

Abstract
We performed a drug repurposing screening of a US Food and Drug Administration (FDA)-approved drug compound library and identified Anagrelide (ANA), a known phosphodiesterase 3A (PDE3A) inhibitor, that selectively and potently inhibited the growth of cancer cells. However, inactivation of PDE3A or knocking-down its gene expression did not inhibit cancer cell growth. It was the interaction of ANA with PDE3A that created a new function of PDE3A to alter the activities of another unknown function protein SLFN12 to cause the inhibition of cancer cell growth. The expressions of both PDE3A and SLFN12 were required for ANA to inhibit cancer cell growth. Depletion of PDE3A or SLFN12 led to ANA resistance. Furthermore, the effects of ANA on different cancer cells were different depending on the expression levels of PDE3A and SLFN12, causing G0/G1 cell cycle arrest in the cells expressing lower levels of SLFN12, but apoptosis in the cells expressing higher levels of the two proteins. Further investigation into the molecular mechanisms of the ANA-induced cell cycle arrest and apoptosis identified a set of cell cycle and apoptosis-related genes whose expressions were altered by ANA treatment. ANA also synergized with the cell death-inducing cytokines IFN-α, IFN-γ, TNF-α, or TRAIL, which regulated the same set of genes as ANA did, to induce apoptosis of the cancer cells. Our study uncovered new activities, functions, and mechanisms of ANA and SLFN12 and provided a diagnosis method to precisely use ANA as an anti-cancer drug. It also revealed PDE3A and SLFN12 as new anti-cancer drug targets for developing novel anti-cancer therapies.
Keywords: Anagrelide, PDE3A, SLFN12, cancer, cytokine
Introduction
Cancer is an individualized disease characterized by specific genetic alterations coupled with activations of specific complex cell growth signaling networks in specific cancer cells [1,2]. Therefore, targeted therapies based on specific genetic mutations of oncogenes and specific alterations of signaling pathways in specific cancer cells have been rapidly developed [3-6]. There have been many efforts made to develop new drugs targeting key drivers of carcinogenesis for personalized treatment of cancers [7-10]. One of the most efficient and economical ways to identify a reagent that can be put into clinic use directly is drug repositioning, i.e., the discoveries of new bioactivities of old drugs against new targets and pathways or generating new activities from the same target involved in other diseases. This strategy provides a promising way to find new therapeutic applications for old drugs which have been fully assessed for clinical safety and bioavailability [11-15]. The most recognizable example of drug repositioning is Sildenafil, which was originally developed as an anti-hypertensive drug and now has been repurposed for the treatments of pulmonary arterial hypertension and erectile dysfunction [16,17].
Anagrelide (ANA) is a cyclic nucleotide phosphodiesterase 3 (PDE3) inhibitor and a marketed drug used for the treatment of essential thrombocytosis (essential thrombocythemia), or overproduction of blood platelets by inhibiting the maturation of megakaryocytes into platelets [18,19]. The link between the inhibition of PDE3 and the inhibition of maturation of megakaryocytes has not been fully understood. It also has been used in the treatment of chronic myeloid leukemia, but the mechanisms are unknown [20,21]. Its use in the treatment of solid cancers has not been reported.
Cyclic nucleotide phosphodiesterase (PDE) family proteins, composed of 11 structurally related but functionally distinct members (PDE1 to PDE11), regulate intracellular concentrations of cyclic nucleotides by catalyzing the hydrolyses of second messengers cyclic AMP (cAMP) and cyclic GMP (cGMP), orchestrating many important intracellular signal transductions and cellular activities [22-24].
PDE3 is one member of the PDE family and has two subtypes, PDE3A and PDE3B [25]. PDE3A is mainly expressed in heart, platelet, vascular smooth muscle, and oocytes and is involved in oocyte maturation and platelet aggregation [26,27]. Its inhibitors, such as cilostamide and milrinone, have been used to treat heart failure, intermittent claudication, and platelet aggregation [28,29]. PDE3B is mostly expressed in white and brown adipose cells, hepatocytes, renal collecting duct epithelium, and developing spermatocytes [30] and is important for the regulation of glucose and lipid metabolism [31]. Recently, the PDE3 inhibitors have also been found effective against malignant tumors [32]. Cilostamide, for example, a PDE3-specific inhibitor, inhibits the proliferation of human squamous cell carcinoma KB cells [33]. Another PDE3 inhibitor DNMDP selectively inhibits cancer cell growth through promoting interactions between PDE3A and Schlafen 12 (SLFN12) [34].
SLFN12 belongs to the schlafen family and was firstly identified as a regulator for thymocyte maturation [35,36]. SLFN members are classified into three different groups based on their sizes and domain compositions. All of the SLFN members share a common N-terminal AAA domain, which is involved in GTP/ATP binding, and a SLFN-box. However, group II and III contain a unique motif, consisting of a Ser-Trp-Ala-Asp-Leu sequence. Group III has an additional C-terminal domain homologous to the DNA/RNA helicases of superfamily. There are 5 human SLFN isoforms: SLFN5, SLFN11, SLFN12, SLFN13, and SLFN14. Human SLFN12 belongs to the group II category and is the only human cytoplasmic SLFN. The remaining human SLFNs harbor a helicase domain as well as a nuclear localization signal, placing them in group III [37,38]. Although the SLFNs have been identified over 20 years, their precise biological functions remain largely unknown. It has been reported that human SLFN5 negatively regulates anchorage-independent growth and invasion of malignant melanoma cells [39]. In the recent years, SLFN11 has become a new research hotspot. It has been shown to regulate the activities of DNA-damaging agents in cancer cells [40,41], inhibit human immunodeficiency virus 1 (HIV-1) [42], and function in drug resistances in human cancers [43,44]. The functions of SLFN12, however, are largely unknown. One study reported that SLFN12 modulated differentiation of prostate cancer cells by increasing DPP4 expressing while suppressing PSA expression [45]. Recently, SLFN12 was reported to be a key protein involved in the growth inhibition of cancer cells by a PDE3 inhibitor DNMDP [46,47]. SLFN12 could be co-immunoprecipitated with PDE3A after DNMDP treatment [47].
In this study, we performed a drug repurposing screening of an US FDA-approved drug library and identified anagrelide (ANA) as a potent and selective inhibitor of specific types of cancer cells. Its interaction with PDE3A generated a new function of PDE3A to interact with SLFN12 to alter the expressions of a set of genes related to cell cycle progression and cell survival/death, leading to cell cycle arrest and apoptosis of cancer cells expressing the two proteins. Furthermore, ANA synergized with cell death-inducing cytokines IFN-α, IFN-γ, or TRAIL to induce the deaths of the cancer cells. Our study uncovered new functions and mechanisms of ANA, PDE3A, and SLFN12 and provided a basis for using ANA as an anti-cancer drug. It also revealed PDE3A and SLFN12 as new anti-cancer drug targets and a new strategy to develop novel anti-cancer drugs.
Results
PDE3 inhibitor anagrelide potently and selectively inhibited growth of cancer cells
To identify anticancer compounds from existing drugs that selectively inhibit cancer cell growth, we screened 640 compounds from an US FDA-approved drug library against a panel of 31 different types of cancer cell lines at a single dose of 10 μM. Two compounds, Anagrelide (ANA) and Zardaverine (ZARD), were identified that selectively inhibited the growth of a subset of the human cancer cell lines (Figure 1A). The selectivity of ANA in cancer cells was shown in Figure 1B and in Supplementary Figure 1A. The IC50s of ANA for the sensitive cell lines were shown in Figure 1C, ranging from 5 nM to 50 nM.
Figure 1.
