

Abstract
High rates of relapse are a chronic and debilitating obstacle to effective treatment of alcohol use disorder (AUD); however, no effective treatments are available to treat symptoms induced by protracted abstinence. In the first part of this two-part review series, we examine the literature supporting the effects of alcohol exposure within the extended amygdala (EA) neural circuitry. In part two, we focus in on a potential way to combat negative affect associated with AUD, by exploring the therapeutic potential of the endogenous cannabinoid (eCB) system. The eCB system is a potent modulator of neural activity in the brain, and its ability to mitigate stress and negative affect has long been an area of interest for developing novel therapeutics. This review details the recent advances in our understanding of eCB signaling in two key regions of the EA, the central nucleus of the amygdala (CeA) and the bed nucleus of the stria terminalis (BNST), and their role in regulating negative affect. Despite an established role for EA eCB signaling in reducing negative affect, few studies have examined the potential for eCB-based therapies to treat AUD-associated negative affect. In this review, we present an overview of studies focusing on eCB signaling in EA and cannabinoid modulation on EA synaptic activity. We further discuss studies suggesting dysregulation of eCB signaling in models of AUD and propose that pharmacological augmentation of eCB could be a novel approach to treat aspects of AUD. Lastly, future directions are proposed to advance our understanding of the relationship between AUD-associated negative affect and the EA eCB system that could yield new pharmacotherapies targeting negative affective symptoms associated with AUD.
INTRODUCTION
Alcohol use disorder (AUD) is a chronic, relapsing disorder that has been characterized by compulsion to seek and take alcohol, the loss of control over alcohol intake and emergence of negative emotional states and craving during abstinence. AUD is highly comorbid with psychiatric disorders such as generalized anxiety and major depression (Oliveira et al., 2018, Black et al., 2015, Hasin and Grant, 2002). Affective disorders and negative affective states can drive initial alcohol use, and can highly predict a transition from use to dependence (Koob and Schulkin, 2018). While negative affect contributes to all phases of addiction, it is highly prevalent during abstinence, as patients with AUD commonly report heightened anxiety, irritability, dysphoria, and negative emotional states as potent triggers of cravings and relapse (Fox et al., 2017, Litt et al., 1990, Cooney et al., 1997). This negative affective state can last from a few weeks (early abstinence) to several months (protracted abstinence) and is distinct from acute physical withdrawal symptoms (Heilig et al., 2010). Indeed, a major obstacle in the alcohol addiction recovery is the susceptibility to relapse, even after protracted abstinence (Willinger et al., 2002, Driessen et al., 2001). Although negative affective symptoms associated with protracted alcohol withdrawal are major causes of alcohol craving and relapse (Mathew et al., 1979, Mason et al., 2009, Driessen et al., 2001, Willinger et al., 2002), no effective treatments are available as use of traditional antidepressants has yielded inconsistent results and in some cases can even increase alcohol drinking (Nunes and Levin, 2004, Pettinati, 2004).
The endocannabinoid (eCB) system has attracted attention for its role in stress- and fear-related behaviors and as a therapeutic target in neuropsychiatric disease state, including anxiety- and depression-related disorders (Patel et al., 2017, Hill et al., 2018, Lutz et al., 2015). The extended amygdala (EA) plays an important role in several stress-related components of drug withdrawal (Koob, 2003), and is a site where the eCB system seems to be critical for modulating the influence of stress on the addiction cycle. The EA is an anatomically and neurochemically interconnected brain structure in the basal forebrain that mainly consist of central nucleus of amygdala (CeA) and bed nucleus of the stria terminals (BNST), but also the shell of the nucleus accumbens (Cassell et al., 1999). The CeA and the BNST exhibit overlapping cellular compositions and patterns of connectivity and both have been implicated in mediating alcohol-related behaviors (Koob et al., 1998). While extensive research has implicated the EA in modulating several facets of AUD, and parallel lines of research have established a critical role for the EA eCB system in modulating affective states, very few studies have combined these two lines of research to fully understand the neurobiological substrates of eCB-AUD interactions or the therapeutic potential of the EA eCB system for treating AUD. In Part 1 of this two-part review, we discuss in detail how chronic ethanol use causes dysregulation of the EA neurocircuitry and overactivation of the brain stress system within the EA. The current review will provide an overview of preclinical studies on EA-eCB signaling and its involvement in ethanol actions and AUD. Specifically, we will focus on eCB signaling in EA and its role in stress responsivity and anxiety-related behaviors, and further, we will review ethanol-induced eCB dysregulation in EA and effects of pharmacological manipulation of the EA-eCB system on the ethanol withdrawal induced anxiety- and stress-related behaviors. Finally, we will discuss future directions needed to advance our understanding of the eCB signaling in EA neurocircuitry governing negative affective symptoms associated with AUD, highlighting the potential therapeutic implications of targeting EA-eCB signaling.
METHODS
The EA includes the BNST, CeA, and nucleus accumbens shell (NAcc shell), and the transition zone, substantia innominate (SI). For the scope of this review, we will exclude the NAcc shell and the SI, and instead focus on what many consider to be the two key defining areas of the EA, the CeA and the BNST. We identified preclinical studies through queries of PubMed database. The initial PubMed searches were undertaken on November 1, 2018 using following terms, with a final updated search date of March 22, 2019:
Endocannabinoids and the extended amygdala
(endocannabinoid OR cannabinoid OR CB1 OR anandamide OR 2-arachidonoylglycerol OR faah OR MGL) AND (“extended amygdala” OR “central amygdala” OR “bed nucleus stria terminalis” OR bnst OR CeA) NOT (“substantia innominata”) NOT (“nucleus accumbens”). This search yielded 83 articles. After excluding reviews, human studies, and papers deemed irrelevant to the topic of this review, we reviewed 60 articles.
Endocannabinoids, alcohol, and the extended amygdala
(endocannabinoid OR cannabinoid OR CB1 OR anandamide OR 2-arachidonoyl glycerol OR “cannabinoid receptors” OR FAAH OR MGL) AND (alcohol OR ethanol OR etoh) AND (“extended amygdala” OR “central amygdala” OR “bed nucleus stria terminalis” OR BNST OR CeA) NOT (“substantia innominata”) NOT (“nucleus accumbens”). This search yielded 21 articles. After excluding reviews, human studies, and papers deemed irrelevant to the topic of this review, we reviewed 7 articles.
The above search criteria were used to summarize extant literature; however, additional citations not present in the search results, but deemed relevant were included to support the topics covered in this review.
INTRODUCTION TO THE ENDOCANNABINOID SYSTEM
The eCB system is a versatile neuromodulatory system expressed widely throughout the central nervous system and is involved in numerous fundamental physiological processes. The eCB signaling system is composed of cannabinoid receptors, the endogenous ligands and enzymes involved in synthesis, degradation and transportation of eCBs. The cannabinoid receptors are G protein-coupled receptors activated by endogenous or exogenous cannabinoids. There are two types of cannabinoid receptors: cannabinoid receptor type- 1 (CB1) and type-2 (CB2) (Matsuda et al., 1990, Munro et al., 1993). CB1 receptors are expressed almost everywhere in the body but most abundantly in the central nervous system (Mackie, 2005, Freund et al., 2003). CB2 receptors are mainly expressed in peripheral immune cells (Liu et al., 2009), although emerging evidence suggests the presence of CB2 receptors in brain under some circumstances as well (Buckley et al., 2000, Lanciego et al., 2011). CB1 receptors are predominantly expressed on the axon terminals within the brain (Howlett et al., 2002, Katona et al., 1999). Activation of CB1 receptors results in inhibiting the release of various neurotransmitters such as glutamate, GABA, serotonin, dopamine, and noradrenalin (Piomelli, 2003, Katona and Freund, 2012).
Anandamide (AEA) and 2-arachidonoylglycerol (2-AG) are the most well-studied eCBs (Devane et al., 1992, Mechoulam et al., 1995, Sugiura et al., 1995). These are retrograde lipid messengers synthesized primarily on-demand in postsynaptic neurons from membrane phospholipids (Lu and Mackie, 2016). They travel to presynaptic neurons to activate CB1 receptors and inhibit neurotransmitter release (Castillo et al., 2012). eCBs have been shown to mediate multiple forms of short-term plasticity in the form of depolarization-induced suppression of inhibition/excitation (DSE/DSI) and long-term depression (LTD) depending on the brain region and synapses (Kreitzer and Regehr, 2001, Lu and Mackie, 2016, Ohno-Shosaku et al., 2001). AEA signaling extends beyond eCB signaling and acts as an endogenous ligand for transient receptor potential channels (TRPV1), peroxisome proliferator activated receptor (PPAR) alpha, PPAR gamma and L-type Ca+2 cannels (O’Sullivan, 2007, Bouaboula et al., 2005, Di Marzo et al., 2002, Toth et al., 2009).
AEA is synthesized from its membrane phospholipid precursor N-arachidonoyl phosphatidylethanolamine (NAPE) through hydrolysis by a phospholipase D (NAPE-PLD), and degraded by fatty acid amide hydrolase (FAAH) (Liu et al., 2008). 2-AG is synthesized from membrane phospholipids via sequential activation of phospholipase C (PLC) and diacylglycerol lipase (DAGL) (Murataeva et al., 2014) and primarily degraded by monoacylglycerol lipase (MAGL). Pharmacological modulation of eCB signaling, via inhibition of AEA and 2-AG degradation represents a novel approach for the treatment of a variety of psychiatric disorders (Clapper et al., 2018) and several compounds targeting FAAH and MAGL are in clinical trials (Huggins et al., 2012, Kerbrat et al., 2016, van Esbroeck et al., 2017). The FAAH inhibitor, PF-04457845 has shown promise for the treatment of cannabis dependence (D’Souza et al., 2019), while several MAGL inhibitor trials are in early phases trails but have yet to be tested in substance use or related disorders (Granchi et al., 2017).
