

Abstract
Background:
The hippocampus is particularly vulnerable to the teratogenic effects of prenatal ethanol exposure (PNEE), and hippocampal structural and functional deficits are thought to contribute to the learning and memory deficits that are a hallmark feature of fetal alcohol spectrum disorders (FASDs).
Method:
Sprague-Dawley dams were exposed to a liquid diet that contained EtOH (35.5% EtOH-derived calories) throughout gestation, and then PNEE juvenile (P21-28) male and female offspring were used for in vitro electrophysiological recordings. We examined long-term potentiation (LTP), long-term depression (LTD) and depotentiation in the medial perforant path (MPP) input to the dentate gyrus (DG) to determine the impact of PNEE on the dynamic range of bidirectional synaptic plasticity in both sexes.
Results:
PNEE reduced the responsiveness of the DGs of male but not in female offspring, and this effect was no longer apparent when GABAergic signalling was inhibited. There was also a sex-specific LTD impairment in males, but increasing the duration of the conditioning stimulus could overcome this deficit. The magnitude of LTP was also reduced, but in both sexes following PNEE. This appears to be an increase in the threshold for induction, not in capacity, as the level of LTP induced in PNEE animals was increased to control-levels when additional conditioning stimuli were administered.
Conclusions:
These data are the first to describe, in a single study, the impact of PNEE on the dynamic range of bidirectional synaptic plasticity in the juvenile DG in both males and in females. The data suggest that PNEE increases the threshold for LTP in the DG in both sexes, but produces a sex-specific increase in the threshold for LTD in males These alterations reduce the dynamic range for synaptic plasticity in both sexes.
Introduction
Fetal alcohol spectrum disorders (FASDs) result from the consumption of alcohol during pregnancy and are the most common preventable cause of mental disability in the modern world. The rate of FASD in North America has been estimated at 1-5% although this number may underestimate the true prevalence (May et al., 2018; Popova et al., 2017). Surveys indicate that approximately 50% of women aged 15-44 consume alcohol, with reports of ~23% binge alcohol consumption, and 48% of pregnancies in this cohort are reported as unintended (Ahrnsbrak et al., 2016; Moos et al., 2008; Singh et al., 2010). Thus, alcohol consumption can occur when individuals are unaware of their pregnancy, and any alcohol exposure during a period of rapid fetal development has the potential to lead to persistent morphological and physiological impairments. These can include facial dysmorphology, as well as difficulties in social, emotional, and spatial processing (Guerri et al., 2009; Hannigan and Riley, 1988; Mattson and Riley, 1998; Patten et al., 2014; Riley et al., 2011; Riley and McGee, 2005). The presentation of FASD can be exceedingly complex as they are not only influenced by the pattern and amount of alcohol exposure, but also by maternal health and diet (Benz et al., 2009; May and Gossage, 2011).
Difficulties with learning and remembering are a key diagnostic feature of FASDs (Cook et al., 2016; Mattson and Riley, 1998), and this not only impacts academic performance, but can also lead to downstream difficulties in life. Learning and memory impairments have been replicated in various animal models of PNEE and have been related to teratogen-induced damage to the hippocampal region of the brain (Berman and Hannigan, 2000; Patten et al., 2014). Indeed there are reports of reduced cell numbers, dendritic atrophy, impairments in synaptic plasticity and altered neurogenesis in this area following PNEE (Gil-Mohapel et al., 2010). As such, the hippocampal network is an area of intense investigation in pre-clinical studies of FASDs, and restoration of its healthy structure and function by therapeutic intervention. Synaptic plasticity is the main biological candidate for a cellular mechanism underlying behavioural learning and memory processes. Long-term potentiation (LTP) is a form of synaptic plasticity where synaptic connections to become stronger following the application of high frequency stimuli (HFS). Our previous work has provided evidence for sex differences in synaptic plasticity in the dentate gyrus (DG) subregion of the rat hippocampus. In both adolescent rats (Titterness and Christie, 2012), and adults (Patten et al., 2013c, 2013b), male PNEE offspring have reliably shown deficits in LTP in vivo. Conversely, changes have only been observed in adolescent females, where paradoxically, LTP has been shown to be enhanced (Patten et al., 2013d, 2013a; Titterness and Christie, 2012). Long-term changes in synaptic plasticity can also involve a weakening of synaptic efficacy, and this is referred to as long-term depression (LTD). Currently there is a paucity of data on how PNEE impacts LTD, which is a form of synaptic plasticity more readily observed using in vitro electrophysiology than in vivo (Pierrefiche and Olivier, 2017; Pinar et al., 2017). In the current study we use in vitro electrophysiology to study bidirectional synaptic plasticity (both LTP and LTD) in juvenile rats (Rattus norvegicus) following PNEE. We show for the first time that PNEE not only impacts LTD, but that it more significantly reduces bidirectional plasticity in the dentate gyrus of males than females.
METHODS
Animals & Breeding
All procedures were performed in accordance with the University of Victoria Institutional Animal Care Committee following the standards set by the Canadian Council for Animal Care.
Adult male and female Sprague-Dawley rats (Rattus norvegicus) were obtained from Charles River Laboratories (Quebec, Canada). Animals were housed in standard cages in colony rooms kept at 21ºC and maintained on a 12-hour light-dark cycle. All animals had as libitum access to standard solid rat chow and drinking water except when being administered Ethanol (EtOH) liquid diets. Nulliparous adult female rats were paired with adult male rats until sperm was present in a daily vaginal smear. Upon detection of sperm the female was immediately re-housed under standard, non-enriched conditions and randomly assigned to one of three prenatal diet conditions: PNEE, Pair-fed (PF) or Control. The date of sperm detection was designated as Gestational Day (GD) 1. All dams were weighed regularly on GD1, 7, 14 and 21. All animals had ad libitum access to drinking water regardless of prenatal diet condition.
Ethanol Diet Condition:
Animals received ad libitum access to a nutritionally fortified liquid diet (Weinberg/Keiver high-protein liquid diet-experimental, Dyets Inc., No. 710324) containing 35.5% ethanol-derived calories (EDC). Dams were gradually introduced to this diet from GD1-3 and consumed the EtOH liquid diet until GD21, or the day before birth. Fresh diet was provided approximately 2 hours prior to the onset of the dark phase in the light cycle throughout this period. Following this animals were returned to a solid control chow diet throughout the remainder of parturition.
Pair-fed Diet Condition:
Controlled access to a nutritionally fortified liquid diet (Weinberg/Keiver high protein liquid diet-control, no. 710109, Dyets Inc., Bethlehem, PA, USA) identical to the ethanol diet with the EDC isocalorically substituted with maltose-dextrin. In order to control for the stress associated with the consumption of a liquid diet in the ethanol group, dams assigned to the pair-fed (PF) condition received the same amount of food in g/kg/day as their weight-matched ethanol-exposed dams. PF dams were supplied with the pair-fed liquid diet from GD1-21, where on GD22, the liquid diet was replaced with standard solid rat chow which was provided throughout parturition. The PF diet group is a common control for the liquid diet model of PNEE, although the irregular feeding pattern of this group can lead to undue stress effects on the developing offspring and may lead to effects that make these animals an imperfect control. The results from these offspring are compared against the ad libitum control group and are included as supplementary data as supplementary table 2 and supplementary figure 1 and 2.
Control Diet Condition:
Animals had ad libitum access to standard solid rat chow throughout the gestational and parturition period.