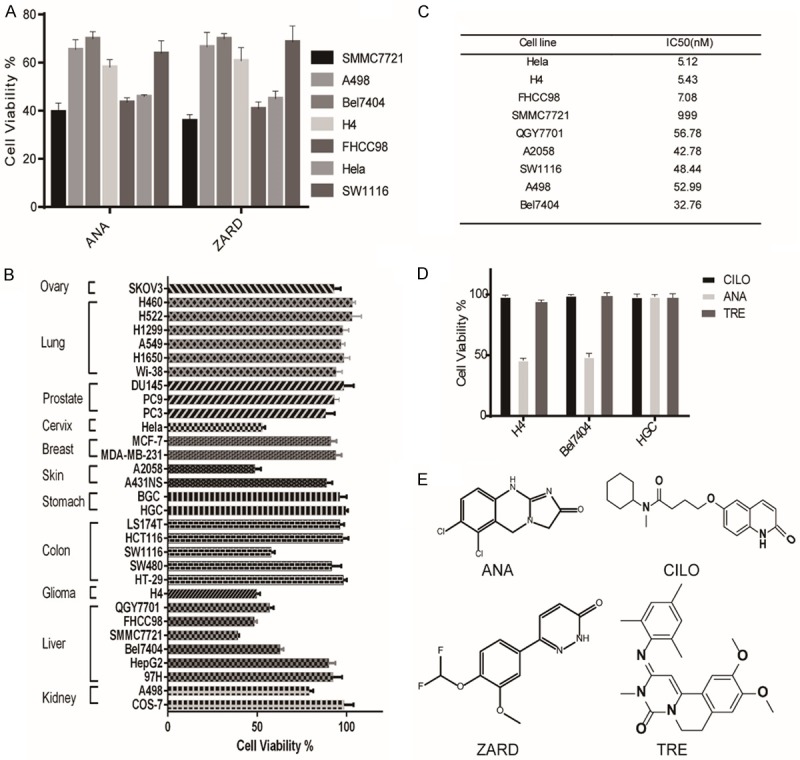
Anagrelide (ANA) selectively inhibited growth of cancer cells from different tissues. A. Effects of ANA and ZARD on the growth of different types of cancer cells. The indicated cancer cell lines were cultured with 10 µM ANA or ZARD for 72 h and cell growths were measured by the MTT assay. B. Effects of ANA (100 nM) on the growth of different types of cancer cell lines. 31 indicated cancer cell lines were cultured with 100 nM ANA for 72 h and the cell growths were measured by the MTT assay. C. IC50 values of ANA in the ANA-sensitive cell lines. All data were presented as mean ± s.d. (n = 3). D. Comparison of the effects of PDE3 inhibitors Anagrelide (ANA), Trequinsin (TRE), Cilostamide (CILO) on the growths of the cancer cells. The cells were cultured with the indicated PDE3 inhibitors (1 μM) for 72 h and cell growths were measured by the MTT assay. E. The chemical structures of PDE3 inhibitors.
Both ANA and ZARD are US FDA-approved PDE3 inhibitors. Inhibition of the cancer cell growth by both of the PDE3 inhibitors suggested that inhibition of PDE3 might be the key to inhibit the cancer cell growth. We therefore examined two additional PDE3 inhibitors, Trequinsin (TRE) and Cilostamide (CILO), for their effects on the cancer cell growth. Surprisingly, these two PDE3 inhibitors, although inhibit PDE3 with similar potency as that of ANA and ZARD, did not inhibit the cancer cell growth even at much higher concentrations (Figure 1D). These data suggested that inhibition of PDE3 enzymatic activity may not be the major reason to inhibit cancer cell growth.
ANA selectively induced G1 cell cycle arrest and apoptosis in cancer cells
To determine the nature of the ANA-induced cell growth inhibition, we analyzed the effects of ANA on cell cycle and cell death of the various cancer cells. We first analyzed the effects of ANA on cell morphologies under the microscope. Exposure of the cancer cells to ANA for 36 hours led to complete cell death in the H4 (Figure 2A), SMMC7721, Hela, FHCC98, A2058, and QGY7701 cells (data not shown) and reduced cell number in the Bel7404 (Figure 2A), SW1116, and A498 cells (data not shown), but had no obvious effects on the BGC cells (Figure 2A), H460, MDA-MB-231, and SKOV3 cells (data not shown). Similar results were obtained by using the Real-Time Cell Analyzer (RTCA) analyses (Figure 2B). The 31 cancer cell lines we analyzed were classified into three categories, the ANA death-sensitive cells such as H4, SMMC7721, Hela, FHCC98, A2058, and QGY7701 cell lines, in which the cell numbers were dramatically reduced, the ANA growth arrest-sensitive cells such as Bel7404, SW1116, and A498 cell lines, in which the cell number increase rates were reduced, or the ANA-insensitive cells such as H460, MDA-MB-231, SKOV3, and BGC cell lines, on which ANA had no effects (Figure 2B and Supplementary Figure 1B).
Figure 2.

ANA induced G1 cell cycle arrest in some cancer cells but apoptosis in others. (A) Effects of ANA on cell morphologies of different types of human cancer cells (× 200 magnifications). (B) RTCA analyses of the effects of ANA on the growths of different types of cancer cells. The indicated cancer cells were cultured with 100 nM ANA (green) or DMSO (red) and the cell growths were recorded continuously by RTCA. (C) Flow cytometry analyses of the effects of ANA on cell cycle progressions of the cancer cells. The indicated cells were treated with ANA (100 nM) for indicated time periods followed by Flow cytometry analyses. The percentage of cells in different phases of cell cycle were indicated. (D) Immunoblotting analyses of the effects of ANA on apoptosis of the cancer cells. The indicated cells were treated with ANA (100 nM) for indicated time periods and the whole cell lysates were processed for immunoblotting analyses using an anti-PARP antibody. The anti-tubulin antibody was used as a control for equal protein loading.
We then examined the effects of ANA on cell cycle progression and apoptosis of the cancer cells. We found that ANA blocked cell cycle at G0/G1 for both of the ANA death-sensitive and ANA growth arrest-sensitive cells, and induced apoptosis in the ANA death-sensitive cells, but had no effects on either cell cycle or apoptosis of the ANA-insensitive cells (Figure 2C and 2D). ANA also induced a slight amount of apoptosis in the growth-sensitive Bel7404 cells (Figure 2D), suggesting that it was a quantitative rather than a qualitative difference for the two types of cell lines.
PDE3A was necessary but not sufficient for ANA to inhibit cancer cell growth
The above data that some PDE3 inhibitors inhibited cancer cell growth while others did not raised the question whether PDE3 were the target for mediating the growth inhibitory effects of ANA. The PDE3 family has two subtypes, PDE3A and PDE3B. To understand the roles of the PDE3 in the ANA-induced cell growth inhibition, we firstly analyzed the expressions of PDE3A and PDE3B and their correlations with the sensitivities of the cells to ANA in various cell lines. 12 of the 31 cell lines analyzed expressed detective levels of PDE3A (Figure 3A). 9 of the 12 PDE3A-positive cell lines were ANA-sensitive, while all of the PDE3A-negative cells were ANA-insensitive. However, there were 3 cell lines which were PDE3A-positive but insensitive to ANA treatment, suggesting that PDE3A expression may be necessary but not sufficient for ANA to cause growth inhibition. PDE3B protein was detected in all of the cell lines we analyzed but had no correlation with the sensitivity to ANA (Figure 3A), suggesting that PDE3B was not involved in the ANA-induced cell growth inhibition.
Figure 3.
