

Abstract
Background
In mammals, two distinct Leydig cell populations, fetal Leydig cells (FLCs) and adult Leydig cells (ALCs), appear in the prenatal and postnatal testis, respectively. Although the functional differences between these cell types have been well described, the developmental relationship between FLCs and ALCs has not been fully understood. In this review, I focus on the cellular origins of FLCs and ALCs as well as the developmental and functional links between them.
Methods
I surveyed previous reports about FLC and/or ALC development and summarized the findings.
Main findings
Fetal Leydig cells and ALCs were identified to have separate origins in the fetal and neonatal testis, respectively. However, several studies suggested that FLCs and ALCs share a common progenitor pool. Moreover, perturbation of FLC development at the fetal stage induces ALC dysfunction in adults, suggesting a functional link between FLCs and ALCs. Although the lineage relationship between FLCs and ALCs remains controversial, a recent study suggested that some FLCs dedifferentiate at the fetal stage, and that these cells serve as ALC stem cells.
Conclusion
Findings obtained from animal studies might provide clues to the causative mechanisms of male reproductive dysfunctions such as testicular dysgenesis syndrome in humans.
Keywords: adult Leydig cell, androgen, cell lineage, differentiation, fetal Leydig cell
1. INTRODUCTION
1.1. Development of fetal and adult Leydig cells in mammals
Androgens play pivotal roles in the development as well as homeostasis of male reproductive functions. Although both major steroidogenic tissues, the adrenal gland and testis, produce androgens, testicular Leydig cells are well known as the main source of physiologically active androgens. In mammals, at least two types of Leydig cell, fetal Leydig cells (FLCs) and adult Leydig cells (ALCs), sequentially develop in the fetal and adult testis, respectively,1 and show distinct characteristic features. For example, electron microscopy analyses of the rat testis revealed that FLCs have numerous lipid droplets, while lipid droplets are not abundant in ALCs.2 Moreover, several genes are expressed in ALCs but not in FLCs,3 and recent transcriptomic analyses have extended this knowledge by showing distinct gene expression profiles in FLCs and ALCs.4 In particular, 17β‐hydroxysteroid dehydrogenase type 3 (HSD17B3) is expressed in ALCs but not in FLCs, and therefore, FLCs produce androstenedione, while ALCs produce testosterone.5
As FLCs rapidly decrease in number after birth, FLCs are supposed to die or degenerate and be replaced by newly developed ALCs in the postnatal testis.1, 6, 7, 8 However, morphometric analyses of the fetal and postnatal testis suggested that FLCs persist even in the adult testis.9, 10 Several researchers recently waded into this controversy by adopting a lineage tracing technique and clearly revealed that FLCs persist in the postnatal testis, at least in mice.11, 12 Although these analyses clarified the fate of FLCs, the origins of FLCs and ALCs remained a matter of debate. Intensive studies have identified progenitor cells for FLCs and ALCs in the fetal testis and neonatal testis, respectively,13, 14, 15 and these results suggested that FLCs and ALCs have distinct origins. However, a recent study instead suggested that FLCs and ALCs share a common origin,16 and also that perturbation of FLC differentiation in the fetal period affects ALC differentiation later in puberty. These results suggested that the development and function of ALC are closely associated with those of FLCs. Although the lineage relationship between FLCs and ALCs is still controversial, Shima et al recently performed rigorous lineage tracing of FLCs by using the FLC‐specific regulatory region (Fetal Leydig Enhancer, FLE) of the Nr5a1 gene to specifically label FLCs. Their results strongly suggested that some FLCs dedifferentiate at the fetal to neonatal stages and that these dedifferentiated cells serve as ALC progenitor cells.17
In this review, I focus on the cellular origins of FLCs and ALCs and present an overview of recent knowledge about the relationship between FLCs and ALCs.
2. MORPHOLOGICAL AND FUNCTIONAL DIFFERENCES BETWEEN FETAL AND ADULT LEYDIG CELLS
2.1. Morphology of fetal and adult Leydig cells
Leydig cells were initially described in 1850 as testicular interstitial cells containing characteristic lipid droplets.18 Since then, numerous researchers have observed Leydig cells using electron microscopy; these cells were identified as the testicular interstitial cells, which have abundant smooth endoplasmic reticulum and lipid droplets, mitochondria with tubular cristae, and crystals of Reinke.19 Most of these morphological features of Leydig cells match those of other steroidogenic cells such as adrenocortical cells, and one study supported the hypothesis that Leydig cells are the main source of androgens.20
Most morphological studies of the testis have used the rat as a model animal, and some of these studies demonstrated that FLCs have numerous lipid droplets whereas ALCs contain a small number of them. However, this morphological difference between these two cell types was not apparent in the case of mice. Therefore, recent studies attempted to identify the genes that show distinctive expression patterns between FLCs and ALCs, and to date, several molecular markers of FLCs and ALCs have been reported.
