

Abstract
Alzheimer disease (AD) and dementia are becoming increasingly prevalent due to the aging of the global populations. Currently available treatment options, including acetylcholinesterase inhibitors and memantine, only have symptomatic effects and no drugs with disease-modifying properties are available. Research on the amyloid cascade indicates that amyloid-β (Aβ) clearance from the brain may be the main pathophysiological change in late-onset AD and the key driver of neurodegeneration, which ultimately results in progressive cognitive deterioration and dementia. Most new AD drug candidates target different aspects of Aβ clearance, eg, using passive anti-Aβ immunization, but so far, all efforts to develop more effective drugs have failed. In parallel, nonpharmacological prevention trials are being conducted to modify dementia risk associated with known epidemiological risk factors. Some initial results are promising, but replication across independent cohorts remains a challenge.
Keywords: Alzheimer disease, biomarker, clinical trial, dementia, epidemiology, mild cognitive impairment, prevention, prognosis, public health, treatment
Introduction
Due to the aging of the global populations, chronic disorders are becoming increasingly prevalent. This includes dementia, which in 2015 affected 47 million people worldwide and is expected to affect twice as many people 20 years from now.1 Most late-onset dementia cases are related to Alzheimer disease (AD) pathology, but mixed etiologies are more prevalent in older populations. Amyloid-β (Aβ) plaques and tau neurofibrils, the two pathological hallmarks of AD, have less impact on cognitive performance in the oldest-old compared with younger individuals.2 and “pure” AD cases are also less frequent in older patients, who mostly have additional neuropathological lesions, including vascular changes. Also, significant Aβ pathology is not only found in individuals with dementia but is also prevalent in cognitively intact individuals, and age is a main predictor of Aβ plaques, even in the absence of relevant cognitive decline.3
In neuropsychiatric tradition, AD was conceptualized as a clinicopathological duality, ie, a diagnosis was only possible in the presence of an amnestic-type progressive dementia syndrome and the exclusion of alternative etiologies. This simple set of criteria is not particularly sensitive for early changes nor is it specific enough for AD pathophysiology. Therefore, in the last 10 years evolving sets of new AD criteria were proposed by different international expert groups, aiming to steer the dementia field towards a more biologically oriented disease concept, similar to other areas of medicine, such as cancer, where biomarkers are used to define diseases. This paradigm shift is fuelled by the urgent desire to develop more effective, ie, disease-modifying rather than purely symptomatic, treatment options. Being able to identify a disease in its pre-symptomatic or prodromal stage would open new opportunities to prevent or slow pathophysiological processes rather than merely trying to retard the worsening of symptoms. The development of biomarkers which are more sensitive for the early stages of AD is a prerequisite for a successful transformation of the diagnostic approaches.
Individuals at risk of cognitive deterioration and dementia, who are still asymptomatic, would probably benefit most from intervention strategies aimed at prevention of further neuronal loss.4 If no or only minimal symptoms are present, effective disease modification could help the target population to maintain their independence, fulfilment of social roles, and ability to work for a longer period of time.
Biomarkers of AD pathophysiology may improve diagnosis and prognosis in early AD cases; also, surrogate end points are required to ensure that only individuals with a high a priori likelihood of the target pathology are included in interventional studies and to avoid exposing individuals to potentially significant side effects who are unlikely to benefit from the treatment. Biomarker-based endophenotypes are also required to improve the feasibility of large-scale prevention studies aimed at exploring the effectiveness of factors derived from epidemiological research, such as obesity, diabetes mellitus, and physical inactivity.5 Due to the decades-long silent, pre-clinical stage of AD, prevention trials would have to be conducted over extended periods to be able to reliably assess their effectiveness. Using imaging or other biomarkers to identify eligible study participants and to demonstrate effectiveness of the new intervention would significantly reduce sample size and study length. However, so far it has not been possible to replace traditional clinical study end points by biomarker-derived surrogates.6
Early diagnosis
Early diagnosis is essential in chronic disorders if disease-modifying treatment options are available, which can alter disease trajectories, and which offer meaningful benefits to the affected individuals. For AD, all available treatments ie, cholinesterase inhibitors and memantine, only have symptomatic effects, which on average offer comparably small clinical benefits. Since all efforts to develop more effective, disease-modifying drugs have so far been unsuccessful,7 the future availability of these drugs is currently a far-fetched argument in support of the early recognition of AD. In contrast, providing individuals and their families with reliable information about the underlying causes of cognitive and functional decline and behavioral change can reduce uncertainty and conflicts and may therefore be a valid reason for early diagnosis in certain situations. Furthermore, individuals at increased risk for short-term cognitive deterioration may benefit from early diagnosis if it enables them to make important decisions affecting their future lives, such as making a will, as long as their decision-making capacity is still preserved.8,9 However, only a few patients seem to use the early diagnostic information to initiate advance care planning, indicating that there is either no great demand for this information, or that improved counselling is required in these situations.10
There are also certain disadvantages and risks associated with receiving information about a diagnosis of early AD. Without effective treatment options, affected individuals are confronted with the prospect of inevitable loss of cognitive abilities and independence, while no countermeasure is available. The ambiguous prognosis associated with currently available biomarkers may have significant psychological consequences, including stress, despair, depression, or even suicide in extreme cases.11 Negative effects on employment or interpersonal relationships may also occur.