Dams gave birth on GD22 (date of birth: postnatal day (P) 0) and were left undisturbed for 24 hours. Litters were then culled to 12 (6 male and 6 female, where possible) on P1 and dam and pup weight were recorded regularly throughout postnatal development at P1, 4, 9, 13, 17 and 21. On P21 pups were weaned into same-sex cages or 2-3 littermates until experimental use. Information about diets and electrophysiological supplies can be found in supplementary table 1.
In vitro electrophysiology
Male and female offspring (P21-28) from each prenatal diet condition were used to study bidirectional DG synaptic plasticity. Animals were deeply anesthetized with inhaled isoflurane and rapidly decapitated. Brains were quickly removed and cut into transverse slices (400μm) using a vibratome in cold artificial cerebrospinal fluid (aCSF; 125mM NaCl, 2.5mM KCl, 1.25mM NaH2PO4, 25mM NaHCO3, 2mM CaCl2, 1.3mM MgCl2 and 1.4mM Dextrose) equilibrated with carbogen (95% Oxygen, 5% Carbon dioxide). Slices were then left to recover in warmed (32 ± 0.5 ºC), carbogenated aCSF for a minimum of 1hr prior to being transferred to the recording chamber for experimentation.
In recording chambers slices were continuously perfused with fresh, cabogenated and warmed (30 ± 0.5 ºC) aCSF and visualized with an upright microscope (Olympus BX5OWI). Extracellular field excitatory postsynaptic potentials (fEPSPs) were evoked by lowering a concentric bipolar stimulating electrode (FHC, Bowdoinham, ME) in the medial perforant path (MPP) fibres located in the middle third of the molecular layer in the DG. The resulting fEPSPs were recorded by an electrode bathed in aCSF and housed in a glass micropipette placed in the dendritic arbour of the DG granule cells. fEPSPs we collected using an Axon Multiclamp 700B amplifier, digitized by an Axon Digidata 1440 and recorded using Clampex 10.5 software (Molecular Devices, CA).The electrode placement was optimized on a slice-by-slice basis using test pulses delivered every 5s. A minimum fEPSP amplitude of 0.7 mV was required for all slices. The stimulation intensity was reduced to 50% of that required to obtain the maximal response for all LTP experiments, and 70% of the maximal response for LTD experiments. For LTP experiments 10μM bicuculline methiodide (BIC; MJS Biolynx) was also included in the aCSF in order to block GABAA receptors. The fEPSPs were evoked by stimulation pulses every 15s to establish a baseline level of responsiveness in the immediate slope of the fEPSP. Stability in the fEPSP slope (< 10% variation) was required for at least 20 minutes before conditioning stimuli were applied.
Measurement of Synaptic Efficacy
In order to determine whether PNEE affects presynaptic neurotransmitter release or fEPSP responsiveness, paired-pulse (PP) and input-output (I/O) experiments were conducted respectively. For paired-pulse experiments, two pulses were delivered in rapid succession (50ms) five times (15s inter-pair-interval). The average slope of the second fEPSP was divided by that of the first fEPSP slope to generate a ratio of the responses to the two pulses. I/O experiments consisted of measurements in the immediate fEPSP slopes in response to increasing stimulation intensity in the form of increasing pulse widths (from 30-300μs; 30s inter-stimulus intervals).
Long-Term Potentiation
LTP-inducing high-frequency stimulation (HFS) was delivered to the slice (4 trains of 50 pulses at 100Hz; 30s inter-train intervals; 0.24ms pulse width) and the fEPSP response was recorded for 60 minutes following HFS. The magnitude of post-tetanic potentiation (PTP) was evaluated as the average fEPSP slope for the first minute following HFS and the magnitude of LTP was determined as the average fEPSP slope for the last 5 minutes of the post-conditioning recording (55-60 min). In some experiments, LTP was saturated by delivering the HFS protocol twice.
Long-Term Depression
LTD was induced using low-frequency stimulation (LFS; 900 × 1Hz; 0.24ms pulse width) and the fEPSP response was then recorded for 60 minutes. The magnitude of short-term depression (STD) was evaluated as the average fEPSP slope for the first minute following LFS and the magnitude of LTD was determined as the average fEPSP slope for the last 5 minutes of the post-conditioning recording (55-60 min). For LTD saturation experiments the LFS was administered twice so that a total of 1800 pulses was given at 1 Hz.
Depotentiation
PNEE has been documented to affect the magnitude of depotentiation of previously induced LTP in other areas of the brain (An et al., 2013; An and Zhang, 2013; Kervern et al., 2015), however depotentiation has not been studied in the DG. For these experiments, LTP was induced as described above, and then LFS (900 × 1Hz) was delivered 60 min following the induction of LTP. The magnitude of depotentiation was calculated as the difference between the average fEPSP slope at 55-60 min post-HFS and the slope recorded 25-30 min following application of the LFS.
Data & Statistical Analyses
Electrophysiological data were recorded with a Multiclamp 700B (Molecular Devices) and acquired and analyzed with pClamp 10.5 and Clampfit 10.5 (Axon Instruments) respectively. I/O curves were analyzed by a repeated measures 2-way ANOVA followed by Bonferroni multiple comparisons. For all recordings, the average fEPSP slope is depicted graphically for each minute (average of 4 responses). Student’s t-tests were used to statistically compare magnitudes of PTP, STD, LTP, LTD and depotentiation by control and PNEE groups within each sex. All data are represented as the mean ± the standard error of the mean (SEM). Effect sizes are represented as Cohen’s d.
Results
Metrics for Dams and Litters
A total of 10 litters of Control offspring and 7 litters of Ethanol-exposed offspring were used for this study. Dams in the ethanol diet condition consumed on average 66.25g ± 3.75g per day of the liquid diet, which equated to on average 13.57 ± 0.39 g/kg/day ethanol consumed. The ethanol consumption values ranged between 11.58 g/kg/day to a maximum of 14.96 g/kg/day. This pattern and type of diet administration typically achieves blood alcohol concentrations of between 80-180mg/dl (Christie et al., 2005; Patten et al., 2013c, 2013b, 2016; Uban et al., 2010).
As is shown in Table 1, there was a transient sex specific reduction in the body weight of male animals at P1 (p = 0.0066) and P3 (p = 0.014). This weight reduction was not observed in female offspring at any postnatal day.
Table 1. Prenatal ethanol exposure transiently reduces body weight in males in the early postnatal period.