THE ANATOMY OF ENDOCANNABINOID SIGNALING IN THE EXTENDED AMYGDALA
CeA
In the CeA, eCB ligands, receptors, and enzymes are all expressed, albeit at varying levels. While CB1 mRNA is uniformly expressed in the CeA (Matsuda et al., 1993, Marsicano and Lutz, 1999, Chhatwal et al., 2005, Hermann and Lutz, 2005), CB1 protein expression is more intense in the lateral subdivision of the CeA (CeAL) than the centromedial amygdala (Kamprath et al., 2011, McDonald and Mascagni, 2001, Ramikie and Patel, 2012, Tsou et al., 1998). Our group revealed a detectable CB1 mRNA signal within the CeA and abundant expression of CB1 mRNA in basolateral amygdala (BLA) neurons, which project glutamatergic afferents to CeAL. Consistent with this, CB1 receptors were found to be heavily expressed in the presynaptic boutons forming asymmetric synapses onto dendritic shafts and spines within the CeAL. Figure 1 depicts a simplified illustration of the eCB signaling in the CeA. CB1 receptors are also present in astrocytes in the CeA, and can be activated by eCBs released from neurons in the medial subdivision of the CeA (CeAM) (Martin-Fernandez et al., 2017). Activation of astrocytic CB1 receptor increases astrocytic calcium levels, enhancing inhibitory synaptic transmission at CeAL-CeAM synapses and depressing excitatory synaptic transmission at BLA-CeAM synapses (Figure 1). The CeA sends inhibitory efferent projections to the BNST, periaqueductal gray (PAG), locus coeruleus, and parabrachial nucleus (PBN) (Janak and Tye, 2015, Dong et al., 2001). These projections play important role in mediating emotional, fear-related responses and alcohol-related behaviors (Pomrenze et al., 2015, Janak and Tye, 2015). CeA-BNST inhibitory projections are regulated by the eCB signaling (Lange et al., 2017). However, it is still unclear whether eCB signaling regulates long-range CeA efferent projections. The 2-AG synthesizing enzyme DAGLα is expressed postsynaptically in the CeA in dendritic shafts and spine heads forming asymmetric synapses in the CeAL (Patel et al., 2009, Ramikie et al., 2014, Yoshida et al., 2011). The 2-AG- and AEA-degrading enzymes MAGL and FAAH are also expressed in the CeA, albeit at relatively low levels (Dinh et al., 2002, Gulyas et al., 2004, Thomas et al., 1997). In summary, the eCB machinery is highly expressed in the CeA. The CB1 receptors are mainly present on the presynaptic boutons of glutamatergic neurons but also GABAergic terminals and astrocytes at lower levels.
Figure 1. Endocannabinoid signaling in the central nucleus of the amygdala (CeA).
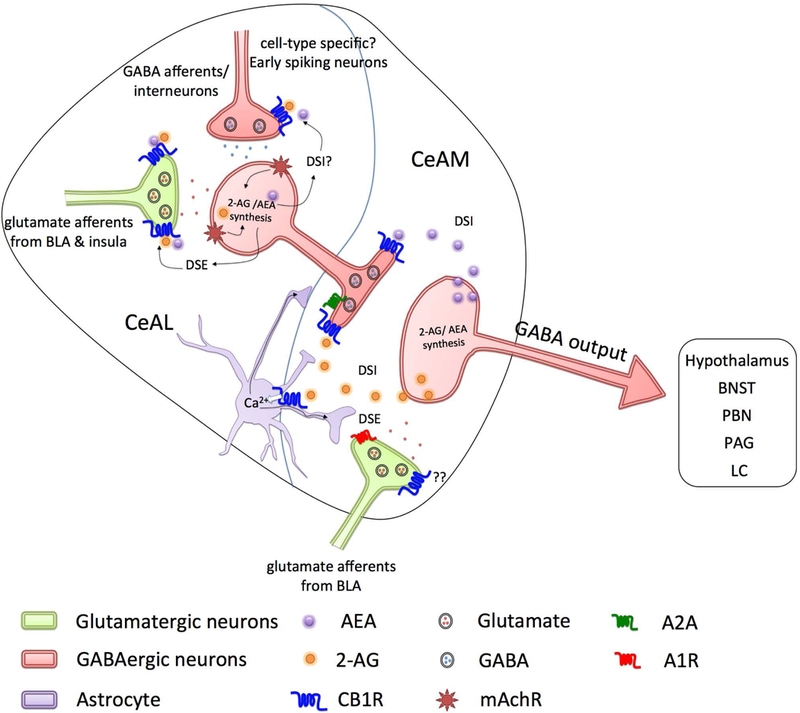
Glutamatergic inputs (light green) from the basolateral amygdala (BLA) and the insula project onto GABAergic neurons (light red) in the lateral subdivision of the CeA (CeAL). These GABAergic neurons send axon terminals to the medial subdivision of the CeA (CeAM). Both inputs are strongly regulated by the endocannabinoid (eCB) system via presynaptic CB1 receptors. In the CeAL, activation of mAChRs initiates 2-arachidonoyl glycerol (2-AG) and anandamide (AEA) production, and subsequent activation of presynaptic CB1 receptors by eCBs at excitatory inputs inhibits glutamate release (depolarization-induced suppression of excitation, DSE). Tonic eCB signaling in the CeAM inhibits the GABA release onto the GABAergic projection neurons (depolarization-induced suppression of inhibition, DSI), regulating CeA output. eCBs also regulate synaptic neurotransmission in the CeAM through activation of CB1 receptors on astrocytes (light violet). Activation of astrocytic CB1 receptors increases astrocytic calcium and results in increasing synaptic vesicle release probability in the CeL-CeM through activation of A2A receptors and decreases synaptic probability of release through activation of A1 receptors in BLA-CeAM synapses. CeA sends inhibitory efferent projections to lateral hypothalamus, the bed nucleus of stria terminalis (BNST), parabrachial nucleus (PBN), periaqueductal gray (PAG) and locus coeruleus (LC).
BNST
Uniform CB1 mRNA expression and moderate protein levels have been reported in the BNST (Chhatwal et al., 2005, Hermann and Lutz, 2005, Marsicano and Lutz, 1999, Massi et al., 2008, Matsuda et al., 1993, Puente et al., 2011, Tsou et al., 1998). CB1 receptor expression is detected within the BNST on the axon terminals arising from the infralimbic cortex (Massi et al., 2008). Particularly intense CB1 labeling is found in anterodorsal, anteroventral and anterolateral parts of BNST (Puente et al., 2010). . CB1 receptors are localized in the both excitatory and inhibitory-like boutons synapsing onto BNST neurons (Figure 2) (Puente et al., 2011, Puente et al., 2010); 55% excitatory and 64% of inhibitory synaptic terminals showed CB1 immunolabeling in anterolateral BNST (Puente et al., 2010). Interestingly 22% of Corticotrophin releasing hormone (CRH)-positive cells co-express CB1 mRNA (Cota et al., 2007). The BNST receives inputs from variety of cortical structures such as the infralimbic cortex, insula, CeA, BLA and medial amygdala. Glutamatergic inputs from infralimbic cortex, insula, and BLA, and GABAergic inputs from the CeA are CB1 sensitive (Centanni et al., 2018, Lange et al., 2017, Massi et al., 2008). Moreover, BNST neurons send dense projections to the PBN, lateral hypothalamus, PAG, and ventral tegmental area (VTA) (Dong and Swanson, 2004, Daniel and Rainnie, 2016). Although BNST efferent projections are important in mediating the behaviors related to stress, reward and emotional processing, it is not clear whether these projections are regulated by eCB signaling in the downstream structures. DAGLα is expressed in the postsynaptic dendritic and spine compartment away from postsynaptic densities in BNST neurons (Puente et al., 2011). In contrast, NAPE-PLD is expressed in the perisynaptic region of dendrites and spine membranes contacted by excitatory synaptic terminals (Puente et al., 2011). In terms of degradation enzymes, the BNST contains relatively low levels of FAAH and MAGL (Gulyas et al., 2004). Figure 2 depicts a simplified illustration of eCB signaling in the BNST. In summary, the eCB machinery is moderately expressed in BNST. CB1 receptors are expressed on both afferent GABAergic and glutamatergic neurons. NAPE-PLD is expressed in the perisynaptic region while DAGLα is expressed away from postsynaptic densities in postsynaptic spines and dendrites.
Figure 2. Endocannabinoid signaling in the bed nucleus of the stria terminalis (BNST).
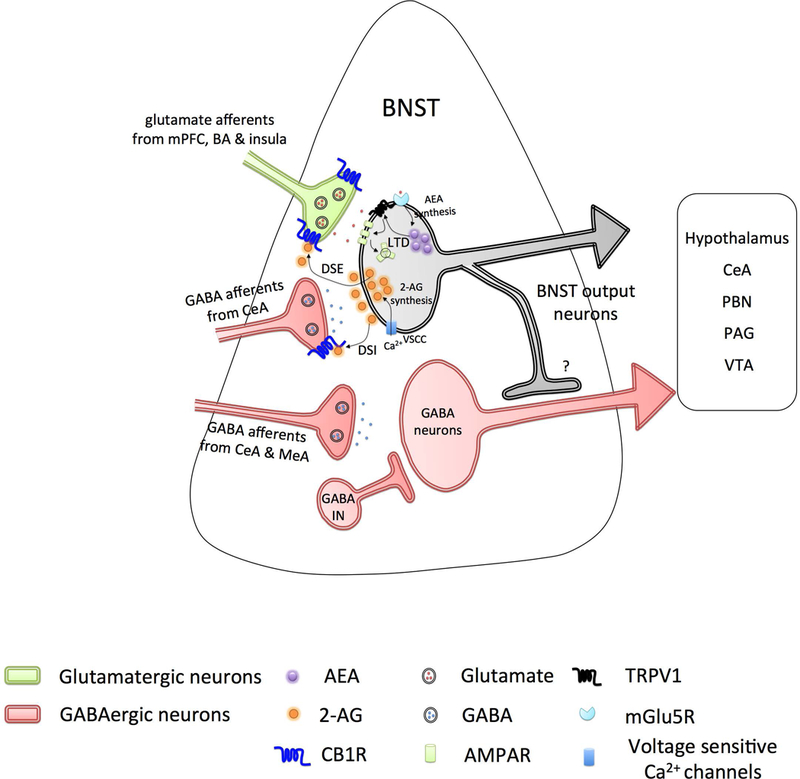
Excitatory inputs (light green) from medial prefrontal cortex (mPFC), basal amygdala (BA) and insula, and inhibitory inputs (light red) from central nucleus of the amygdala (CeA) project onto BNST neurons and are regulated by the endocannabinoid system via presynaptic CB1 receptors. Inhibitory inputs from CeA and medial amygdala (MeA) projecting onto GABAergic BNST neurons (light red) are devoid of CB1 receptors. Activity at excitatory inputs evokes postsynaptic depolarization of BNST neurons, subsequent Ca2+ entry via voltage sensitive calcium channels (VSCC), initiates the production of 2-arachidonoyl glycerol (2-AG), activation of presynaptic CB1 receptors at both excitatory and inhibitory inputs, in turn, resulting in short-term suppression of glutamate (depolarization-induced suppression of excitation, DSE) and GABA (depolarization-induced suppression of inhibition, DSI) release. Activation of mGluR5 receptor initiates the production of anandamide (AEA), which further activates the postsynaptic TRPV1 channels in an autocrine manner. TRPV1 triggers internalization of postsynaptic AMPA receptors and induce long-term depression (LTD). BNST neurons send dense projections to the lateral hypothalamus, CeA, parabrachial nucleus (PBN), periaqueductal gray (PAG), and ventral tegmental area (VTA).