2.2. Androgen production in fetal and adult Leydig cells
Androgens produced by FLCs induce masculinization of the fetus: the development of external genitalia such as the scrotum and penis; the development of the accessory sex organs such as the epididymis, deferent ducts, and seminal vesicles; and male‐specific neuronal network formation in the brain. Testosterone, the most potent androgen in mammals, is synthesized from cholesterol via multiple reactions mediated by a set of steroidogenic enzymes. FLCs express most of these enzymes such as steroidogenic acute regulatory protein (StAR), cholesterol side‐chain cleavage P450 (P450SCC or CYP11A1), 3β‐hydroxysteroid dehydrogenase/∆5‐∆4 isomerase (3β‐HSD or HSD3B), and 17β‐hydroxylase/17,20‐lyase P450 (CYP17A1). However, 17β‐hydroxysteroid dehydrogenase type 3 (HSD17B3), an enzyme that mediates the final reaction of testosterone synthesis, is not expressed in FLCs.21 Therefore, the major androgen produced by FLCs is not testosterone but androstenedione. Although Sertoli cells are accepted as nonsteroidogenic cells, they express HSD17B3 only in the fetal period. Shima et al examined the activities of steroidogenic enzymes in FLCs, fetal Sertoli cells, and ALCs, with the obtained results supporting the conclusion that androstenedione produced by FLCs is transferred to fetal Sertoli cells and then converted to testosterone, whereas ALCs are capable of producing testosterone by themselves because they express HSD17B3 as well as other steroidogenic enzymes.5
There are multiple subtypes of 17β‐HSD in both mouse and human, one of which, 17β‐HSD type 1 (HSD17B1), plays a central role in ovarian steroidogenesis in mice.22 Recently, Hakkarainen et al reported that HSD17B1 is expressed in the fetal and neonatal Sertoli cells in mice; moreover, male mice with Hsd17b1 gene knockout showed morphologically abnormal spermatozoa and compensated upregulation of HSD17B3, suggesting that HSD17B1 contributes to testosterone synthesis in the fetal and neonatal testis.23
There are also multiple subtypes of 3β‐HSD in both mouse and humans. Interestingly, 3β‐HSD type 1 (HSD3B1) is expressed in both FLCs and ALCs, whereas 3β‐HSD type 6 (HSD3B6) is expressed only in ALCs in mice. Therefore, most previous studies used HSD3B6 as an ALC‐specific marker. Recently, Yokoyama et al generated HSD3B1‐specific antibody and performed double immunostaining of the adult mouse testis with this antibody and a previously generated HSD3B6‐specific antibody.24 They showed that there are three distinct Leydig cell populations in the adult testis: HSD3B1(+)/HSD3B6(−) cells, HSD3B1(+)/HSD3B6(+) cells, and HSD3B1(−)/HSD3B6(+) cells.25 Although these results suggested that ALCs consist of heterogeneous subpopulations, it seems certain that FLCs do not express HSD3B6.
2.3. Actions of LH and androgen on fetal and adult Leydig cells
Luteinizing hormone (LH) is secreted from the pituitary gonadotrope and stimulates testicular Leydig cells to produce testosterone. Lack of LH or LHR has been shown to result in markedly reduced testosterone production and the arrest of postnatal sexual development in male mice, indicating that androgen production in ALC is solely dependent on LH.26, 27, 28 However, the disruption of LHβ or LHR did not affect fetal masculinization, suggesting that androgen production in FLCs does not depend on LH, although LHR starts to be expressed in the FLCs at embryonic day 16 (E16)29 and fetal testes are capable of responding to LH stimulation and produce testosterone.30
Androgens exert their physiological actions by binding to androgen receptor (AR). Ar gene mutation or disruption leads to the failure of ALC functional maturation, and these mice fail to produce enough testosterone to induce puberty and sperm maturation.3, 31 Ar gene mutation in humans induces androgen insensitivity syndrome (AIS). AIS patients exhibit symptoms quite similar to those of mice with Ar gene disruption, such as female external genitalia, female breast development, blind vagina, and undescended testes. These observations provide evidence that AR plays an essential role in postnatal masculinization in both mice and humans.
Interestingly, FLCs do not express AR, and fetal masculinization is not affected by systemic Ar gene mutation or disruption in mice. Shima et al recently generated mice with FLC‐specific Ar gene disruption, which can be used to analyze the cell‐autonomous roles of AR in FLCs. Their results reconfirmed that FLC differentiation does not depend on AR and moreover that FLCs persist as an AR‐independent cell population even in the adult testis.11 Although AR is not expressed in FLCs, the interstitial cells surrounding FLCs express AR.11, 32 As these interstitial cells are supposed to be Leydig cell progenitors, it can be assumed that androgen signaling might play an important role in maintaining the Leydig progenitor population in the fetal testis. Kilcoyne et al hypothesized that the AR‐positive interstitial cells in the fetal testis serve as ALC progenitor cells, and they actually showed that low androgen exposure at fetal stages resulted in a decreased number of Leydig progenitor cells and the functional failure of ALCs in adulthood.33
2.4. Nr5a1 gene regulation in fetal and adult Leydig cells
NR5A1 (also known as Ad4BP or SF‐1) is an orphan nuclear receptor expressed in various tissues such as the ventromedial hypothalamus, pituitary gonadotrope, adrenal cortex, spleen, as well as testis and ovary. Systemic disruption of the Nr5a1 gene resulted in complete gonadal and adrenal agenesis, indicating that this factor is essential for the development of the adrenal gland and gonad.34, 35 This factor was initially identified as a transcriptional activator of steroidogenic genes,36 and accordingly, NR5A1 is strongly expressed in the adrenocortical cells, testicular Leydig cells, and ovarian theca cells.37 Buaas et al generated mice with steroidogenic cell‐specific Nr5a1 gene disruption by mating Cyp11a1‐iCre mice and Nr5a1 flox mice 38; the phenotypes of these mice reconfirmed the functional importance of NR5A1 in steroidogenic cells including FLCs and ALCs.39
Nr5a1 gene expression in each tissue is strictly regulated by tissue‐specific enhancer. Transgenic analyses have revealed that the Nr5a1 gene promoter induces reporter gene expression in the undifferentiated gonad.40 In addition, intensive transgenic studies have identified the enhancers for fetal adrenal cortex,41 ventromedial hypothalamus,42 pituitary gonadotrope,43 and FLCs.44 Identification of the FLE (FLC‐specific enhancer of Nr5a1 gene) was followed by another study which confirmed that FLE induces reporter gene expression in FLCs but not in ALCs,11 and simultaneously suggested that Nr5a1 gene expression in ALCs is regulated by other enhancer(s). Intriguingly, the deletion of FLE resulted in the complete disappearance of FLCs as well as ALCs,17 indicating that FLE is essential for Nr5a1 gene expression in both FLCs and ALCs. Taking these lines of evidence into consideration, Shima et al proposed a putative model of Nr5a1 gene regulation in FLCs and ALCs17 (Figure 1).
Figure 1.