Average offspring weight gain separated by sex starting on the day after birth (P1) to weaning an experimental use (P21). On P1 litters were culled to 12 pups consisting of 6 pups of each sex where possible. A two-tailed student’s t-test was used to evaluate the effect of prenatal diet treatment within sex and age. PNEE had no effect on offspring weight at any age in female pups, however it significantly reduced male pup weights at P1 and P3 but at no other age. The average weights are expressed below ± the SEM. Statistical significance was considered when p < 0.05 and is indicated using distinct symbols (* or Δ), indicating statistical significance from the values indicated with the identical symbol within this table.
| Postnatal day (P) |
Average Control Pup Weight (g) |
Average Ethanol Pup Weight (g) |
||
|---|---|---|---|---|
| Male | Female | Male | Female | |
| P1 | 8.13 ± 0.16* | 6.93 ± 0.49 | 7.20 ± 0.24* | 6.58 ± 0.29 |
| P3 | 10.69 ± 0.36Δ | 8.94 ± 0.63 | 9.29 ± 0.35Δ | 8.79 ± 0.37 |
| P7 | 16.83 ± 0.67 | 16.45 ± 0.55 | 15.48 ± 0.83 | 14.15 ± 0.91 |
| P14 | 29.71 ± 2.57 | 30.85 ± 1.39 | 29.70 ± 1.37 | 28.18 ± 1.30 |
| P21 | 55.52 ± 3.72 | 48.69 ± 5.31 | 48.34 ± 2.53 | 44.43 ± 2.41 |
Measures of Synaptic Efficacy
PNEE had no effect on the PP ratio in males (PNEE: 0.997 ± 0.0646; Controls: 1.027 ± 0.0315; nControl = 12 slices, 5 animals, 2 litters; nPNEE = 11 slices, 5 animals, 3 litters; p = 0.568) or females (PNEE: 1.044 ± 0.0271; Controls: 1.112 ± 0.0206; nControl = 14 slices, 6 animals, 2 litters; nPNEE = 9 slices, 7 animals, 3 litters; p = 0.0647) in normal aCSF (Figure 1). We also examined the PP ratio in aCSF that contained the GABAA receptor antagonist BIC (10mM) and again found there was no difference in either males (PNEE: 1.199 ± 0.0496; Controls: 1.238 ± 0.0470; nControl = 8 slices, 5 animals, 3 litters; nPNEE = 8 slices, 5 animals, 3 litters; p = 0.572) or females (PNEE: 1.265 ± 0.0512; Control: 1.193 ± 0.0562; nControl = 7 slices, 5 animals, 2 litters; nPNEE = 11 slices, 5 animals, 3 litters; p = 0.367).
Figure 1. Paired pulse plasticity is unaffected by prenatal ethanol exposure.

The ratios of the slopes of the second pulse relative to the slopes of the first pulses are unaffected by PNEE in females (A1) nor in males (A2) nor when GABAARs are blocked by BIC as in LTP recordings in females (B1) or males (B2). Changes to paired-pulse plasticity can provide insight on presynaptic neurotransmitter release probability from the MPP fibres. Bars represent average paired pulse ratios and error bars represent standard error of the mean (SEM).
I/O curves were constructed by comparing the fEPSP slope relative to the pulse width of stimulation (0-300 μs). For all groups, there was a main effect of pulse width where increasing pulse width led to increased fEPSP slopes. As is shown in Figure 2, PNEE had an impact on the I/O curves in males when regular aCSF was used (Fig 2. A2) but not when BIC was used (Fig 2. B2). In normal aCSF for male slices, there was a significant main effect of prenatal diet (F(1,16) = 8.219, p = 0.0112; nControl = 12 slices, 5 animals, 2 litters; nPNEE = 11 slices, 5 animals, 3 litters). In normal aCSF, fEPSP slopes were reduced at pulse widths of 120 μs (p = 0.0500), 150 μs (p = 0.0347), 180 μs (p = 0.0499), 210 μs (p = 0.0226), 240 μs (p = 0.0321), 270 μs (p = 0.0463) and 300 ms μs (p = 0.0529) pulse widths. There were no main effect of prenatal diet in regular aCSF in females (F(1,21) = 0.2159, p = 0.6470; nControl = 14 slices, 6 animals, 2 litters; nPNEE = 9 slices, 7 animals, 3 litters) or in either males (F(1,14) = 0.0002206, p = 0.9884; nControl = 8 slices, 5 animals, 3 litters; nPNEE = 8 slices, 5 animals, 3 litters) or females (F(1,16) = 0.00001620, p = 0.9968; nControl = 7 slices, 5 animals, 2 litters; nPNEE = 11 slices, 5 animals, 3 litters) when BIC was present in the aCSF.
Figure 2. Prenatal ethanol exposure impairs postsynaptic responsiveness in males to increasing stimulation.

Postsynaptic fEPSP size to increasing pulse widths is reduced by PNEE in males (A2) but not in females (A1) when GABAA is not inhibited by bicuculline methiodide (BIC; 10μM) prior to LTD recordings. Under BIC conditions, the input-output curve is unaffected by PNEE in both females (B1) and males (B2). Points represent average fEPSP slopes by group and error bars represent the standard error of the mean (SEM). * represent p < 0.05 relative to control levels at the same pulse width.
Long-Term Potentiation
The magnitude of PTP (post-conditioning minute 0-1) immediately after 1xHFS was unchanged by PNEE in either sex however the magnitudes of LTP (post-conditioning minutes 55-60) were reduced in both females (LTP: 29.67 ± 7.66 %; n = 11 slices, 5 animals, 3 litters; p = 0.000756) and males (LTP: 34.89 ± 7.98 %; n = 8 slices, 5 animals, 3 litters; p = 0.0067) compared to same-sex controls (LTPFemale: 74.82 ± 7.53 %; n = 7 slices, 5 animals, 2 litters; LTPMale: 91.81 ± 17.18 %; n = 8 slices, 5 animals, 3 litters; Figure 3). The effect size for the reduction in the magnitude of LTP was found to be large in females (d = 1.92) and in males (d = 1.06).
Figure 3. The magnitude of long-term potentiation is reduced in both sexes by prenatal ethanol exposure.

(A) Average long-term potentiation (LTP) recordings from the beginning of baseline to the end of the post-conditioning recording. Dots represent the average percentage of change in the field excitatory postsynaptic potential (fEPSP) slope relative to baseline and the error bars represent the SEM. Representative traces for each group are indicated above with 1 indicating the average fEPSP at baseline and 2 indicating the average fEPSP at the end of the post-conditioning recording. (B) Short-term post-tetanic potentiation (PTP) is measured as the percentage of change in the fEPSP slope for the first minute immediately following the HFS and is unaffected by PNEE. (C) LTP was measured as the average percentage of change in the fEPSP slope relative to baseline for minutes 55-60 following delivery of the high-frequency stimulation (HFS; indicated as black arrow in A). Following PNEE the magnitude of LTP was reduced in both sexes by approximately 50%. Bars represent the average percentage of change in the fEPSP slope for PTP (B) and LTP (C) with points representing the average PTP and LTP for each individual slice in this dataset. Experimental n: LTPControl Female: 74.82 ± 7.53 %; n = 7 slices, 5 animals, 2 litters; LTPPNEE Female: 29.67 ± 7.66 %; n = 11 slices, 5 animals, 3 litters; LTPControl Male: 91.81 ± 17.18 %; n = 8 slices, 5 animals, 3 litters; LTPPNEEMale: 34.89 ± 7.98 %; n = 8 slices, 5 animals, 3 litters. * p < 0.05.
In order to assess whether the deficits in LTP could be a result of PNEE-induced changes in the threshold necessary for the induction of LTP, we delivered a second bout of HFS (2xHFS) in PNEE slices (Figure 4). The use of 2xHFS led to a significant increase in the magnitude of LTP in PNEE females (LTP2xHFS: 63.59 ± 7.92 %; n = 16 slices, 5 animals, 2 litters; p = 0.0051) and in males (LTP2xHFS: 94.76 ± 12.43 %; n = 13 slices, 7 animals, 3 litters; p = 0.0230) as compared to the magnitude of LTP induced by 1xHFS in these offspring. The effect size of the increase in the magnitude of LTP by 2xHFS was large in PNEE females (d = 2.17) and in PNEE males (d = 1.15). The magnitude of LTP induced by 2xHFS in PNEE offspring of both sexes did not differ from the magnitude of 1xHFS LTP in control females (p = 0.318) and males (p = 0.920).