CANNABINOID MODULATION OF SYNAPTIC SIGNALING IN THE EXTENDED AMYGDALA
CeA
Pharmacologically activating cannabinoid receptors in acute CeA slices decreases GABAergic transmission and increases the paired-pulse ratio (PPR) in a CB1-dependent manner (Roberto et al., 2010). In contrast, acute CB1 blockade elicites a marked increase in inhibitory postsynaptic current (IPSC) amplitude and decreases PPR suggesting a tonic eCB/CB1 tone actively inhibits GABAergic transmission in the CeA (Neu et al., 2007, Roberto et al., 2010). Moreover, CB1 receptors in the CeA mediate short-term synaptic plasticity, namely DSI and DSE. Inhibitory inputs from CeAL to CeAM are susceptible to DSI and excitatory inputs from BLA to CeAM are susceptible to DSE (Figure 1) (Kamprath et al., 2011). Both DSI and DSE are blocked by a CB1 antagonist and absent in CB1 KO mice (Kamprath et al., 2011).
CB1 receptor activation also significantly depresses evoked excitatory postsynaptic current (eEPSC) amplitude to ∼50% of baseline (Ramikie et al., 2014). Further, CeAL neurons express prototypic 2-AG-mediated eCB signaling (i.e., DSE) mediated via a calcium-dependent, 2-AG synthesis inhibitor tetrahydrolipstatin (THL)-sensitive, and CB1-dependent mechanism (Ramikie et al., 2014). However, the effects of the CB1 agonist CP55940 on GABAergic transmission were smaller and more variable relative to the effects on glutamatergic transmission (Ramikie et al., 2014). Interestingly, acute activation of mAChRs by Oxo-M increases AEA-mediated depression of glutamatergic transmission (Ramikie et al., 2014), while prolonged activation of mAChRs results in increases tonic and phasic 2-AG-mediated depression of CeA glutamatergic synapses (Ramikie et al., 2014). Recently, Hou et al. examined eCB signaling within two major electrophysiologically distinct CeAL neuron classes; early spiking and late spiking neurons (Hou et al., 2016). Robust DSI is exclusively present at the synapses with presynaptic early spiking cells. DSI indeed only occurred at the early spiking→ late spiking or early spiking→ early spiking synapses but not at late spiking→ early spiking or late spiking→ late spiking synapses suggesting that CB1 receptors might be expressed only on early spiking axon terminals (Hou et al., 2016). These data reveal CeA activity to be regulated by tonic and phasic forms of eCB signaling.
The CeA is a principal output nucleus of amygdala containing rich local GABAergic microcircuits and long range projections influencing the excitability of downstream nuclei. The fine-tuning of GABAergic signaling is a prerequisite in controlling CeA output neurons. In CeAL, eCB released from GABAergic neurons will activate CB1 receptors on glutamatergic afferents and dampen glutamate release onto inhibitory CeAL neurons resulting in disinhibition of CeAM. In CeAM, a tonic eCB tone will dampen local GABA release and consequently increase activity of CeAM neurons. Both mechanisms could increase in inhibition of downstream nuclei. Conversely, CB1 blockade could increase glutamate release in CeAL and GABA release in CeAM, dampening CeA output and relieving inhibition of downstream structures. Functional consequences of these proposed circuit-modulatory mechanisms are currently under investigation.
BNST
CB1 receptor and eCB signaling have been demonstrated to modulate both glutamatertic and GABAergic transmission in the BNST. For example, in vivo extracellular electrophysiology experiments showed specific effects of a CB1 agonism at glutamatergic synapses between infralimbic cortex and BNST neurons. Microinfusion of the cannabinoid agonist WIN 55,212–2 into the BNST inhibited excitation of BNST neurons evoked by infralimbic cortex stimulation and these effects were mediated by CB1 receptors (Massi et al., 2008). Bath application of a cannabinoid agonist WIN55,212,2 to acute BNST brain slices inhibited field-evoked excitatory postsynaptic potentials (fEPSPs) and inhibitory postsynaptic potentials (IPSPs) in BNST neurons. This depression was completely reversed by application of CB1 antagonist SR141716A. The depression of evoked release induced by CB1 activation was accompanied by an increase in PPR, suggested a presynaptic site of action (Puente et al., 2010).
eCBs have also been shown to modulate short-term depression (STD) and LTD in the BNST. For example, Manzoni and co-workers showed that STD and LTD can be induced sequentially in the same BNST neuron without occluding each other, independently of the order at which they were induced (Puente et al., 2011). The CB1 antagonist AM251 prevented both STD and LTD. However, the DAGL inhibitor prevented, while the MAGL inhibitor JZL184 enhanced, depolarization-induced STD in BNST neurons (Puente et al., 2011), outlining a potentially role for 2-AG signaling in the meditation of STD at excitatory synapses in the (Puente et al., 2011). LTD in the BNST can also be mediated by eCB signaling, as activating group I mGlu receptors induces CB1-dependent acute depression in the BNST (Grueter et al., 2006). Further, the TRPV1 antagonists capsazepine or AMG9810 completely prevented the induction of LTD in the BNST (Puente et al., 2011). AEA is an endogenous ligand for TRPV1 (Toth et al., 2009) and postsynaptic activation of TRPV1 triggers internalization of postsynaptic AMPA receptors resulting LTD in hippocampus, for example (Chavez et al., 2010). A robust LTD was induced in slices pretreated with the FAAH inhibitors URB597 or JNJ-1661010 further implicating AEA signaling in BNST LTD (Puente et al., 2011). Thus, the eCB system appears to modulate synaptic transmission and plasticity in the BNST. In BNST neurons (both interneurons and projection neurons), the eCB system mediates short-term suppression i.e. DSE or DSI via 2-AG acting on presynaptic CB1 receptors, whereas LTD of excitatory inputs is mediated via AEA acting on postsynaptic TRPV1 receptors.
THE ROLE OF EXTENDED AMYGDLA ENDOCANNABINOID SIGNALING IN STRESS, ANXIETY, AND FEAR
Several clinical studies have pointed the link between the stress-related disorders and cannabis use. Posttraumatic disorder (PTSD) patients are more likely to use cannabis (Bonn-Miller et al., 2007), suggesting the comorbidity between PTSD and cannabis use. Many patients with PTSD cite the ability of cannabis to promote relaxation and sleep, and reduce anxiety symptoms as motives for continued use (Betthauser et al., 2015, Bonn-Miller et al., 2007). In contrast, blockade of CB1 receptor has reported to produce depressive-like and anxiogenic effects in humans, which ultimately led to the withdrawal of CB1 antagonist rimonabant from European market (Doggrell, 2008, Marco et al., 2011). These data strongly implicate cannabinoid signaling in the regulation of anxiety and stress responsivity in humans. In the following section, we will discuss preclinical studies examining the eCB signaling in the CeA and BNST in the modulation of anxiety-related behaviors and stress responsivity.
CeA
Stress sensitivity is increased in animals that had received chronic treatment of a high dose of the cannabinoid agonist HU210, as these mice showed an exaggerated corticosterone secretion in response to restraint stress compared to control mice (Hill and Gorzalka, 2006). Importantly, robust expression of the immediate early gene c-fos was found in the CeA in response to acute stress in animals chronically HU-210 treated mice. This early study suggested the possibility that cannabinoid sensitization of acute stress effects could involve the CeA. This increased stress sensitivity might be due to the downregulation of CB1 receptors on glutamatergic axon terminals, which in turn could reduce threshold for the activation of the CeA by incoming excitatory afferents stimulated by stressful stimuli.
Activation of CB1 receptors in the CeA by a CB1 agonist reduces anxiety-like behaviors (Zarrindast et al., 2008), and conversely, systemic administration of the CB1 antagonist SR141716 precipitates a negative emotional state in rats withdrawn for 4 days from chronic intermittent access to a highly palatable food. This withdrawal period increased levels of 2-AG and CB1 receptor protein and mRNA in the CeA of mice (Blasio et al., 2013). Thus, the 2-AG/CB1 receptor system within the CeA appears to be recruited during abstinence from palatable diet cycling as a compensatory mechanism to dampen anxiety. In contrast, CB1 mRNA expression is decreased in the CeA of rats with heighted anxiety after nicotine withdrawal (Aydin et al., 2012), collectively suggesting abstinence from drugs of abuse and palatable food causes dysregulation of CeA eCB signaling, with the direction of changes in eCB signaling depending on the nature of the withdrawal.
Microinfusion of the CB1 antagonist AM251 in the CeA acutely increases fear responses in an auditory fear-conditioning paradigm (Kamprath et al., 2011). The CeA also plays an important role in both active and passive shock avoidance. Post-training bilateral injection of the cannabinoid agonist WIN 55, 212–2 into the CeA significantly decreased latency to enter the dark compartment (paired with footshock) in a dose-dependent manner on an inhibitory avoidance task (Hasanein and Sharifi, 2015, Zarrindast et al., 2012). In sum, activation of CeA eCB system reduces anxiety and fear-related responses. 2-AG-CB1 signaling system is recruited during abstinence from palatable food to compensate abstinence-induced anxiety. Blockade of CeA CB1 receptors could precipitate negative emotional state and increase fear responses.
BNST
The BNST has been associated with stress, negative emotional state and fear-related behaviors (Ulrich-Lai and Herman, 2009). The presence of eCB signaling machinery in the BNST opens the possibility that eCBs might be playing an important role in BNST function and related behavioral responses. Systemic treatment with a CB1 receptor antagonist AM251 increases BNST neuronal activation induced by aversive stimulus, indicating that eCB signaling in BNST is activated during stressful events (Newsom et al., 2012). At the synaptic level, the BNST receives a major input from the medial prefrontal cortex (mPFC), and in vivo recording from BNST neurons revealed that 1h of restraint stress induced a switch form LTD to LTP of mPFC afferents to the BNST (Massi et al., 2008). The eCB system, through the stimulation of CB1 receptors present on glutamatergic terminals, plays a key role in this plasticity shift (Glangetas et al., 2013). This was confirmed by the fact that the stress-induced plasticity shift was absent in CB1−/− mice and wild-type mice that received bilateral microinjections of the CB1 antagonist AM251 in the BNST. The absence of stress-induced plasticity shift in conditional knockout Glu-CB1−/− (mice lacking CB1 expression on forebrain glutamatergic neurons), revealed that stress-elicited shift in plasticity is fully controlled by CB1 receptors located on glutamatergic terminals (Glangetas et al., 2013). It is not clear exactly how eCB signaling on glutamatergic neurons is involved in stress-elicited shift in plasticity in the BNST neurons, thus future studies into the cellular mechanisms underlying these effects are of high importance.