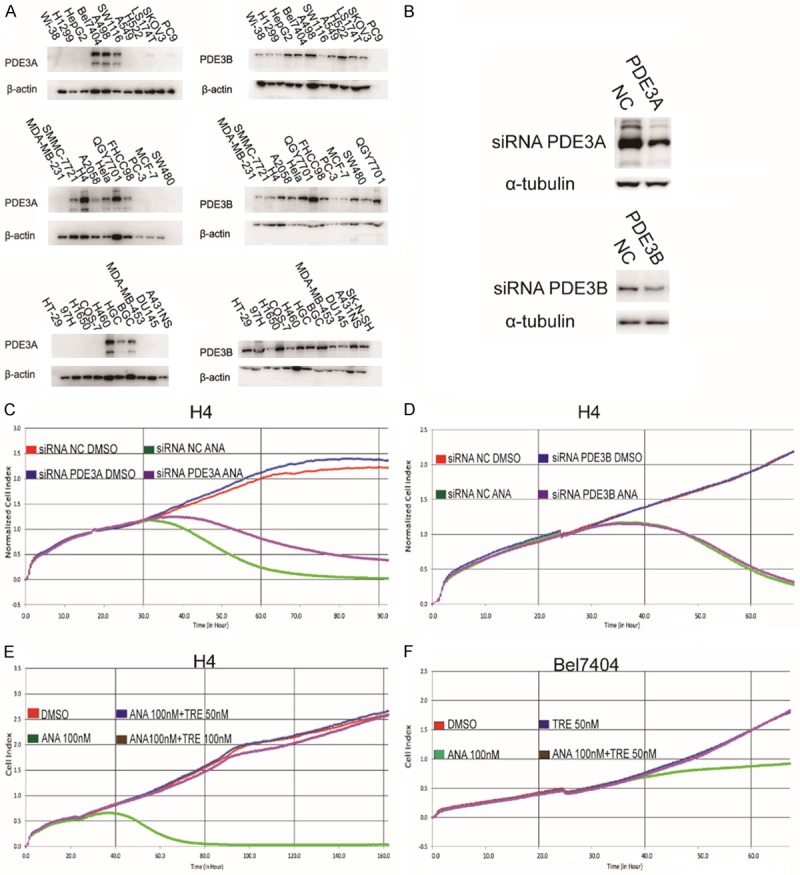
PDE3A was necessary but not sufficient for ANA to induce growth inhibition of cancer cells. (A) Immunoblotting analyses of PDE3A and PDE3B expressions in different cancer cell lines. The whole cell lysates of the indicated cells were analyzed by immunoblotting using the anti-PDE3A or anti-PDE3B antibody. The anti-tubulin or β-actin antibody was used as a control for equal protein loading. (B) Effects of siRNA knockdown on the protein expressions of PDE3A and PDE3B. H4 cells were transiently transfected with a PDE3A-specific siRNA, a PDE3B-specific siRNA, or a non-specific scrambled control siRNA. Cells were harvested 24 h later and the cell lysates were processed for immunoblotting using an anti-PDE3A, PDE3B, or α-tubulin antibody. (C and D) RTCA analyses of the effects of knocking-down PDE3A (C) or PDE3B (D) on the ANA-induced cell growth inhibition. H4 cells were transfected with PDE3A or PDE3B siRNA for 24 h, and the cells were then re-plated into 96-well plates and cultured with ANA (100 nM) or DMSO. Cell growths were recorded by RTCA. (E and F) RTCA analyses of the effects of combinational treatments of the PDE3 inhibitors on the growths of the cancer cells. The ANA death-sensitive cell line H4 and the ANA cell cycle arrest-sensitive cell line Bel7404 were treated with vehicle, ANA, TRE, or combination of ANA with TRE at the indicated doses, followed by RTCA analyses.
To confirm above observations, we knocked down the expression of PDE3A or PDE3B in the ANA death-sensitive cell line H4 by RNA interference (RNAi), in which the level of PDE3A or PDE3B was reduced about 50% (Figure 3B). The reduction of PDE3A or PDE3B expression did not influence the growth of the cells (Figure 3C and 3D). However, the growth inhibitory effects of ANA were alleviated by knocking down PDE3A, but not PDE3B (Figure 3C and 3D), further suggesting that PDE3A, but not PDE3B, was the mediator of the ANA-induced cell growth inhibition. ANA could not inhibit cancer cell growth without PDE3A.
To understand the functions of the other two PDE3 inhibitors that did not inhibit the tumor cell growth, we combined them respectively with ANA to treat the cancer cells. Interestingly, TRE or CILO completely blocked the growth inhibitory effects of ANA in both the death-sensitive H4 cells (Figure 3E and Supplementary Figure 2A) and the cell cycle arrest-sensitive Bel7404 cells (Figure 3F and Supplementary Figure 2B), suggesting that TRE or CILO might compete with ANA to interact with PDE3A. Excess TRE or CILO might block ANA to interact with PDE3A. It also confirmed that PDE3A was required for ANA to inhibit the cancer cell growth. In addition, ANA upregulated cAMP levels in three types of cancer cells (Supplementary Figure 2C), suggesting that the inhibition of PDE3A enzymatic activities and the induction of cAMP were not the mechanisms for the ANA-induced growth inhibition and apoptosis.
SLFN12 was required for the ANA-induced growth inhibition of cancer cells
SLFN12, an unknown function protein, has been reported to be involved in the cell growth inhibition by DNMDP, another PDE3 inhibitor. SLFN12 could be co-immunoprecipitated with PDE3A after DNMDP treatment, but not after TRE treatment [47]. To determine the roles of SLFN12 in the ANA-induced growth inhibition, we first analyzed SLFN12 mRNA expression in the 13 PDE3A-positive cancer cell lines. SLFN12 was highly expressed in the ANA death-sensitive cell lines and moderately expressed in the ANA cell cycle arrest-sensitive cell lines, but was not expressed in the ANA-insensitive cell lines. It seemed that the expression of SLFN12 strongly correlated with the sensitivity to ANA in the PDE3A-possitive cancer cell lines (Figure 4A). We then analyzed the effects of blocking SLFN12 expression on the ANA-induced growth inhibition by knocking down SLFN12 with its siRNAs in the ANA death-sensitive H4 cells (Figure 4B) and the ANA cell cycle arrest-sensitive Bel7404 cells (Supplementary Figure 3A-D). Similar to the results of reducing PDE3A expression, reduction of SLFN12 could also alleviate the growth inhibitory effects of ANA and abrogated the G1 arrest in the cell cycle arrest-sensitive cell lines (Figure 4C and Supplementary Figure 3E). In addition, reduction of PDE3A and SLFN12 had a synergetic effect on the alleviation (Figure 4D). Knocking-down SLFN12 expression had no obvious effects on PDE3A expression, and vice versa (Supplementary Figure 3F and 3G).
Figure 4.
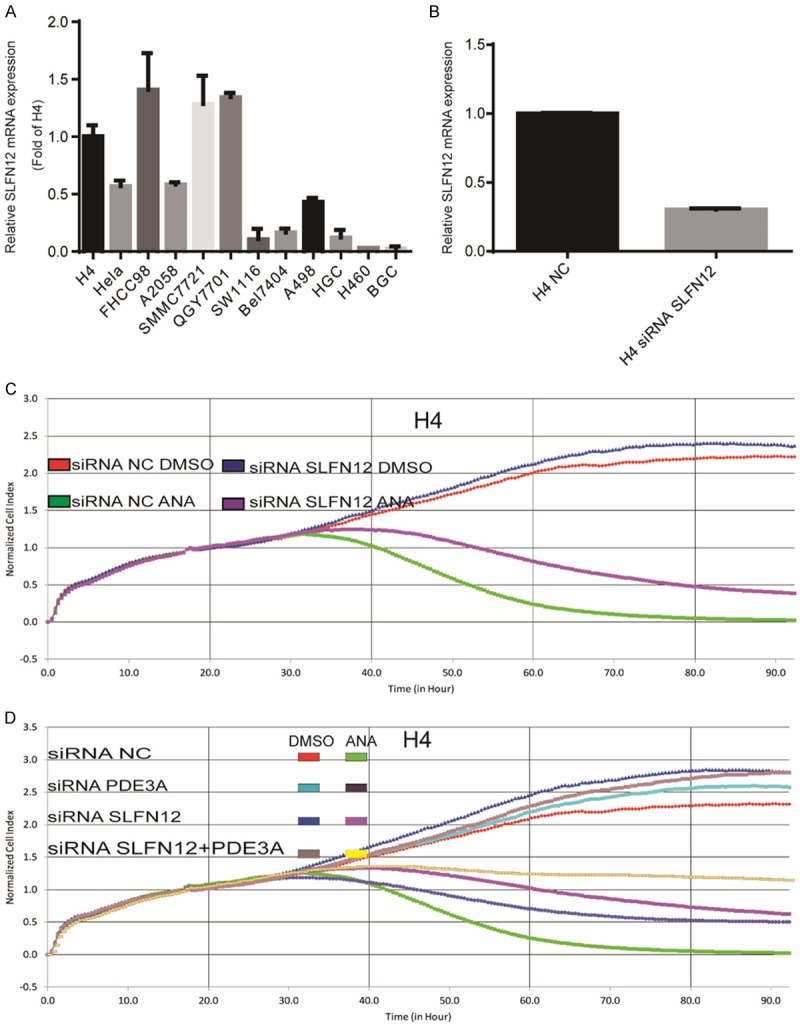
SLFN12 was required for the ANA-induced growth inhibition of cancer cells. A. Quantitative Real-Time PCR analyses of SLFN12 mRNA expression in 13 cancer cell lines. The mRNA levels were normalized to the GAPDH mRNA expression. B. Effects of SLFN12 siRNA on the mRNA expression of SLFN12. The H4 cells were transiently transfected with the SLFN12-specific siRNAs targeting three different sequences or non-specific scrambled siRNA. SLFN12 mRNA expression was measured by real-time PCR 48 h after siRNA transfection. C. RTCA analyses of the effects of knocking down SLFN12 expression on the ANA-induced growth inhibition. The siRNA-transfected H4 cells were cultured with ANA (100 nM) or DMSO and the growth of the cells were recorded by RTCA. D. The H4 cells were transiently transfected with the SLFN12-specific siRNA, PDE3A-specific siRNA, or a combination of the two with or without ANA treatments and the cell growths were recorded by RTCA.
