMP. At these concentrations, c-di-GMP is bound to Vc2 and TfoY production is reduced (10). Prior studies had predicted that Vc2 regulates translation of tfoY (18, 19), leading us to hypothesize that the 3′ ends of P1-tfoY transcripts would be located downstream of the tfoY gene. In total, we recovered 66 sequences which mapped to the VC1721-tfoY intergenic region (Fig. 1B). We did not recover any transcripts with a 3′-end mapping downstream of the tfoY stop codon; however, we did recover multiple transcripts whose 3′ ends were dispersed within the tfoY coding sequence, suggesting large amounts of mRNA degradation.
Surprisingly, the 3′ end of the majority of sequences was located upstream of the tfoY translational start site. The greatest number of 3′ ends we observed, 38% of all recovered transcripts, mapped to the region −110 to −102 relative to the start of the tfoY coding sequence, which is the region immediately adjacent to the 3′ end of the P1 stem-loop that forms the base of the Vc2 riboswitch (20). An additional 17% of the sequences contained a 3′ end located further upstream, between −227 and −225 relative to the start of the tfoY coding sequence. This region correlates with the 3′ end of a putative rho-independent terminator located between the P2-tfoY and P3-tfoY transcriptional start sites (marked as “term” in Fig. 1A), and it is also the location of the identified VpsR binding sites (10, 21). Of all the transcripts recovered, 30% featured 3′-end tails containing between 1 and 4 extragenomic nucleotides, almost exclusively adenines, and the majority of these tailed transcripts mapped to the aforementioned sites of the Vc2 aptamer or the upstream rho-independent terminator. These results led us to hypothesize that most of the transcripts initiated from P1-tfoY and P2-tfoY exist as noncoding RNAs with the Vc2 riboswitch aptamer located at their 3′ ends, and these transcripts did not contain a full-length tfoY sequence, explaining the results of our previous primer extension assay.
Multiple putative sRNAs are transcribed from the VC1721-tfoY intergenic region.
To confirm that the noncoding RNA species predicted by our 3′-RACE analysis were produced by the upstream tfoY promoters in V. cholerae, we performed a series of Northern blots with probes encompassing different segments of the VC1721-tfoY intergenic region against RNA harvested from WT V. cholerae (Fig. 2). Four RNA probes were used that are complementary to the following regions: probe P1 (−337 to −272, between the P1-tfoY and P2-tfoY start sites), probe P2 (−272 to −191, between the P2-tfoY and P3-tfoY start sites), probe P1 + 2 (−337 to −191, a larger sequence overlapping the P1-tfoY to P3-tfoY start sites), and probe Vc2 (−211 to −94, the sequence of the Vc2 riboswitch).
FIG 2.
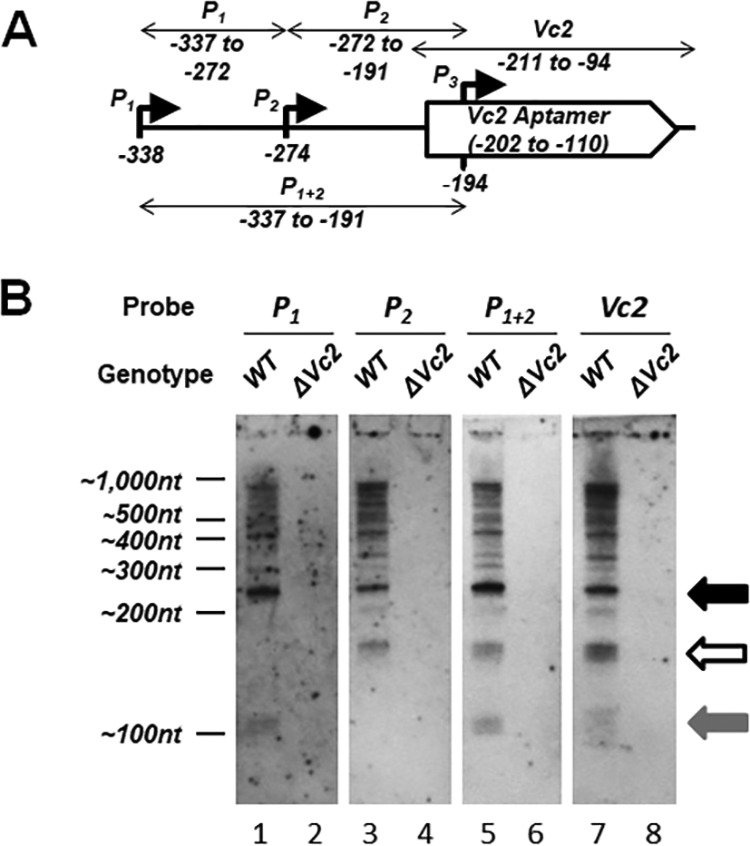
Northern blot analysis of VC1721-tfoY intergenic region. (A) The location of the four RNA probes used for the Northern blot in panel B and Fig. 3. (B) Northern blot analysis of RNA extracted from either the WT or the ΔVc2 mutant V. cholerae. The locations of the ladder bands are shown to the left (nt, nucleotides). The black arrow shows P1-Vc2, the white arrow indicates P2-Vc2, and the gray arrow indicates the transcript from P1-tfoY to the terminator.
Based on the results of the 3′-RACE assay, we expected to observe specific putative sRNA transcripts of ∼231 nucleotides and ∼167 nucleotides in size originating from the P1-tfoY and P2-tfoY promoters, respectively, with both ending at the immediate 3′ edge of the Vc2 aptamer. The presence of the ∼231-nucleotide band in all four blots (Fig. 2B, lanes 1, 3, 5, and 7, black arrow) and the presence of the ∼167-nucleotide band in all but the probe P1 blot (Fig. 2, lanes 3, 5, and 7, white arrow) confirms the location of these putative sRNA species relative to the promoters and the riboswitch aptamer. We refer to these ∼231-nucleotide and ∼167-nucleotide transcripts here as P1-Vc2 and P2-Vc2, respectively. We also expected to observe a noncoding RNA ∼112 nucleotides in size initiated from the P1-tfoY promoter and ending at the predicted intrinsic terminator immediately downstream of the P2-tfoY start site. The appearance of this band in the probe P1 and P1 + 2 blots and its absence from the probe P2 blot confirmed the presence of this transcript as well (Fig. 2, lanes 1 and 5, gray arrow).
All of the probes detected a large number of longer RNA species extending into the tfoY coding sequence. This is consistent with our 3′-RACE results identifying multiple 3′ RNA ends in this region (Fig. 1B), and it further suggests that transcription from P1-tfoY and P2-tfoY can extend into the tfoY ORF. All the signals in the Northern blot analysis from every probe used were lost in the ΔVc2 mutant, which has a deletion from −359 to −43 on the genome of V. cholerae, showing that these RNA species are specific to the Vc2 riboswitch locus. It is also worth noting that the coding sequence of VC1721, the nearest gene upstream of the riboswitch, is 1,011 nucleotides in length. Therefore, based on their size, all the transcripts detected here should have originated downstream from the VC1721 ORF and do not represent runoff transcription from upstream VC1721 mRNA. These data show that the tfoY upstream region produces multiple putative sRNA species.
The Vc2 riboswitch and c-di-GMP regulate the abundance of the putative sRNAs.
To investigate the effect of changes in c-di-GMP on Vc2 riboswitch-containing transcripts in vivo, we performed a higher-resolution Northern blot of V. cholerae RNA extracts with probe Vc2 (Fig. 3). An initial observation across all of our samples is that there are many different Vc2 riboswitch-containing transcripts between 300 and 1,000 nucleotides in length (Fig. 3). The annotated coding sequence of tfoY is 588 nucleotides; therefore, the minimum size of full-length tfoY mRNAs should be 926 nucleotides if originating from P1-tfoY or 862 nucleotides if originating from P2-tfoY. This implies that a significant number of the transcripts detected contain only a fraction of the annotated tfoY ORF, again consistent with data from our 3′-RACE analysis (Fig. 1). An integrity analysis of rRNA from our samples using an Agilent Bioanalyzer confirmed that our RNA was of high quality with little to no degradation occurring during extraction. From this we conclude that most Vc2 riboswitch-containing transcripts are inherently unstable and subject to a high rate of degradation in vivo.
FIG 3.