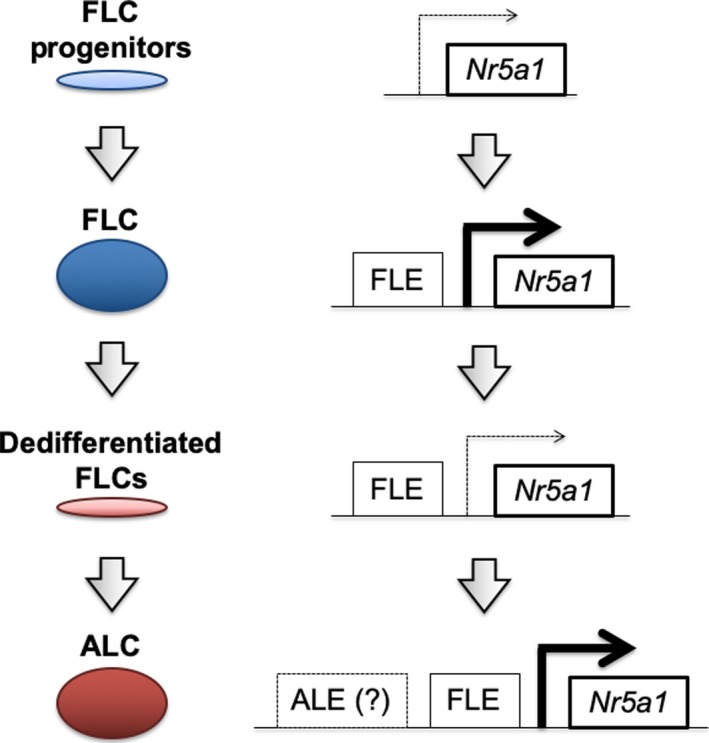
Nr5a1 gene regulation in FLCs and ALCs. Shima et al proposed a model of Nr5a1 gene regulation in FLCs and ALCs based on their observations.17 Fetal Leydig enhancer (FLE) of the Nr5a1 gene induces NR5A1 expression in the FLC progenitor cells, which differentiate into FLCs. Thereafter, FLCs undergo dedifferentiation at the fetal to neonatal stages. NR5A1 is downregulated in the dedifferentiated cells, although the mechanism behind this is unclear. The dedifferentiated cells start to redifferentiate at the prepubertal stage, and eventually contribute to ALCs in adult testis. FLE is essential for Nr5a1 gene expression in both FLCs and ALCs. However, since FLE induces Nr5a1 gene expression only in FLCs,11 it was suggested that other regulatory element(s) of the Nr5a1 gene (adult Leydig enhancer, ALE) might be involved in the Nr5a1 gene expression in ALCs
3. ORIGIN OF LEYDIG CELLS
3.1. Origin of fetal Leydig cells
The testis originates from the genital ridge, a thickening of the ventral coelomic epithelium. The genital ridge at the early fetal stage is undifferentiated and has the potential to develop both testis and ovary. However, activation of the Sry‐Sox9 genetic cascade induces Sertoli cell differentiation and the male gonad is prompted to develop into the testis. Sertoli cells surround the germ cells, which have migrated from the extraembryonic ectoderm through the mesenterium, and the testis is divided into two compartments: the testis cord and the interstitium.
Sertoli cells produce desert hedgehog (DHH) and platelet‐derived growth factor (PDGF) to induce FLC differentiation,45, 46 and FLCs initially appear in the interstitial region at E12.5 in mice. Thereafter, FLCs increase in number throughout the fetal period. As functionally differentiated FLCs are mitotically inactive,2, 47, 48 it was assumed that FLC progenitors reside within the interstitial compartment of the fetal testis, and these progenitor cells continually give rise to FLCs throughout the fetal period. NR5A1 is essential for steroidogenic cell differentiation,35, 49 and this factor is expressed in the adrenogonadal primordium (AGP), which thereafter divides and gives rise to the adrenal primordium and genital ridge.37 The adrenocortical cells and FLCs commonly express steroidogenic genes; these two cell populations are considered to originate from NR5A1‐positive cells in AGP. At E12.5 onward, NR5A1 is strongly and weakly expressed in the FLCs and other interstitial cells, respectively,50 and the interstitial cells expressing NR5A1 weakly are expected to include Leydig progenitor cells. Recently, mRNA‐sequence analyses of bulk sorted cells or single‐cell transcriptomic analyses of fetal testis identified the interstitial Leydig progenitor cells that express NR5A1 weakly.51, 52 Furthermore, Inoue et al isolated the interstitial cell population that expresses NR5A1 weakly, and demonstrated that these cells, at least in part, serve as FLC progenitor cells by performing testis reconstruction experiments.13
3.2. Adult Leydig progenitor cells in neonatal testis
The number of FLCs declines after birth and thereafter another Leydig cell population, ALCs, appears and rapidly increases in number during the pubertal period. However, once ALCs have occupied the interstitial space of the adult testis, there is a low rate of ALC turnover. Therefore, it is difficult to observe differentiating ALCs in the adult testis.
An alkylating reagent, ethane dimethanesulfonate (EDS), has been identified as a Leydig cell‐specific killer. EDS treatment induces ALC death at 1‐3 days after the treatment (Days 1‐3), and almost all of the ALCs have disappeared by Days 7‐14. Thereafter, ALCs start to regenerate from Day 21 to reach the control level by Day 49.53 Davidoff et al hypothesized that this regeneration process mimics ALC differentiation in the pubertal period, and closely observed the regenerating ALCs. They revealed that vascular pericytes transdifferentiate into ALCs, suggesting the possibility that the vascular pericytes can serve as ALC stem cells.54 In contrast, Ge et al hypothesized that ALC stem cells reside within the neonatal testis, and these cells contribute to the rapid increase in ALCs at the pubertal stage. They isolated peritubular cells from the neonatal testis that were HSD3B1‐negative, LHR‐negative, and PDGF receptor α (PDGFRα)‐positive. These cells started to express HSD3B1 and produce androgens upon culture under adequate conditions; moreover, these cells contributed to ALCs when transplanted into the interstitial space of recipient testis.15 These results strongly suggested that the peritubular nonsteroidogenic cells in the neonatal testis serve as ALC stem cells, and subsequent studies reconfirmed the potential of these peritubular cells to differentiate into ALCs via in vitro seminiferous tubule culture experiments.55, 56 Moreover, Li et al reported that the peritubular cells in the neonatal testis differentiate into ALCs in response to DHH, TGF‐β, and FGF2.57
3.3. Common progenitor pool for FLCs and ALCs in fetal testis
As described in previous sections, FLC progenitors and ALC stem cells were separately identified in the fetal and neonatal testis, respectively; these results suggested that FLCs and ALCs have distinct origins (Figure 2A). However, several studies have instead raised the possibility that these two cell types share a common progenitor pool.