Most studies concur that currently available AD biomarkers perform remarkably well in situations when prodromal AD cases need to be separated from “normal” aging or when progression from minor cognitive complaints to full-blown dementia has to be predicted. However, even in specialist settings, in which AD cases are preselected and relatively “pure,” at least 30% of individuals with full-blown AD-type dementia have normal biomarker findings. Current biomarkers show better sensitivity than specificity; therefore, ruling in AD as the main cause of cognitive complaints is more difficult and inaccurate than ruling it out.12
Biomarkers with improved specificity and sensitivity for early AD may be available in the near future. Ideally, they will not only offer better diagnostic and prognostic value, but will also be less invasive than current biomarkers, which require a lumbar puncture for CSF protein assessment or the application of a radioactive tracer for positron emission tomography (PET) studies. Blood-based markers, for example, would facilitate large-scale screening efforts and the inclusion of a large number of individuals at risk for future cognitive decline in prevention studies. An improved biomarker could potentially be used in a two-stage screening approach, with an initial pre-screening step using affordable, low-invasive techniques to identify individuals with Aβ pathology, followed by more expensive and invasive testing only in a subset of participants. Using traditional technology such as enzyme-linked immune sorbent assays (ELISA) to measure Aβ in blood was associated with insufficient accuracy.13 Using a new generation of high-sensitivity technology, more accurate measurement of blood biomarkers appears to be possible.14,15
Pathomechanisms
The most influential model of AD pathophysiology, and therefore the most frequently targeted mechanisms in AD drug development, is the amyloid cascade hypothesis, which was originally derived from findings in rare autosomal dominant mutation carriers.16 Briefly, amyloid precursor protein (APP) is cleaved by either α-secretase, resulting in soluble non-amyloidogenic products, or β- and γ-secretases, which results in the Aβ peptide, which initially aggregates to form soluble oligomers, and subsequently insoluble fibrils, later found in the typical senile plaques. The soluble Aβ aggregates may be the main drivers of synaptic and neuronal loss, rather than the insoluble, fibrillar deposits.17
The importance of soluble forms of Aβ in the pathological AD cascade is underlined by the toxic effects of small peptide complexes on synapses and mitochondria.18 Respiratory chain defects and autophagic degradation are central mitochondria-related pathomechanisms in AD,19 which contribute to the release of toxic oxygen species, impair energy production and related axonal transport, and interfere with calcium homeostasis.17 Aβ also leads to a local inflammatory response, which involves microglia clusters, upregulated acute phase proteins and other mediators of an inflammatory response.20 Microglia activation and neuroinflammation may be beneficial and neuroprotective in the early stages of AD, but over-activation of the cerebral immune system may be a harmful driver of neurodegeneration in later disease stages.21
It was repeatedly demonstrated that the number of senile plaques is not the main structural correlate of cognitive deficits in AD; loss of synapses and neurons in the neocortex and hippocampus is much closer correlated with the deterioration of cognitive performance.22 An imbalance of Aβ is most likely the main driver of protein accumulation in the brain. Overproduction of Aβ is likely the key pathomechanism in early onset autosomal dominant cases, but Aβ production in late-onset sporadic disease is only slightly higher than in age-matched control subjects.23 Therefore, Aβ clearance from the brain is probably a major contributor to cerebral peptide accumulation.
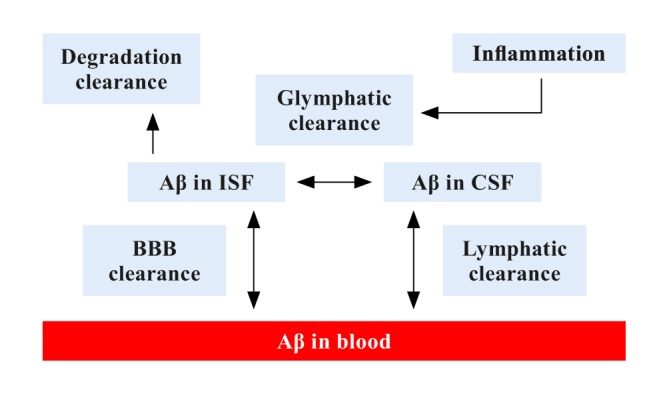
A number of different mechanisms for Aβ clearance have been described. This includes active, receptor-mediated transport across the blood-brain barrier (BBB) by the receptor for advanced glycation end products (RAGE) from blood to brain24 and by a soluble form of the low-density lipoprotein receptor related protein 1 (LRP1) from brain to blood.23 Until recently, transport across the BBB was considered to be the main mechanism for Aβ removal from the brain; however, findings from the past few years support the existence of additional important mechanisms.25 Bulk-flow of interstitial fluid (ISF) mediated by astroglia (ie, the glymphatic system) and recently discovered lymphatic vessels in the meninges may also make a meaningful contribution to Aβ clearance.26 The degradation of Aβ by cleaving enzymes such as neprilysin and insulin-degrading enzyme may also play an important role.27 Since the different clearance systems probably act together to drive Aβ from the brain, any change in their function could contribute to AD. It is therefore key to improve our mechanistic understanding of Aβ clearance, which may be used to develop improved approaches to reduce excess Aβ deposits and delay, or prevent AD onset.