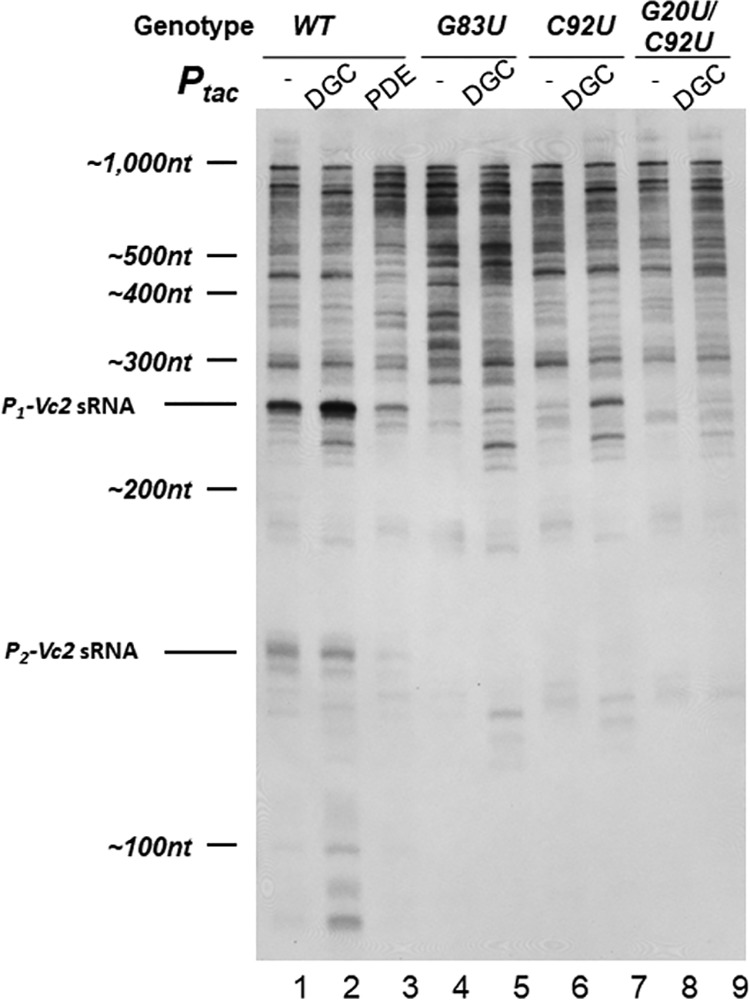
Northern blot analysis of V. cholerae riboswitch mutants. Lanes are as described in the main text. Shown is a Northern blot of the four V. cholerae strains indicated at the top of the blot with the Vc2 probe show in Fig. 2A. It is representative of multiple experiments. The film shown here is overexposed in order to aid the visualization of bands. Values reported in the text were quantified from blots exposed to film over a linear range of detection. DGC, overexpression of QrgB; PDE, overexpression of VC1086.
Regardless of the transcriptional processes occurring downstream of Vc2, under wild-type conditions P1-Vc2 is the most abundant single RNA species produced that contains the Vc2 sequence (Fig. 3, lane 1). This is consistent with the disparity we had previously observed between the high activity of the P1-tfoY promoter and its low contribution to total tfoY mRNA (10). To determine if this putative sRNA is impacted by changing c-di-GMP levels, we expressed from plasmids the diguanylate cyclase (DGC) enzyme QrgB to increase c-di-GMP concentrations or the phosphodiesterase (PDE) enzyme VC1086 to decrease c-di-GMP concentrations. We have previously demonstrated these enzymes generate physiologically relevant concentrations of c-di-GMP in V. cholerae (10). Upon DGC induction, we observed a 2.5-fold increase in the abundance of P1-Vc2 (Fig. 3, lane 2). Our previous results show that the transcription of P1-tfoY is not significantly affected by induction of QrgB (10), implying that the increase in P1-Vc2 observed here is the result of a posttranscriptional mechanism. On the other hand, induction of PDE, which decreased c-di-GMP, led to decreased P1-Vc2 (Fig. 3, lane 3).
The P2-Vc2 putative sRNA that we previously observed was also evident as a 167-nucleotide band, although it was not as abundant as P1-Vc2. The abundance of P2-Vc2 was slightly decreased upon DGC induction (Fig. 3, lane 2). This decrease was expected and can be attributed to c-di-GMP-mediated repression of the P2-tfoY promoter that we have previously reported (10). P2-Vc2 was nearly lost upon induction of PDE (Fig. 3, lane 3).
We hypothesized that the posttranscriptional mechanism responsible for the increase in P1-Vc2 during changes in c-di-GMP concentrations is dependent on c-di-GMP binding to the Vc2 aptamer. To test this, we generated several chromosomal V. cholerae mutations in Vc2 to assess its function. We first constructed a G83U chromosomal mutation in the Vc2 riboswitch, as this has been shown to disrupt its structure. When this mutation is present, P1-Vc2 is no longer visible at wild-type levels of c-di-GMP and is only very faint when DGC is induced (Fig. 3, lanes 4 and 5).
We next mutated the C92 and G20 bases in Vc2 to thymines on the V. cholerae chromosome, generating uracils in the riboswitch to assess the role of c-di-GMP binding in formation of these sRNAs. The G20 and C92 bases of the Vc2 riboswitch are the sites which directly interact with the guanosine bases of c-di-GMP, and substitutions of uracil at either of these sites significantly disrupts ligand binding without generally altering the ligand-free structure (20, 22). Accordingly, in strains with either single or double mutations at the ligand binding sites, we observed loss of P1-Vc2 (Fig. 3, lanes 6 and 8). In the C92U mutant, the reduction in abundance from the wild type was 30-fold, and in the G20U/C92U mutant the amount of P1-Vc2 remaining was below the limit of detection. We also observed complete loss of P2-Vc2 in both of these mutant backgrounds. Expression of the DGC QrgB in the C92U and G20U/C92U strains led to partial recovery of the abundance of P1-Vc2 in the C92U mutant but virtually no recovery in the G20U/C92U double mutant (Fig. 3, lanes 7 and 9). The relative strength of this recovery in each strain was consistent with the previous biochemical data that the single binding site mutant retains a higher c-di-GMP affinity than the double mutant (20).
The Vc2 riboswitch is not sufficient for transcription termination in vitro or in vivo.
The observation that short transcripts are produced which contain the Vc2 aptamer at the 3′ end could be explained if the expression platform of the Vc2 riboswitch functions as a rho-independent transcriptional terminator. No readily apparent terminator structure is located adjacent to the Vc2 riboswitch, and bioinformatic analysis with rho-independent terminator prediction software indicates that the best putative terminator nearby is located at the +193 to +235 region of the tfoY coding sequence, which is more than 300 nucleotides downstream of the 3′ end of the Vc2 aptamer (13, 21, 23). Furthermore, recent publications suggested that Vc2 regulates tfoY by controlling translation, not transcription read-through (18, 19). Nevertheless, to experimentally test if the Vc2 aptamer stimulates transcription termination upon binding to c-di-GMP, we conducted an in vitro transcription termination assay with Escherichia coli RNA polymerase complexed with σ70 using a linear PCR template encompassing the genomic region −535 to +120 relative to the tfoY coding sequence.
Because our transcription template included all of the tfoY promoters, we expected to see full-length transcription products of 458, 394, 314, and 166 nucleotides corresponding to the observed transcriptional start sites of the P1-tfoY, P2-tfoY, P3-tfoY, and P4-tfoY promoters, respectively, and indeed all of those transcripts were observed (Fig. 4, lane 1). Only two other major transcripts were detected, one of >600 nucleotides, likely generated from end-to-end transcription of the template, and one at the ∼180-nucleotide size range. Most notably, we did not observe any major transcript in the size range of the P1-Vc2 putative sRNA, which is the most prominent riboswitch-containing transcript generated in vivo.
FIG 4.
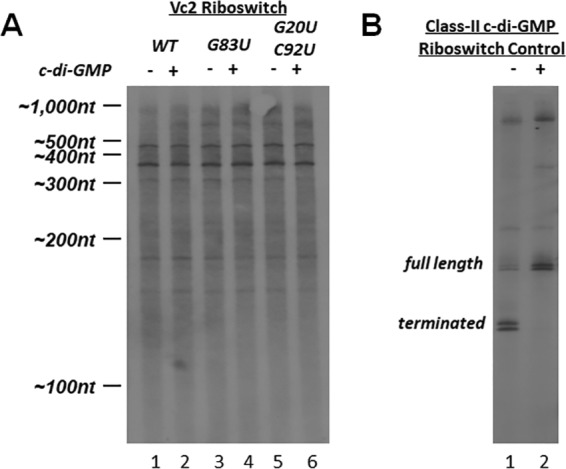
In vitro termination assay with c-di-GMP. (A) An in vitro transcription reaction with E. coli RNA polymerase and sigma 70 was performed on a DNA template containing the sequence upstream of tfoY and part of the tfoY open reading frame. Mutations to the riboswitch are indicated. (B) A similar in vitro transcription reaction was performed with a class II riboswitch from Clostridium difficile. c-di-GMP was added at a concentration of 0.1 mM.