Figure 2.
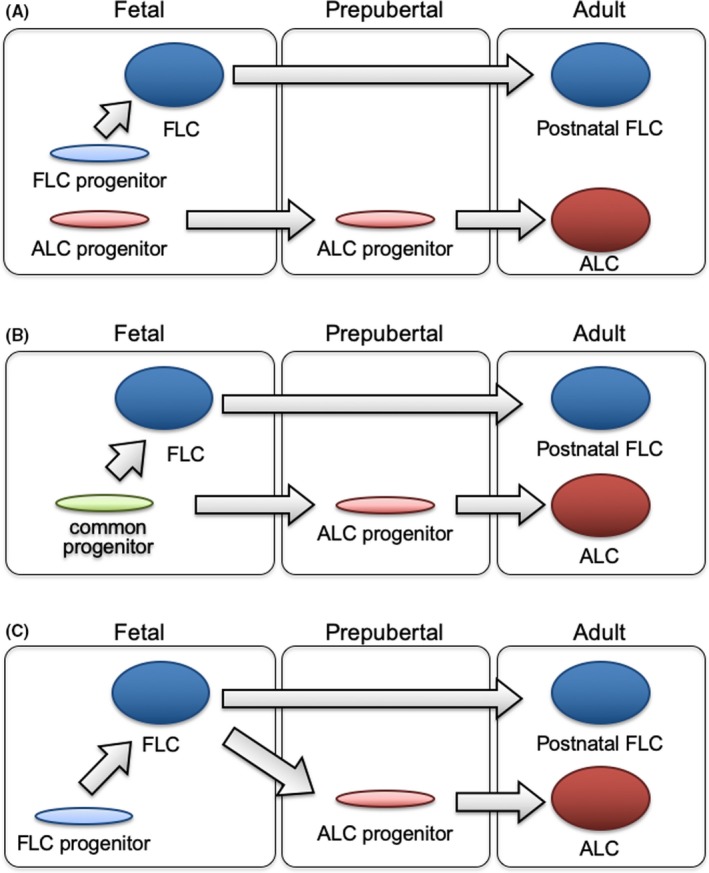
Proposed models of Leydig cell development. Three distinct models of Leydig cell development have been proposed. A, FLCs differentiate from FLC progenitors in the fetal testis. In contrast, there is another progenitor population (ALC progenitors) that starts to differentiate into ALCs at the prepubertal stage. Recent studies confirmed that FLCs do not disappear but persist in the postnatal testis (postnatal FLCs). B, There is a common Leydig cell progenitor pool in the fetal testis, and this population gives rise to both FLCs and ALCs, although it is still unclear how each progenitor is destined to become FLCs or ALCs. C, FLCs originate from FLC progenitors in the fetal testis. FLCs then dedifferentiate at the fetal to neonatal stages, and these dedifferentiated cells start to redifferentiate at the prepubertal stage, and contribute to ALC formation in the adult testis
Barsoum et al noticed that not all NR5A1‐positive progenitor cells in the fetal testis differentiate into FLCs, and hypothesized that these progenitor cells have the potential to differentiate into both FLCs and ALCs. They reported that constitutive activation of the hedgehog pathway resulted in an increased number of FLCs as well as a reduced number of progenitors, and moreover, ALCs were decreased in the prepubertal period. They interpreted these results as evidence that FLCs and ALCs share a progenitor population in the fetal testis 16 (Figure 2B). If FLCs and ALCs arise from a common progenitor pool, there should be mechanisms that determine the fate of each progenitor cell to differentiate into FLC or ALC. Although such mechanisms remain unknown, several studies have suggested putative regulators of FLC and ALC differentiation, such as NR5A1,58 COUP‐TFII,59 WT1 expression in Sertoli cells,60 androgen signaling in Sertoli cells,61 and hedgehog signaling from Sertoli cells to the Leydig progenitors.62, 63
4. FATE OF FLCS IN THE POSTNATAL TESTIS
As Leydig cells show characteristic features such as abundant smooth endoplasmic reticulum, mitochondria with tubular cristae, and lipid droplets, this cell type has been subjected to numerous histological studies. In particular, electron microscopy analyses of the rat testis revealed that FLCs contain numerous lipid droplets, whereas ALCs have a small number of them.2 Most researchers focused on the cell density and serum testosterone concentration, and they concluded that FLCs completely regress and are replaced by ALCs.1, 6, 7, 8 In contrast, several researchers performed morphometric analyses of FLCs in the prenatal and postnatal testis, and their results suggested that FLCs persist in the postnatal testis.2, 9, 10
In recent studies, the Cre‐loxP system was used to label and trace the FLCs. Kaftanovskaya et al used mice in which Cre recombinase is expressed under the control of the Retinoic acid receptor 2 (Rarb) gene promoter, and reported that FLCs persist in the adult testis.12 In another study, Shima et al generated FLC‐specific CreERT mice by using FLE of the Nr5a1 gene and crossed these mice with CAG‐CAT‐EGFP reporter mice.64 Using the double transgenic mice, they labeled FLCs with EGFP at E14.5 by tamoxifen treatment and analyzed their fate in the adult testis. The results showed that EGFP‐labeled cells persisted in the adult testis, and these cells did not express HSD3B6 and HSD17B3, marker proteins for ALCs. These lines of evidence strongly support the conclusion that FLCs persist in the adult testis, at least in mice.