Previously, it was thought that about 75% of Aβ is cleared by BBB transport and only 10% by the glymphatic system. However, recent photon imaging studies in mice, using microscopy with fluorescent tracers, have suggested that the glymphatic system contributes to a larger part to Aβ clearance than previously thought.28 Water channels called aquaporin 4 (AQP4) on the vascular end feet of astrocytes facilitate convective flow out of the para-arterial space and into the interstitial space. Mislocation of AQP4 water channels may contribute to neurodegenerative disease progression.29
During wakefulness, the interstitial volume is more contracted, which increases resistance to convective flow and cerebrospinal fluid (CSF) movement. During sleep, the interstitial space increases in volume, which facilitates convective flow, CSF-to-ISF turnover and thus glymphatic clearance.30 Choroid plexus functioning and arterial pulsatility determine the CSF dynamics and for instance regulate CSF influx through the perivascular space and are the driving forces of the glymphatic system.31 Factors influencing arterial pulsatility, such as vessel stiffness and heart rate, affect the amount of waste clearance. Arterial pulsatility decreases due to processes such as metabolic syndrome, hypertension, hyperlipidemia, diabetes, or aging. Arterial stiffness and increased pulsatility of cerebral blood flow potentially damage small vessels and may indirectly affect glymphatic clearance and can possibly lead to neurodegeneration.32
Besides BBB disruption, inflammation can also contribute to glymphatic dysfunction. Inflammation slows down the convective flow, decreases CSF-to-ISF turnover, and impairs glymphatic clearance.33 A schematic representation of clearance pathways is provided in Figure 1.
Figure 1. Schematic representation of key amyloid-β clearance pathways (modified from ref 70: Tarasoff-Conway JM, Carare RO, Osorio RS, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11(8):457-470. © Springer Nature Publishing, 2015). Aβ, amyloid-β; CSF, cerebrospinal fluid; ISF: interstitial fluid; BBB, blood-brain barrier.
The exact pathophysiological link between Aβ and the second pathological hallmark of AD, hyperphosphorylated tau, is still largely unknown. At the same time, there is a growing body of evidence suggesting that tau hyperphosphorylation and formation of neurofibrils are secondary events, which are catalysed by Aβ.34 In vitro experiments using different cell types (for example, primary cortical or hippocampal neurons and hippocampal organotypic cultures) support the notion of Aβ-induced changes of tau. For example, there is evidence that Aβ oligomers promote tau phosphorylation35 and induce oxidative damage.36 An increasing number of animal experiments are also in support of tau pathology induced by Aβ. Even though initial AD mouse models expressing mutant APP without overexpression of tau did not have significant tau aggregation or neurofibrils, subtle changes of endogenous tau related to increased Aβ included hyperphosphorylated tau. Moreover, mouse AD models with high senile plaque load consistently showed dystrophic neurites with hyperphosphorylated tau around the plaque edges.37
Current treatment options
Treatments used for specific disorders reflect the pharmacological and pathophysiological understanding of their time, and AD is no exception in this regard. Initial attempts to treat AD were not related to any of the currently known core pathophysiological abnormalities, but were merely targeting unspecific suspected disease mechanisms, including brain metabolism and perfusion.38 Subsequently, drugs were developed, constituting the current generation of treatment options, which aim at rectifying the biochemical consequences of nerve cell loss in specific neuron populations, still without any impact on the underlying pathophysiology.
Neurodegeneration in AD involves subcortical brain regions, which includes the locus coeruleus,39 the dorsal raphe nuclei, and the basal forebrain40; the associated loss of neurons results in deficits of the neurotransmitters acetylcholine, serotonin, and norepinephrine, contributing to the progressive impairment of higher functions such as memory, attention, behavior, and mood. Currently, two symptomatic treatment approaches are available for AD. Acetylcholinesterase inhibitors (tacrine, donepezil, galantamine, and rivastigmine) aim to improve the cortical concentration of acetylcholine, which is reduced due to loss of neurons in the basal forebrain nuclei (nucleus basalis Meynert).41 Similar effects are seen in nicotinic acetylcholine receptor sensitizers (galantamine).42 The symptomatic effect of memantine is ascribed to a different mechanism of action, which targets the excessive release of glutamate occurring as a result of cortical neuronal loss, by improving the signal to noise ratio of glutamalergic transmission and potentially protecting neurons to a certain degree from the toxic effects of chronically increased exposition to glutamate.43 The clinical benefits of these drugs are related to a delay of progression of symptoms over several months, but an inconsistent impact on everyday function44 and other relevant outcome measures such as behavioral and psychological symptoms of dementia (BPSD).45 Furthermore, effectiveness has only been shown in the dementia stage of AD, not in prodromal stages.46 New symptomatic drugs are still being developed and a combination of disease-modifying and symptomatic treatments may be an effective means in the future in individuals already suffering from minor AD symptoms. A review of the 2018 drug development pipeline for AD showed that most compounds currently being scrutinized in clinical trials have disease-modifying properties (63% across phases 1 to 3), followed by cognitive enhancers (23%), and drugs targeting BPSD (12%).