BNST eCB signaling ahs also been implicated in the physiological responses to acute stress. Microinjection of the CB1 antagonist AM251 into the BNST enhances tachycardia associated with restraint stress (Gomes-de-Souza et al., 2016). Conversely, augmenting AEA signaling in BNST via FAAH inhibition (URB597) dampenes restraint-stress evoked tachycardia and microinjection of the MAGL inhibitor JZL184 into the BNST decreased restraint stress-evoked increase in heart rate in CB1 dependent manner (Gomes-de-Souza et al., 2016). These data indicate involvement of BNST eCB signaling in cardiovascular adjustments during emotion stress that could be mediated by local release of AEA and/or 2-AG.
CB1 receptors in BNST were recently shown to be critical for the fear response to an unpredictable threat, as local BNST infusion of the CB1 antagonist AM251 prevented a shift from phasic fear to sustained fear in response to an unpredictable threat (Lange et al., 2017). Electrophysiology and optogenetics were used to identify putative glutamatergic neurons of the BNST that receive basal amygdala (BA) glutamatergic and CeA GABAergic afferent inputs regulated by the eCB system via presynaptic CB1 receptors, while the inputs from medial amygdala to BNST are not regulated by CB1 receptors (Lange et al., 2017). Mice holding a loxP-flank transcriptional stop-cassette upstream of the CB1-coding region (Stop-CB1), which prevents CB1 expression, displayed rapidly declining phasic freezing suggesting CB1 receptor function in the CeA/BA projections to alBNST is necessary to express sustained fear. Cre-dependent rescue of CB1 receptor function in BA/CeA neurons in Stop-CB1 mice reinstated sustained fear. Using conditional knock out mice, CB1 receptor function in both CeA GABAergic and BA glutamatergic inputs to alBNST are necessary to express sustained fear. These results suggest stimulation of CB1 receptors, on BA and CeA projections to BNST, are necessary and sufficient for the shift from phasic to sustained fear in response to unpredictable threat stimuli. However, CB1 receptors on these inputs are not involved in mediating phasic fear responses to predictable threat (Lange et al., 2017). Although the brain regions important for producing unconditioned anxiety and conditioned fear partially overlap, the aforementioned studies indicated an opposite role for the eCB system in mediating these behaviors. One possible explanation for this apparent difference is that different neural circuits regulate conditioned fear and unconditioned anxiety. It is also possible that eCB augmentation exerts opposing effects on fear and anxiety behaviors due to its action on specific glutamatergic circuits that differentially promote or inhibit the expression of fear and anxiety phenotypes.
EXTENDED AMYGDALA ENDOCANNABINOID SIGNALING IN ALCOHOL DEPENDENCE
Altered extended amygdala endocannabinoid signaling after alcohol exposure
The EA is critical for the reinforcing effects of ethanol and the transition to dependence (Koob et al., 1998, Koob, 2003). Ethanol perfusion increases GABAergic transmission in CeA by acting on both presynaptic and postsynaptic neurons (Roberto et al., 2003). Subsequent activation of CB1 receptors together with ethanol significantly decreases IPSC amplitudes by ∼30 % compared to ethanol-only. Interestingly, bath perfusion of the CB1 antagonists AM251 or SR141716 revealed a tonic eCB/CB1 control of GABA release in CeA neurons of ethanol naïve rats (Roberto et al., 2010). Moreover, ethanol and AM251 or SR141716 increased GABA transmission in an additive manner (Roberto et al., 2010), indicating that CB1 signaling and ethanol-induced modulation of GABAergic transmission occur independently. Acute ethanol perfusion also decreases glutamatergic transmission in rats with no change in PPR, indicating a postsynaptic site of action of ethanol. No tonic eCB signaling was found at glutamatergic synapses in CeA (Kirson et al., 2018). The exact molecular mechanisms involved in ethanol and cannabinoid effects on GABA release are still unclear. It is possible that both ethanol and CB1 receptors are acting on common signaling pathways to regulate GABA release. Protein Kinase A (PKA) and adenylate cyclase (AC) antagonists inhibit the ethanol-induced increases in spontaneous GABA release (Kelm et al., 2008), while CB1 receptors inhibit AC and decrease PKA activity (Katona and Freund, 2008), therefore the AC/PKA pathway in the CeA may be a common mechanism targeted in opposite ways by ethanol and eCBs. Interestingly, chronic in vivo ethanol exposure abolishes tonic eCB/CB1 influence on mIPSC, but not sIPSC, frequency (Varodayan et al., 2016). This suggests that chronic ethanol exposure could decrease CB1 receptor function at GABAergic synapses in CeA.
Role of extended amygdala endocannabinoid signaling in ethanol abstinence-induced anxiety-like behavior and negative affective states
Early and protracted abstinence from alcohol reduces social interaction, increases motor stereotypies, and exacerbates anxiety and depressive-like behaviors (Becker et al., 2017, Holleran et al., 2016, Serrano et al., 2018). Ethanol-preferring rats display increased co-morbid signs of anxiety and excessive ethanol drinking. A major contributor to excessive ethanol intake in this rat model relates to attenuation of anxiety-like behaviors, consistent with negative-reinforcement-driven alcohol consumption (Natividad et al., 2017, Stopponi et al., 2018). Ethanol-preferring rats display aberrations in stress signaling systems including upregulation of CRF1 receptor in the CeA and medial amygdala that is linked to the single nucleotide polymorphism in the promoter region of the gene (Hansson et al., 2007). FAAH activity is also increased in the CeA of ethanol-preferring rats. CRF-induced anxiety relies on modulation of eCBs and activation of CRF1 receptors, as CRF evokes a rapid induction of FAAH activity which reduces AEA (Gray et al., 2015). This increased FAAH activity is accompanied by reductions in AEA dialysate levels in the rat CeA of ethanol-preferring rats (Natividad et al., 2017). Voluntary ethanol self-administration downregulates CRF1 receptor transcripts in CeA and MeA and CRF1 blockade normalizes elevated FAAH expression in ethanol-preferring rats suggesting a mechanism whereby CRF1-mediated FAAH activity-dependent AEA deficiency drives anxiety-like behavior and excessive drinking in this rat line (Natividad et al., 2017). Ethanol-preferring rats also exhibit elevated baseline glutamatergic transmission in the CeA which could be a consequence of AEA deficiency and could contribute to the behavioral phenotypes.
2-AG has also been implicated with withdrawal-induced anxiety in rodent models. Pharmacological augmentation of 2-AG reduces early ethanol abstinence-induced neuronal activity and glutamatergic transmission in the BNST (Centanni et al., 2018). The insula provides interoceptive cues through neuronal projections to cortical and subcortical brain regions, including dense projections to BNST. Insula afferents onto BNST CRF+ neurons are eCB-sensitive and a possible source of abstinence-induced increases in glutamatergic neurotransmission in BNST. Chemogenetic inhibition of insula neurons reduced early abstinence-induced increase in neuronal activity and glutamatergic transmission in BNST and decreased early abstinence-induced negative affect (Centanni et al., 2018), thus mimicking the effects of activating the Gi-coupled CB1 receptors and providing evidence for a potential role of insula-BNST eCB signaling in abstinence-induced negative affect. Accordingly, chemogenetic activation of the BNST neurons receiving insular projections produces a negative affective phenotype. This suggests a distinct role of insula-BNST circuits in mediating negative affect in early ethanol abstinence and that enhancing 2-AG is sufficient to prevent abstinence-induced hyperactivity in the BNST.
Ethanol abstinence-induced changes in expression of endocannabinoid signaling components in the extended amygdala
Acute ethanol withdrawal significantly changes mRNA expression for various components of eCB signaling system in EA. Specifically, FAAH and MAGL mRNA is significantly reduced after 24h of ethanol withdrawal. These changes are accompanied by decreased CB1 and CB2 receptors mRNA expression (Serrano et al., 2012). Interestingly, the first withdrawal after continuous ethanol exposure primarily alters gene expression related to AEA biosynthesis and clearance, whereas repeated withdrawals induced by chronic intermittent ethanol (CIE) exposure decreased gene expression for the 2-AG degrading enzyme MAGL and cannabinoid receptors (CB1 and CB2). Further, CIE significantly downregulates CB1 receptor function in the CeA (Varodayan et al., 2016), and alters behavior and physiological responses to CB1 activation (Vinod et al., 2006, Vinod et al., 2012, Mitrirattanakul et al., 2007). Postmortem studies in AUD report disruptions of CB1 receptor expression in ventral striatum and cortical regions (Vinod et al., 2010). Imaging studies have reported lower CB1 receptor availability in heavy drinking alcoholics that persisted for 1 month of abstinence (Ceccarini et al., 2014, Hirvonen et al., 2013). Overall the alterations in mRNA expression for various components of eCB signaling system were more prominent in rats exposed to CIE compared to continuous access exposed rats (Serrano et al., 2012). Abstinent rats showed alteration in gene expression of FAAH, MAGL, and DAGL in the CeA, suggesting sensitivity to the intermittent nature of ethanol exposure and post-ethanol abstinence period (Serrano et al., 2012). A recent report by the same group specifically studied ethanol-abstinence-induced gene expression in CeA. After 12h of ethanol withdrawal, the abstinence group showed an overall increase in the mRNA expression of MAGL, FAAH and DAGL enzymes related to the eCB signaling in CeA (Serrano et al., 2018). These discrepant findings could be the result of different time points and brain regions examined. Collectively, these studies provide evidence of ethanol-induced dysregulation of the eCB system.
CIE reduces baseline dialysate 2-AG levels in the CeA, and this persisted up to 7 days into abstinence, consistent with the increased gene expression of several enzymes involved in synthesis and degradation of 2-AG observed in CeA after 12h of ethanol withdrawal (Serrano et al., 2018). CIE and subsequent protracted abstinence did not alter AEA levels in the CeA. Interestingly, the reductions in CeA 2-AG levels in ethanol dependent rats were enhanced during acute abstinence, but 2-AG levels were normalized by re-exposure to ethanol (Serrano et al., 2018). As expected, increased GABA and glutamate dialysate levels in CeA accompanied the reduced 2-AG baseline levels (Serrano et al., 2018). These results suggest 2-AG may be responsible for eCB-mediated neuroadaptations in the CeA in response to CIE. Pharmacological and genetic 2-AG deficiency has been shown to generate anxiety-like and sex-specific anhedonic phenotypes (Bedse et al., 2017, Shonesy et al., 2014) suggesting ethanol abstinence-induced 2-AG signaling deficiency in the amygdala could contribute to the increased anxiety-like and anhedonic phenotypes. Furthermore, ethanol reinstatement could normalize the eCB deficiency suggesting 2-AG deficiency may represent a novel mechanism contributing to negative reinforcement-driven ethanol drinking.