ANA altered the expressions of genes regulating cell cycle and death through SLFN12
In order to understand the events and mechanisms downstream of the ANA-PDE3A-SLFN12 interactions, we examined the expressions of key cell cycle and apoptosis-related genes after ANA treatment. We found that the expression of p21 was upregulated while the expression of cyclin D1 was downregulated by ANA in both the ANA death-sensitive H4 cells and the ANA cell cycle arrest-sensitive Bel7404 cells. On the contrary, ANA had no effects on the expressions of the two genes in the ANA-insensitive BGC cells (Figure 5A), demonstrating that the cell cycle arrest induced by ANA correlated with the alterations of the cell cycle regulating gene expressions. Similarly, the expressions of the key cell death regulating genes, Bcl-2, Bcl-xl, TNF-α, DR4, and DR5 were all altered (Figure 5B-E), caspase8 and caspase9 were activated, and PARP was cleaved by ANA treatment in the ANA cell death-sensitive H4 cells (Figure 5F). Importantly, all of the ANA-induced alterations of the cell growth/death regulating genes were abrogated by knocking-down SLFN12 gene expression (Figure 5B-F). On the other hand, ANA had no effects on the cell growth/death genes in the ANA-insensitive BGC cells (Figure 5E and data not shown). Taken together, these data strongly suggested that the cell growth/death regulating genes were genes regulated by SLFN12 upon ANA treatment and were responsible for the ANA-induced cell growth inhibition. ANA induced cell cycle arrest and apoptosis by altering the expressions of key cell cycle and cell death regulating genes.
Figure 5.
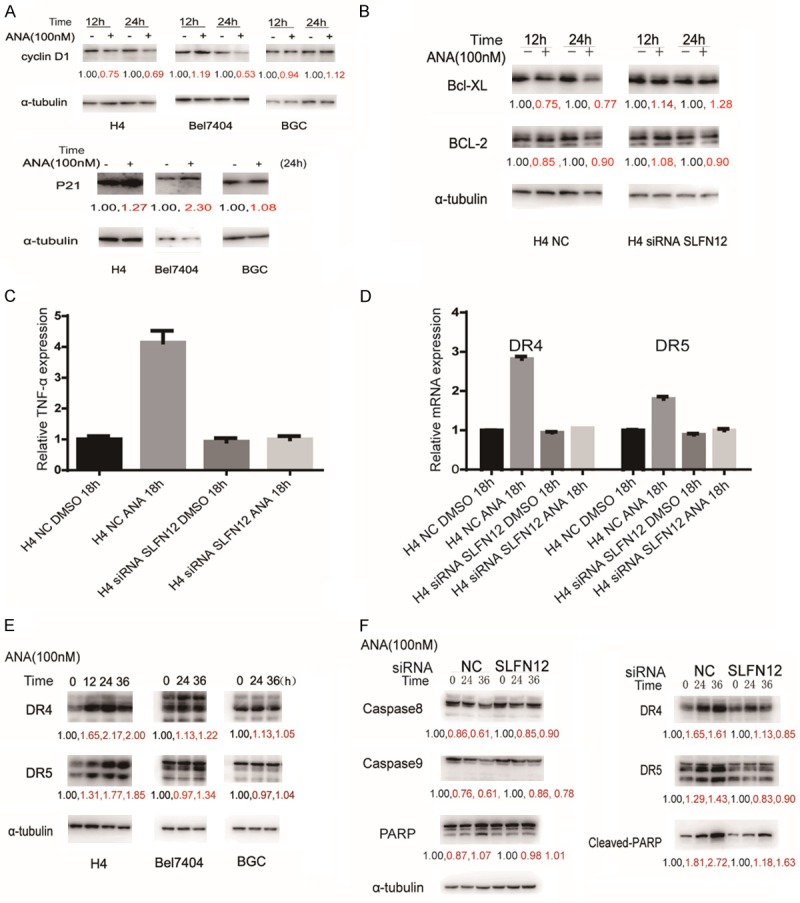
Effects of ANA on the expressions of cell cycle and cell death regulating genes. (A) Immunoblotting analyses of effects of ANA on cyclinD1 and P21 protein expression in the three types of cells. The cells were treated with ANA for the indicated time periods and the cell lysates were processed for immunoblotting with indicated antibodies. α-tubulin antibody was used as a control for equal protein loading. (B) Effects of ANA on the expressions of Bcl-XL and Bcl-2 proteins (left) and the effects of SLFN12 siRNA on the ANA-induced protein level changes of Bcl-XL and Bcl-2 (right). H4 cells were treated with or without ANA (100 nM), or with or without SLFN12 siRNA as indicate for 12 or 24 hours, and the cell lysates were processed for immunoblotting analyses using antibodies as indicated. (C and D) Effects of ANA on the expressions of TNF-α, DR4, and DR5 mRNAs and the effects of SLFN12 siRNA on the ANA-induced mRNA level changes of TNF-α, DR4, and DR5 in H4 cells. The H4 cells were treated with or without ANA (100nM), or with or without the SLFN12 siRNA as indicated for 18 h, and the cell lysates were processed and the mRNA levels of TNF-α (C), DR4, DR5 (D) were measured by quantitative RT-PCR analyses. (E) Effects of ANA on DR4 and DR5 protein expressions in the three types of cells. The cells were treated with ANA (100 nM) for indicated time periods and the cell lysates were processed for immunoblotting analyses using the indicated antibodies. Anti-tubulin antibody was used as a control for equal protein loading. (F) Effects of ANA or ANA plus SLFN12 siRNA on the activation of caspase 8 and caspase 9, protein expressions of DR4 and DR5, and cleavage of PARP. The H4 cells were treated with ANA, a combination of ANA and SLFN12 siRNA, for the indicated time periods, and the cells were processed for immunoblotting analyses using antibodies as indicated. Anti-tubulin antibody was used as a control for equal protein loading.
Cell death inducing cytokines IFN-α, IFN-γ, or TRAIL synergized with ANA to induce apoptosis of cancer cells
Above data suggested that SLFN12 and its level were critical to determine the effects of ANA on the cancer cells. It has been reported that the expressions of SLFNs were regulated by interferons (IFNs) [48]. We therefore analyzed the regulation of SLFN12 expressions by IFN-α or IFN-γ. We found that both IFN-α and IFN-γ induced a time-dependent increase of the SLFN12 mRNA expression (Figure 6A and Supplementary Figure 4A).
Figure 6.