Novel drugs
A recent analysis of studies available on www.clinicaltrials.gov shows that in 2018 the current AD drug development pipeline across all study phases encompasses 112 compounds7, including 23 agents in 25 phase 1 trials, 63 agents in 75 phase 2 trials, and 26 agents in 35 phase 3 trials. This includes eight new drugs in phase 1, 14 in phase 2, and four in phase 3 compared with an analysis conducted in 2017.47 Biomarkers, mainly indicating Aβ status, are increasingly being used as study entry criterion, particularly for studies on disease-modifying agents. There is also a trend towards targeting earlier disease stages, ie, prodromal or preclinical AD. Most studies (14 phase 3 trials in 2018) target Aβ, but an increase of non-Aβ mechanisms of action of compounds in earlier stages of development is noted.
Among the different approaches targeted at Aβ, immunotherapy remains the best developed strategy, particularly passive immunization with monoclonal antibodies. Other strategics are being explored, including efforts to inhibit the activity of the APP cleaving enzymes β- and γ-secretase or Aβ aggregation, amongst others. Immunization trials had an ill-fated start with AN1792 (active immunization with full-length Aβ42), for which the development was terminated prematurely because of T cell mediated meningoencephalitis in 6% of the treated study population.48 Second-generation active vaccines use antibodies restricted to the N-terminus of Aβ, avoiding T cell epitopes at the C-terminus. So far only CAD106 has advanced to phase 3 clinical development and is being studied in the Alzheimer Prevention Initiative study in homozygous carriers of the apolipoprotein E (APOE) eε risk allele.49
Because of the better risk-benefit profile, passive anti-Aβ immunization appears to be more promising. Passive immunization allows antibody titers to be more stable and treatment can be stopped if severe adverse events occur. However, monoclonal antibodies are relatively expensive to produce and have to be administered repeatedly.50 Bapineuzumab (AAB-001) was the first monoclonal antibody that entered human studies, and the first passive immunization attempt that failed. Bapineuzumab is a humanized immunoglobulin G1 anti-Aβ monoclonal antibody, which binds the five N-terminal residues and clears both fibrillar and soluble Aβ. Bapineuzumab was generally safe and well-tolerated, but some participants receiving higher doses developed transient vasogenic edema, now referred to as ARIA (amyloid-related imaging abnormalities).51 All bapineuzumab trials were terminated in 2012 because no clinical effectiveness could be shown,52 despite reduction of fibrillar Aβ in PET and CSF studies in patients with AD receiving the drug.53 Further failures of monoclonal antibody studies followed in the subsequent years (eg, solanezumab).54 supporting the assumption that treatment should have been commenced earlier in the disease course, administered in higher doses, or that the wrong Aβ species were targeted. Ongoing research tries to remedy the shortcomings of previous studies by, for example, targeting preclinical AD populations to prevent neurodegeneration and associated symptoms.49 Improved Aβ clearance from the brain is a major aim of most ongoing trials.25
In addition to Aβ, tau remains an important treatment target for disease modification. Early attempts to influence tau aggregation largely failed, but some are currently being reevaluated.55 Similar to immunization targeted at Aβ, immunotherapy studies against tau have reached early clinical development phases. Unknowns remain, including which tau epitope to target, the question of extravs intracellular tau and the level of required target engagement.56 A growing number of compounds are aiming at targets related to tau, including neurofibrillary tangles consisting of microtubule associated hyperphosphorylated tau aggregates. The effectiveness of these strategies remains to be shown.
Prevention trials
A number of large cohorts have been established over the last decades in Europe and globally to prospectively study the effects of aging and neurodegeneration.57 Many of these studies collect important data on potentially modifiable risk factors of cognitive impairment and dementia, which are supported by epidemiological evidence. 5,58 The available information includes lifestyle and clinical information and in some cases neuroimaging and other biomarker data and biomaterial from large numbers of relevant individuals collected at various points in life. Continuing to follow up these individuals and harmonize data collection and analysis for relevant outcome measures (eg, genome-wide data, environmental exposures, neuroimaging data, blood and CSF proteins, etc) across individual cohorts is a powerful approach to fill existing knowledge gaps related to risk and protective factors and how they can be influenced by new intervention and treatment strategies. Examples of large-scale initiatives to effectively integrate information from diverse cohorts include the European Medical Information Framework for AD, the Dementias Platform UK, and the EU Joint Program for Neurodegenerative Disease Research (JPND) longitudinal cohort studies action group.58
Given the complex structure of risk and etiological factors of AD, a multimodal prevention approach seems advisable. Thirty-five percent of the dementia risk is attributable to the nine most important environmental risk factors, which are all potentially modifiable.5 Several of these factors have been studied in prevention trials aiming to reduce risk associated with physical inactivity.59 suboptimal diets.60 and limited cognitive stimulation,61 for example. Overall results were mixed, and the field has since moved to investigating the effectiveness of complex interventions, addressing several risk factors concurrently in the same study.