Figure 4. Delivery of additional high-frequency stimuli overcomes the deficit in long-term potentiation following prenatal ethanol exposure in both sexes.

(A) Average long-term potentiation (LTP) recordings from the beginning of baseline to the end of the post-conditioning recording. Dots represent the average percentage of change in the field excitatory postsynaptic potential (fEPSP) slope relative to baseline and the error bars represent the SEM. The arrow indicates the delivery of 2 bouts of high-frequency stimulation (B) LTP was measured as the average percentage of change in the fEPSP slope relative to baseline for minutes 55-60 following delivery of the 2xHFS. The delivery of additional HFS restored the magnitude of LTP to control levels (as indicated by the dashed grey bar in B). Bars represent the average percentage of change in the fEPSP slope for and LTP with points representing the average LTP for each individual slice in this dataset. Experimental n: LTPEtOH Female 1xHFS: 29.67 ± 7.66 %; n = 11 slices, 5 animals, 3 litters; LTPEtOH Female 2xHFS: 63.59 ± 7.92 %; n = 16 slices, 5 animals, 2 litters; LTPEtOH Male 1xHFS: 34.89 ± 7.98 %; n = 8 slices, 5 animals, 3 litters; LTPEtOH Male 2xHFS: 94.76 ± 12.43 %; n = 13 slices, 7 animals, 3 litters. * p < 0.05.
Long-Term Depression
The magnitude of STD (post-conditioning minute 0-1) was unaffected by PNEE in either sex, however the magnitude of LTD (post-conditioning minutes 55-60) was reduced by PNEE in male offspring (LTDcontrol: −24.89 ± 4.76 %; n = 12 slices, 5 animals, 2 litters; LTDPNEE: −10.83 ± 3.33 %; n = 11 slices, 5 animals, 3 litters; p = 0.0257; Figure 5). The effect size of the reduced magnitude of LTD in PNEE males was large (d = 0.999). In contrast there was no effect of PNEE on LTD in females (LTDcontrol: −28.46 ± 3.56 %; n = 14 slices, 6 animals, 2 litters; LTDPNEE: −27.90 ± 5.46 %; n = 9 slices, 7 animals, 3 litters; p = 0.932). The prolonged LFS (1800 × 1Hz) did elicit greater magnitudes of LTD in PNEE males (LTD1800: −32.66 ± 6.86 %; n = 5 slices, 3 animals, 3 litters; Figure 6) as compared to the magnitude of LTD elicited by 900 × 1Hz in this same condition (p = 0.0288).
Figure 5. Prenatal ethanol exposed male offspring exhibit reduced long-term depression.

(A) Dots represent the average percentage of change in the field excitatory postsynaptic potential (fEPSP) slope relative to baseline and the error bars represent the SEM. Representative traces for each group are indicated above with 1 indicating the average fEPSP at baseline and 2 indicating the average fEPSP at the end of the post-conditioning recording. (B) Short-term depression (STD) is measured as the percentage of change in the fEPSP slope for the first minute immediately following the low-frequency stimulation (LFS; 900×1Hz) and is unaffected by PNEE. (C) LTD was measured as the average percentage of change in the fEPSP slope relative to baseline for minutes 55-60 following delivery of the LFS (indicated as black bar in A). The magnitude of LTD was reduced only in male offspring following PNEE. Bars represent the average percentage of change in the fEPSP slope for STD (B) and LTD (C) with points representing the average PTP and LTP for each individual slice in this dataset. Experimental n: LTDControl Female: −28.46 ± 3.56 %; n = 14 slices, 6 animals, 2 litters; LTDPNEE Female: −27.90 ± 5.46 %; n = 9 slices, 7 animals, 3 litters; LTDControl Male: −24.89 ± 4.76 %; n = 12 slices, 5 animals, 2 litters; LTDPNEE Male: −10.83 ± 3.33 %; n = 11 slices, 5 animals, 3 litters * p < 0.05.
Figure 6. Prolonged low-frequency stimulation induced long-term depression is unaffected by prenatal ethanol exposure.

(A) Average long-term depression (LTD) recordings from the beginning of baseline to the end of the post-conditioning recording. Dots represent the average percentage of change in the field excitatory postsynaptic potential (fEPSP) slope relative to baseline and the error bars represent the SEM. (B) LTD was measured as the average percentage of change in the fEPSP slope relative to baseline for minutes 55-60 following delivery of the LFS (1800×1Hz; indicated as black bar in A). The magnitude of LTD was reduced only in male offspring following PNEE. Bars represent the average percentage of change in the fEPSP slope for LTD (C) with points representing the average STD and LTD for each individual slice in this dataset. Experimental n: LTDControl Female: −36.64 ± 5.44 %; n = 14 slices, 6 animals, 4 litters; LTDPNEE Female: −28.83 ± 5.67 %; n = 7 slices, 5 animals, 3 litters; LTDControl Male: −36.82 ± 5.12 %; %; n = 8 slices, 4 animals, 2 litters; LTDPNEE Male: −32.66 ± 6.86 %; n = 5 slices, 3 animals, 3 litters. * p < 0.05.
Depotentiation
Depotentiation is the reversal of HFS-induced LTP by LFS. The magnitude of depotentiation was not found to affected by PNEE in females (Control: 45.19 ± 11.94 %; n = 7 slices, 6 animals, 2 litters; PNEE: 37.68 ± 8.81 %; n = 9 slices, 8 animals, 2 litters; p = 0.622; Figure 7) nor in males (Control: 56.39 ± 9.65 %; n = 6 slices, 4 animals, 2 litters; PNEE: 48.04 ± 6.89 %; n = 8 slices, 6 animals, 3 litters; p = 0.501; Figure 4).
Figure 7. Depotentiation is unaffected by prenatal ethanol exposure in either sex.
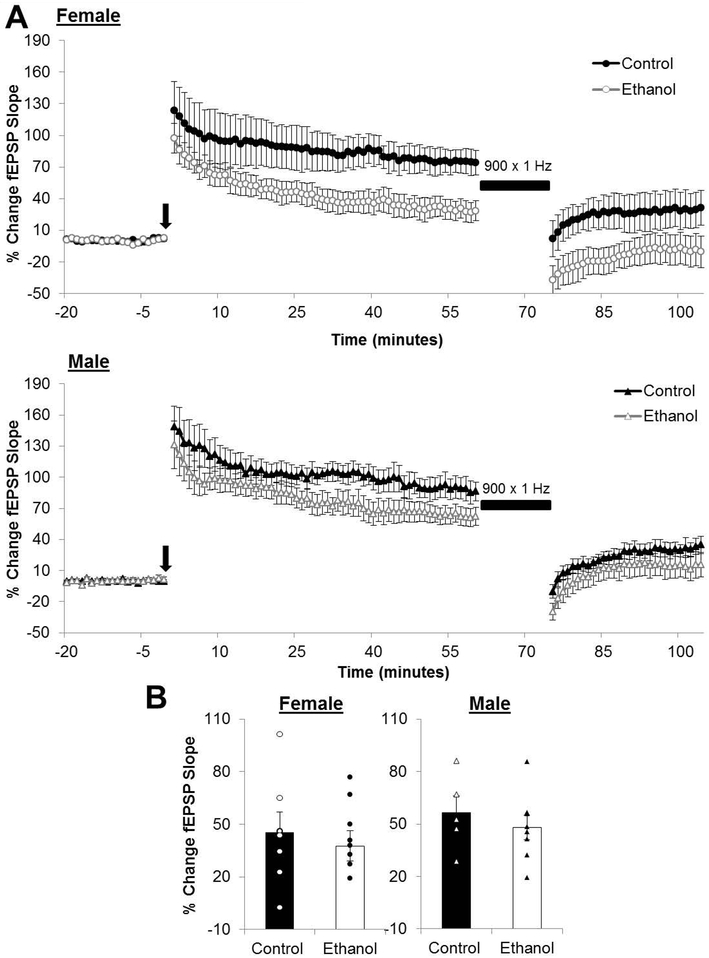
(A) Average depotentiation recordings from the beginning of baseline to the end of the post-conditioning recordings. The black arrow indicates the delivery of high-frequency stimulation and the black bar represents the low-frequency stimulation (900 × 1Hz). (A) Magnitude of depotentiation in control and ethanol-exposed offspring. Bars represent the average percentage of change in the field excitatory postsynaptic potential (fEPSP) slope calculated as the average percentage change in the fEPSP slope of minutes 100-105 subtracted from that of minutes 55-60, defined as the magnitude of depotentiation. Points represent the average magnitude of depotentiation for each individual slice in this dataset. Experimental n: ControlFemale: 45.19 ± 11.94 %; n = 7 slices, 6 animals, 2 litters; PNEEFemale: 37.68 ± 8.81 %; n = 9 slices, 8 animals, 2 litters; ControlMale: 56.39 ± 9.65 %; n = 6 slices, 4 animals, 2 litters; PNEEMale: 48.04 ± 6.89 %; n = 8 slices, 6 animals, 3 litters.