5. DEVELOPMENTAL LINK BETWEEN FETAL AND ADULT LEYDIG CELLS
5.1. Influence of fetal Leydig cell dysfunction on adult Leydig cell development
Although FLCs and ALCs have been considered to be independent cell populations, several studies have reported that FLC dysfunction resulted in the functional failure of ALC. van den Driesche et al attempted to suppress androgen production in the rat fetal testis by treating fetuses with dibutyl phthalate (DBP) and analyzed the effects at later stages. They found that DBP treatment induced reduction of anogenital distance (AGD) and compensated ALC failure. Interestingly, these effects at later stages were observed when fetuses were treated in a limited time window (they called this period the masculinization programming window).65 In another study, Su et al eliminated FLCs in the rat testis by EDS treatment at neonatal stages and analyzed the effect of FLC loss on ALC function in the pubertal and adult periods. They found that testosterone production was dramatically decreased at P56, suggesting that FLC dysfunction attenuates ALC development in the adult stage.66 Kilcoyne et al attempted to obtain mechanistic insights into the developmental link between FLCs and ALCs by focusing on the Leydig progenitor cells in fetal testis. Since the Leydig progenitor cells in fetal testis express AR, they hypothesized that the FLC‐derived androgen signal is indispensable for the maintenance of ALC stem cells. According to their conclusion, ALC dysfunction during puberty might be attributable to susceptibility to androgen concentration in the fetal period.33
5.2. Dedifferentiation or degeneration of fetal Leydig cells in human fetal testis
Most previous animal studies appeared to support the conclusion that FLCs and ALCs are independent cell populations, and it has been generally accepted that FLCs do not contribute to ALCs.67, 68 However, observation of human fetuses conversely suggested the possibility that FLCs dedifferentiate and contribute to the postnatal Leydig cell population.
One of the largest differences between rodent FLCs and human FLCs is LH dependence. As noted in the previous section, FLCs in mice develop independently of LH throughout the fetal period.26, 27, 69 In contrast, human fetuses are exposed to hCG secreted from the placenta, and the FLC population is dependent on hCG.70 Serum hCG decreases at the second trimester of pregnancy, and the number of FLCs decreases in this period accordingly.71 Moreover, several histological features suggesting FLC degeneration or dedifferentiation, such as cell volume reduction, electron‐dense cells, and lipid droplets in the degenerating cells, were observed.71, 72, 73 Thereafter, the hypothalamic‐pituitary‐gonadal axis was shown to be temporarily activated and Leydig cells were found to reappear in the neonatal testis.74 As this Leydig cell population, neonatal Leydig cells, shows morphological features similar to FLCs, these cells are considered to be redifferentiated FLCs.74, 75 After a sharp increase in serum testosterone, this level decreases again and remains low until prepubertal ALC differentiation. Although neonatal Leydig cells are not observed in rodents’ testis, these observations in human testis might provide clues to clarify the lineage relationship between FLCs and ALCs in other animals.
5.3. FLCs dedifferentiate and thereby redifferentiate to ALCs in mouse testis
Recently, Shima et al adopted the powerful genetic tool lineage tracing analysis to reveal the fate of FLCs in mice. In this analysis, FLE of the Nr5a1 gene was used to label FLCs. FLE induced CreER expression in FLCs, and tamoxifen treatment induced the recombination of loxP sites in CAG‐CAT‐EGFP, and resulted in FLC‐specific labeling by EGFP in a limited time window. These analyses clearly revealed that some FLCs undergo dedifferentiation in the fetal to neonatal period. Moreover, some FLCs contribute to peritubular myoid cells and vascular pericytes, and consistent with previous studies, FLC‐derived peritubular myoid cells and vascular pericytes showed the potential to differentiate into ALCs (Figure 2C). They also generated two mouse models: FLC‐specific Nr5a1 gene‐disrupted mice and mice lacking FLE of the Nr5a1 gene. Both of these mouse models demonstrated the disappearance of FLCs as well as the marked decline or complete loss of ALCs, supporting the conclusion that a considerable proportion of ALCs originate from dedifferentiated FLCs in mice.17
6. CONCLUDING REMARKS
Since Leydig cells were first described as characteristic testicular interstitial cells, many researchers have attempted to reveal their morphological and functional features in the prenatal and postnatal testis. These intensive studies revealed morphological and functional differences in FLCs and ALCs as well as the fate of FLCs, and many researchers are now focusing on the functional link between FLCs and ALCs. Although progenitor/stem cells for FLCs and ALCs were distinctively identified in the prenatal and neonatal testis, respectively, several recent studies suggested that FLCs and ALCs share a common progenitor pool in the fetal testis. Moreover, several studies provided evidence, suggesting that FLCs play pivotal roles in ALC stem cell maintenance as well as the functional differentiation of ALCs in the pubertal period.
Hutchison et al reported that fetal exposure to DBP induces the focal dysgenesis of seminiferous tubules and intratubular Leydig cells in rats, both of which are known as characteristic symptoms of testicular dysgenesis syndrome (TDS) in humans.76 TDS is known as one of the causes of male infertility, and epidemiological studies suggested that its pathogenesis is attributable to a low testosterone level in the fetal period.77, 78 Further analyses of animal models might provide clues to clarify the causative mechanisms underlying the pathogenesis of TDS in humans.
Finally, the lineage relationship between FLCs and ALCs is still a matter of debate. Observation of human fetuses suggested that FLCs dedifferentiate to give rise to postnatal Leydig cells.79 In addition, recent lineage tracing analyses strongly suggested that FLCs dedifferentiate and redifferentiate to ALCs also in mice.17 However, it is still unclear whether all of the ALCs are derived from FLCs or some of them differentiate from their own progenitors. It is anticipated that future studies will resolve this important issue.
CONFLICTS OF INTEREST
The author has no conflicts of interest to disclose.
HUMAN/ANIMAL RIGHTS
This article does not contain any studies with human and animal subjects performed by the author.
ACKNOWLEDGMENTS
We thank Edanz (www.edanzediting.co.jp) for editing the English text of a draft of this manuscript.