Self-administration of ethanol increases interstitial CeA 2-AG levels, and this increase is absent in ethanol dependent rats (Serrano et al., 2018). Although dependent rats showed elevated ethanol self-administration, there were no alterations in CeA AEA levels in dependent or control rats. A 30 min of restraint increased CeA 2-AG levels in control rats. However, in ethanol dependent rats stress exposure did not alter the CeA 2-AG levels. In contrast, CeA AEA levels were lowered in response to stress in both control and ethanol dependent rats. The initial decline in brain AEA levels appears to enable the manifestation of the stress response and the subsequent increase in 2-AG appears to terminate the stress response and HPA-axis activation. The reduced interstitial 2-AG levels observed during acute abstinence were accompanied by increased anxiety-like behaviors in ethanol dependent rats. Collectively, dysregulation of 2-AG, but not AEA signaling, appears critical for anxiety-like behaviors in ethanol dependent rats. It is possible that reduced CeA 2-AG levels that result from ethanol dependence mediate the negative affective state during withdrawal and that could contribute to the excessive ethanol consumption. Figure 3 depicts a proposed mechanism for ethanol-induced dysregulation of eCB signaling in the EA.
Figure 3. A proposed mechanism of ethanol-induced dysregulation of endocannabinoid signaling in extended amygdala.
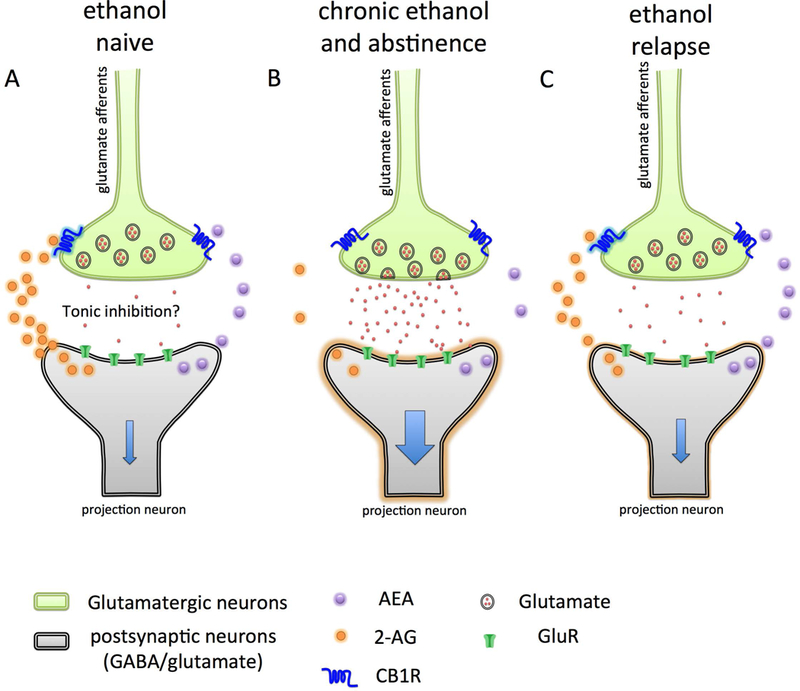
In ethanol naïve conditions (A) endocannabinoid signaling within the central nucleus of amygdala (CeA) and bed nucleus of stria terminalis (BNST) regulates the presynaptic release of glutamate through activation of CB1 receptors on glutamatergic inputs (light green), which dampens the activation of neurons of CeA and BNST (light gray). In ethanol abstinence, the levels of 2-arachidonoyl glycerol (2-AG) decreases, possibly through enhanced degradation by monoacylglycerol lipase (MAGL). The reduced 2-AG signaling at CB1 disinhibits glutamatergic inputs to CeA and BNST, resulting in increased glutamate release and firing of CeA and BNST neurons. Activation of CeA and BNST neurons further activates downstream brain nuclei involved in the brain stress system regulating the negative affective state. (C) Ethanol re-exposure increases the 2-AG levels through an unknown mechanism and temporarily alleviates negative emotional state. This model also suggests pharmacological inhibition of MAGL, which would increase 2-AG signaling, could reduce abstinence-induced EA over activation and negative affective states.
PHARMACOLOGICAL AUGMENTATION OF ENDOCANNABINOID SIGNALING FOR TREATMENT OF ETHANOL ABSTINENCE-INDUCED NEGATIVE EMOTIONAL STATES
A number of studies have demonstrated anxiolytic potential of pharmacological eCB augmentation approaches in variety of preclinical models of negative affect (Hill et al., 2018, Patel et al., 2017). Pharmacological inhibition of eCB-degrading enzymes, FAAH and MAGL, elicit promising anxiolytic effects in rodent models with limited adverse effects (Bedse et al., 2017, Bedse et al., 2018, Bedse et al., 2014, Bedse et al., 2015, Bluett et al., 2014, Patel et al., 2017, Zhong et al., 2014, Lim et al., 2016). With regard to modulation of alcohol-abstinence-induced negative affect, MAGL inhibition (with JZL184 or MJN110) reverses early abstinence-induced anxiety in mice (Holleran et al., 2016, Serrano et al., 2018), and the CB1 antagonist rimonabant blocked these effects (Holleran et al., 2016). FAAH inhibition with PF-3845 alleviated stress-induced increases in glutamatergic transmission and attenuated the excessive anxiety-like behavior in in alcohol preferring rats (Natividad et al., 2017). Intra-CeA injection of FAAH inhibitor URB597 attenuated ethanol self-administration in ethanol-preferring rats, and this may be dependent on the ability of the drug to decrease anxiety in ethanol-preferring rats. Accordingly, URB597 at the same dose that produced maximal effects on ethanol self-administration reduced anxiety-like effects in ethanol-preferring rats (Stopponi et al., 2018). These data demonstrate that FAAH inhibition in CeA alleviates co-morbid signs of anxiety and excessive ethanol drinking, which is driven in part by chronic dysregulation of CRF signaling. The restoration of eCB signaling with the inhibition of MAGL or FAAH could be a promising therapeutic approach to treat ethanol early abstinence-induced anxiety-like phenotype. However, the efficacy of eCB augmentation on protracted abstinence-induced anxiety need to be evaluated.
In summary, eCB signaling in the EA is altered in models of ethanol dependence, which likely plays an important role in negative affect-driven relapse and excessive drinking. eCB signaling in stress responsive brain circuits is blunted in protracted withdrawal suggesting abstinence-induced deficiencies in eCB signaling could constitute a breakdown of important homeostatic mechanism that dampens physiological and behavioral response to stress. Therefore, augmentation of eCB signaling by inhibiting the degradation of eCBs could be used to decrease the anxiety-like phenotype during alcohol abstinence.
FUTURE DIRECTIONS AND CONCLUSIONS
Using our search criteria, we identify a major gap in research focusing on the relationship between eCB signaling in the EA and ethanol exposure. The scope of research highlighting the role of the EA in stress, anxiety, and depression coupled with the ability of eCB to modulate neuronal signaling and behavioral output in the EA highlights the growing therapeutic potential of the eCB system for treating negative affect associated with AUD. To maximize this potential, future research needs to further characterize the complex intricate neural signaling of the eCB system in the EA, and specifically how ethanol exposure and withdrawal impacts eCB signaling in these brain areas. In this vein, how eCB signaling is directly impacted by ethanol exposure and if ethanol directly alters eCB levels in the EA needs to be better studied.
Cell-type specificity
eCB signaling can modulate both excitatory and inhibitory neurotransmission underscoring the broad impact of this system on neurotransmission. However, the ability of eCBs to modulate both excitatory and inhibitory neurotransmitter release speaks to the difficulty in predicting the net effect on the excitation and inhibition of EA nuclei. More research needs to be conducted to understand the fundamental effects of eCB modulation on neurotransmission of specific cell-types in the EA, and moreover, how this modulation is altered after acute ethanol exposure, chronic ethanol exposure, acute withdrawal, and protracted abstinence. The effects of ethanol on several target cell -types have been discussed in the first review in this series, including neuropeptide markers such as CRF and NPY, and cell-types characterized based on their electrophysiological profiles; however, much less is known about eCB signaling at specific EA synapses and cell-types in the context of alcohol actions and neuropeptide signaling.
Input specificity
The dense expression of eCB signaling in numerous brain regions, coupled with the presynaptic actions of eCB signaling has further impeded progress in understanding of the circuit-level mechanisms by which eCBs could regulate effects of alcohol and abstinence-related negative affect. These issues can be addressed by conducting more studies exploring the various inputs to the CeA and BNST, and how these inputs drive negative affect during abstinence. With the rapid advancement of virus-guided neural tracing, genetic manipulation, and brain imaging techniques, studying the effects of ethanol on specific inputs is more feasible than it has ever been. Understanding how ethanol exposure effects the various inputs to the CeA and BNST, and moreover, how eCB can modulate these inputs, will provide invaluable insight to guide our understanding of the circuitry driving negative affect and reveal new insights into the pathophysiology of alcohol abstinence-induced affective changes.
Therapeutic Implications and Conclusions
As indicated throughout this review, several compounds targeting the enzymatic machinery driving eCB degradation, such as FAAH and MAGL, have the potential to reduce alcohol abstinence-induced negative affect state. Continued examination of FAAH and MAGL-inhibition in the various models of AUD and withdrawal-induced negative affect could help provide a solid preclinical base to facilitate eCB-based drug development for AUD. However, to produce a truly successful eCB-based pharmacotherapeutic for treating negative affect associated with AUD, we must use the strategies outlined above to gain a better understanding of how these compounds, when administered systemically, impact specific neural circuits and cell-types. It is, in theory, possible that eCB augmentation could induce adverse cannabimimetic adverse effects on motor function or cognition and carry some abuse liability. However, many studies to date have demonstrated preclinical efficacy of eCB degradation inhibitors without overt cannabimimetic side effects (Hruba et al., 2015, Bedse et al., 2018, Long et al., 2009, Ignatowska-Jankowska et al., 2014, Cravatt and Lichtman, 2003, Ghosh et al., 2015, Booker et al., 2012, Curry et al., 2018, Wilkerson et al., 2016). Indeed, these cannabimimetic side-effects are most apparent during blockade of both FAAH and MAGL concomitantly (Wise et al., 2012, Hruba et al., 2015, Long et al., 2009), this using single enzyme inhibition represents the most likely path forward for eCB-based therapeutics development.
While the topic of this review focuses on eCB signaling in the EA, it should be noted that exciting and ongoing research examining eCB modulation of several other brain regions and neural circuits including the dopamine reward system in theVTA, striatum, and nucleus accumbens could have important implication for developing eCB-based treatments for AUD. Continuing parallel lines of research focused on eCB modulation of multiple aspects of AUD will be essential for ultimate development of novel treatments for AUD.