We tested the possibility that the Vc2 riboswitch could induce termination by adding exogenous c-di-GMP to the transcription reaction (Fig. 4, lane 2), yet no changes in the size or abundance of transcripts were observed. We also tested the effect of disrupting the structural integrity of the riboswitch by using transcription templates containing the same mutations in the aptamer sequence which had earlier been investigated in vivo. Neither a G83U structural mutation that disrupts tertiary structure of the riboswitch nor the G20U/C92U binding site mutations described earlier had any detectable effect on the production of transcripts in vitro. Therefore, the riboswitch had no impact on transcription of this region in vitro with or without c-di-GMP.
To ensure that our reaction conditions were sufficient for detecting potential riboswitch-mediated termination, we tested a transcriptional template comprising the ompR-associated class II c-di-GMP riboswitch of Clostridium difficile, for which a transcription termination mechanism had previously been demonstrated in vitro (24). For this control experiment, addition of c-di-GMP inhibited transcription termination, consistent with the known mechanism of regulation of this riboswitch (Fig. 4B). Since we observed c-di-GMP regulation of termination with the control riboswitch but not with the Vc2 riboswitch, we conclude that the Vc2 riboswitch does not generate the P1-Vc2 and P2-Vc2 putative sRNAs through rho-independent termination.
We also considered the possibility that binding of c-di-GMP to Vc2 could induce transcriptional termination in vivo in a way that could not be recapitulated in this in vitro assay. Arguing against this possibility, we had previously demonstrated using a primer extension assay that the size and amount of transcript from the P1-tfoY promoter did not change when c-di-GMP was elevated (10). To further test for in vivo transcription termination, we created a gfp transcriptional fusion of the entire tfoY upstream regulatory region, including all four promoters, and measured production of GFP upon induction of the DGC QrgB, the PDE VC1086, or their active-site mutant derivatives (Fig. 5). Our previous studies had determined that the P1-tfoY promoter and P2-tfoY promoter, which both include transcripts containing the full-length Vc2 aptamer (Fig. 1A), exhibit the highest levels of transcription (10). Therefore, if binding of Vc2 terminated transcription arising from these promoters, we would expect to see decreased transcription of gfp at higher concentrations of c-di-GMP. However, our results were the opposite of this prediction, and we observed significantly increased transcription of gfp at elevated concentrations of c-di-GMP and decreased transcription of gfp at lower concentrations of c-di-GMP (Fig. 5). This finding is consistent with our previous finding of transcriptional upregulation of tfoY (10). These results further argue against c-di-GMP binding to Vc2 inducing transcription termination to generate P1-Vc2 and P2-Vc2.
FIG 5.
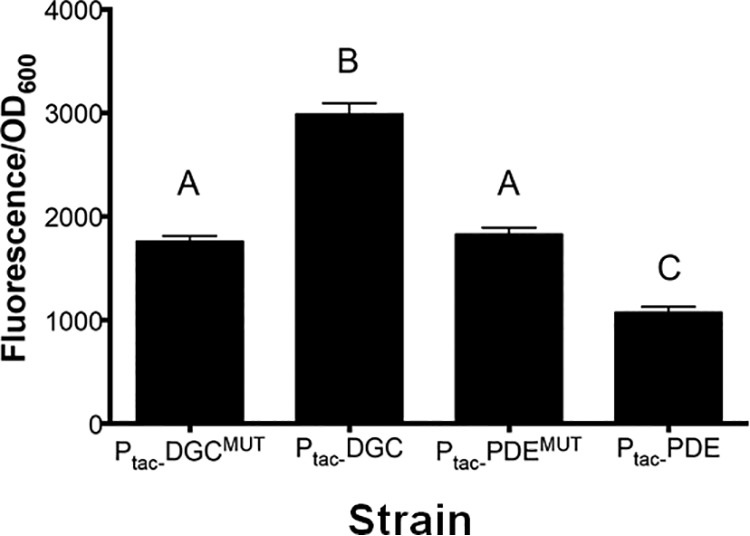
Cyclic di-GMP positively regulates tfoY transcription. P1-tfoY transcriptional reporter (pBRP37) activity was measured in high (DGC), unaltered (DGCMUT and PDEMUT), and low (PDE) levels of cyclic di-GMP. Data are averages and standard deviations from three biological replicates. Bars with different letters indicate statistically significant differences determined by one-way analysis of variance followed by Tukey’s multiple-comparison test (P < 0.05).
Stability of P1-Vc2 is c-di-GMP dependent.
Because Vc2 does not appear to regulate transcription termination and we observe significant RNA degradation in this region, we hypothesized that the Vc2 aptamer controls the posttranscriptional stability of the Vc2 sRNAs. To test this hypothesis, we added rifampin to halt transcription at mid-log-phase growth of V. cholerae cells under conditions of both wild-type and elevated c-di-GMP and subsequently extracted RNA from the cells over a series of multiple time intervals (Fig. 6). These experiments were not informative when a PDE was expressed or with the Vc2 mutants, as these conditions had levels of the P1-Vc2 sRNA that were too low to perform this analysis. Transcripts containing the Vc2 aptamer sequence were detected by Northern blotting with probe Vc2 (Fig. 2A), and we then quantified the amount of P1-Vc2 remaining at each time point after the addition of rifampin relative to the amount of P1-Vc2 at time zero (Fig. 6B).
FIG 6.

RNA stability assay with rifampin. (A) A single exposure of a Northern blot from one of the replicates of the RNA stability assay. Time is indicated as minutes after rifampin treatment. Measurements of RNA abundance were performed from a series of exposures in order to quantify the intensity of each band on the blot within its appropriate linear range. (B) Stability assays were performed in triplicate for cells grown under both wild-type and elevated c-di-GMP conditions, and the mean percentage of P1-Vc2 sRNA transcript remaining at each time point is depicted. Error bars indicate the standard deviations among three biological replicates. (C) From the same data shown in panel A, the half-life of the P1-Vc2 sRNA was calculated for the period between each pair of time points.
Our analysis revealed that the P1-Vc2 and P2-Vc2 putative sRNA species are significantly more stable than all other transcripts originating from the VC1721-tfoY intergenic locus (Fig. 6A). Surprisingly, this analysis also revealed that P1-Vc2 and P2-Vc2 are not single uniform RNA species but instead are both represented by a doublet of transcripts with a 5- to 10-nucleotide difference in size (Fig. 6A). Although it was not determined in this specific assay, these transcripts may represent the most prominent 3′-tailed and nontailed sRNA species we observed in our 3′-RACE experiments. For the purposes of our quantitative analysis of the amount of transcript remaining, both bands of P1-Vc2 were quantified together as the total amount of P1-Vc2.
Our RNA stability assay indicated that the individual members of the P1-Vc2 population, on average, persist longer in the cell when c-di-GMP is elevated (Fig. 6B). This difference in stability between conditions of intermediate and high c-di-GMP is a sufficient mechanism to explain the difference in the abundance of P1-Vc2 between those same conditions. Interestingly, the observed degradation of P1-Vc2 does not adhere to a simple one-phase decay model. Instead, this putative sRNA appears to be degraded rapidly at early time points and more slowly at later time points. To compare the stability of P1-Vc2 transcripts over time, we calculated the effective half-lives observed for the transcripts relative to each set of time points (Fig. 6C). At the first two time points under wild-type conditions, the half-life of the P1-Vc2 putative sRNA is less than half a minute, while at later time points this half-life had risen to the range of 4 to 8 min. Under elevated c-di-GMP conditions, the half-life of P1-Vc2 starts at more than twice the length of the wild-type conditions but then settles into a similar range of 4 to 8 min (Fig. 6C).
The observed pattern of increasing half-lives is consistent with there being multiple versions of P1-Vc2 with different rates of decay. Because the P1-Vc2 putative sRNA contains a functional Vc2 aptamer domain, we hypothesize these different versions of transcript to be c-di-GMP-bound and -unbound forms of P1-Vc2. In this way, the average half-life of P1-Vc2 is changing over time because at early time points many molecules of P1-Vc2 do not contain a ligand-bound riboswitch and are rapidly degraded, but at later time points all the unbound transcripts have been degraded and only stable ligand-bound P1-Vc2 remains. From this interpretation, the 4- to 8-min half-life measurement at later time points represents the closest approximation to the actual half-life of the c-di-GMP-bound form of P1-Vc2, and the actual half-life of the c-di-GMP-unbound form of P1-Vc2 is less than the lowest measured half-life at the earliest time point, i.e., less than 15.6 s. Compared to studies on global RNA stability in other bacteria, this shift in the stability of P1-Vc2 is quite dramatic, as measurements in E. coli indicate the half-life of most RNAs to be in the range of 2 to 8 min (25–28).