Shima Y. Development of fetal and adult Leydig cells. Reprod Med Biol. 2019;18:323–330. 10.1002/rmb2.12287
REFERENCES
- 1. Roosen‐Runge EC, Anderson D. The development of the interstitial cells in the testis of the albino rat. Acta Anat (Basel). 1959;37:125‐137. [DOI] [PubMed] [Google Scholar]
- 2. Kerr JB, Knell CM. The fate of fetal Leydig cells during the development of the fetal and postnatal rat testis. Development. 1988;103(3):535‐544. [DOI] [PubMed] [Google Scholar]
- 3. O'Shaughnessy PJ, Johnston H, Willerton L, Baker PJ. Failure of normal adult Leydig cell development in androgen‐receptor‐deficient mice. J Cell Sci. 2002;115(Pt 17):3491‐3496. [DOI] [PubMed] [Google Scholar]
- 4. Miyabayashi K, Shima Y, Inoue M, et al. Alterations in fetal Leydig cell gene expression during fetal and adult development. Sex Dev. 2017;11(2):53‐63. [DOI] [PubMed] [Google Scholar]
- 5. Shima Y, Miyabayashi K, Haraguchi S, et al. Contribution of Leydig and Sertoli cells to testosterone production in mouse fetal testes. Mol Endocrinol. 2013;27(1):63‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lording DW, De Kretser DM. Comparative ultrastructural and histochemical studies of the interstitial cells of the rat testis during fetal and postnatal development. J Reprod Fertil. 1972;29(2):261‐269. [DOI] [PubMed] [Google Scholar]
- 7. Hardy MP, Zirkin BR, Ewing LL. Kinetic studies on the development of the adult population of Leydig cells in testes of the pubertal rat. Endocrinology. 1989;124(2):762‐770. [DOI] [PubMed] [Google Scholar]
- 8. Zirkin BR, Ewing LL. Leydig cell differentiation during maturation of the rat testis: a stereological study of cell number and ultrastructure. Anat Rec. 1987;219(2):157‐163. [DOI] [PubMed] [Google Scholar]
- 9. Ariyaratne HB, Chamindrani M‐H. Changes in the testis interstitium of Sprague Dawley rats from birth to sexual maturity. Biol Reprod. 2000;62(3):680‐690. [DOI] [PubMed] [Google Scholar]
- 10. Mendis‐Handagama SM, Risbridger GP, de Kretser DM. Morphometric analysis of the components of the neonatal and the adult rat testis interstitium. Int J Androl. 1987;10(3):525‐534. [DOI] [PubMed] [Google Scholar]
- 11. Shima Y, Matsuzaki S, Miyabayashi K, et al. Fetal Leydig Cells persist as an androgen‐independent subpopulation in the postnatal testis. Mol Endocrinol. 2015;29(11):1581‐1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaftanovskaya EM, Lopez C, Ferguson L, Myhr C, Agoulnik AI. Genetic ablation of androgen receptor signaling in fetal Leydig cell lineage affects Leydig cell functions in adult testis. FASEB J. 2015;29(6):2327‐2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Inoue M, Shima Y, Miyabayashi K, et al. Isolation and characterization of Fetal leydig progenitor cells of male mice. Endocrinology. 2016;157(3):1222‐1233. [DOI] [PubMed] [Google Scholar]
- 14. Stevant I, Nef S. Single cell transcriptome sequencing: a new approach for the study of mammalian sex determination. Mol Cell Endocrinol. 2018. [DOI] [PubMed] [Google Scholar]
- 15. Ge RS, Dong Q, Sottas CM, Papadopoulos V, Zirkin BR, Hardy MP. In search of rat stem Leydig cells: identification, isolation, and lineage‐specific development. Proc Natl Acad Sci USA. 2006;103(8):2719‐2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barsoum IB, Kaur J, Ge RS, Cooke PS, Yao HH. Dynamic changes in fetal Leydig cell populations influence adult Leydig cell populations in mice. FASEB J. 2013;27(7):2657‐2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shima Y, Miyabayashi K, Sato T, et al. Fetal Leydig cells dedifferentiate and serve as adult Leydig stem cells. Development. 2018;145(23):dev169136. [DOI] [PubMed] [Google Scholar]
- 18. Leydig F. Zur Anatomie der Männlichen Geschlechtsorgane und Analdrüsen der Säugethiere. Z Wiss Zool. 1850;2:1‐57. [Google Scholar]
- 19. Reinke F. Betiräge zur Histologie des Menschen. I. Ueber Krystalloidbildungen in den interstitiellen Zellen des menschlichen. Hodens. Archiv f. Mikrosk. Anat. u. Entwicklungsmech. 1896;47:34‐44.
- 20. Bouin P, Ancel P. Recherches sur les cellules interstitielles du testicule des mammifères. Arch de Zoöl Exp et Gén. 1903;4(1):437‐523. [Google Scholar]
- 21. O'Shaughnessy PJ, Baker PJ, Heikkila M, Vainio S, McMahon AP. Localization of 17beta‐hydroxysteroid dehydrogenase/17‐ketosteroid reductase isoform expression in the developing mouse testis–androstenedione is the major androgen secreted by fetal/neonatal leydig cells. Endocrinology. 2000;141(7):2631‐2637. [DOI] [PubMed] [Google Scholar]
- 22. Hakkarainen J, Jokela H, Pakarinen P, et al. Hydroxysteroid (17beta)‐dehydrogenase 1‐deficient female mice present with normal puberty onset but are severely subfertile due to a defect in luteinization and progesterone production. FASEB J. 2015;29(9):3806‐3816. [DOI] [PubMed] [Google Scholar]
- 23. Hakkarainen J, Zhang FP, Jokela H, et al. Hydroxysteroid (17beta) dehydrogenase 1 expressed by Sertoli cells contributes to steroid synthesis and is required for male fertility. FASEB J. 2018;32(6):3229‐3241. [DOI] [PubMed] [Google Scholar]
- 24. Doi M, Takahashi Y, Komatsu R, et al. Salt‐sensitive hypertension in circadian clock‐deficient Cry‐null mice involves dysregulated adrenal Hsd3b6. Nat Med. 2010;16(1):67–74. [DOI] [PubMed] [Google Scholar]
- 25. Yokoyama C, Chigi Y, Baba T, et al. Three populations of adult Leydig cells in mouse testes revealed by a novel mouse HSD3B1‐specific rat monoclonal antibody. Biochem Biophys Res Commun. 2019;511(4):916‐920. [DOI] [PubMed] [Google Scholar]
- 26. Lei ZM, Mishra S, Zou W, et al. Targeted disruption of luteinizing hormone/human chorionic gonadotropin receptor gene. Mol Endocrinol. 2001;15(1):184‐200. [DOI] [PubMed] [Google Scholar]