Discussion
This study elucidates how PNEE impacts the dynamic range of bidirectional synaptic plasticity in the DG in vitro in both males and female juvenile offspring. We show that bidirectional synaptic plasticity in the DG is differentially impacted in juvenile males and females by PNEE. Our initial findings in our I/O experiments indicate a sex-specific reduction in postsynaptic responsiveness in males that is overcome by inhibition of GABAA receptors. This suggests that following PNEE there may be less excitation, or more inhibition, in the DG of male animals. As with our previous studies using the liquid diet model of PNEE, we report reduced magnitudes of LTP in the DG for males, however in the present study we found that both male and female PNEE offspring display 50% less LTP than sex-matched controls. These LTP impairments were however overcome when an additional bout of HFS was used to induce LTP in both sexes, indicating a change to the threshold necessary to induce LTP in PNEE offspring. With respect to the LTD experiments we provide the first report of a sex-specific reduction in the magnitude of LTD in males in the DG. As with the LTP evaluated in this study, the PNEE-induced changes to LTD could be overcome by delivering prolonged LFS (1800 × 1Hz). LTD was unaffected by PNEE in females regardless of the duration of the LFS. While depotentiation was unaffected by PNEE in either sex, together this study suggests a sensitivity of the induction mechanisms to teratogens in utero.
Changes in the (I/O) curve to MPP stimulation in the male PNEE offspring suggest that there is a sex-specific decrease postsynaptic excitability in these animals. This effect of PNEE in the males is no longer apparent under GABAA inhibition by BIC (Fig 2B2) which together indicate that PNEE may lead to a change in the delicate balance between excitation and inhibition in males only. Following ethanol gavage (6g/kg/day) from GD8-20 there is a report of a similar reduction in the I/O relationship in young (P25-32) male PNEE rat offspring (Krahl et al., 1999) in the CA1 region. In this study, there was no significant effect of PNEE at a lower ethanol dose (4g/kg/day), nor in adult (P63-77) animals. Curiously, the altered I/O relationship was not accompanied by a LTP deficit in the slices generated from young male offspring. A similar result was obtained in the CA1 region of adult male guinea pig offspring after PNEE (gavage ethanol at 4g/kg/day from GD2-67) where increasing stimulus intensities resulted in reductions in the resulting fEPSP amplitude in vivo (Richardson et al., 2002). In this study however PNEE prevented the induction of CA1 LTP. The effect in males in normal aCSF (e.g. LTD experiments) has significant implications for the rest of this body of work because it shows that DG slices from male PNEE offspring respond differently to increasing stimulus durations than do slices from Control and female PNEE animals. The effect of PNEE on the I/O relationship does not account for the deficits in LTP observed in both males and females, as these experiments are carried out under GABAA inhibition, and in these conditions the I/O response curve for males identical to that observed in Control animals.
As we have recently noted in a review (Fontaine et al., 2016), unlike in the CA1 region, PNEE consistently impairs LTP in the DG and the data from the present study further supports this assessment. It is important to note that unlike some of our previous work in vivo (Patten et al., 2013d, 2013a; Titterness and Christie, 2012), we did not see sex differences in DG LTP in vitro. It is interesting that the DG LTP observed in vivo in slightly older female juveniles (P30-35) following PNEE was enhanced. It is also notable that in vivo, LTP in control females was reduced in this study compared to control males (Titterness and Christie, 2012). This effect that has also been observed in studies by others (Maren, 1995; Maren et al., 1994). Damage to the structure (Hughes et al., 1998; Nixon et al., 2004) and function the NMDAR (Lee et al., 1994; Savage et al., 1991, 1992; Spuhler-Phillips et al., 1997) by PNEE is reported in the literature and likely plays a role in the reduction of LTP observed in the present study, as this HFS paradigm is routinely recognized as inducing NMDAR-dependent plasticity in the DG. The magnitude of LTP in both sexes was restored to control-levels by the delivery of an additional HFS, indicating that PNEE increases the threshold for LTP in the DG. Indeed, a lack of sex differences in the effect of PNEE on learning and memory behaviours is not uncommon (Patten et al., 2016), although the absence of sex specific impacts following PNEE likely depends on the task used, metrics analyzed and age of the offspring (Berman and Hannigan, 2000; Patten et al., 2014).
While the use of juvenile offspring is not only important in the world of clinical FASDs, it also allows a unique window in the study of synaptic plasticity pertaining to LTD. The magnitude of LTD is classically age-dependent in rats (as reviewed in (Pinar et al., 2017)) making it challenging to study in adult animals. As such, this is the first study to assess sex differences in DG LTD in juvenile offspring following PNEE. Similar to our previous work in vivo, juvenile males were vulnerable to the effects of PNEE with respect to DG LTD. This type of LTD in the DG has been previously found to not dependent on NMDARs (Pöschel and Manahan-Vaughan, 2007; Trommer et al., 1996; Wang et al., 1997). Given that this LTP is likely NMDAR-dependent and we see no sex differences whereas in LTD we see specific impairments in males, it is likely that this LTD is also not NMDAR-dependent. As with LTP, the LTD deficits in male PNEE offspring could be overcome by prolonged LFS, evidence of a sex-specific shift for LTD induction in males and that the saturation point for LTD is unaffected by this teratogen exposure.