ACKNOWLEDGEMENTS
These studies were supported by NIH grants AA026186 (S.P.) and AA019455 (D.G.W.) and Brain Behavior Research Foundation NARSAD Young Investigator Awards 26024 (G.B.) and 27172 (S.W.C.).
Footnotes
DISCLOSURES
S.P. has received research funding from H. Lundbeck A/S within the past three years and is a scientific consultant for Psy Therapeutics and Atlas Ventures.
References
- Aydin C, Oztan O, Isgor C (2012) Long-term effects of juvenile nicotine exposure on abstinence-related social anxiety-like behavior and amygdalar cannabinoid receptor 1 (CB1R) mRNA expression in the novelty-seeking phenotype. Behav Brain Res 228:236–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker JAJ, Kieffer BL, Le Merrer J (2017) Differential behavioral and molecular alterations upon protracted abstinence from cocaine versus morphine, nicotine, THC and alcohol. Addict Biol 22:1205–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedse G, Bluett RJ, Patrick TA, Romness NK, Gaulden AD, Kingsley PJ, Plath N, Marnett LJ, Patel S (2018) Therapeutic endocannabinoid augmentation for mood and anxiety disorders: comparative profiling of FAAH, MAGL and dual inhibitors. Transl Psychiatry 8:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedse G, Colangeli R, Lavecchia AM, Romano A, Altieri F, Cifani C, Cassano T, Gaetani S (2014) Role of the basolateral amygdala in mediating the effects of the fatty acid amide hydrolase inhibitor URB597 on HPA axis response to stress. Eur Neuropsychopharmacol 24:1511–1523. [DOI] [PubMed] [Google Scholar]
- Bedse G, Hartley ND, Neale E, Gaulden AD, Patrick TA, Kingsley PJ, Uddin MJ, Plath N, Marnett LJ, Patel S (2017) Functional Redundancy Between Canonical Endocannabinoid Signaling Systems in the Modulation of Anxiety. Biol Psychiatry 82:488–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedse G, Romano A, Tempesta B, Lavecchia MA, Pace L, Bellomo A, Duranti A, Micioni Di Bonaventura MV, Cifani C, Cassano T, Gaetani S (2015) Inhibition of anandamide hydrolysis enhances noradrenergic and GABAergic transmission in the prefrontal cortex and basolateral amygdala of rats subjected to acute swim stress. J Neurosci Res 93:777–787. [DOI] [PubMed] [Google Scholar]
- Betthauser K, Pilz J, Vollmer LE (2015) Use and effects of cannabinoids in military veterans with posttraumatic stress disorder. Am J Health Syst Pharm 72:1279–1284. [DOI] [PubMed] [Google Scholar]
- Black JJ, Clark DB, Martin CS, Kim KH, Blaze TJ, Creswell KG, Chung T (2015) Course of alcohol symptoms and social anxiety disorder from adolescence to young adulthood. Alcohol Clin Exp Res 39:1008–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasio A, Iemolo A, Sabino V, Petrosino S, Steardo L, Rice KC, Orlando P, Iannotti FA, Di Marzo V, Zorrilla EP, Cottone P (2013) Rimonabant precipitates anxiety in rats withdrawn from palatable food: role of the central amygdala. Neuropsychopharmacology 38:2498–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluett RJ, Gamble-George JC, Hermanson DJ, Hartley ND, Marnett LJ, Patel S (2014) Central anandamide deficiency predicts stress-induced anxiety: behavioral reversal through endocannabinoid augmentation. Transl Psychiatry 4:e408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonn-Miller MO, Vujanovic AA, Feldner MT, Bernstein A, Zvolensky MJ (2007) Posttraumatic stress symptom severity predicts marijuana use coping motives among traumatic event-exposed marijuana users. J Trauma Stress 20:577–586. [DOI] [PubMed] [Google Scholar]
- Booker L, Kinsey SG, Abdullah RA, Blankman JL, Long JZ, Ezzili C, Boger DL, Cravatt BF, Lichtman AH (2012) The fatty acid amide hydrolase (FAAH) inhibitor PF-3845 acts in the nervous system to reverse LPS-induced tactile allodynia in mice. Br J Pharmacol 165:2485–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaboula M, Hilairet S, Marchand J, Fajas L, Le Fur G, Casellas P (2005) Anandamide induced PPARgamma transcriptional activation and 3T3-L1 preadipocyte differentiation. Eur J Pharmacol 517:174–181. [DOI] [PubMed] [Google Scholar]
- Buckley NE, McCoy KL, Mezey E, Bonner T, Zimmer A, Felder CC, Glass M, Zimmer A (2000) Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB(2) receptor. Eur J Pharmacol 396:141–149. [DOI] [PubMed] [Google Scholar]
- Cassell MD, Freedman LJ, Shi C (1999) The intrinsic organization of the central extended amygdala. Ann N Y Acad Sci 877:217–241. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chavez AE, Hashimotodani Y (2012) Endocannabinoid signaling and synaptic function. Neuron 76:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarini J, Hompes T, Verhaeghen A, Casteels C, Peuskens H, Bormans G, Claes S, Van Laere K (2014) Changes in cerebral CB1 receptor availability after acute and chronic alcohol abuse and monitored abstinence. J Neurosci 34:2822–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centanni SW, Morris BD, Luchsinger JR, Bedse G, Fetterly TL, Patel S, Winder DG (2018) Endocannabinoid control of the insular-bed nucleus of the stria terminalis circuit regulates negative affective behavior associated with alcohol abstinence. Neuropsychopharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez AE, Chiu CQ, Castillo PE (2010) TRPV1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat Neurosci 13:1511–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatwal JP, Davis M, Maguschak KA, Ressler KJ (2005) Enhancing cannabinoid neurotransmission augments the extinction of conditioned fear. Neuropsychopharmacology 30:516–524. [DOI] [PubMed] [Google Scholar]
- Clapper JR, Henry CL, Niphakis MJ, Knize AM, Coppola AR, Simon GM, Ngo N, Herbst RA, Herbst DM, Reed AW, Cisar JS, Weber OD, Viader A, Alexander JP, Cunningham ML, Jones TK, Fraser IP, Grice CA, Ezekowitz RAB, O’Neill GP, Blankman JL (2018) Monoacylglycerol Lipase Inhibition in Human and Rodent Systems Supports Clinical Evaluation of Endocannabinoid Modulators. J Pharmacol Exp Ther 367:494–508. [DOI] [PubMed] [Google Scholar]
- Cooney NL, Litt MD, Morse PA, Bauer LO, Gaupp L (1997) Alcohol cue reactivity, negative-mood reactivity, and relapse in treated alcoholic men. J Abnorm Psychol 106:243–250. [DOI] [PubMed] [Google Scholar]
- Cota D, Steiner MA, Marsicano G, Cervino C, Herman JP, Grubler Y, Stalla J, Pasquali R, Lutz B, Stalla GK, Pagotto U (2007) Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology 148:1574–1581. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Lichtman AH (2003) Fatty acid amide hydrolase: an emerging therapeutic target in the endocannabinoid system. Curr Opin Chem Biol 7:469–475. [DOI] [PubMed] [Google Scholar]
- Curry ZA, Wilkerson JL, Bagdas D, Kyte SL, Patel N, Donvito G, Mustafa MA, Poklis JL, Niphakis MJ, Hsu KL, Cravatt BF, Gewirtz DA, Damaj MI, Lichtman AH (2018) Monoacylglycerol Lipase Inhibitors Reverse Paclitaxel-Induced Nociceptive Behavior and Proinflammatory Markers in a Mouse Model of Chemotherapy-Induced Neuropathy. J Pharmacol Exp Ther 366:169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza DC, Cortes-Briones J, Creatura G, Bluez G, Thurnauer H, Deaso E, Bielen K, Surti T, Radhakrishnan R, Gupta A, Gupta S, Cahill J, Sherif MA, Makriyannis A, Morgan PT, Ranganathan M, Skosnik PD (2019) Efficacy and safety of a fatty acid amide hydrolase inhibitor (PF-04457845) in the treatment of cannabis withdrawal and dependence in men: a double-blind, placebo-controlled, parallel group, phase 2a single-site randomised controlled trial. Lancet Psychiatry 6:35–45. [DOI] [PubMed] [Google Scholar]
- Daniel SE, Rainnie DG (2016) Stress Modulation of Opposing Circuits in the Bed Nucleus of the Stria Terminalis. Neuropsychopharmacology 41:103–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258:1946–1949. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, De Petrocellis L, Fezza F, Ligresti A, Bisogno T (2002) Anandamide receptors. Prostaglandins Leukot Essent Fatty Acids 66:377–391. [DOI] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D (2002) Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci U S A 99:10819–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doggrell SA (2008) Is rimonabant efficacious and safe in the treatment of obesity? Expert Opin Pharmacother 9:2727–2731. [DOI] [PubMed] [Google Scholar]
- Dong HW, Petrovich GD, Swanson LW (2001) Topography of projections from amygdala to bed nuclei of the stria terminalis. Brain Res Brain Res Rev 38:192–246. [DOI] [PubMed] [Google Scholar]
- Dong HW, Swanson LW (2004) Organization of axonal projections from the anterolateral area of the bed nuclei of the stria terminalis. J Comp Neurol 468:277–298. [DOI] [PubMed] [Google Scholar]
- Driessen M, Meier S, Hill A, Wetterling T, Lange W, Junghanns K (2001) The course of anxiety, depression and drinking behaviours after completed detoxification in alcoholics with and without comorbid anxiety and depressive disorders. Alcohol Alcohol 36:249–255. [DOI] [PubMed] [Google Scholar]
- Fox HC, Milivojevic V, Angarita GA, Stowe R, Sinha R (2017) Peripheral immune system suppression in early abstinent alcohol-dependent individuals: Links to stress and cue-related craving. J Psychopharmacol 31:883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D (2003) Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83:1017–1066. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Kinsey SG, Liu QS, Hruba L, McMahon LR, Grim TW, Merritt CR, Wise LE, Abdullah RA, Selley DE, Sim-Selley LJ, Cravatt BF, Lichtman AH (2015) Full Fatty Acid Amide Hydrolase Inhibition Combined with Partial Monoacylglycerol Lipase Inhibition: Augmented and Sustained Antinociceptive Effects with Reduced Cannabimimetic Side Effects in Mice. J Pharmacol Exp Ther 354:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glangetas C, Girard D, Groc L, Marsicano G, Chaouloff F, Georges F (2013) Stress switches cannabinoid type-1 (CB1) receptor-dependent plasticity from LTD to LTP in the bed nucleus of the stria terminalis. J Neurosci 33:19657–19663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes-de-Souza L, Oliveira LA, Benini R, Rodella P, Costa-Ferreira W, Crestani CC (2016) Involvement of endocannabinoid neurotransmission in the bed nucleus of stria terminalis in cardiovascular responses to acute restraint stress in rats. Br J Pharmacol 173:2833–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granchi C, Caligiuri I, Minutolo F, Rizzolio F, Tuccinardi T (2017) A patent review of Monoacylglycerol Lipase (MAGL) inhibitors (2013–2017). Expert Opin Ther Pat 27:1341–1351. [DOI] [PubMed] [Google Scholar]