- 27. Zhang FP, Poutanen M, Wilbertz J, Huhtaniemi I. Normal prenatal but arrested postnatal sexual development of luteinizing hormone receptor knockout (LuRKO) mice. Mol Endocrinol. 2001;15(1):172‐183. [DOI] [PubMed] [Google Scholar]
- 28. Ma X, Dong Y, Matzuk MM, Kumar TR. Targeted disruption of luteinizing hormone beta‐subunit leads to hypogonadism, defects in gonadal steroidogenesis, and infertility. Proc Natl Acad Sci USA. 2004;101(49):17294‐17299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. O'Shaughnessy PJ, Baker P, Sohnius U, Haavisto AM, Charlton HM, Huhtaniemi I. Fetal development of Leydig cell activity in the mouse is independent of pituitary gonadotroph function. Endocrinology. 1998;139(3):1141‐1146. [DOI] [PubMed] [Google Scholar]
- 30. Pointis G, Mahoudeau JA. Responsiveness of foetal mouse testis to gonadotrophins at various times during sexual differentiation. J Endocrinol. 1977;74(1):149‐150. [DOI] [PubMed] [Google Scholar]
- 31. Murphy L, Jeffcoate IA, O'Shaughnessy PJ. Abnormal Leydig cell development at puberty in the androgen‐resistant Tfm mouse. Endocrinology. 1994;135(4):1372‐1377. [DOI] [PubMed] [Google Scholar]
- 32. Majdic G, Millar MR, Saunders PT. Immunolocalisation of androgen receptor to interstitial cells in fetal rat testes and to mesenchymal and epithelial cells of associated ducts. J Endocrinol. 1995;147(2):285‐293. [DOI] [PubMed] [Google Scholar]
- 33. Kilcoyne KR, Smith LB, Atanassova N, et al. Fetal programming of adult Leydig cell function by androgenic effects on stem/progenitor cells. Proc Natl Acad Sci USA. 2014;111(18):E1924‐E1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luo X, Ikeda Y, Parker KL. A cell‐specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77(4):481‐490. [DOI] [PubMed] [Google Scholar]
- 35. Honda S, Morohashi K, Nomura M, Takeya H, Kitajima M, Omura T. Ad4BP regulating steroidogenic P‐450 gene is a member of steroid hormone receptor superfamily. J Biol Chem. 1993;268(10):7494‐7502. [PubMed] [Google Scholar]
- 36. Lala DS, Rice DA, Parker KL. Steroidogenic factor I, a key regulator of steroidogenic enzyme expression, is the mouse homolog of fushi tarazu‐factor I. Mol Endocrinol. 1992;6(8):1249‐1258. [DOI] [PubMed] [Google Scholar]
- 37. Hatano O, Takakusu A, Nomura M, Morohashi K. Identical origin of adrenal cortex and gonad revealed by expression profiles of Ad4BP/SF‐1. Genes Cells. 1996;1(7):663‐671. [DOI] [PubMed] [Google Scholar]
- 38. Zhao L, Bakke M, Krimkevich Y, et al. Steroidogenic factor 1 (SF1) is essential for pituitary gonadotrope function. Development. 2001;128(2):147‐154. [DOI] [PubMed] [Google Scholar]
- 39. Buaas FW, Gardiner JR, Clayton S, Val P, Swain A. In vivo evidence for the crucial role of SF1 in steroid‐producing cells of the testis, ovary and adrenal gland. Development. 2012;139(24):4561‐4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wilhelm D, Englert C. The Wilms tumor suppressor WT1 regulates early gonad development by activation of Sf1. Genes Dev. 2002;16(14):1839‐1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zubair M, Ishihara S, Oka S, Okumura K, Morohashi K. Two‐step regulation of Ad4BP/SF‐1 gene transcription during fetal adrenal development: initiation by a Hox‐Pbx1‐Prep1 complex and maintenance via autoregulation by Ad4BP/SF‐1. Mol Cell Biol. 2006;26(11):4111‐4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shima Y, Zubair M, Ishihara S, et al. Ventromedial hypothalamic nucleus‐specific enhancer of Ad4BP/SF‐1 gene. Mol Endocrinol. 2005;19(11):2812‐2823. [DOI] [PubMed] [Google Scholar]
- 43. Shima Y, Zubair M, Komatsu T, et al. Pituitary homeobox 2 regulates adrenal4 binding protein/steroidogenic factor‐1 gene transcription in the pituitary gonadotrope through interaction with the intronic enhancer. Mol Endocrinol. 2008;22(7):1633‐1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shima Y, Miyabayashi K, Baba T, et al. Identification of an enhancer in the Ad4BP/SF‐1 gene specific for fetal Leydig cells. Endocrinology. 2012;153(1):417‐425. [DOI] [PubMed] [Google Scholar]
- 45. Yao HH, Whoriskey W, Capel B. Desert hedgehog/patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev. 2002;16(11):1433‐1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brennan J, Tilmann C, Capel B. Pdgfr‐alpha mediates testis cord organization and fetal Leydig cell development in the XY gonad. Genes Dev. 2003;17(6):800‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Orth JM. Proliferation of Sertoli cells in fetal and postnatal rats: a quantitative autoradiographic study. Anat Rec. 1982;203(4):485‐492. [DOI] [PubMed] [Google Scholar]
- 48. Miyabayashi K, Katoh‐Fukui Y, Ogawa H, et al. Aristaless related homeobox gene, Arx, is implicated in mouse fetal Leydig cell differentiation possibly through expressing in the progenitor cells. PLoS ONE. 2013;8(6):e68050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lala DS, Ikeda Y, Luo X, Baity LA, Meade JC, Parker KL. A cell‐specific nuclear receptor regulates the steroid hydroxylases. Steroids. 1995;60(1):10‐14. [DOI] [PubMed] [Google Scholar]
- 50. Zhao L, Wang C, Lehman ML, et al. Transcriptomic analysis of mRNA expression and alternative splicing during mouse sex determination. Mol Cell Endocrinol. 2018;478:84‐96. [DOI] [PubMed] [Google Scholar]
- 51. McClelland KS, Bell K, Larney C, et al. Purification and transcriptomic analysis of mouse fetal Leydig cells reveals candidate genes for specification of gonadal steroidogenic cells. Biol Reprod. 2015;92(6):145. [DOI] [PubMed] [Google Scholar]
- 52. Stévant I, Neirijnck Y, Borel C, et al. Deciphering cell lineage specification during Male sex determination with single‐cell RNA sequencing. Cell Rep. 2018;22(6):1589‐1599. [DOI] [PubMed] [Google Scholar]