Theoretically developing male and female offspring have an equal likelihood to be exposed to ethanol in the gestating female rat, which raises the question as to why we observe different effects of PNEE on bidirectional synaptic plasticity between the sexes as juveniles. There are differences between the sexes in circulating sex hormones during early development and sex hormones can be synthesized in a variety of brain regions, including the hippocampus (see (McCarthy, 2008, 2009) for review). It is possible that the aromatization of testosterone to estradiol involved in masculinizing the male brain may play a role in vulnerability to EtOH-exposure during this time. In immature neurons in the developing brain GABA plays a dramatically different role than it does in mature neurons of the adult brain. In immature neurons, the chemical gradient drives chloride ions outwards, leading to depolarization of the membrane and opening of voltage-gated receptors such as those for calcium, when GABA receptors are activated (Rivera et al., 1999). Estradiol in fact enhances these GABA responses by increasing calcium flux through voltage-gated LTCCs, which is important in normal neural development for cell differentiation and synaptic integration (Ganguly et al., 2001). Given that EtOH is a positive allosteric modulator of the GABA receptor (specifically of GABAA), and is delivered throughout the gestation period it is possible that to coincidence of the actions of estradiol and EtOH on GABA receptors in the male brain in particular could contribute to the apparent vulnerability of this sex to impairments in bidirectional synaptic plasticity in the DG and possibly reduced excitability of this area as suggested by this work. Direct agonism of GABAA by muscimol combined with estradiol has been shown to lead to increased hippocampal cell death, an effect which could be prevented by blockade of LTCCs (Nuñez et al., 2003; Nunez and McCarthy, 2003).
Unlike previous studies we found no effect of PNEE on the magnitude of depotentiation in either sex. In the CA1 region, PNEE has been reported to lead to imbalanced bidirectional synaptic plasticity, resulting in reduced magnitudes of depotentiation in adolescent (P35) male rats (An et al., 2013), and enhanced depotentiation in females (An and Zhang, 2013) in vivo. The molecular mechanisms underpinning depotentiation are thought to overlap somewhat with those of LTD from a baseline condition, or de novo LTD, and potentially involve different phosphorylation states of the gluA1 subunit of the AMPAR (Lee et al., 2000, 2003, 2010). While we did not directly examine the mechanisms responsible for depotentiation, we would anticipate that if the same systems were responsible for de novo LTD that PNEE males would display deficits in the form of plasticity, which was not the case.
The PF group has commonly been used as a second control group in the study of PNEE with liquid diets, though it is clear that these animals go through gestation under dramatically different conditions. Dams in the EtOH condition have ad libitum access to the liquid diet and may freely consume as much as desired in 24 hours before fresh diet is supplied by the experimenter. The amount of diet consumed by these dams is tracked daily and is yoked to paired PF dams, taking into account any differences in their relative body weights. The goal of the PF group is to control for the effects of reduced food intake by the PNEE group. In reality, PF dams typically eat all of their allotted diet within short period of time after delivery (1-2 hours) and are then are fasting for the following 22-23 hours (Gallo and Weinberg, 1981; Rao and Larkin, 1987). Furthermore, EtOH consumption itself alters nutrient absorption including placental function and the delivery of nutrients and vitamins to the developing fetus (Bode and Bode, 2003; Dreosti, 1993; Weinberg, 1984). This change to the pattern but not the amount of diet consumed or the function of the digestive and placental systems can lead to unanticipated changes in hippocampal outcomes that are likely to be mechanistically different to those caused by PNEE (Helfer et al., 2014; Morgane et al., 2002). In the present study, we found that pair-feeding did not significantly affect postnatal weights in either males or females however these offspring did display deficits in LTP but not in LTD. In slightly older animals (P30-35) we have shown that PF offspring show disproportionately greater magnitudes of CA1 LTD as compared to both PNEE and ad libitum controls in males and show an altered pattern of this LTD in response to stress in a sex-specific fashion (Titterness and Christie, 2008). Pair-feeding is a complex form of undernutrition, fasting and prenatal stress that is becoming recognized as an imperfect dietary control in the study of PNEE with liquid diets as it can lead to unanticipated impairments distinct from those caused by PNEE.
The present study is the first to directly examine the dynamic range of bidirectional synaptic plasticity in the juvenile DG following PNEE. We found that male LTP and LTD are reduced in magnitude by PNEE while in females only LTP was affected. The delivery of additional CS to examine the range of plasticity allow the interpretation that these deficits are due to shifts in the thresholds for bidirectional plasticity in males and in synaptic potentiation alone in females, highlighting the need for this added level of depth in the study of synaptic plasticity following PNEE.
Supplementary Material
{kind=link}
Acknowledgements
The authors thank other members of the laboratory that aided in the raising of dams and experimental preparations. In particular we thank Erin Gräfe, Ryann Tansey, Tarndeep Chahal, Celine Chaudhary, Matthew Noseworthy, Ben Preston, Courtney Zoschke, Connor Mabbott and Christine Chiu. We also thank Dr. Pedro Grandes and Dr. Joanne Weinberg for their helpful discussion and input on this work. This research is supported by grants from NIH, NSERC and CIHR to B.R.C. C.J.F was supported by a Vanier CGS (NSERC). C.P is supported by the Agusti Pedro I Pons Foundation. W.Y, A.P,K.S. & J.C. were each supported by the NSERC USRA program.
REFERENCES
- Ahrnsbrak R, Bose J, Hedden SL, Lipari RN, Park-Lee E, and Tice P (2016). National Survey on Drug Use and Health Key Substance Use and Mental Health Indicators in the United States: Results from the 2016 National Survey on Drug Use and Health Results from the 2016 National Survey on Drug Use and Health. [Google Scholar]
- An L, Yang Z, and Zhang T (2013). Imbalanced synaptic plasticity induced spatial cognition impairment in male offspring rats treated with chronic prenatal ethanol exposure. Alcohol. Clin. Exp. Res. 37, 763–70. doi: 10.1111/acer.12040. [DOI] [PubMed] [Google Scholar]
- An L, and Zhang T (2013). Spatial cognition and sexually dimorphic synaptic plasticity balance impairment in rats with chronic prenatal ethanol exposure. Behav. Brain Res. 256, 564–74. doi: 10.1016/j.bbr.2013.09.017. [DOI] [PubMed] [Google Scholar]