Because Vc2 aptamer mutants that could not bind c-di-GMP were difficult to detect using Northern blot analysis, we alternatively used quantitative reverse transcriptase PCR (qRT-PCR) and P1-Vc2-specific primers to compare the stability of P1-Vc2 RNA encoding a wild-type Vc2 sequence versus P1-Vc2 RNA encoding the G20U/C92U mutations in the Vc2 aptamer domain that were unable to bind to c-di-GMP. To reduce the amount of signal originating from larger transcripts, we also performed these experiments in a ΔtfoY deletion mutant. Cells were grown to mid-log phase and treated with rifampin, and RNA was harvested at 0, 0.5, and 1 min. Beyond this time, RNA levels were difficult to reliably quantify with this method, as the signal approached our limit of detection. Consistent with our model, we observed more rapid decay of P1-Vc2 that encoded the Vc2 riboswitch mutant unable to bind to c-di-GMP (see Fig. S1 in the supplemental material).
High levels of P1-Vc2 do not impact biofilm formation but enhance motility.
Accumulation of the P1-Vc2 putative sRNA occurs in response to increased c-di-GMP. We therefore tested if overproduction of this RNA impacted the c-di-GMP-regulated phenotypes of biofilm formation and motility. To do this, we engineered an overexpression plasmid containing P1-Vc2 followed by a 3′-factor-independent transcriptional terminator from the E. coli rrnB gene. Expression of this construct was driven from the Ptac promoter. Expression of P1-Vc2 in wild-type V. cholerae containing a functional vps system did not impact biofilm formation, as measured by a crystal violet (CV) assay, either at unaltered or elevated concentrations of c-di-GMP (Fig. 7A). We next expressed the P1-Vc2 RNA in the same wild-type V. cholerae strain and measured flagellum-based motility. These experiments were performed in agar plates containing 10% of the nutrients typically used in LB, as we have previously shown this condition induces dispersive motility (10). Induction of P1-Vc2 in this experiment significantly induced motility 2.4-fold, suggesting this is a regulatory target of P1-Vc2 (Fig. 7B).
FIG 7.
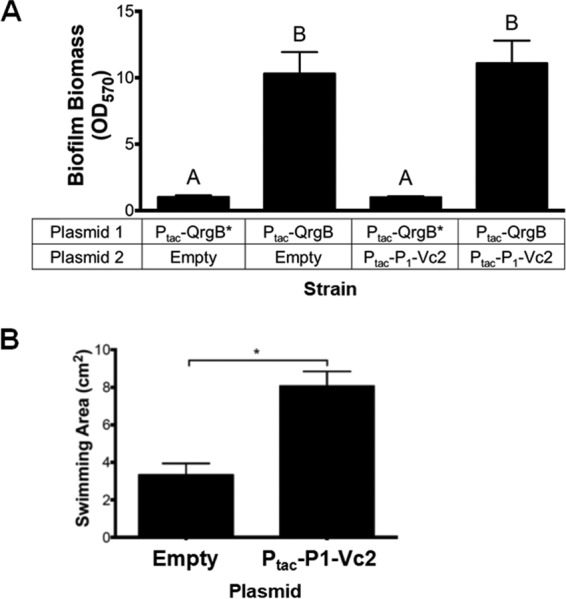
Impact of P1-Vc2 expression in wild-type V. cholerae on biofilm formation and motility. (A) Biofilm formation was measured while expressing the P1-Vc2 RNA or the empty vector control along with high (Ptac-DGC) or unaltered (Ptac-DGCMUT) levels of c-di-GMP after 24 h. Data are averages and standard deviations from six biological replicates. Bars with different letters indicate statistically significant differences determined by one-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparison test (P < 0.05). (B) The motility of V. cholerae in low-nutrient LB plates with 0.35% agar was determined at 23 h. Data are the averages and standard deviations from three biological replicates, each grown on three independent plates. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple-comparison test (*, P < 0.05).
DISCUSSION
In this report, we describe a novel regulatory mechanism whereby binding of a ligand to a 3′ riboswitch regulates the stability of the upstream noncoding RNA. Our findings arose from the observation that the vast majority of Vc2 riboswitch-containing transcripts do not ultimately contain tfoY mRNA. Instead, the upstream tfoY promoters produce two major putative sRNA species, the most prominent of which, designated P1-Vc2, originates at the P1-tfoY promoter and features the Vc2 riboswitch at its 3′ terminus. Our results are consistent with the results of multiple transcriptomic studies in V. cholerae, which have reported numerous sRNAs that map to the VC1721-tfoY intergenic region (see Fig. S2 in the supplemental material) (29–33). Importantly, a transcriptome sequencing (RNA-Seq) analysis of noncoding RNAs by Papenfort et al. identified P1-Vc2, supporting our findings here (33). However, none of these other studies report P2-Vc2, which is consistent with our observations that P1-Vc2 is present at higher levels than P2-Vc2 (Fig. 3). This result is likely due to the P1-tfoY promoter initiating more transcription than the P2-tfoY promoter (10).
Superficially, the observation that increased c-di-GMP leads to production of short transcripts gives an impression that the Vc2 riboswitch serves as a transcriptional terminator. However, our results do not support that conclusion, as we did not observe transcription termination mediated by the Vc2 riboswitch in vitro. Furthermore, our evidence does not support the conclusion that transcription is being terminated in vivo. Finally, termination at a site downstream of the riboswitch would not explain how P1-Vc2 becomes truncated at the immediate 3′ end of the P1 stem that forms the base of the Vc2 aptamer structure. Riboswitches that function by intrinsic termination employ an additional RNA structure, termed the expression platform, which is adjacent to, but separate from, the aptamer domain (34). For the Vc2 riboswitch, no such structure has been identified downstream of the aptamer domain (13, 23). Finally, both primer extension analysis and Northern blotting indicate that transcription initiated at the P1-tfoY promoter is regularly able to proceed past the riboswitch (10). No evidence, therefore, supports the hypothesis that the Vc2 riboswitch functions by controlling transcription termination.
We previously demonstrated that Vc2 bound to c-di-GMP inhibits production of TfoY, leading to decreased dispersive motility (10). Here, we demonstrate that a second phenotype for Vc2 is the changing abundance of the P1-Vc2 and P2-Vc2 putative sRNAs. When intracellular c-di-GMP concentrations are elevated, more P1-Vc2 is present, and mutation of Vc2 to disrupt its structure or ability to bind c-di-GMP leads to less P1-Vc2. P2-Vc2 does not increase at higher concentrations of c-di-GMP due to repression of transcription of the P2-tfoY promoter (10), but its stability is still dependent on Vc2 binding to c-di-GMP. Because the activity of the P1-tfoY promoter is not significantly affected by changes in c-di-GMP, the changing abundance of P1-Vc2 must be the consequence of posttranscriptional regulation. Our data indicate that the c-di-GMP-unbound P1-Vc2 putative sRNA is subject to very rapid turnover by the RNA degradosome, but c-di-GMP-bound P1-Vc2 is significantly more resistant to degradation. Thus, we conclude that the Vc2 riboswitch functions to control the cellular levels of the upstream RNA by ligand-mediated aptamer protection from 3′ degradation. Considering that a characteristic of aptamer domains is to fold into a compact nucleotide structure around the target ligand, we expect that this form of riboswitch-mediated gene regulation is widespread.
Riboswitches typically consist of two elements, a ligand binding aptamer domain and a cis-encoded expression platform that differentially interacts with the aptamer based on whether it is bound to its cognate ligand. Two important papers have concluded that Vc2 regulates tfoY by altering translation (18, 19). These authors proposed a structural model in which the P1 stem-loop that is formed upon binding of c-di-GMP to Vc2 leads to sequestration of the tfoY RBS. Alternatively, when c-di-GMP concentrations are low, the P1 loop is not formed but rather interacts with downstream sequence to prevent formation of the anti-RBS structure. Although these structural models are not yet fully validated in the native Vc2 sequence, it seems clear that binding of c-di-GMP to Vc2 impacts formation of a downstream expression platform to inhibit translation. However, when considering the upstream sequence, our data suggest that the Vc2 riboswitch prevents its degradation by stabilizing the 3′ ends of putative sRNAs. This raises the intriguing possibility that for the upstream P1-Vc2 and P2-Vc2 putative sRNAs, the aptamer domain itself dually functions in both ligand binding and as the expression platform. It is also possible that sequences located 3′ of Vc2 that are impacted by c-di-GMP binding are important in regulating RNA degradation.