- Gray JM, Vecchiarelli HA, Morena M, Lee TT, Hermanson DJ, Kim AB, McLaughlin RJ, Hassan KI, Kuhne C, Wotjak CT, Deussing JM, Patel S, Hill MN (2015) Corticotropin-releasing hormone drives anandamide hydrolysis in the amygdala to promote anxiety. J Neurosci 35:3879–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter BA, Gosnell HB, Olsen CM, Schramm-Sapyta NL, Nekrasova T, Landreth GE, Winder DG (2006) Extracellular-signal regulated kinase 1-dependent metabotropic glutamate receptor 5-induced long-term depression in the bed nucleus of the stria terminalis is disrupted by cocaine administration. J Neurosci 26:3210–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, Freund TF (2004) Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci 20:441–458. [DOI] [PubMed] [Google Scholar]
- Hansson AC, Cippitelli A, Sommer WH, Ciccocioppo R, Heilig M (2007) Region-specific down-regulation of Crhr1 gene expression in alcohol-preferring msP rats following ad lib access to alcohol. Addict Biol 12:30–34. [DOI] [PubMed] [Google Scholar]
- Hasanein P, Sharifi M (2015) GABA(A) receptors in the central amygdala are involved in memory retention deficits induced by cannabinoids in rats. Pharmacol Biochem Behav 138:26–31. [DOI] [PubMed] [Google Scholar]
- Hasin DS, Grant BF (2002) Major depression in 6050 former drinkers: association with past alcohol dependence. Arch Gen Psychiatry 59:794–800. [DOI] [PubMed] [Google Scholar]
- Heilig M, Egli M, Crabbe JC, Becker HC (2010) Acute withdrawal, protracted abstinence and negative affect in alcoholism: are they linked? Addict Biol 15:169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann H, Lutz B (2005) Coexpression of the cannabinoid receptor type 1 with the corticotropin-releasing hormone receptor type 1 in distinct regions of the adult mouse forebrain. Neurosci Lett 375:13–18. [DOI] [PubMed] [Google Scholar]
- Hill MN, Campolongo P, Yehuda R, Patel S (2018) Integrating Endocannabinoid Signaling and Cannabinoids into the Biology and Treatment of Posttraumatic Stress Disorder. Neuropsychopharmacology 43:80–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Gorzalka BB (2006) Increased sensitivity to restraint stress and novelty-induced emotionality following long-term, high dose cannabinoid exposure. Psychoneuroendocrinology 31:526–536. [DOI] [PubMed] [Google Scholar]
- Hirvonen J, Zanotti-Fregonara P, Umhau JC, George DT, Rallis-Frutos D, Lyoo CH, Li CT, Hines CS, Sun H, Terry GE, Morse C, Zoghbi SS, Pike VW, Innis RB, Heilig M (2013) Reduced cannabinoid CB1 receptor binding in alcohol dependence measured with positron emission tomography. Mol Psychiatry 18:916–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holleran KM, Wilson HH, Fetterly TL, Bluett RJ, Centanni SW, Gilfarb RA, Rocco LE, Patel S, Winder DG (2016) Ketamine and MAG Lipase Inhibitor-Dependent Reversal of Evolving Depressive-Like Behavior During Forced Abstinence From Alcohol Drinking. Neuropsychopharmacology 41:2062–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou WH, Kuo N, Fang GW, Huang HS, Wu KP, Zimmer A, Cheng JK, Lien CC (2016) Wiring Specificity and Synaptic Diversity in the Mouse Lateral Central Amygdala. J Neurosci 36:4549–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG (2002) International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 54:161–202. [DOI] [PubMed] [Google Scholar]
- Hruba L, Seillier A, Zaki A, Cravatt BF, Lichtman AH, Giuffrida A, McMahon LR (2015) Simultaneous inhibition of fatty acid amide hydrolase and monoacylglycerol lipase shares discriminative stimulus effects with Delta9-tetrahydrocannabinol in mice. J Pharmacol Exp Ther 353:261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins JP, Smart TS, Langman S, Taylor L, Young T (2012) An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 153:1837–1846. [DOI] [PubMed] [Google Scholar]
- Ignatowska-Jankowska BM, Ghosh S, Crowe MS, Kinsey SG, Niphakis MJ, Abdullah RA, Tao Q, ST ON, Walentiny DM, Wiley JL, Cravatt BF, Lichtman AH (2014) In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: antinociceptive activity without cannabimimetic side effects. Br J Pharmacol 171:1392–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janak PH, Tye KM (2015) From circuits to behaviour in the amygdala. Nature 517:284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamprath K, Romo-Parra H, Haring M, Gaburro S, Doengi M, Lutz B, Pape HC (2011) Short-term adaptation of conditioned fear responses through endocannabinoid signaling in the central amygdala. Neuropsychopharmacology 36:652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Freund TF (2008) Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat Med 14:923–930. [DOI] [PubMed] [Google Scholar]
- Katona I, Freund TF (2012) Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci 35:529–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF (1999) Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci 19:4544–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelm MK, Criswell HE, Breese GR (2008) The role of protein kinase A in the ethanol-induced increase in spontaneous GABA release onto cerebellar Purkinje neurons. J Neurophysiol 100:3417–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbrat A, Ferre JC, Fillatre P, Ronziere T, Vannier S, Carsin-Nicol B, Lavoue S, Verin M, Gauvrit JY, Le Tulzo Y, Edan G (2016) Acute Neurologic Disorder from an Inhibitor of Fatty Acid Amide Hydrolase. N Engl J Med 375:1717–1725. [DOI] [PubMed] [Google Scholar]
- Kirson D, Oleata CS, Parsons LH, Ciccocioppo R, Roberto M (2018) CB1 and ethanol effects on glutamatergic transmission in the central amygdala of male and female msP and Wistar rats. Addict Biol 23:676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF (2003) Neuroadaptive mechanisms of addiction: studies on the extended amygdala. Eur Neuropsychopharmacol 13:442–452. [DOI] [PubMed] [Google Scholar]
- Koob GF, Roberts AJ, Schulteis G, Parsons LH, Heyser CJ, Hyytia P, Merlo-Pich E, Weiss F (1998) Neurocircuitry targets in ethanol reward and dependence. Alcohol Clin Exp Res 22:3–9. [PubMed] [Google Scholar]
- Koob GF, Schulkin J (2018) Addiction and stress: An allostatic view. Neurosci Biobehav Rev. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29:717–727. [DOI] [PubMed] [Google Scholar]
- Lanciego JL, Barroso-Chinea P, Rico AJ, Conte-Perales L, Callen L, Roda E, Gomez-Bautista V, Lopez IP, Lluis C, Labandeira-Garcia JL, Franco R (2011) Expression of the mRNA coding the cannabinoid receptor 2 in the pallidal complex of Macaca fascicularis. J Psychopharmacol 25:97–104. [DOI] [PubMed] [Google Scholar]
- Lange MD, Daldrup T, Remmers F, Szkudlarek HJ, Lesting J, Guggenhuber S, Ruehle S, Jungling K, Seidenbecher T, Lutz B, Pape HC (2017) Cannabinoid CB1 receptors in distinct circuits of the extended amygdala determine fear responsiveness to unpredictable threat. Mol Psychiatry 22:1422–1430. [DOI] [PubMed] [Google Scholar]
- Lim J, Igarashi M, Jung KM, Butini S, Campiani G, Piomelli D (2016) Endocannabinoid Modulation of Predator Stress-Induced Long-Term Anxiety in Rats. Neuropsychopharmacology 41:1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litt MD, Cooney NL, Kadden RM, Gaupp L (1990) Reactivity to alcohol cues and induced moods in alcoholics. Addict Behav 15:137–146. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang L, Harvey-White J, Huang BX, Kim HY, Luquet S, Palmiter RD, Krystal G, Rai R, Mahadevan A, Razdan RK, Kunos G (2008) Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology 54:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QR, Pan CH, Hishimoto A, Li CY, Xi ZX, Llorente-Berzal A, Viveros MP, Ishiguro H, Arinami T, Onaivi ES, Uhl GR (2009) Species differences in cannabinoid receptor 2 (CNR2 gene): identification of novel human and rodent CB2 isoforms, differential tissue expression and regulation by cannabinoid receptor ligands. Genes Brain Behav 8:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF (2009) Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci U S A 106:20270–20275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HC, Mackie K (2016) An Introduction to the Endogenous Cannabinoid System. Biol Psychiatry 79:516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz B, Marsicano G, Maldonado R, Hillard CJ (2015) The endocannabinoid system in guarding against fear, anxiety and stress. Nat Rev Neurosci 16:705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K (2005) Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb Exp Pharmacol:299–325. [DOI] [PubMed] [Google Scholar]
- Marco EM, Garcia-Gutierrez MS, Bermudez-Silva FJ, Moreira FA, Guimaraes F, Manzanares J, Viveros MP (2011) Endocannabinoid system and psychiatry: in search of a neurobiological basis for detrimental and potential therapeutic effects. Front Behav Neurosci 5:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Lutz B (1999) Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci 11:4213–4225. [DOI] [PubMed] [Google Scholar]
- Martin-Fernandez M, Jamison S, Robin LM, Zhao Z, Martin ED, Aguilar J, Benneyworth MA, Marsicano G, Araque A (2017) Synapse-specific astrocyte gating of amygdala-related behavior. Nat Neurosci 20:1540–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason BJ, Light JM, Williams LD, Drobes DJ (2009) Proof-of-concept human laboratory study for protracted abstinence in alcohol dependence: effects of gabapentin. Addict Biol 14:73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massi L, Elezgarai I, Puente N, Reguero L, Grandes P, Manzoni OJ, Georges F (2008) Cannabinoid receptors in the bed nucleus of the stria terminalis control cortical excitation of midbrain dopamine cells in vivo. J Neurosci 28:10496–10508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew RJ, Claghorn JL, Largen J (1979) Craving for alcohol in sober alcoholics. Am J Psychiatry 136:603–606. [PubMed] [Google Scholar]
- Matsuda LA, Bonner TI, Lolait SJ (1993) Localization of cannabinoid receptor mRNA in rat brain. J Comp Neurol 327:535–550. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–564. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F (2001) Localization of the CB1 type cannabinoid receptor in the rat basolateral amygdala: high concentrations in a subpopulation of cholecystokinin-containing interneurons. Neuroscience 107:641–652. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, et al. (1995) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50:83–90. [DOI] [PubMed] [Google Scholar]