- Benz J, Rasmussen C, and Andrew G (2009). Diagnosing fetal alcohol spectrum disorder: History, challenges and future directions. Paediatr. Child Health 14, 231–7. Available at: http://www.ncbi.nlm.nih.gov/pubmed/20357921 [Accessed May 9, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RF, and Hannigan JH (2000). Effects of prenatal alcohol exposure on the hippocampus: spatial behavior, electrophysiology, and neuroanatomy. Hippocampus 10, 94–110. doi:. [DOI] [PubMed] [Google Scholar]
- Bode C, and Bode CJ (2003). Effect of alcohol consumption on the gut. Best Pract. Res. Clin. Gastroenterol. 17, 575–592. doi: 10.1016/S1521-6918(03)00034-9. [DOI] [PubMed] [Google Scholar]
- Christie BR, Swann SE, Fox CJ, Froc D, Lieblich SE, Redila V, et al. (2005). Voluntary exercise rescues deficits in spatial memory and long-term potentiation in prenatal ethanol-exposed male rats. Eur J Neurosci 21, 1719–1726. [DOI] [PubMed] [Google Scholar]
- Cook JL, Green CR, Lilley CM, Anderson SM, Baldwin ME, Chudley AE, et al. (2016). Fetal alcohol spectrum disorder: a guideline for diagnosis across the lifespan. CMAJ 188, 191–7. doi: 10.1503/cmaj.141593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreosti IE (1993). Nutritional Factors Underlying the Expression of the Fetal Alcohol Syndrome. Ann. N. Y. Acad. Sci. 678, 193–204. doi: 10.1111/j.1749-6632.1993.tb26122.x. [DOI] [PubMed] [Google Scholar]
- Fontaine CJCJ, Patten ARAR, Sickmann HMHM, Helfer JLJL, and Christie BRBR (2016). Effects of pre-natal alcohol exposure on hippocampal synaptic plasticity: Sex, age and methodological considerations. Neurosci. Biobehav. Rev. 64, 12–34. doi: 10.1016/j.neubiorev.2016.02.014. [DOI] [PubMed] [Google Scholar]
- Gallo PV, and Weinberg J (1981). Corticosterone rhythmicity in the rat: interactive effects of dietary restriction and schedule of feeding. J. Nutr. 111, 208–218. doi: 10.1093/jn/111.2.208. [DOI] [PubMed] [Google Scholar]
- Ganguly K, Schinder AF, Wong ST, and Poo M (2001). GABA Itself Promotes the Developmental Switch of Neuronal GABAergic Responses from Excitation to Inhibition. Cell 105, 521–532. doi: 10.1016/S0092-8674(01)00341-5. [DOI] [PubMed] [Google Scholar]
- Gil-Mohapel J, Boehme F, Kainer L, and Christie BR (2010). Hippocampal cell loss and neurogenesis after fetal alcohol exposure: Insights from different rodent models. Brain Res. Rev. 64, 283–303. doi: 10.1016/j.brainresrev.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Guerri C, Bazinet A, and Riley EP (2009). Foetal Alcohol Spectrum Disorders and alterations in brain and behaviour. Alcohol Alcohol 44, 108–14. doi: 10.1093/alcalc/agn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannigan JH, and Riley EP (1988). Prenatal ethanol alters gait in rats. Alcohol 5, 451–454. doi: 10.1016/0741-8329(88)90081-X. [DOI] [PubMed] [Google Scholar]
- Helfer JL, White ER, and Christie BR (2014). Prenatal ethanol (EtOH) exposure alters the sensitivity of the adult dentate gyrus to acute EtOH exposure. Alcohol. Clin. Exp. Res. 38, 135–43. doi: 10.1111/acer.12227. [DOI] [PubMed] [Google Scholar]
- Hughes PD, Kim Y-N, Randall PK, and Leslie SW (1998). Effect of Prenatal Ethanol Exposure on the Developmental Profile of the NMDA Receptor Subunits in Rat Forebrain and Hippocampus. Alcohol. Clin. Exp. Res. 22, 1255–1261. doi: 10.1111/j.1530-0277.1998.tb03906.x. [DOI] [PubMed] [Google Scholar]
- Kervern M, Silvestre de Ferron B, Alaux-Cantin S, Fedorenko O, Antol J, Naassila M, et al. (2015). Aberrant NMDA-dependent LTD after perinatal ethanol exposure in young adult rat hippocampus. Hippocampus 25, 912–923. doi: 10.1002/hipo.22414. [DOI] [PubMed] [Google Scholar]
- Krahl SE, Berman RF, and Hannigan JH (1999). Electrophysiology of Hippocampal CA1 Neurons After Prenatal Ethanol Exposure. Alcohol 17, 125–131. doi: 10.1016/S0741-8329(98)00043-3. [DOI] [PubMed] [Google Scholar]
- Lee H-K, Takamiya K, Han J-S, Man H, Kim C-H, Rumbaugh G, et al. (2003). Phosphorylation of the AMPA Receptor GluR1 Subunit Is Required for Synaptic Plasticity and Retention of Spatial Memory. Cell 112, 631–643. doi: 10.1016/S0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Lee H-K, Takamiya K, He K, Song L, and Huganir RL (2010). Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J. Neurophysiol. 103, 479–89. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, and Huganir RL (2000). Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405, 955–9. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Lee YH, Spuhler-Phillips K, Randall PK, and Leslie SW (1994). Effects of prenatal ethanol exposure on N-methyl-D-aspartate-mediated calcium entry into dissociated neurons. J. Pharmacol. Exp. Ther. 271. [PubMed] [Google Scholar]
- Maren S (1995). Sexually dimorphic perforant path long-term potentiation (LTP) in urethane-anesthetized rats. Neurosci. Lett. 196, 177–180. doi: 10.1016/0304-3940(95)11869-X. [DOI] [PubMed] [Google Scholar]
- Maren S, De Oca B, and Fanselow MS (1994). Sex differences in hippocampal long-term potentiation (LTP) and Pavlovian fear conditioning in rats: positive correlation between LTP and contextual learning. Brain Res. 661, 25–34. doi: 10.1016/0006-8993(94)91176-2. [DOI] [PubMed] [Google Scholar]
- Mattson SN, and Riley EP (1998). A Review of the Neurobehavioral Deficits in Children with Fetal Alcohol Syndrome or Prenatal Exposure to Alcohol. Alcohol. Clin. Exp. Res. 22, 279–294. doi: 10.1111/j.1530-0277.1998.tb03651.x. [DOI] [PubMed] [Google Scholar]
- May PA, Chambers CD, Kalberg WO, Zellner J, Feldman H, Buckley D, et al. (2018). Prevalence of Fetal Alcohol Spectrum Disorders in 4 US Communities. JAMA 319, 474. doi: 10.1001/jama.2017.21896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PA, and Gossage JP (2011). Maternal risk factors for fetal alcohol spectrum disorders: not as simple as it might seem. Alcohol Res. Health 34, 15–26. [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM (2008). Estradiol and the Developing Brain. Physiol. Rev. 88, 91–134. doi: 10.1152/physrev.00010.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM (2009). The Two Faces of Estradiol: Effects on the Developing Brain. Neurosci. 15, 599–610. doi: 10.1177/1073858409340924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moos M-K, Dunlop AL, Jack BW, Nelson L, Coonrod DV, Long R, et al. (2008). Healthier women, healthier reproductive outcomes: recommendations for the routine care of all women of reproductive age. Am. J. Obstet. Gynecol. 199, S280–S289. doi: 10.1016/J.AJOG.2008.08.060. [DOI] [PubMed] [Google Scholar]
- Morgane PJ, Mokler DJ, and Galler JR (2002). Effects of prenatal protein malnutrition on the hippocampal formation. Neurosci. Biobehav. Rev. 26, 471–483. doi: 10.1016/S0149-7634(02)00012-X. [DOI] [PubMed] [Google Scholar]
- Nixon K, Hughes PD, Amsel A, and Leslie SW (2004). NMDA receptor subunit expression after combined prenatal and postnatal exposure to ethanol. Alcohol. Clin. Exp. Res. 28, 105–12. doi: 10.1097/01.ALC.0000106311.88523.7B. [DOI] [PubMed] [Google Scholar]