Examples for large ongoing dementia prevention initiatives, which apply a multidomain approach, include preDIVA (Prevention of Dementia by Intensive Vascular Care),62 MAPT (Multidomain Alzheimer Preventive Trial),63 FINGER (Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability),64 and HATICE (Healthy Ageing Through Internet Counselling in the Elderly).65 While, for example, FINGER showed positive effects of a 2-year multidomain intervention (consisting of diet, exercise, cognitive training, and vascular risk monitoring) on cognitive performance compared with general health advice, in preDIVA intensive vascular care was not proven to effectively prevent dementia, except in a posthoc analysis of a subgroup with untreated hypertension, and in MAPT a multimodal lifestyle intervention combined with a nutritional supplement for dementia prevention was unsuccessful as well. It still has to be clarified if certain lifestyle interventions are biologically ineffective or if lack of efficacy is caused by inappropriate methods, particularly related to participant selection, intervention intensity, and adherence.
Conclusions
Despite the disappointment associated with the failed AD treatment trials, the field has learned significant lessons in the last few years. It is now clear that monoclonal antibodies vary considerably in how they interact with Aβ forms. These characteristics impact on Aβ clearance across the BBB and are also important for potential side effects such as ARIA. Starting trials earlier in the disease course, when neurodegenerative damage is absent or minimal, will open a window of opportunity for preventive interventions; this approach will be aided by biomarkers with improved sensitivity in preclinical disease stages, which are ideally associated with less invasive procedures and lower costs. Initial encouraging results with high dose Aducanumab indicate that higher antibody doses may offer promise. Clinical effects of titration dosing seem to be equivalent to fixed dosing, but with a better safety profile with lower incidence of ARIA.66
A concern with the current AD drug development pipeline is that the strong focus on the early disease stages has resulted in a lack of new compounds targeting the later (dementia) stages.7 Only a few ongoing studies allow the inclusion of individuals with moderate AD dementia. The availability of disease-modifying drugs will not completely eliminate the need for symptomatic treatment options, which can also be used in more advanced disease stages. Otherwise, millions of people worldwide will be left to suffer from dementia without any prospect for improved care.
The lack of appropriate surrogate biomarker end points, which could be used to substitute clinical end points, is a major problem both for pharmacological and nonpharmacologic intervention studies. Misdiagnosis rates in AD trials can reach 20% of all included cases,67 which have probably contributed to previous failed trials. A surprisingly high percentage of currently recruiting treatment trials do not employ biomarker inclusion criteria or biomarker (secondary) end points. Several new research frameworks for AD classification have recently been proposed, which all have a strong emphasis on biomarkers. However, the clinical utility of those criteria has yet to be proven.68 Due to the decades-long preclinical stage of AD, surrogate biological end points are also urgently needed to make lifestyle intervention studies more feasible, which would otherwise have to be conducted over many years and result in high costs.
The success of treatment trials depends on the effectiveness of recruitment strategies, and the requirement for effective recruitment has become even more acute for large-scale prevention studies, which include large numbers of cognitive intact individuals followed longitudinally. Different types of clinical readiness registries, which collect different amounts of health-related data, have been established in the last few years.69 The over-arching purpose of these registries is to identify interested individuals who can be assessed for appropriateness for clinical trials and enlist them if they have the prespecified biomarker profile required for trial participation. Optimizing the use of registries to enhance trial recruitment will be among the important lessons from studying the current registries.
Nonpharmacological prevention studies use more sophisticated methods than in the past, including evidence-based multimodal intervention programs. However, results so far have been mixed and it is not clear if reduced vascular risk scores, for example, also reduce risk for cognitive decline and dementia. Ongoing research on the biological effects of the studied interventions may help to improve the design of the prevention trials.
Acknowledgments
The author reports no conflicts of interest and no sources of funding.