Our model of how the Vc2 riboswitch regulates RNA processing is unique from known mechanisms of riboswitch-mediated RNA degradation. The best-characterized examples of riboswitches that both produce small RNA fragments and regulate RNA processing include the glmS riboswitch and ribozyme, the yitJ riboswitch and RNase Y, and the lysC riboswitch and RNase E (35–37). In all those systems, the effect of ligand binding is destabilizing because it promotes degradation of riboswitch-containing transcripts. The Vc2 riboswitch differs from those previous examples, as c-di-GMP binding inhibits degradation of the upstream putative sRNAs. The only previous example of 3′-end riboswitch-regulated RNA degradation comes from the plant domain. In that case, a thiamine pyrophosphate riboswitch that is located downstream of a thiamine biosynthetic gene controls RNA processing of the 3′-UTR in a manner that determines the expression level of the mature mRNA (38). However, again, whether ligand bound or not, the structure of the riboswitch aptamer is sacrificed in either of the processing outcomes, and the mature transcript does not contain a functional riboswitch (38).
Likewise, only a few riboswitch-containing sRNAs have been described. An S-adenosylmethionine riboswitch in Listeria monocytogenes induces transcription termination upon ligand binding, and upon discarding the ligand, the bases of the aptamer rearrange and interact through complementarity with a target mRNA (39). Two B12-binding riboswitches regulate noncoding RNAs in Listeria monocytogenes. A riboswitch located 3′ and antisense to the pocR gene controls the termination of the countertranscribed RNA AspocR such that in the absence of B12, full-length AspocR is produced, inhibiting expression of pocR (40). In addition, a B12-binding riboswitch regulates the length of the noncoding ril55 sRNA in L. monocytogenes by controlling a factor-dependent transcriptional terminator, which interestingly regulates the downstream gene by binding to a response regulator (41). A type I c-di-GMP-binding riboswitch in Bdellovibrio bacteriovorus was found to be more highly represented in an RNA-Seq data set than all other non-rRNA, non-tRNA transcripts and was hypothesized to function as a storage bank for c-di-GMP that controls the transition between the growth and attack phases of the cell, although this hypothesis was never directly tested (42). A highly abundant sRNA in Rhodobacter sphaeroides is derived from a 5′-UTR that encoded a potential riboswitch, although neither the function of the riboswitch nor its role in regulating the sRNA was shown (43). Likewise, an RNA-Seq study focusing on regulatory RNAs in C. difficile included an analysis of the expression of genes downstream of various riboswitches that identified a number of potential sRNAs, but none have been characterized yet (44). However, Vc2 differs from all these previous examples, as it is located at the 3′ end of the P1-Vc2 and P2-Vc2 putative sRNAs and regulates their levels in the cell by impacting RNA stability.
Stem-loop structures are common features at the 3′ end of bacterial mRNAs and have long been understood to provide protection against the 3′ to 5′ exonuclease activity of the RNA degradosome, which has a preference for segments of single-stranded RNA (45). The initial biochemical work on the Vc2 riboswitch provides clues about how the aptamer structure could serve a role in 3′-end stabilization. Specifically, the P1 stem of the Vc2 riboswitch is predicted to be the actor in Vc2 riboswitch function because it is the most labile region of the aptamer structure that is stabilized in the c-di-GMP-bound state, and proper closure of the P1 stem requires ligand binding (13, 20). In fact, the other two helical stems of the Vc2 aptamer undergo relatively little structural change during ligand binding, because their preorganization is a requirement for formation of the c-di-GMP binding pocket (46). It is no coincidence, then, that the 3′ end of P1-Vc2 is located immediately adjacent to the base of the P1 stem and that this was also the site of polyadenylation for the RNAs we recovered (Fig. 1). The doublet sizes of P1-Vc2 transcripts and the pattern of their degradation we observed in our rifampin stability assay are consistent with distinct populations of 3′-tailed and nontailed transcripts, which is a hallmark of the 3′-end degradative processes described for many regulatory RNAs in bacteria (Fig. 6A) (47). We hypothesize that closure of the P1 stem of the Vc2 aptamer during a c-di-GMP binding event increases the stability of P1-Vc2 by making the nucleotides at the 3′ end of the riboswitch transcript inaccessible to 3′ to 5′ exonucleases.
Previous work with the Vc2 riboswitch has made note of the exceptionally strong, picomolar affinity of the c-di-GMP aptamer for its ligand as measured in vitro, despite the observation that c-di-GMP concentrations in vivo reside in the nanomolar to low-micromolar range (20, 48, 49). Specifically, ligand binding at the Vc2 aptamer experiences an unusually slow off rate, so slow, in fact, that each c-di-GMP binding event should effectively be irreversible over the lifetime of the RNA transcript in the cell (20, 48). For canonical riboswitches that employ transcriptional mechanisms of regulation, the on rate of ligand binding is the more important feature of aptamer kinetics, because a switching decision must be made in the brief window of time that transcription of the expression platform is occurring (50). After initial ligand binding occurs, long-term retention of the ligand by the riboswitch aptamer is unimportant in transcriptional regulation because the RNA polymerase has long passed and a final decision about the on or off state of downstream gene expression has already been made. Biochemical study of the G20 and C92 sites of the Vc2 aptamer showed that the effect of those mutations was minimal on the c-di-GMP binding on rate but had a great effect on the off rate (20, 48). The slow off rate of the Vc2 riboswitch is consistent with c-di-GMP binding serving a continued function throughout the lifetime of the P1-Vc2 putative sRNA in the cell by maintaining the Vc2 aptamer in a structure that would make the nucleotides of the aptamer inaccessible to RNases.
The intracellular concentration of P1-Vc2 increases directly with c-di-GMP, which suggests it has a regulatory function at high c-di-GMP. Based on its expression pattern, we predicted that the P1-Vc2 sRNA could regulate traits associated with the biofilm lifestyle of V. cholerae. Interestingly, we found that overexpression of P1-Vc2 did not impact biofilm formation but did significantly induce flagellum-based swimming motility of V. cholerae (Fig. 7). Whether this impact on motility is a direct regulation of target transcripts by P1-Vc2 or an indirect effect via sequestration of c-di-GMP remains to be elucidated.
MATERIALS AND METHODS
Strains and growth conditions.
The wild-type V. cholerae strain used in this study is CW2034, an El Tor biotype C6706str2 derivative containing a deletion of the vpsL gene (51) (Table 1). Use of a vpsL-negative strain facilitates accurate spectrophotometric readings during transcriptional and translational reporter assays due to decreased biofilm formation. For the experiments presented in Fig. 7, wild-type C6706str2 with an intact vpsL was used, as this gene is required for biofilm formation and VPS impacts bacterial motility (9). Propagation of DNA for genetic manipulation and mating of reporter plasmids was conducted in E. coli S-17-λpir (52). All growth of bacteria was performed at 35°C in Miller LB broth (Acumedia) or on LB agar plates, with ampicillin (100 μg/ml), kanamycin (100 μg/ml), polymyxin B (10 IU/ml), streptomycin (500 μg/ml), and isopropyl-β-d-thiogalactoside (IPTG) at 100 μM when appropriate. Liquid cultures in tubes or flasks were continuously shaken at 220 rpm; microplate cultures were continuously shaken at 150 rpm.
TABLE 1.
Strain list
Genetic manipulations.
V. cholerae mutant strains were constructed using allelic exchange with vectors derived from pKAS32 (53). Vectors containing mutant riboswitch alleles were generated with the QuikChange site-directed mutagenesis kit (Agilent) and mated into V. cholerae so as to create markerless strains with single-point mutations on the genome.
3′-RACE.
The 3′-RACE method was adapted from reference 54. A DNA oligonucleotide adapter was synthesized with a monophosphate at the 5′ end and an inverted thymidine base at the 3′ end (IDT). Five hundred pmol of adapter was ligated to 10 μg V. cholerae RNA at 37°C with 20 U of T4 RNA ligase (NEB) in a 20-μl reaction mixture containing 1× T4 RNA ligase buffer, 1 mM ATP, 10% dimethyl sulfoxide (DMSO), and 20 U RNasin (Promega). Ligation products were reverse transcribed into cDNA and amplified with the Access RT-PCR kit (Promega) using a primer complementary to the 3′ adapter and a primer specific for the 5′-end sequence of transcripts initiated at the P1-tfoY promoter (Table 2). The products generated were electrophoresed on an agarose gel, and DNA was excised and purified from all size ranges and recovered by TOPO TA cloning into pCR2.1 (Invitrogen).