- Nuñez JL, Alt JJ, and McCarthy MM (2003). A new model for prenatal brain damage: I. GABAA receptor activation induces cell death in developing rat hippocampus. Exp. Neurol. 181, 258–269. doi: 10.1016/S0014-4886(03)00053-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez JL, and McCarthy MM (2003). Sex Differences and Hormonal Effects in a Model of Preterm Infant Brain Injury. Ann. N. Y. Acad. Sci. 1008, 281–284. doi: 10.1196/annals.1301.032. [DOI] [PubMed] [Google Scholar]
- Patten AR, Brocardo PS, Sakiyama C, Wortman RC, Noonan A, Gil-Mohapel J, et al. (2013a). Impairments in hippocampal synaptic plasticity following prenatal ethanol exposure are dependent on glutathione levels. Hippocampus 23, 1463–75. doi: 10.1002/hipo.22199. [DOI] [PubMed] [Google Scholar]
- Patten AR, Brocardo PS, Sakiyama C, Wortman RC, Noonan A, Gil-Mohapel J, et al. (2013b). Impairments in hippocampal synaptic plasticity following prenatal ethanol exposure are dependent on glutathione levels. Hippocampus. doi: 10.1002/hipo.22199. [DOI] [PubMed] [Google Scholar]
- Patten AR, Fontaine CJ, and Christie BR (2014). A comparison of the different animal models of fetal alcohol spectrum disorders and their use in studying complex behaviors. Front. Pediatr. 2, 93. doi: 10.3389/fped.2014.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten AR, Sawchuk S, Wortman RC, Brocardo PS, Gil-Mohapel J, and Christie BR (2016). Prenatal ethanol exposure impairs temporal ordering behaviours in young adult rats. Behav. Brain Res. 299, 81–89. doi: 10.1016/J.BBR.2015.11.032. [DOI] [PubMed] [Google Scholar]
- Patten AR, Sickmann HM, Dyer RA, Innis SM, and Christie BR (2013c). Omega-3 fatty acids can reverse the long-term deficits in hippocampal synaptic plasticity caused by prenatal ethanol exposure. Neurosci. Lett. 551, 7–11. doi: 10.1016/j.neulet.2013.05.051. [DOI] [PubMed] [Google Scholar]
- Patten AR, Sickmann HM, Dyer RA, Innis SM, and Christie BR (2013d). Omega-3 Fatty Acids can Reverse the Long-Term Deficits in Hippocampal Synaptic Plasticity Caused by Prenatal Ethanol Exposure. Neurosci Lett. doi:S0304-3940(13)00498-9 [pii] 10.1016/j.neulet.2013.05.051. [DOI] [PubMed] [Google Scholar]
- Pierrefiche O, and Olivier (2017). Long Term Depression in Rat Hippocampus and the Effect of Ethanol during Fetal Life. Brain Sci. 7, 157. doi: 10.3390/brainsci7120157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinar C, Fontaine CJ, Triviño-Paredes J, Lottenberg CP, Gil-Mohapel J, and Christie BR (2017). Revisiting the flip side: Long-term depression of synaptic efficacy in the hippocampus. Neurosci. Biobehav. Rev. 80, 394–413. doi: 10.1016/j.neubiorev.2017.06.001. [DOI] [PubMed] [Google Scholar]
- Popova S, Lange S, Probst C, Parunashvili N, and Rehm J (2017). Prevalence of alcohol consumption during pregnancy and Fetal Alcohol Spectrum Disorders among the general and Aboriginal populations in Canada and the United States. Eur. J. Med. Genet 60, 32–48. doi: 10.1016/J.EJMG.2016.09.010. [DOI] [PubMed] [Google Scholar]
- Pöschel B, and Manahan-Vaughan D (2007). Persistent (>24h) long-term depression in the dentate gyrus of freely moving rats is not dependent on activation of NMDA receptors, L-type voltage-gated calcium channels or protein synthesis. Neuropharmacology 52, 46–54. doi: 10.1016/j.neuropharm.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Rao GA, and Larkin EC (1987). Nutritional inadequacy of lieber-decarli alcohol diet. Hepatology 7, 416–417. doi: 10.1002/hep.1840070248. [DOI] [PubMed] [Google Scholar]
- Richardson DP, Byrnes ML, Brien JF, Reynolds JN, and Dringenberg HC (2002). Impaired acquisition in the water maze and hippocampal long-term potentiation after chronic prenatal ethanol exposure in the guinea-pig. Eur. J. Neurosci. 16, 1593–1598. doi: 10.1046/j.1460-9568.2002.02214.x. [DOI] [PubMed] [Google Scholar]
- Riley EP, Infante MA, Warren KR, Court A, Diego S, and Warren KR (2011). Fetal Alcohol Spectrum Disorders: An Overview. Neuropsychol. Rev. 21, 73–80. doi: 10.1007/s11065-011-9166-x.Fetal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley EP, and McGee CL (2005). Fetal Alcohol Spectrum Disorders: An Overview with Emphasis on Changes in Brain and Behavior. Exp. Biol. Med. 230, 357–365. doi: 10.1177/15353702-0323006-03. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, et al. (1999). The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Savage DD, Montano CY, Otero MA, and Paxton LL (1991). Prenatal ethanol exposure decreases hippocampal NMDA-sensitive [3H]-Glutamate binding site density in 45-day-old rats. Alcohol 8, 193–201. doi: 10.1016/0741-8329(91)90806-8. [DOI] [PubMed] [Google Scholar]
- Savage DD, Queen SA, Sanchez CF, Paxton LL, Mahoney JC, Goodlett CR, et al. (1992). Prenatal ethanol exposure during the last third of gestation in rat reduces hippocampal NMDA agonist binding site density in 45-day-old offspring. Alcohol 9, 37–41. doi: 10.1016/0741-8329(92)90007-W. [DOI] [PubMed] [Google Scholar]
- Singh S, Sedgh G, and Hussain R (2010). Unintended Pregnancy: Worldwide Levels, Trends, and Outcomes. Stud. Fam. Plann. 41, 241–250. doi: 10.1111/j.1728-4465.2010.00250.x. [DOI] [PubMed] [Google Scholar]
- Spuhler-Phillips K, Lee Y-H, Hughes P, Randoll L, and Leslie SW (1997). Effects of Prenatal Ethanol Exposure on Brain Region NMDA-Mediated Increase in Intracellular Calcium and the NMDAR1 Subunit in Forebrain. Alcohol. Clin. Exp. Res. 21, 68–75. doi: 10.1111/j.1530-0277.1997.tb03730.x. [DOI] [PubMed] [Google Scholar]
- Titterness AK, and Christie BR (2008). Long-term depression in vivo: effects of sex, stress, diet, and prenatal ethanol exposure. Hippocampus 18, 481–91. doi: 10.1002/hipo.20407. [DOI] [PubMed] [Google Scholar]
- Titterness AK, and Christie BR (2012). Prenatal ethanol exposure enhances NMDAR-dependent long-term potentiation in the adolescent female dentate gyrus. Hippocampus 22, 69–81. doi: 10.1002/hipo.20849. [DOI] [PubMed] [Google Scholar]
- Trommer BL, Liu YB, and Pasternak JF (1996). Long-term depression at the medial perforant path-granule cell synapse in developing rat dentate gyrus. Brain Res. Dev. Brain Res. 96, 97–108. [DOI] [PubMed] [Google Scholar]
- Uban KA, Sliwowska JH, Lieblich S, Ellis LA, Yu WK, Weinberg J, et al. (2010). Prenatal alcohol exposure reduces the proportion of newly produced neurons and glia in the dentate gyrus of the hippocampus in female rats. Horm. Behav. 58, 835–843. doi: 10.1016/j.yhbeh.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Rowan MJ, and Anwyl R (1997). Induction of LTD in the Dentate Gyrus In Vitro Is NMDA Receptor Independent, but Dependent on Ca 2+ Influx via Low-Voltage–Activated Ca 2+ Channels and Release of Ca 2+ From Intracellular Stores. J. Neurophysiol. 77, 812–825. doi: 10.1152/jn.1997.77.2.812. [DOI] [PubMed] [Google Scholar]
- Weinberg J (1984). Nutritional issues in perinatal alcohol exposure. Neurobehav. Toxicol. Teratol. 6, 261–9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/6514087 [Accessed April 19, 2018]. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.