REFERENCES
- 1.World Alzheimer report - the global impact of dementia. Alzheimer's Disease International, London; 2015 [Google Scholar]
- 2.Imhof A, Kovari E, von Gunten A, et al. Morphological substrates of cognitive decline in nonagenarians and centenarians: a new paradigm? J Neuro Sci. 2007;257(1-2):72–79. doi: 10.1016/j.jns.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 3.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Hard Perspect Med. 2011;1(1):a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foster JK, Verdile G, Bates KA, Martins RN. Immunization in Alzheimer's disease: naive hope or realistic clinical potential? Mol Psychiatry. 2009;14(3):239–251. doi: 10.1038/mp.2008.115. [DOI] [PubMed] [Google Scholar]
- 5.Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113):2673–2734. doi: 10.1016/S0140-6736(17)31363-6. [DOI] [PubMed] [Google Scholar]
- 6.Kemppainen N, Johansson J, Teuho J, et al. Brain amyloid load and its associations with cognition and vascular risk factors in FINGER Study. Neurology. 2018;90(3):e206–e213. doi: 10.1212/WNL.0000000000004827. [DOI] [PubMed] [Google Scholar]
- 7.Cummings J, Lee G, Ritter A, Zhong K. Alzheimer's disease drag development pipeline: 2018. Alzheimers Dement (N Y). 2018;4:195–214. doi: 10.1016/j.trci.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamann J, Bronner K, Margull J, et al. Patient participation in medical and social decisions in Alzheimer's disease. J Am Geriatr Soc. 2011;59(11):2045–2052. doi: 10.1111/j.1532-5415.2011.03661.x. [DOI] [PubMed] [Google Scholar]
- 9.Holt GR. Timely diagnosis and disclosure of Alzheimer disease gives patients opportunities to make choices. South Med J. 2011;104(12):779–780. doi: 10.1097/SMJ.0b013e3182389599. [DOI] [PubMed] [Google Scholar]
- 10.Garand L, Dew MA, Lingler JH, DeKosky ST. Incidence and predictors of advance care planning among persons with cognitive impairment. Am J Geriatr Psychiatry. 2011;19(8):712–720. doi: 10.1097/JGP.0b013e3181faebef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erlangsen A, Zarit SH, Conwell Y. Hospital-diagnosed dementia and suicide: a longitudinal study using prospective, nationwide register data. Am J Geriatr Psychiatry. 2008;16(3):220–228. doi: 10.1097/JGP.0b013e3181602a12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ritchie C, Smailagic N, Noel-Storr AH, Ukoumunne O, Ladds EC, Martin S. CSF tau and the CSF tau/ABeta ratio for the diagnosis of Alzheimer's disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst Rev. 2017;3:CD010803. doi: 10.1002/14651858.CD010803.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta-analysis. Lancet Neurol. 2016;15(7):673–684. doi: 10.1016/S1474-4422(16)00070-3. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid-beta biomarkers for Alzheimer's disease. Nature. 2018;554(7691):249–254. doi: 10.1038/nature25456. [DOI] [PubMed] [Google Scholar]
- 15.Verberk IMW, Slot RE, Verfaillie SCI, et al. Plasma amyloid as pre-screener for the earliest Alzheimer's pathological changes. Ann Neurol. 2018 doi: 10.1002/ana.25334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hardy J. The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem. 2009;110:1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- 17.Jakob-Roetne R, Jacobsen H. Alzheimer's disease: From pathology to therapeutic approaches. Angew Chem Int Ed. 2009;48:3030–3059. doi: 10.1002/anie.200802808. [DOI] [PubMed] [Google Scholar]
- 18.Nimmrich V, Ebert U. Is Alzheimer's disease a result of presynaptic failure? Synaptic dysfunctions induced by oligomeric beta-amyloid. Rev Neurosci. 2009;20:1–12. doi: 10.1515/revneuro.2009.20.1.1. [DOI] [PubMed] [Google Scholar]
- 19.Chaturvedi RK, Beal MF. Mitochondrial approaches for neuroprotection. Ann NY Acad Sci. 2008;1147:395–412. doi: 10.1196/annals.1427.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eikelenboom P, Veerhuis R, Scheper W, Rozemuller AJM, van-Gool WA, Hoozemans JJM. The significance of neuroinflammation in understanding Alzheimer's disease. J Neural Transm. 2006;113:1685–1695. doi: 10.1007/s00702-006-0575-6. [DOI] [PubMed] [Google Scholar]
- 21.Fan Z, Brooks DJ, Okello A, Edison P. An early and late peak in microglial activation in Alzheimer's disease trajectory. Brain. 2017;140(3):792–803. doi: 10.1093/brain/aww349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arendt T. Synaptic degeneration in Alzheimer's disease. Acta Neuropathol. 2009;118(1):167–179. doi: 10.1007/s00401-009-0536-x. [DOI] [PubMed] [Google Scholar]
- 23.Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer's disease. CNS Neurol Dis Drug Targets. 2009;8:16–30. doi: 10.2174/187152709787601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Srikanth V, Maczurek A, Phan T, et al. Advanced glycation end products and their receptor RAGE in Alzheimer's disease. Neurobiol Aging. 2009;May21 doi: 10.1016/j.neurobiolaging.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 25.Kurz A, Perneczky R. Amyloid clearance as a treatment target against Alzheimer's disease. J Alzheimers Dis. 2011;24 Suppl 2:61–73. doi: 10.3233/JAD-2011-102139. [DOI] [PubMed] [Google Scholar]