- 53. Jackson AE, O'Leary PC, Ayers MM, de Kretser DM. The effects of ethylene dimethane sulphonate (EDS) on rat Leydig cells: evidence to support a connective tissue origin of Leydig cells. Biol Reprod. 1986;35(2):425‐437. [DOI] [PubMed] [Google Scholar]
- 54. Davidoff MS, Middendorff R, Enikolopov G, Riethmacher D, Holstein AF, Muller D. Progenitor cells of the testosterone‐producing Leydig cells revealed. J Cell Biol. 2004;167(5):935‐944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stanley E, Lin C‐Y, Jin S, et al. Identification, proliferation, and differentiation of adult Leydig stem cells. Endocrinology. 2012;153(10):5002‐5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Odeh HM, Kleinguetl C, Ge R, Zirkin BR, Chen H. Regulation of the proliferation and differentiation of Leydig stem cells in the adult testis. Biol Reprod. 2014;90(6):123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li X, Wang Z, Jiang Z, et al. Regulation of seminiferous tubule‐associated stem Leydig cells in adult rat testes. Proc Natl Acad Sci U S A. 2016;113(10):2666‐2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Karpova T, Ravichandiran K, Insisienmay L, Rice D, Agbor V, Heckert LL. Steroidogenic factor 1 differentially regulates fetal and adult leydig cell development in male mice. Biol Reprod. 2015;93(4):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. van den Driesche S, Walker M, McKinnell C, et al. Proposed role for COUP‐TFII in regulating fetal Leydig cell steroidogenesis, perturbation of which leads to masculinization disorders in rodents. PLoS ONE. 2012;7(5):e37064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wen Q, Zheng Q‐S, Li X‐X, et al. Wt1 dictates the fate of fetal and adult Leydig cells during development in the mouse testis. Am J Physiol Endocrinol Metab. 2014;307(12):E1131‐E1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hazra R, Jimenez M, Desai R, Handelsman DJ, Allan CM. Sertoli cell androgen receptor expression regulates temporal fetal and adult Leydig cell differentiation, function, and population size. Endocrinology. 2013;154(9):3410‐3422. [DOI] [PubMed] [Google Scholar]
- 62. Defalco T, Saraswathula A, Briot A, Iruela‐Arispe ML, Capel B. Testosterone levels influence mouse fetal Leydig cell progenitors through notch signaling. Biol Reprod. 2013;88(4):91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Park SY, Tong M, Jameson JL. Distinct roles for steroidogenic factor 1 and desert hedgehog pathways in fetal and adult Leydig cell development. Endocrinology. 2007;148(8):3704‐3710. [DOI] [PubMed] [Google Scholar]
- 64. Kawamoto S, Niwa H, Tashiro F, et al. A novel reporter mouse strain that expresses enhanced green fluorescent protein upon Cre‐mediated recombination. FEBS Lett. 2000;470(3):263‐268. [DOI] [PubMed] [Google Scholar]
- 65. van den Driesche S, Kilcoyne KR, Wagner I, et al. Experimentally induced testicular dysgenesis syndrome originates in the masculinization programming window. JCI Insight. 2017;2(6):e91204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Su DM, Feng Y, Wang L, Wu YL, Ge RS, Ma X. Influence of fetal Leydig cells on the development of adult Leydig cell population in rats. J Reprod Dev. 2018;64(3):223‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Griswold SL, Behringer RR. Fetal Leydig cell origin and development. Sex Dev. 2009;3(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Svingen T, Koopman P. Building the mammalian testis: origins, differentiation, and assembly of the component cell populations. Genes Dev. 2013;27(22):2409‐2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. O'Shaughnessy PJ, Fowler PA. Endocrinology of the mammalian fetal testis. Reproduction. 2011;141(1):37‐46. [DOI] [PubMed] [Google Scholar]
- 70. Huhtaniemi I, Lautala P. Stimulation of steroidogenesis in human fetal testes by the placenta during perifusion. J Steroid Biochem. 1979;10(1):109‐113. [DOI] [PubMed] [Google Scholar]
- 71. Holstein AF, Wartenberg H, Vossmeyer J. Cytology of the prenatal development of human gonads. 3. Development of leydig cells in the testis of embryos and fetuses. Z Anat Entwicklungsgesch. 1971;135(1):43‐66. [PubMed] [Google Scholar]
- 72. Pelliniemi LJ, Niemi M. Fine structure of the human foetal testis. I. The interstitial tissue. Z Zellforsch Mikrosk Anat. 1969;99(4):507‐522. [DOI] [PubMed] [Google Scholar]
- 73. Codesal J, Regadera J, Nistal M, Regadera‐Sejas J, Paniagua R. Involution of human fetal Leydig cells. An immunohistochemical, ultrastructural and quantitative study. J Anat. 1990;172:103‐114. [PMC free article] [PubMed] [Google Scholar]
- 74. Nistal M, Paniagua R, Regadera J, Santamaria L, Amat P. A quantitative morphological study of human Leydig cells from birth to adulthood. Cell Tissue Res. 1986;246(2):229‐236. [DOI] [PubMed] [Google Scholar]
- 75. Prince FP. Ultrastructural evidence of mature Leydig cells and Leydig cell regression in the neonatal human testis. Anat Rec. 1990;228(4):405‐417. [DOI] [PubMed] [Google Scholar]
- 76. Hutchison GR, Sharpe RM, Mahood IK, et al. The origins and time of appearance of focal testicular dysgenesis in an animal model of testicular dysgenesis syndrome: evidence for delayed testis development? Int J Androl. 2008;31(2):103‐111. [DOI] [PubMed] [Google Scholar]
- 77. Juul A, Almstrup K, Andersson A‐M, et al. Possible fetal determinants of male infertility. Nat Rev Endocrinol. 2014. [DOI] [PubMed] [Google Scholar]
- 78. Wohlfahrt‐Veje C, Main KM, Skakkebaek NE. Testicular dysgenesis syndrome: foetal origin of adult reproductive problems. Clin Endocrinol (Oxf). 2009;71(4):459‐465. [DOI] [PubMed] [Google Scholar]
- 79. Teerds KJ, Huhtaniemi IT. Morphological and functional maturation of Leydig cells: from rodent models to primates. Hum Reprod Update. 2015;21(3):310‐328. [DOI] [PubMed] [Google Scholar]