TABLE 2.
Vector and primer lista
| Name | Description | Sequence | Reference or source |
|---|---|---|---|
| pEVS141 | Protein overexpression control | 56 | |
| pCMW75 | Ptac-QrgB (pEVS141 backbone) | 51 | |
| pCMW98 | Ptac-QrgB active-site mutant (pEVS141 backbone) | 51 | |
| pCMW121 | Ptac-VC1086 (pEVS141 backbone) | 51 | |
| pCMW121 | Ptac-VC1086 active-site mutant (pEVS141 backbone) | 51 | |
| pBRP37 | P1-4-gfp transcriptional fusion | Fwd-5′-GCAGAATACTTCTCTCCACC-3′ | This study |
| Rev-5′-GTTGTACAGTTCATCCAT-3′ | |||
| pBRP50 | P3-RNA probe P2 template | Fwd-5′-ATCAGGTACCGCTCATTCTCACATTTGAAATA-3′ | 10 |
| Rev-5′-ATCACTCGAGCTGTGCGTGACATTTTTCCTG-3′ | |||
| pBRP71 | P2-RNA probe P1 template, EMSA probe P3 template | Fwd-5′-ATCAGGTACCTATATTTGAAAGCTTGTCAC-3′ | 10 |
| Rev-5′-ATCACTCGAGCCCTACTAATAATACTCGCAC-3′ | |||
| pBRP78 | PCR template for in vitro transcription of C. difficile class II c-di-GMP riboswitch | Fwd-5′-ATCAGGTACCATCTTATATCTAAGAATATGGAAATATTG-3′ | This study |
| Rev-5′-ATCACTCGAGCTAATACTCTTATTTTCAAATTTTGCAAC-3′ | |||
| pBRP86 | RNA probe P1 + 2 template | Fwd-5′-ATCAGGTACCTATATTTGAAAGCTTGTCAC-3′ | This study |
| Rev-5′-ATCACTCGAGCTGTGCGTGACATTTTTCCTG-3′ | |||
| pBRP91 | P3-RNA probe P2 template | Fwd-5′-ATCAGGTACCGTGAGAATGACCCAAGAATG-3′ | 10 |
| Rev-5′-ATCACTCGAGCTGTGCGTGACATTTTTCCTG-3′ | |||
| pBRP172 | Allelic exchange vector, Vc2 (WT); PCR template for in vitro transcription | Fwd-5′-ATCAGGTACCTCGCGACCAAATATC-3′ | This study |
| Rev-5′-ATCAGAGCTCCAATGTTTACCTTCGATTGC-3′ | |||
| pBRP177 | Allelic exchange vector, Vc2 (G83U); PCR template for in vitro transcription | Mutagenesis-5′-TGGTAGGTATCGGGGTTAC-3′ | This study |
| pBRP178 | Allelic exchange vector, Vc2 (C92U) | Mutagenesis-5′-AGCGGGGTTATCGATGGCAA-3′ | 10 |
| pBRP179 | Allelic exchange vector, Vc2 (G20U, C92U); PCR template for in vitro transcription | Mutagenesis-5′-ACGCACAGTGCAAACCATTC-3′ | 10 |
| pBRP269 | P1-Vc2 RNA overexpression vector | Fwd-5′-GCCTATATTTGAAAGCTTGTCA-3′ | This study |
| Rev-5′-GTATGCATTTTGCCATCGGTA-3′ | |||
| qRT-PCR primers | CMW2946 | GACAAAACATCCCACTCTTTCGAAT | This study |
| CMW2947 | GGTTTTATGTAGTGCGAG |
For each vector constructed for this study, sequences are given for primers used in PCR to generate vector inserts. For primer sequences, restriction endonuclease sites are indicated by underlining.
Northern blotting.
RNA was extracted from mid-log-phase cultures using TRIzol reagent (Invitrogen). Samples were normalized to the same concentration using spectrophotometric measurement and evaluated for rRNA integrity using a Bioanalyzer (Agilent). RNA was separated by PAGE, transferred by semidry blotting onto a positively charged nylon membrane, and baked in a vacuum oven. The membrane was hybridized with the probe at 65°C for at least 4 h in ULTRAhyb buffer (Ambion) and washed three times with 0.1× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) at 65°C. The probe was detected by chemiluminescence using the Phototope-Star detection kit (NEB) and autoradiographic film. Film images were digitized by scanning at high resolution with a Typhoon FLA 9500 Imager (GE Healthcare), and quantitative measurements were performed using the image analysis software Fiji (55).
In all, four RNA probes, named probe P1, probe P2, probe P1 + 2, and probe Vc2, which were complementary to the genomic regions −337 to −272, −272 to −191, −337 to −191, and −211 to −94, respectively, relative to the coding sequence of tfoY, were used (Fig. 2A). Probes were labeled with Bio-11-UTP during in vitro transcription with the MAXIscript T7 kit (Ambion) from PCR-derived DNA templates containing a consensus T7 promoter fused to the appropriate sequence.
(i) In vitro transcription.
DNA templates for in vitro transcription were generated by PCR from the vectors used for construction of V. cholerae genomic riboswitch mutants. Templates encompassed the genomic region from nucleotides −535 to +120 relative to the tfoY coding sequence. The template for the C. difficile class II riboswitch encompassed the region from nucleotides −761 to −505 relative to the coding sequence of the gene CD3267 of C. difficile 630, the same region investigated by Lee et al. (24). Transcription reaction mixtures included 150 ng of DNA template, 2 mM dithiothreitol, 0.25 mM nucleoside triphosphates, 0.1 mM c-di-GMP when required, and 0.3 U of σ70-saturated E. coli RNA polymerase holoenzyme (Epicentre) in the transcription buffer recommended by the manufacturer. Bio-11-UTP (Ambion) was used in reaction mixtures at a concentration of 20% of total UTP to label transcripts after it had been determined not to interfere with the generation of transcription products (data not shown).
(ii) Transcriptional reporter assays.
Three individual colonies of CW2034 harboring pCMW75, pCMW98, pCMW121, and pCMW126 were inoculated into 2 ml LB with the appropriate antibiotics in 10- by 150-mm borosilicate test tubes. Independent overnight cultures were diluted 1:5,000 in 1 ml LB with appropriate antibiotics and IPTG at 100 μM final concentration. Two hundred microliters of the subculture was delivered into technical triplicate wells in a black 96-well plate (Costar). This was repeated for each individual culture for each strain. The inoculated 96-well plate was incubated at 37°C with shaking at 200 rpm until the cultures reached mid-log phase (optical density at 600 nm [OD600] near 0.450). Fluorescence was measured using the Perkin Elmer Envision multiplate reader with an emission wavelength of 395 nm and an excitation wavelength of 500 nm. Fluorescence data were normalized for cell growth using OD600 measurement.
RNA stability assay.
Two hundred-milliliter cultures were grown with shaking in baffled flasks after being started with a 1:10,000 dilution of overnight culture into fresh media. When the optical density reached ∼0.400 to 0.500, 10 ml of culture was withdrawn for a time zero reading and rifampin was added to the culture for a final concentration of 250 μg/ml. An additional 10 ml of culture was removed at each subsequent time point, stabilized with 1 ml of RNA stop solution (10% phenol in 95% ethanol), and placed on ice, as described by Bernstein et al. (26). RNA was promptly extracted from the cells using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. RNA samples were normalized, electrophoresed, blotted, and detected as described above. For quantitative analysis, a series of film exposures over multiple lengths of time were collected, and once digitized, images were only compared between images for which the film exposure was not saturated.
For the qRT-PCR measurement of RNA stability, overnight cultures were inoculated in triplicate from freezer stocks into LB medium and incubated for 16 h at 37°C with 210-rpm shaking. Cultures were diluted 1:1,000 into 25 ml of fresh medium and grown at 37°C with 210-rpm shaking to an OD600 of 0.4 to 0.5. One milliliter of culture (time 0) was removed, and global transcription of the remaining bulk culture was arrested with the addition of 250 μg/ml rifampin. One-milliliter culture aliquots were also collected at 0.5 and 1 min following the addition of rifampin. Culture samples were immediately pelleted by 30 s of centrifugation at 15,000 × g. Supernatants were rapidly removed by aspiration, cell pellets were resuspended in TRIzol reagent (Invitrogen), and RNA purification was performed by following the manufacturer’s instructions. Five nanograms of total RNA was treated with TURBO DNase (Thermo Fischer Scientific) and reverse transcribed to cDNA using SuperScript III first-strand synthesis (Life Technologies) per the manufacturer’s instructions. SYBR green PCR master mix (Thermo Fisher Scientific), with a final primer concentration of 250 nM, was used to perform qRT-PCRs on a StepOnePlus real-time PCR system (Thermo Fisher Scientific). The relative abundance of P1-Vc2 transcripts was calculated by comparing the sample threshold cycle (CT) value to a CT standard curve of purified pBRP269 plasmid (encoding a single P1-Vc2 locus per plasmid) in a 1:10 serial dilution ranging from a final concentration of 1.0 to 1.0 × 10−8 ng/μl.