- 26.Nedergaard M. Neuroscience. Garbage truck of the brain. Science. 2013;340(6140):1529–1530. doi: 10.1126/science.1240514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jha NK, Jha SK, Kumar D, et al. Impact of insulin degrading enzyme and neprilysin in Alzheimer's disease biology: characterization of putative cognates for therapeutic applications. J Alzheimers Dis. 2015;48(4):891–917. doi: 10.3233/JAD-150379. [DOI] [PubMed] [Google Scholar]
- 28.Eide PK, Vatnehol SAS, Emblem KE, Ringstad G. Magnetic resonance imaging provides evidence of glymphatic drainage from human brain to cervical lymph nodes. Sci Rep. 2018;8(1):7194. doi: 10.1038/s41598-018-25666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rainey-Smith SR, Mazzucchelli GN, Villemagne VL, et al. Genetic variation in Aqua porin-4 moderates the relationship between sleep and brain Abeta-amyloid burden. Transl Psychiatry. 2018;8(1):47. doi: 10.1038/s41398-018-0094-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Musiek ES, Holtzman DM. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science. 2016;354(6315):1004–1008. doi: 10.1126/science.aah4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iliff JJ, Wang M, Zeppenfeld DM, et al. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci. 2013;33(46):18190–18199. doi: 10.1523/JNEUROSCI.1592-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kress BT, Iliff JJ, Xia M, et al. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 2014;76(6):845–861. doi: 10.1002/ana.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muszynski P, Kulczynska-Przybik A, Borawska R, et al. The relationship between markers of inflammation and degeneration in the central nervous system and the blood-brain barrier impairment in Alzheimer's disease. J Alzheimers Dis. 2017;59(3):903–912. doi: 10.3233/JAD-170220. [DOI] [PubMed] [Google Scholar]
- 34.Duyckaerts C, Hauw JJ. Prevalence, incidence and duration of Braak's stages in the general population: Can we know? Neurobiol Aging. 1997;18:362–369. doi: 10.1016/s0197-4580(97)00047-x. [DOI] [PubMed] [Google Scholar]
- 35.De-Felice FG, Wu D, Lambert MP, et al. Alzheimer's disease-type neuronal tau hyperphosphorylation induced by Abeta oligomers. Neurobiol Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sultana R, Peruigi M, Butterfield DA. Oxidatively modified proteins in Alzheimer's disease (AD), mild cognitive impairment and animal models of AD: role of Abeta in pathogenesis. Ada Neuropathol. 2009;118:131–150. doi: 10.1007/s00401-009-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis J, Dickson DW, Lin WL, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293(5534):1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 38.Waters C. Cognitive enhancing agents: current status in the treatment of Alzheimer's disease. Can J Neurol Sci. 1988;15:249–256. doi: 10.1017/s0317167100027694. [DOI] [PubMed] [Google Scholar]
- 39.Weinshenker D. Functional consequences of locus coeruleus degeneration in Alzheimer's disease. Curr Alzheimer Res. 2008;5:342–345. doi: 10.2174/156720508784533286. [DOI] [PubMed] [Google Scholar]
- 40.Schliebs R, Arendt T. The significance of the cholinergic system in the brain during aging and in Alzheimer's disase. J Neural Transm. 2006;113:1625–1644. doi: 10.1007/s00702-006-0579-2. [DOI] [PubMed] [Google Scholar]
- 41.Bartus RT, Dean RL, Beer B, Lippa AS. The cholinergic hpothesis of geriatric memory dysfunction. Science. 1982;217:408–417. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- 42.Maelicke A. Allosteric modulation of nicotinic receptors as a treatment strategy for Alzheimer's disease. Dement Geriatr Cogn Disord. 2000;11(suppl 1):11–18. doi: 10.1159/000051227. [DOI] [PubMed] [Google Scholar]
- 43.Greenamyre JT, Maragos EF, Albin RL, Penney JB, Young AB. Glutamate transmission and toxicity in Alzheimer's disease. Prog Neuro-Psych Biol Psych. 1988;12:421–430. doi: 10.1016/0278-5846(88)90102-9. [DOI] [PubMed] [Google Scholar]
- 44.Farlow MR, Pejovic MLMV. Treatment options in Alzheimer's disease: maximizing benefit, managing expectations. Dement Geriatr Cogn Disord. 2008;25:408–422. doi: 10.1159/000122962. [DOI] [PubMed] [Google Scholar]
- 45.Grimmer T, Kurz A. Effects of cholinestorase inhibitors on behavioural disturbances in Alzheimer's disease. A systematic review. Drugs Aging. 2006;23:957–967. doi: 10.2165/00002512-200623120-00003. [DOI] [PubMed] [Google Scholar]