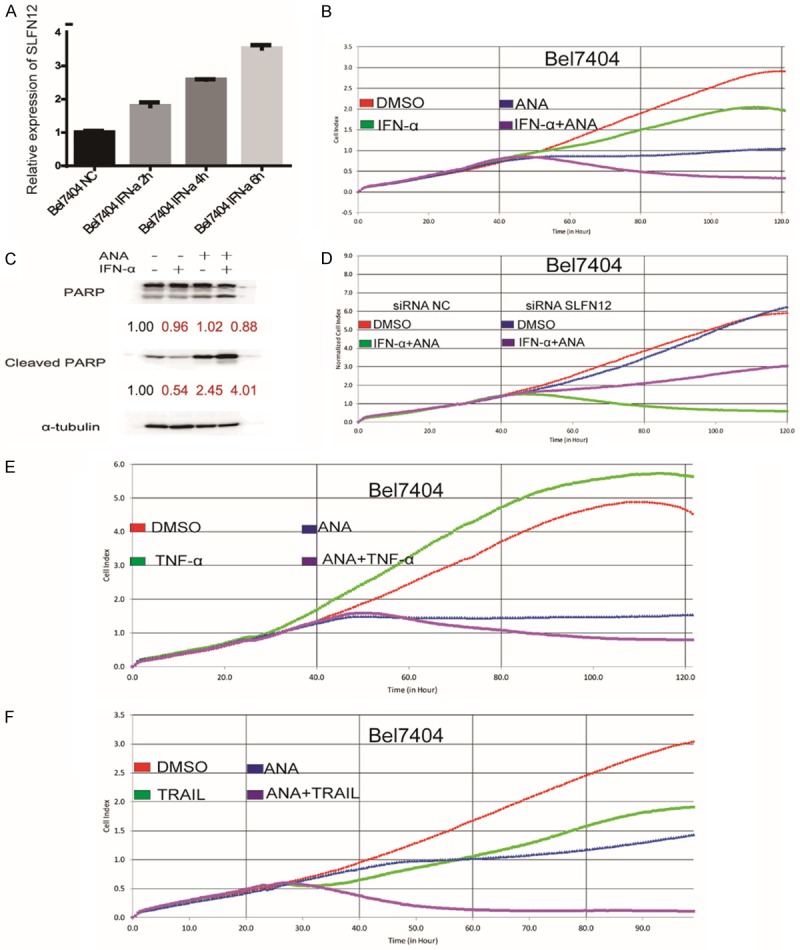
TRAIL or interferons synergized with ANA to inhibit cancer cell growth. A. Effects of IFN-α on SLFN12 mRNA expression. Bel7404 cells were stimulated with IFN-α (3000 IU/mL) for indicated time periods and the SLFN12 mRNA expression was measured by Quantitative Real-Time PCR. B. Effects of ANA or ANA+ IFN-α on the growth of Bel7404 cells. Bel7404 cells were stimulated with IFN-α (3000 IU/mL), ANA (100 nM), or a combination of the two and the cell growths were analyzed by RTCA. C. Effects of IFN-α, ANA or ANA+ IFN-α on cell death. Bel7404 cells were treated with IFN-α (3000 IU/mL), ANA (100 nM), or a combination of the two, and the cell death were analyzed by immunoblotting for PARP cleavage. The anti-tubulin antibody was used as a control for equal protein loading. D. Effects of SLFN12 siRNA-knockdown on the IFN-α and ANA combination treatment. The Bel7404 cells were transfected with either a control siRNA or the SLFN12 siRNA for 24 hours, then the siRNA-transfected H4 cells were cultured with ANA (100 nM) plus IFN-α or DMSO and the growth of the cells were measured by RTCA. E. Effects of TNF-α or TRAIL on the ANA-induced cell growth inhibition. The Bel7404 cells were treated with TNF-α (10 ng/mL), ANA, or a combination of TNF-α and ANA. The cell growths were monitored by the RTCA analyzer. F. Effects of TRAIL on the ANA-induced cell growth inhibition. The Bel7404 cells were treated with TRAIL (100 ng/mL), ANA, or a combination of TRAIL and ANA. The cell growths were monitored by the RTCA analyzer.
We next investigated the possibility to sensitize the cells with IFNs treatment to the ANA-induced cell death and to transform the ANA cell cycle arrest-sensitive cells into the ANA death-sensitive cells. We pre-treated the ANA growth arrest-sensitive Bel7404 cells and the ANA-insensitive BGC cells with IFN-α or IFN-γ for 4 hours before addition of ANA. IFN-α or IFN-γ significantly enhanced ANA to induce apoptosis in the Bel7404 cells (Figure 6B, 6C and Supplementary Figure 4B) but not in the ANA-insensitive BGC cells (Supplementary Figure 4D). Although SLFN12 was also stimulated by IFN-α in the BGC cells, but the induction was much lower than that of the Bel7404 cells (Supplementary Figure 4C).
To confirm that SLFN12 was the key mediator of the IFNs-ANA synergistic effects, we blocked SLFN12 expression with the SLFN12-specific siRNAs and analyzed its effects on the IFNs-ANA combination-induced cell death. We found that blocking SLFN12 expression alleviated the combination treatment-induced cell death (Figure 6D), confirming that SLFN12 was the key player in the ANA-IFNs synergism.
We also found that ANA induced the expressions of the cell death signaling pathway genes TNF-α, DR4, and DR5. We therefore investigated possible synergies of ANA with TNF-α or TRAIL, the ligand of DR4 and DR5. We treated the Bel7404 cells with ANA in combination with TNF-α or TRAIL and indeed found that the combination of ANA with TNF-α or TRAIL also synergistically induced apoptosis of the Bel7404 cells (Figure 6E and 6F).
ANA inhibited the growth of tumor xenograft in vivo
To evaluate the potential of ANA to be an anti-cancer drug, we examined the effects of ANA on tumor growth in a mouse cancer xenograft model. Human cancer H4 cells were transplanted into nude mice. ANA was orally administered for 15 consecutive days. ANA dramatically inhibited the growth of the xenografts in nude mice (Figure 7A and 7C-E), with marginal effects on the mice body weights (Figure 7B), suggesting that ANA had a good potential to be a safe and effective anti-cancer drug.
Figure 7.
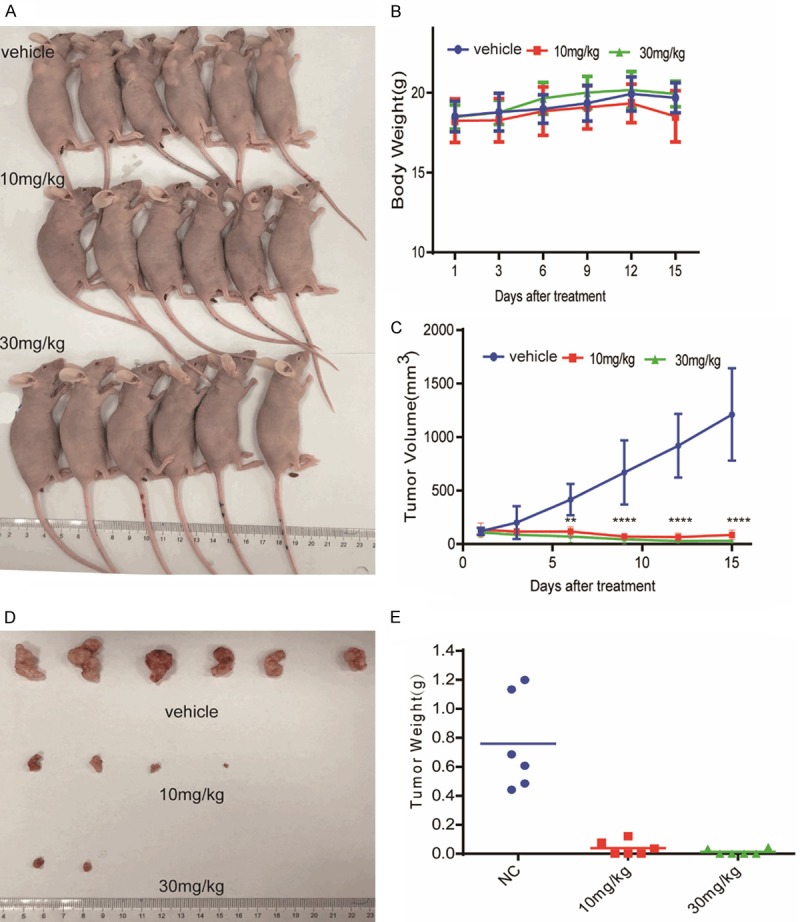
ANA inhibited the growth of tumor xenograft in vivo. Human tumor xenografts of H4 cells were established by subcutaneously inoculating the cells into female BALB/c-nude mice. The mice were orally administered with ANA (10 mg/kg or 30 mg/kg) or vehicle daily for 15 days when the tumor volumes reached 100 mm3. A. Photograph of the tumor-burdened mice after 15 days of ANA treatment. B. Body weight curves of the tumor-burdened mice. C. The tumor growth curves of the tumor-burdened mice. The tumors were measured and analyzed by two-way ANOVA. Data were presented as means ± s.d. (n = 6), ** P < 0.01, **** P < 0.0001. D. Photograph of the tumors from the control and ANA-treated mice. Mice were sacrificed after 15 days ANA treatment and the tumors were dissected and photographed. E. The weights of the individual tumors and their means.