- Mitrirattanakul S, Lopez-Valdes HE, Liang J, Matsuka Y, Mackie K, Faull KF, Spigelman I (2007) Bidirectional alterations of hippocampal cannabinoid 1 receptors and their endogenous ligands in a rat model of alcohol withdrawal and dependence. Alcohol Clin Exp Res 31:855–867. [DOI] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–65. [DOI] [PubMed] [Google Scholar]
- Murataeva N, Straiker A, Mackie K (2014) Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br J Pharmacol 171:1379–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natividad LA, Buczynski MW, Herman MA, Kirson D, Oleata CS, Irimia C, Polis I, Ciccocioppo R, Roberto M, Parsons LH (2017) Constitutive Increases in Amygdalar Corticotropin-Releasing Factor and Fatty Acid Amide Hydrolase Drive an Anxious Phenotype. Biol Psychiatry 82:500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu A, Foldy C, Soltesz I (2007) Postsynaptic origin of CB1-dependent tonic inhibition of GABA release at cholecystokinin-positive basket cell to pyramidal cell synapses in the CA1 region of the rat hippocampus. J Physiol 578:233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsom RJ, Osterlund C, Masini CV, Day HE, Spencer RL, Campeau S (2012) Cannabinoid receptor type 1 antagonism significantly modulates basal and loud noise induced neural and hypothalamic-pituitary-adrenal axis responses in male Sprague-Dawley rats. Neuroscience 204:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes EV, Levin FR (2004) Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA 291:1887–1896. [DOI] [PubMed] [Google Scholar]
- O’Sullivan SE (2007) Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol 152:576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M (2001) Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 29:729–738. [DOI] [PubMed] [Google Scholar]
- Oliveira LM, Bermudez MB, Macedo MJA, Passos IC (2018) Comorbid social anxiety disorder in patients with alcohol use disorder: A systematic review. J Psychiatr Res 106:8–14. [DOI] [PubMed] [Google Scholar]
- Patel S, Hill MN, Cheer JF, Wotjak CT, Holmes A (2017) The endocannabinoid system as a target for novel anxiolytic drugs. Neurosci Biobehav Rev 76:56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Kingsley PJ, Mackie K, Marnett LJ, Winder DG (2009) Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signaling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology 34:2699–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinati HM (2004) Antidepressant treatment of co-occurring depression and alcohol dependence. Biol Psychiatry 56:785–792. [DOI] [PubMed] [Google Scholar]
- Piomelli D (2003) The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 4:873–884. [DOI] [PubMed] [Google Scholar]
- Pomrenze MB, Millan EZ, Hopf FW, Keiflin R, Maiya R, Blasio A, Dadgar J, Kharazia V, De Guglielmo G, Crawford E, Janak PH, George O, Rice KC, Messing RO (2015) A Transgenic Rat for Investigating the Anatomy and Function of Corticotrophin Releasing Factor Circuits. Front Neurosci 9:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente N, Cui Y, Lassalle O, Lafourcade M, Georges F, Venance L, Grandes P, Manzoni OJ (2011) Polymodal activation of the endocannabinoid system in the extended amygdala. Nat Neurosci 14:1542–1547. [DOI] [PubMed] [Google Scholar]
- Puente N, Elezgarai I, Lafourcade M, Reguero L, Marsicano G, Georges F, Manzoni OJ, Grandes P (2010) Localization and function of the cannabinoid CB1 receptor in the anterolateral bed nucleus of the stria terminalis. PLoS One 5:e8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramikie TS, Nyilas R, Bluett RJ, Gamble-George JC, Hartley ND, Mackie K, Watanabe M, Katona I, Patel S (2014) Multiple mechanistically distinct modes of endocannabinoid mobilization at central amygdala glutamatergic synapses. Neuron 81:1111–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramikie TS, Patel S (2012) Endocannabinoid signaling in the amygdala: anatomy, synaptic signaling, behavior, and adaptations to stress. Neuroscience 204:38–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Cruz M, Bajo M, Siggins GR, Parsons LH, Schweitzer P (2010) The endocannabinoid system tonically regulates inhibitory transmission and depresses the effect of ethanol in central amygdala. Neuropsychopharmacology 35:1962–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR (2003) Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc Natl Acad Sci U S A 100:2053–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano A, Pavon FJ, Buczynski MW, Schlosburg J, Natividad LA, Polis IY, Stouffer DG, Zorrilla EP, Roberto M, Cravatt BF, Martin-Fardon R, Rodriguez de Fonseca F, Parsons LH (2018) Deficient endocannabinoid signaling in the central amygdala contributes to alcohol dependence-related anxiety-like behavior and excessive alcohol intake. Neuropsychopharmacology 43:1840–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano A, Rivera P, Pavon FJ, Decara J, Suarez J, Rodriguez de Fonseca F, Parsons LH (2012) Differential effects of single versus repeated alcohol withdrawal on the expression of endocannabinoid system-related genes in the rat amygdala. Alcohol Clin Exp Res 36:984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonesy BC, Bluett RJ, Ramikie TS, Baldi R, Hermanson DJ, Kingsley PJ, Marnett LJ, Winder DG, Colbran RJ, Patel S (2014) Genetic disruption of 2-arachidonoylglycerol synthesis reveals a key role for endocannabinoid signaling in anxiety modulation. Cell Rep 9:1644–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopponi S, Fotio Y, Domi A, Borruto AM, Natividad L, Roberto M, Ciccocioppo R, Cannella N (2018) Inhibition of fatty acid amide hydrolase in the central amygdala alleviates co-morbid expression of innate anxiety and excessive alcohol intake. Addict Biol 23:1223–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K (1995) 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun 215:89–97. [DOI] [PubMed] [Google Scholar]
- Thomas EA, Cravatt BF, Danielson PE, Gilula NB, Sutcliffe JG (1997) Fatty acid amide hydrolase, the degradative enzyme for anandamide and oleamide, has selective distribution in neurons within the rat central nervous system. J Neurosci Res 50:1047–1052. [DOI] [PubMed] [Google Scholar]
- Toth A, Blumberg PM, Boczan J (2009) Anandamide and the vanilloid receptor (TRPV1). Vitam Horm 81:389–419. [DOI] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM (1998) Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83:393–411. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Herman JP (2009) Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci 10:397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Esbroeck ACM, Janssen APA, Cognetta AB 3rd, Ogasawara D, Shpak G, van der Kroeg M, Kantae V, Baggelaar MP, de Vrij FMS, Deng H, Allara M, Fezza F, Lin Z, van der Wel T, Soethoudt M, Mock ED, den Dulk H, Baak IL, Florea BI, Hendriks G, De Petrocellis L, Overkleeft HS, Hankemeier T, De Zeeuw CI, Di Marzo V, Maccarrone M, Cravatt BF, Kushner SA, van der Stelt M (2017) Activity-based protein profiling reveals off-target proteins of the FAAH inhibitor BIA 10–2474. Science 356:1084–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varodayan FP, Soni N, Bajo M, Luu G, Madamba SG, Schweitzer P, Parsons LH, Roberto M (2016) Chronic ethanol exposure decreases CB1 receptor function at GABAergic synapses in the rat central amygdala. Addict Biol 21:788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinod KY, Kassir SA, Hungund BL, Cooper TB, Mann JJ, Arango V (2010) Selective alterations of the CB1 receptors and the fatty acid amide hydrolase in the ventral striatum of alcoholics and suicides. J Psychiatr Res 44:591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinod KY, Maccioni P, Garcia-Gutierrez MS, Femenia T, Xie S, Carai MA, Manzanares J, Cooper TB, Hungund BL, Colombo G (2012) Innate difference in the endocannabinoid signaling and its modulation by alcohol consumption in alcohol-preferring sP rats. Addict Biol 17:62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinod KY, Yalamanchili R, Xie S, Cooper TB, Hungund BL (2006) Effect of chronic ethanol exposure and its withdrawal on the endocannabinoid system. Neurochem Int 49:619–625. [DOI] [PubMed] [Google Scholar]
- Wilkerson JL, Niphakis MJ, Grim TW, Mustafa MA, Abdullah RA, Poklis JL, Dewey WL, Akbarali H, Banks ML, Wise LE, Cravatt BF, Lichtman AH (2016) The Selective Monoacylglycerol Lipase Inhibitor MJN110 Produces Opioid-Sparing Effects in a Mouse Neuropathic Pain Model. J Pharmacol Exp Ther 357:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willinger U, Lenzinger E, Hornik K, Fischer G, Schonbeck G, Aschauer HN, Meszaros K, European fluvoxamine in alcoholism study g (2002) Anxiety as a predictor of relapse in detoxified alcohol-dependent patients. Alcohol Alcohol 37:609–612. [DOI] [PubMed] [Google Scholar]
- Wise LE, Long KA, Abdullah RA, Long JZ, Cravatt BF, Lichtman AH (2012) Dual fatty acid amide hydrolase and monoacylglycerol lipase blockade produces THC-like Morris water maze deficits in mice. ACS Chem Neurosci 3:369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Uchigashima M, Yamasaki M, Katona I, Yamazaki M, Sakimura K, Kano M, Yoshioka M, Watanabe M (2011) Unique inhibitory synapse with particularly rich endocannabinoid signaling machinery on pyramidal neurons in basal amygdaloid nucleus. Proc Natl Acad Sci U S A 108:3059–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrindast MR, Ghiasvand M, Rezayof A, Ahmadi S (2012) The amnesic effect of intra-central amygdala administration of a cannabinoid CB1 receptor agonist, WIN55,212–2, is mediated by a beta-1 noradrenergic system in rat. Neuroscience 212:77–85. [DOI] [PubMed] [Google Scholar]
- Zarrindast MR, Sarahroodi S, Arzi A, Khodayar MJ, Taheri-Shalmani S, Rezayof A (2008) Cannabinoid CB1 receptors of the rat central amygdala mediate anxiety-like behavior: interaction with the opioid system. Behav Pharmacol 19:716–723. [DOI] [PubMed] [Google Scholar]
- Zhong P, Wang W, Pan B, Liu X, Zhang Z, Long JZ, Zhang HT, Cravatt BF, Liu QS (2014) Monoacylglycerol lipase inhibition blocks chronic stress-induced depressive-like behaviors via activation of mTOR signaling. Neuropsychopharmacology 39:1763–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]