- 46.Raschetti R, Albanese E, Vanacore N, Maggini M. Cholinesterase inhibitors in mild cognitive impairment: A systgematic review of randomised trials. PLoS Med. 2007;4:1818–1828. doi: 10.1371/journal.pmed.0040338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cummings J, Lee G, Mortsdorf T, Ritter A, Zhong K. Alzheimer's disease drug development pipeline: 2017. Alzheimers Dement (N Y). 2017;3(3):367–384. doi: 10.1016/j.trci.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilman S, Koller M, Black RS, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64(9):1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 49.Reiman EM, Langbaum JB, Fleisher AS, et al. Alzheimer's Prevention Initiative: a plan to accelerate the evaluation of prosymptomatic treatments. J Alzheimers Dis. 2011;26 Suppl 3:321–329. doi: 10.3233/JAD-2011-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lemere CA. Immunotherapy for Alzheimer's disease: hoops and hurdles. Mol Neurodegener. 2013;8:36. doi: 10.1186/1750-1326-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sperling RA, Jack CR, Jr., Black SE, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7(4):367–385. doi: 10.1016/j.jalz.2011.05.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N Engl J Med. 2014;370(4):322–333. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu E, Schmidt ME, Margolin R, et al. Amyloid-beta 11C-PiB-PET imaging results from 2 randomized bapineuzumab phase 3 AD trials. Neurology. 2015;85(8):692–700. doi: 10.1212/WNL.0000000000001877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Honig LS, Vellas B, Woodward M, et al. Trial of solanezumab for mild dementia due to Alzheimer's disease. N Engl J Med. 2018;378(4):321–330. doi: 10.1056/NEJMoa1705971. [DOI] [PubMed] [Google Scholar]
- 55.Bakota L, Brandt R. Tau Biology and tau-directed therapies for Alzheimer's disease. Drugs. 2016;76(3):301–313. doi: 10.1007/s40265-015-0529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pedersen JT, Sigurdsson EM. Tau immunotherapy for Alzheimer's disease. Trends Mol Med. 2015;21(6):394–402. doi: 10.1016/j.molmed.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 57.Longitudinal cohort studies in neurodegeneration research: Report of the JPND action group. 2013 [Google Scholar]
- 58.World Alzheimer report - dementia and risk reduction. Alzheimer's Disease International. London; 2014 [Google Scholar]
- 59.Lautenschlager NT, Cox KL, Flicker L, et al. Effect of physical activity on cognitive function in older adults at risk for Alzheimer disease: a randomized trial. JAMA. 2008;300(9):1027–1037. doi: 10.1001/jama.300.9.1027. [DOI] [PubMed] [Google Scholar]
- 60.Soininen H, Solomon A, Visser PJ, et al. 24-month intervention with a specific multinutrient in people with prodromal Alzheimer's disease (LipiDiDiot): a randomised, double-blind, controlled trial. Lancet Neurol. 2017;16(12):965–975. doi: 10.1016/S1474-4422(17)30332-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fotuhi M, Lubinski B, Trullinger M, et al. A personalized 12-wcek “Brain Fitness Program” for improving cognitive function and increasing the volume of hippocampus in elderly with mild cognitive impairment. J Prev Alzheimers Dis. 2016;3(3):133–137. doi: 10.14283/jpad.2016.92. [DOI] [PubMed] [Google Scholar]
- 62.Moll van Charante EP, Richard E, Eurelings LS, et al. Effectiveness ofa 6-year multidomain vascular care intervention to prevent dementia (preDIVA): a cluster-randomised controlled trial. Lancet. 2016;388(10046):797–805. doi: 10.1016/S0140-6736(16)30950-3. [DOI] [PubMed] [Google Scholar]
- 63.Andrieu S, Guyonnet S, Coley N, et al. Effect of long-term omega 3 polyunsaturated fatty acid supplementation with or without multidomain intervention on cognitive function in elderly adults with memory complaints (MAPT): a randomised, placebo-controlled trial. Lancet Neurol. 2017;16(5):377–389. doi: 10.1016/S1474-4422(17)30040-6. [DOI] [PubMed] [Google Scholar]
- 64.Ngandu T, Lehtisalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255–2263. doi: 10.1016/S0140-6736(15)60461-5. [DOI] [PubMed] [Google Scholar]
- 65.Barbera M, Mangialasche F, Jongstra S, et al. Designing an internet-based multidomain intervention for the prevention of cardiovascular disease and cognitive impairment in older adults: The HATICE Trial. J Alzheimers Dis. 2018;62(2):649–663. doi: 10.3233/JAD-170858. [DOI] [PubMed] [Google Scholar]
- 66.Viglietta V, O'Gorman J, Williams L, et al. Titration dosing of aducanumab: results of a 12-month interim analysis from a randomized, double-blind, placebo-controlled Phase 1b study (PRIME) in patients with prodromal or mild Alzheimer's Disease. Neurology. 2017;88(Suppl 16) [Google Scholar]
- 67.Doody RS, Farlow M, Aisen PS. Alzheimer's Disease Cooperative Study Data A, Publication C. Phase 3 trials of solanezumab and bapineuzumab for Alzheimer's disease. N Engl J Med. 2014;370(15):1460. doi: 10.1056/NEJMc1402193. [DOI] [PubMed] [Google Scholar]
- 68.Illan-Gala I, Pegueroles J, Montal V, et al. Challenges associated with biomarker-based classification systems for Alzheimer's disease. Alzheimers Dement (Amst). 2018;10:346–357. doi: 10.1016/j.dadm.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Larsen ME, Curry L, Mastellos N, Robb C, Car J, Middleton LT. Development of the CHARIOT research register for the prevention of Alzheimer's dementia and other late onset neurodegenerative diseases. PLoS One. 2015;10(11):e0141806. doi: 10.1371/journal.pone.0141806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tarasoff-Conway JM, Carare RO, Osorio RS, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11(8):457–470. doi: 10.1038/nrneurol.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]