Discussion
In this study, we described the identification of ANA, a known phosphodiesterase PDE3 inhibitor and a drug for the treatment of essential thrombocytosis, as a novel cell-selective anticancer compound and investigated its mechanisms of action. We present evidences to suggest that the interaction of ANA with PDE3A transformed PDE3A into a regulator of SLFN12 and the ANA-PDE3A-SLFN12 interactions suppressed a set of cell survival/growth-promoting genes and activated a set of death/growth arrest-inducing genes to selectively suppress cell cycle progression and induce apoptosis of the cancer cells that expressed both PDE3A and SLFN12 proteins. Furthermore, ANA synergized with the cytokines that regulated the similar sets of genes to inhibit the cancer cell growth (Figure 8).
Figure 8.
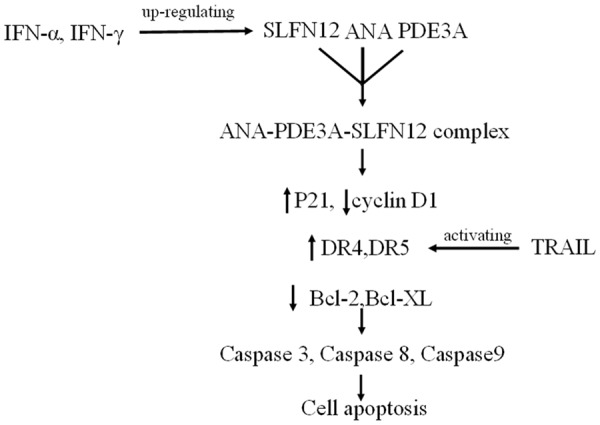
A schematic model of the mechanisms of the ANA-induced apoptosis and its synergies with the cell death inducing cytokines.
Our data, on one hand, demonstrated that PDE3A was the essential and specific target and mediator of ANA to inhibit cancer cell growth, because ANA was only effective in the PDE3A-possitive cells and knocking-down PDE3A expression eliminated the growth suppressive activities of ANA, while knocking down its close relative PDE3B had no such effects. On the other hand, our data also excluded the possibility that inactivation of PDE3A enzymatic activity was the major mechanism for ANA to inhibit cancer cell growth, because other PDE3 inhibitors such as Trequinsin (TRE) and Cilostamide (CILO), although equally inhibit PDE3A enzymatic activity, had no cell growth inhibitory activities. On the contrary, they blocked ANA to inhibit the cancer cell growth, possibly by competing with ANA to interact with PDE3A or by changing the conformation of the ANA-PDE3 complex. Therefore, our data strongly suggested that it was a specific interaction of ANA with PDE3A that generated a new activity deviating PDE3A from its normal phosphodiesterase function to selectively suppress the growth of some of the PDE3A-possitive cancer cells.
Although PDE3A was essential for the ANA-induced cell growth inhibition, the interaction of ANA and PDE3A per se was not sufficient to inhibit the cancer cell growth. Many PDE3A-possitive cells were insensitive to ANA treatment. It appeared that there might be other components involved. The involvement of SLFN12 in the ANA-induced cell growth inhibition was suggested by a recent report that DNMDP, another PDE3 inhibitor, also selectively induced cancer cell death. It was reported that DNMDP induced a physical interaction between PDE3A and SLFN12 and both of which were required for the cell death-inducing activity of DNMDP. SLFN12 could be co-immunoprecipitated with PDE3A after DNMDP treatment [47]. We therefore investigated the possible role of SLFN12 in the ANA-induced cell growth inhibition. Although we were unable to co-immunoprecipitated PDE3A and SLFN12 after ANA treatment, we did find that both PDE3A and SLFN12 were required for ANA to inhibit cancer cell growth and there was a good correlation between the expressions of the two genes and the sensitivities of the cells to ANA treatment. It appeared that an ANA-PDE3A-SLFN12 complex was formed after ANA treatment and this complex altered the activities of SLFN12 leading to cell growth inhibition, similar to that of DNMDP.
The function of SLFN12 is not known. It was reported to be a cytoplasmic protein expressed in many but selective tissues such as endocrine tissues, bone marrow and immune cells, lung, liver, gallbladder, kidney, and urinary bladder cells. It has been shown to modulate differentiation of prostate cancer cells, but the detailed mechanisms are not known [45]. To understand the events downstream of SLFN12 that are responsible for the ANA-induced cell growth inhibition, we analyzed the expressions of a set of key genes that are involved in the regulation of cell cycle progression and cell death. We found that the key cell cycle regulating genes cyclin D1 and p21 and the cell death regulating genes TNF-α, DR4, DR5, Bcl-2, and Bcl-XL were all altered by ANA treatment. Although it is still not entirely clear whether these genes are direct targets of SLFN12 and how these genes are regulated by SLFN12, but knocking-down of SLFN12 abrogated the effects of ANA on these genes suggested that they are all regulated by SLFN12. The most significant and immediate changes in gene expressions were the genes in the TNF family signaling pathways, i.e., TNF-α, DR4, and DR5, suggesting that they might be the most direct targets of SLFN12. This possibility was further supported by the observation that TNF-α and TRAIL, the ligand of DR4 and DR5, synergized with ANA to induce cell death.
The cell growth inhibition caused by ANA was a combination of cell cycle arrest and cell death. The cell cycle arrest was more significant than the cell death in some cells, while the opposite was true in others. There seemed to be a link between the cell cycle arrest and the cell death induced by ANA and the levels of PDE3A and SLFN12, particularly SLFN12, appeared to be a significant factor in determining the cell fate, the more of the SLFN12 expression, the more of cell death. The cell death appeared to be a severer phenotype than the cell cycle arrest caused by ANA treatment, because addition of the cell death-inducing cytokines sensitized the ANA cell cycle arrest-sensitive cells to become the ANA cell death-sensitive cells, either by inducing SLFN12 expression or by inducing the cell death genes directly. Furthermore, overexpression of SLFN12 in all the cancer cells induced cell death (data now shown), further suggesting that the level of SLFN12 played a role in determining the fates of the cancer cells. The exact mechanisms linking the cell cycle arrest and the cell death, however, remain to be revealed.
The inhibition of tumor growth in vivo by ANA was dramatic. 10 mg/kg of ANA almost completely inhibited the cancer cell growth in the xenograft models and the dose we used in the in vivo study did not seem to have visible toxic effects on the mice. The fact that the co-expression of PDE3A and SLFN12 were essential for ANA to inhibit cancer cell growth while the two genes are not co-expressed in most of the normal tissues may also warrant ANA to be a relatively safe drug in clinical use. These data strongly support ANA to be a safe and effective anti-cancer drug.