Motility assay.
Swimming motility was assessed for WT V. cholerae expressing the P1-Vc2 sRNA from a Ptac inducible promoter (pBRP269) or an empty vector control (pEVS141). LB medium supplemented with antibiotics was inoculated in triplicate from freezer stocks and grown for 24 h at 37°C with 210-rpm shaking. Low-nutrient, low-density agar (0.1% [wt/vol] tryptone, 0.05% [wt/vol] yeast extract, 1% [wt/vol] sodium chloride, and 0.35% [wt/vol] agar) containing 100 μM isopropyl-β-d-1-thiogalactopyranoside (IPTG) and antibiotics was aliquoted in 15-ml volumes into 100-mm by 15-mm petri dishes and allowed to solidify for 2 h at room temperature. Plates were surface inoculated with 1.5 μl of liquid culture and left upright for 1 h to allow penetration of the culture into the agar. Plates were inverted, placed into a humidity box, and incubated at 35°C for 23 h. Images of the plates were captured with an AlphaImager HP system (ProteinSimple), and swimming areas were quantitatively measured using the image analysis software Fiji (55).
CV biofilm formation assay.
Three individual colonies of WT V. cholerae harboring plasmids to modulate riboswitch expression (pBRP269 or pEVS141) and intracellular cyclic di-GMP (pBRP1 or pBRP2) were used to inoculate 2 ml LB supplemented with the appropriate antibiotics for overnight cultures. Overnight cultures were then diluted 1:100 in 1 ml LB supplemented with the appropriate antibiotics and IPTG at 100 μM final concentration. Two hundred microliters from each 1-ml subculture was delivered into a clear 96-well plate (Costar) in technical triplicate and was repeated for each individual subculture. The plate was incubated at 37°C with shaking at 200 rpm for 24 h. After 24 h, the planktonic cultures were removed and the wells were washed with 200 μl 1× phosphate-buffered saline (1× PBS) twice. The wells were then filled with 200 μl of crystal violet (CV) (0.41% CV, 12% ethanol) and stained for 15 min. After staining, CV was removed and the wells were washed twice with 1× PBS as before. The adhered CV was eluted by the addition of 200 μl 95% ethanol and was diluted 1:10 prior to absorbance reading at 570 nm using the Perkin Elmer Envision multiplate reader.
Supplementary Material
ACKNOWLEDGMENTS
This material is based in part upon work supported by the National Science Foundation under Cooperative Agreements MCB-1253684 and DBI-0939454 and NIH grants GM109259, GM110444, and AI130554.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00293-19.
REFERENCES
- 1.Silva AJ, Benitez JA. 2016. Vibrio cholerae biofilms and cholera pathogenesis. PLoS Negl Trop Dis 10:e0004330. doi: 10.1371/journal.pntd.0004330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tischler AD, Camilli A. 2004. Cyclic diguanylate (c-di-GMP) regulates Vibrio cholerae biofilm formation. Mol Microbiol 53:857–869. doi: 10.1111/j.1365-2958.2004.04155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lim B, Beyhan S, Meir J, Yildiz FH. 2006. Cyclic-diGMP signal transduction systems in Vibrio cholerae: modulation of rugosity and biofilm formation. Mol Microbiol 60:331–348. doi: 10.1111/j.1365-2958.2006.05106.x. [DOI] [PubMed] [Google Scholar]
- 4.Sloup RE, Konal AE, Severin GB, Korir ML, Bagdasarian MM, Bagdasarian M, Waters CM. 2017. Cyclic di-GMP and VpsR induce the expression of type II secretion in Vibrio cholerae. J Bacteriol 199:e00106-17. doi: 10.1128/JB.00106-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez NL, Srivastava D, Ngouajio AL, Waters CM. 2018. Cyclic di-GMP positively regulates DNA repair in Vibrio cholerae. J Bacteriol 200:e00005-18. doi: 10.1128/JB.00005-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Srivastava D, Waters CM. 2012. A tangled web: regulatory connections between quorum sensing and cyclic Di-GMP. J Bacteriol 194:4485–4493. doi: 10.1128/JB.00379-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krasteva PV, Fong JC, Shikuma NJ, Beyhan S, Navarro MV, Yildiz FH, Sondermann H. 2010. Vibrio cholerae VpsT regulates matrix production and motility by directly sensing cyclic di-GMP. Science 327:866–868. doi: 10.1126/science.1181185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Srivastava D, Harris RC, Waters CM. 2011. Integration of cyclic di-GMP and quorum sensing in the control of vpsT and aphA in Vibrio cholerae. J Bacteriol 193:6331–6341. doi: 10.1128/JB.05167-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srivastava D, Hsieh ML, Khataokar A, Neiditch MB, Waters CM. 2013. Cyclic di-GMP inhibits Vibrio cholerae motility by repressing induction of transcription and inducing extracellular polysaccharide production. Mol Microbiol 90:1262–1276. doi: 10.1111/mmi.12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pursley BR, Maiden MM, Hsieh ML, Fernandez NL, Severin GB, Waters CM. 2018. Cyclic di-GMP regulates TfoY in Vibrio cholerae to control motility by both transcriptional and posttranscriptional mechanisms. J Bacteriol 200:e00578-17. doi: 10.1128/JB.00578-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kariisa AT, Weeks K, Tamayo R. 2016. The RNA domain Vc1 regulates downstream gene expression in response to cyclic diguanylate in Vibrio cholerae. PLoS One 11:e0148478. doi: 10.1371/journal.pone.0148478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith KD, Shanahan CA, Moore EL, Simon AC, Strobel SA. 2011. Structural basis of differential ligand recognition by two classes of bis-(3’-5’)-cyclic dimeric guanosine monophosphate-binding riboswitches. Proc Natl Acad Sci U S A 108:7757–7762. doi: 10.1073/pnas.1018857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sudarsan N, Lee ER, Weinberg Z, Moy RH, Kim JN, Link KH, Breaker RR. 2008. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science 321:411–413. doi: 10.1126/science.1159519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKee RW, Harvest CK, Tamayo R. 2018. Cyclic diguanylate regulates virulence factor genes via multiple riboswitches in Clostridium difficile. mSphere 3:e00423-18. doi: 10.1128/mSphere.00423-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren A, Wang XC, Kellenberger CA, Rajashankar KR, Jones RA, Hammond MC, Patel DJ. 2015. Structural basis for molecular discrimination by a 3’,3’-cGAMP sensing riboswitch. Cell Rep 11:1–12. doi: 10.1016/j.celrep.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelson JW, Sudarsan N, Phillips GE, Stav S, Lunse CE, McCown PJ, Breaker RR. 2015. Control of bacterial exoelectrogenesis by c-AMP-GMP. Proc Natl Acad Sci U S A 112:5389–5394. doi: 10.1073/pnas.1419264112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metzger LC, Matthey N, Stoudmann C, Collas EJ, Blokesch M. 2019. Ecological implications of gene regulation by TfoX and TfoY among diverse Vibrio species. Environ Microbiol 21:2231–2247. doi: 10.1111/1462-2920.14562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inuzuka S, Kakizawa H, Nishimura KI, Naito T, Miyazaki K, Furuta H, Matsumura S, Ikawa Y. 2018. Recognition of cyclic-di-GMP by a riboswitch conducts translational repression through masking the ribosome-binding site distant from the aptamer domain. Genes Cells 23:435–447. doi: 10.1111/gtc.12586. [DOI] [PubMed] [Google Scholar]
- 19.Inuzuka S, Nishimura K, Kakizawa H, Fujita Y, Furuta H, Matsumura S, Ikawa Y. 2016. Mutational analysis of structural elements in a class-I cyclic di-GMP riboswitch to elucidate its regulatory mechanism. J Biochem 160:153–162. doi: 10.1093/jb/mvw026. [DOI] [PubMed] [Google Scholar]