In summary, we have uncovered ANA as a novel cell-selective anti-cancer agent and revealed its molecular mechanisms of action. We identified PDE3A and SLFN12 as the direct targets of ANA and a set of cell cycle and cell death-regulating genes as the key genes altered by ANA and were responsible for the ANA-induced cell cycle arrest and cell death. We also identified a set of cytokines that synergized with ANA to induce cancer cell death. Our studies have provided a novel anti-cancer drug and a concomitant diagnosis method, a synergistic combinational anti-cancer therapy, and new targets for anticancer drugs development.
Materials and method
Reagents
Anagrelide Hydrochloride (ANA) was purchased from J&K Chemical Company (Shanghai, China). Cilostazol (CILO) was purchased from Selleck Chemical Company (Shanghai, China). Trequinsin (TRE) was purchased from Enzo life sciences (Farmingdale, USA). Recombinant Human IFN-α 2A (IFN-α), Recombinant Human TRAIL (TRAIL), Recombinant Human IFN-gamma (IFN-γ) were purchased from Peprotech (London, UK).
Cell culture and cell lines
All cells in the experiment were purchased from the American Type Culture Collection (ATCC). The human liver cancer cell lines (FHCC98, Bel7404), kidney cancer cell line A498, stomach cancer cell lines (HGC, BGC), and human non-small cell lung cancer cell lines (A549, H460, H522) were cultured in RPMI-1640 in the presence of 10% fetal bovine serum (Gibco, Life Technologies). The cervix cancer cell HeLa, liver cancer cell line SMMC7721, glioma cancer cell line H4, and ovarian cancer cell line SKOV3 were cultured in DMEM supplemented with 10% fetal bovine serum. The human colon cancer cell line SW1116 was cultured in Eagle’s MEM with 10% fetal bovine serum. All cells were maintained in a humidified incubator at 37°C with 5% CO2 and passaged before reaching confluence.
Antibodies
Anti-PDE3A (#99236), anti-DR5 (#ab199357), anti-GAPDH (#8245) antibodies were purchased from Abcam; anti-Bcl-2 (#2876), anti-Bcl-XL (#2764), anti-Caspase-8 (#4790), anti-Caspase-9 (#9508), anti-Cleaved-PARP (#9541), anti-Cyclin D1 (#2978) antibodies were purchased from CST; anti-DR4 (#24063-1-AP) antibody was purchased from Proteintech; anti-α-tubulin (#5286) antibody was purchased from Santa Cruz; anti-PDE3B (#138892) antibody was purchased from Absin; anti-PARP (#556494) antibody was purchased from BD.
xCELLigence RTCA analysis
The E96-xCELLigence plates were prepared by addition of complete media (50 µL). After equilibration to 37°C, plates were inserted into the xCELLigence station, and the base-line impedance was measured to ensure that all wells and connections were working within acceptable limits. Cells were harvested and diluted to a seeding density of 5 × 103 cells/well and were seeded to the E-plate in 50 µL volume. 50 μL of each drug dissolved in the medium was added to achieve defined final concentrations. The cell index (CI) was monitored every 5 min by the RTCA DP analyzer (from ACEA Biosciences Inc.).
Animals and Mouse xenografts
Female BALB/c athymic (nu/nu) nude mice (age, 4-6 weeks; weight, 18-20 g) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd, Beijing, China. Animals were housed in controlled conditions of temperature (23 ± 2°C), humidity (50 ± 5%), and a 12 h light/dark cycle. The animals had free access to sterile food and water. All animal experiments were carried out under the guidance of the approved protocol (2012-10-YQ-10) by Shanghai Institute of Materia Medical. Animals were allowed to acclimate for at least 5 days before any handling. For ectopic implantation experiments, 5.5 × 106 cells per mouse were injected subcutaneously into the flanks of nude mice. After tumor volumes reached nearly 100 mm3, the mice were randomly divided into 3 groups (n = 6/group). The mice were orally administered with ANA (10 mg/kg or 30 mg/kg) or vehicle daily for 15 days. Tumor volume and body weight were measured very three days. Tumor volume calculated by the formula V (mm3) = 1/2 (a*b2), where a = length, b = width, all studies were terminated when the tumors of vehicle group reached nearly 1,000 mm3 [49].
Cell viability assay
Cells were seeded into 96-well plates and cultured overnight, the cells were then treated with vehicle control or various concentrations of different compounds for 72 h. 30 μL of MTT solution (5 mg/mL, Sigma Aldrich) was added to each well and incubated at 37°C for 4-6 h. The supernatants were aspirated, and the MTT-formazan crystals were dissolved in 150 μL of dimethyl sulfoxide. The absorbance value was measured by the microplate reader (Synergy H1, Bioteck, Winooski, USA) at a wavelength of 492 nm.
Cell cycle analysis
A flow cytometry assay was used to analyze the cell cycle distribution as previously reported. Briefly, the cells were plated into 6-well plates and incubated overnight. After treatment with the test substances for 24 or 36 h, the cells were harvested and fixed in cold 75% ethanol overnight at -20°C, and then washed with PBS and stained with PI solution (20 mg/mL PI and 20 mg/mL RNaseA in PBS) for 15 min. The cell fluorescence was measured using FACS Cytometry [50].
Immunoblotting analysis
Cells were harvested in cold Laemmli buffer (Sigma-Aldrich). Protein lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes (GE Healthcare), probed with primary antibodies, and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies. Immune complexes were detected by Immobilon Western Chemiluminescent HRP Substrate (Millipore) and photographed using Darkroom Eliminator (C300, AZURE biosystems).
Real time quantified PCR assay
Cells were lysed in Trizol (Invitrogen, Grand Island, NY) incubation and total RNA was extracted for further experiments. The Real-time Quantitative PCR Assay was performed as previously described. The sequences of primers are as follows: GAPDH Forward: 5’-CGA CCA CTT TGT CAA GCT CA-3’, GAPDH Reverse: 5’-AGG GGA GAT TCA GTG TGG TG-3’; TNF-α Forward: 5’-CAG GGC AAT GAT CCC AAA GTA-3’, TNF-α Reverse: 5’-GCA GTC AGA TCA TCT TCT CGA-3’; DR4 Forward: 5’-AGC TCA GCT GCA ACC ATC AA-3’, DR4 Reverse: 5’-CAC AAC CCT GAG CCG ATG CAA-3’; DR5 Forward: 5’-GTC TGC TCT GAT CAC CCA AC-3’; DR5 Reverse: 5’-CTG CAA ACT GTG ACT CCT ATG-3’.
RNAi and transfection
Specific siRNAs and a random sequence control siRNA were purchased from Genepharma (Shanghai, China). SiRNAs were transected into cells using Lipofectamine 2000 (Invitrogen, Grand Island, NY), the sequences of siRNAs are as following: PDE3A-siRNA-1 (sense 5’-CCAGUAUUACUGUGGACAUGG-3’, anti-sense 5’-AUGUCCACAGUAAUACUGGUU-3’), PDE3A-siRNA-2 (sense 5’-GUGGCCGUAUUCUUAGUCAGG-3’, anti-sense 5’-UGACUAAGAAUACGGCCACAU-3’), PDE3B-siRNA-1 (sense 5’-GGGACUAAAUAGGAAUAGU-3’, anti-sense 5’-ACUAUUCCUAUUUAGUCCC-3’), PDE3B-siRNA-2 (sense 5’-GCAGAUGAGAUUCAGGUAA-3’, anti-sense 5’-UUACCUGAAUCUCAUCUGC-3’), SLFN12-siRNA-1 (sense 5’-AGACUAGAGGGAGAUUGUAUU-3’, antisense 5’-UACAAUCUCCCUCUAGUCUUU-3’), SLFN12-siRNA-2 (sense 5’-GACUAGAGGGAGAUUGUAUUU-3’, anti-sense 5’-AUACAAUCUCCCUCUAGUCUU-3’), SLFN12-siRNA-3 (sense 5’-GAGAUUCUCCCUCAAUAUGUU-3’, anti-sense 5’-CAUAUUGAGGGAGAAUCUCUU-3’) NC (sense 5’-UUCUCCGAACGUGUCACGUTT-3’, anti-sense 5’-ACGUGACACGUUCGGAGAATT-3’).