- 20.Smith KD, Lipchock SV, Ames TD, Wang J, Breaker RR, Strobel SA. 2009. Structural basis of ligand binding by a c-di-GMP riboswitch. Nat Struct Mol Biol 16:1218–1223. doi: 10.1038/nsmb.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kingsford CL, Ayanbule K, Salzberg SL. 2007. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol 8:R22. doi: 10.1186/gb-2007-8-2-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kulshina N, Baird NJ, Ferré-D'Amaré AR. 2009. Recognition of the bacterial second messenger cyclic diguanylate by its cognate riboswitch. Nat Struct Mol Biol 16:1212–1217. doi: 10.1038/nsmb.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furukawa K, Gu H, Sudarsan N, Hayakawa Y, Hyodo M, Breaker RR. 2012. Identification of ligand analogues that control c-di-GMP riboswitches. ACS Chem Biol. doi: 10.1021/cb300138n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee ER, Baker JL, Weinberg Z, Sudarsan N, Breaker RR. 2010. An allosteric self-splicing ribozyme triggered by a bacterial second messenger. Science 329:845–848. doi: 10.1126/science.1190713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pedersen S, Reeh S. 1978. Functional mRNA half lives in E. coli. Mol Gen Genet 166:329–336. doi: 10.1007/bf00267626. [DOI] [PubMed] [Google Scholar]
- 26.Bernstein JA, Khodursky AB, Lin PH, Lin-Chao S, Cohen SN. 2002. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc Natl Acad Sci U S A 99:9697–9702. doi: 10.1073/pnas.112318199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selinger DW, Saxena RM, Cheung KJ, Church GM, Rosenow C. 2003. Global RNA half-life analysis in Escherichia coli reveals positional patterns of transcript degradation. Genome Res 13:216–223. doi: 10.1101/gr.912603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen H, Shiroguchi K, Ge H, Xie XS. 2015. Genome-wide study of mRNA degradation and transcript elongation in Escherichia coli. Mol Syst Biol 11:781. doi: 10.15252/msb.20145794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu JM, Livny J, Lawrence MS, Kimball MD, Waldor MK, Camilli A. 2009. Experimental discovery of sRNAs in Vibrio cholerae by direct cloning, 5S/tRNA depletion and parallel sequencing. Nucleic Acids Res 37:e46. doi: 10.1093/nar/gkp080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bradley ES, Bodi K, Ismail AM, Camilli A. 2011. A genome-wide approach to discovery of small RNAs involved in regulation of virulence in Vibrio cholerae. PLoS Pathog 7:e1002126. doi: 10.1371/journal.ppat.1002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mandlik A, Livny J, Robins WP, Ritchie JM, Mekalanos JJ, Waldor MK. 2011. RNA-Seq-based monitoring of infection-linked changes in Vibrio cholerae gene expression. Cell Host Microbe 10:165–174. doi: 10.1016/j.chom.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raabe CA, Hoe CH, Randau G, Brosius J, Tang TH, Rozhdestvensky TS. 2011. The rocks and shallows of deep RNA sequencing: examples in the Vibrio cholerae RNome. RNA 17:1357–1366. doi: 10.1261/rna.2682311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Papenfort K, Forstner KU, Cong JP, Sharma CM, Bassler BL. 2015. Differential RNA-seq of Vibrio cholerae identifies the VqmR small RNA as a regulator of biofilm formation. Proc Natl Acad Sci U S A 112:E766–E775. doi: 10.1073/pnas.1500203112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Serganov A, Nudler E. 2013. A decade of riboswitches. Cell 152:17–24. doi: 10.1016/j.cell.2012.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collins JA, Irnov I, Baker S, Winkler WC. 2007. Mechanism of mRNA destabilization by the glmS ribozyme. Genes Dev 21:3356–3368. doi: 10.1101/gad.1605307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shahbabian K, Jamalli A, Zig L, Putzer H. 2009. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J 28:3523–3533. doi: 10.1038/emboj.2009.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caron M-P, Bastet L, Lussier A, Simoneau-Roy M, Massé E, Lafontaine DA. 2012. Dual-acting riboswitch control of translation initiation and mRNA decay. Proc Natl Acad Sci U S A 109:E3444–E3453. doi: 10.1073/pnas.1214024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wachter A, Tunc-Ozdemir M, Grove BC, Green PJ, Shintani DK, Breaker RR. 2007. Riboswitch control of gene expression in plants by splicing and alternative 3’ end processing of mRNAs. Plant Cell 19:3437–3450. doi: 10.1105/tpc.107.053645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loh E, Dussurget O, Gripenland J, Vaitkevicius K, Tiensuu T, Mandin P, Repoila F, Buchrieser C, Cossart P, Johansson J. 2009. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell 139:770–779. doi: 10.1016/j.cell.2009.08.046. [DOI] [PubMed] [Google Scholar]
- 40.Mellin JR, Tiensuu T, Becavin C, Gouin E, Johansson J, Cossart P. 2013. A riboswitch-regulated antisense RNA in Listeria monocytogenes. Proc Natl Acad Sci U S A 110:13132–13137. doi: 10.1073/pnas.1304795110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mellin JR, Koutero M, Dar D, Nahori MA, Sorek R, Cossart P. 2014. Riboswitches. Sequestration of a two-component response regulator by a riboswitch-regulated noncoding RNA. Science 345:940–943. doi: 10.1126/science.1255083. [DOI] [PubMed] [Google Scholar]
- 42.Karunker I, Rotem O, Dori-Bachash M, Jurkevitch E, Sorek R. 2013. A global transcriptional switch between the attack and growth forms of Bdellovibrio bacteriovorus. PLoS One 8:e61850. doi: 10.1371/journal.pone.0061850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weber L, Thoelken C, Volk M, Remes B, Lechner M, Klug G. 2016. The conserved Dcw gene cluster of R. sphaeroides is preceded by an uncommonly extended 5’ leader featuring the sRNA UpsM. PLoS One 11:e0165694. doi: 10.1371/journal.pone.0165694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soutourina OA, Monot M, Boudry P, Saujet L, Pichon C, Sismeiro O, Semenova E, Severinov K, Le Bouguenec C, Coppee JY, Dupuy B, Martin-Verstraete I. 2013. Genome-wide identification of regulatory RNAs in the human pathogen Clostridium difficile. PLoS Genet 9:e1003493. doi: 10.1371/journal.pgen.1003493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rauhut R, Klug G. 1999. mRNA degradation in bacteria. FEMS Microbiol Rev 23:353–370. doi: 10.1111/j.1574-6976.1999.tb00404.x. [DOI] [PubMed] [Google Scholar]
- 46.Wood S, Ferré-D’Amaré AR, Rueda D. 2012. Allosteric tertiary interactions preorganize the c-di-GMP riboswitch and accelerate ligand binding. ACS Chem Biol 7:920–927. doi: 10.1021/cb300014u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hajnsdorf E, Kaberdin VR. 2018. RNA polyadenylation and its consequences in prokaryotes. Philos Trans R Soc B 373:20180166. doi: 10.1098/rstb.2018.0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith KD, Lipchock SV, Livingston AL, Shanahan CA, Strobel SA. 2010. Structural and biochemical determinants of ligand binding by the c-di-GMP riboswitch. Biochemistry 49:7351–7359. doi: 10.1021/bi100671e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Massie JP, Reynolds EL, Koestler BJ, Cong J, Agostoni M, Waters CM. 2012. Quantification of high specificity signaling. Proc Natl Acad Sci U S A 109:12746–12751. doi: 10.1073/pnas.1115663109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garst AD, Batey RT. 2009. A switch in time: detailing the life of a riboswitch. Biochim Biophys Acta 1789:584–591. doi: 10.1016/j.bbagrm.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waters CM, Lu W, Rabinowitz JD, Bassler BL. 2008. Quorum sensing controls biofilm formation in Vibrio cholerae through modulation of Cyclic Di-GMP levels and repression of vpsT. J Bacteriol 190:2527–2536. doi: 10.1128/JB.01756-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 53.Skorupski K, Taylor RK. 1996. Positive selection vectors for allelic exchange. Gene 169:47–52. doi: 10.1016/0378-1119(95)00793-8. [DOI] [PubMed] [Google Scholar]
- 54.Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EG, Margalit H, Altuvia S. 2001. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol 11:941–950. doi: 10.1016/s0960-9822(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 55.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dunn AK, Millikan DS, Adin DM, Bose JL, Stabb EV. 2006. New rfp- and pES213-derived tools for analyzing symbiotic Vibrio fischeri reveal patterns of infection and lux expression in situ. Appl Environ Microbiol 72:802–810. doi: 10.1128/AEM.72.1.802-810.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.