Statistical analysis
Statistical analyses were performed by using GraphPad Prism 6. Data were graphically represented as mean ± SD. Two-way ANOVA was used in Figure 7, Statistical significance was established for P < 0.05 (* P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81673465, 81373447 to Qiang Yu) and the China National Key Research and Development Program (2018YFC1705505 to Qiang Yu).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Aggarwal S. Targeted cancer therapies. Nat Rev Drug Discov. 2010;9:427–428. doi: 10.1038/nrd3186. [DOI] [PubMed] [Google Scholar]
- 4.Schilsky RL. Personalized medicine in oncology the future is now. Nat Rev Drug Discov. 2010;9:363–6. doi: 10.1038/nrd3181. [DOI] [PubMed] [Google Scholar]
- 5.Gerber DE. Targeted therapies: a new generation of cancer treatments. Am Fam Physician. 2008;77:311–319. [PubMed] [Google Scholar]
- 6.Savai R, Pullamsetti SS, Banat GA, Weissmann N, Ghofrani HA, Grimminger F, Schermuly RT. Targeting cancer with phosphodiesterase inhibitors. Expert Opin Investig Drugs. 2010;19:117–31. doi: 10.1517/13543780903485642. [DOI] [PubMed] [Google Scholar]
- 7.Duffy MJ, Crown J. A personalized approach to cancer treatment: how biomarkers can help. Clin Chem. 2008;54:1770–1779. doi: 10.1373/clinchem.2008.110056. [DOI] [PubMed] [Google Scholar]
- 8.Cai T, Tian L, Wong PH, Wei LJ. Analysis of randomized comparative clinical trial data for personalized treatment selections. Biostatistics. 2010;12:270–282. doi: 10.1093/biostatistics/kxq060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olar A, Aldape KD. Using the molecular classification of glioblastoma to inform personalized treatment. J Pathol. 2014;232:165–177. doi: 10.1002/path.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Touat M, Idbaih A, Sanson M, Ligon KL. Glioblastoma targeted therapy: updated approaches from recent biological insights. Ann Oncol. 2017;28:1457–1472. doi: 10.1093/annonc/mdx106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu M, Lee EM, Wen Z, Cheng Y, Huang WK, Qian X, Tcw J, Kouznetsova J, Ogden SC, Hammack C, Jacob F, Nguyen HN, Itkin M, Hanna C, Shinn P, Allen C, Michael SG, Simeonov A, Huang W, Christian KM, Goate A, Brennand KJ, Huang R, Xia M, Ming GL, Zheng W, Song H, Tang H. Identification of small-molecule inhibitors of zika virus infection and induced neural cell death via a drug repurposing screen. Nat Med. 2016;22:1101–1107. doi: 10.1038/nm.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oprea TI, Bauman JE, Bologa CG, Buranda T, Chigaev A, Edwards BS, Jarvik JW, Gresham HD, Haynes MK, Hjelle B, Hromas R, Hudson L, Mackenzie DA, Muller CY, Reed JC, Simons PC, Smagley Y, Strouse J, Surviladze Z, Thompson T, Ursu O, Waller A, Wandinger-Ness A, Winter SS, Wu Y, Young SM, Larson RS, Willman C, Sklar LA. Drug repurposing from an academic perspective. Drug Discov Today Ther Strateg. 2011;8:61–69. doi: 10.1016/j.ddstr.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strittmatter SM. Overcoming drug development bottlenecks with repurposing: old drugs learn new tricks. Nat Med. 2014;20:590–591. doi: 10.1038/nm.3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roder C, Thomson MJ. Auranofin: repurposing an old drug for a golden new age. Drugs R D. 2015;15:13–20. doi: 10.1007/s40268-015-0083-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lotfi Shahreza M, Ghadiri N, Mousavi SR, Varshosaz J, Green JR. A review of network-based approaches to drug repositioning. Brief Bioinform. 2018;19:878–892. doi: 10.1093/bib/bbx017. [DOI] [PubMed] [Google Scholar]
- 16.Sisignano M, Parnham MJ, Geisslinger G. Drug repurposing for the development of novel analgesics. Trends Pharmacol Sci. 2016;37:172–183. doi: 10.1016/j.tips.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Boolell M, Gepi-Attee S, Gingell JC, Allen MJ. Sildenafil, a novel effective oral therapy for male erectile dysfunction. Br J Urol. 1996;78:257–261. doi: 10.1046/j.1464-410x.1996.10220.x. [DOI] [PubMed] [Google Scholar]
- 18.Espasandin YR, Glembotsky AC, Grodzielski M, Lev PR, Goette NP, Molinas FC, Marta RF, Heller PG. Anagrelide platelet-lowering effect is due to inhibition of both megakaryocyte maturation and proplatelet formation: insight into potential mechanisms. J Thromb Haemost. 2015;13:631–42. doi: 10.1111/jth.12850. [DOI] [PubMed] [Google Scholar]
- 19.Tomer A. Effects of anagrelide on in vivo megakaryocyte proliferation and maturation in essential thrombocythemia. Blood. 2002;99:1602–1609. doi: 10.1182/blood.v99.5.1602. [DOI] [PubMed] [Google Scholar]
- 20.Trapp OM, Beykirch MK, Petrides PE. Anagrelide for treatment of patients with chronic myelogenous leukemia and a high platelet count. Blood Cells Mol Dis. 1998;24:9–13. doi: 10.1006/bcmd.1998.0166. [DOI] [PubMed] [Google Scholar]
- 21.Silver RT. Anagrelide is effective in treating patients with hydroxyurea-resistant thrombocytosis in patients with chronic myeloid leukemia. Leukemia. 2005;19:39–43. doi: 10.1038/sj.leu.2403556. [DOI] [PubMed] [Google Scholar]
- 22.Ahmad F, Degerman E, Manganiello VC. Cyclic nucleotide phosphodiesterase 3 signaling complexes. Horm Metab Res. 2012;44:776–785. doi: 10.1055/s-0032-1312646. [DOI] [PubMed] [Google Scholar]
- 23.Netherton SJ, Maurice DH. Role of cyclic nucleotide phosphodiesterases in migration of endothelial cells; Implications in angiogenesis. Molecular Biology of The Cell. 2002;13:547a. doi: 10.1124/mol.104.004853. [DOI] [PubMed] [Google Scholar]
- 24.Pyne NJ, Furman BL. Cyclic nucleotide phosphodiesterases in pancreatic islets. Diabetologia. 2003;46:1179–1189. doi: 10.1007/s00125-003-1176-7. [DOI] [PubMed] [Google Scholar]
- 25.Liu H, Maurice DH. Expression of cyclic GMP-inhibited phosphodiesterases 3A and 3B (PDE3A and PDE3B) in rat tissues: differential subcellular localization and regulated expression by cyclic AMP. Br J Pharmacol. 1998;125:1501–10. doi: 10.1038/sj.bjp.0702227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W, Colman RW. Thrombin regulates intracellular cyclic AMP concentration in human platelets through phosphorylation/activation of phosphodiesterase 3A. Blood. 2007;110:1475–1482. doi: 10.1182/blood-2006-10-052522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han SJ, Vaccari S, Nedachi T, Andersen CB, Kovacina KS, Roth RA, Conti M. Protein kinase B/Akt phosphorylation of PDE3A and its role in mammalian oocyte maturation. EMBO J. 2006;25:5716–5725. doi: 10.1038/sj.emboj.7601431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar M, Bhattacharya V. Cilostazol: a new drug in the treatment intermittent claudication. Recent Pat Cardiovasc Drug Discov. 2007;2:181–185. doi: 10.2174/157489007782418991. [DOI] [PubMed] [Google Scholar]
- 29.Movsesian MA, Kukreja RC. Phosphodiesterase inhibition in heart failure. Handb Exp Pharmacol. 2011:237–249. doi: 10.1007/978-3-642-17969-3_10. [DOI] [PubMed] [Google Scholar]
- 30.Murata T, Shimizu K, Hiramoto K, Tagawa T. Phosphodiesterase 3 (PDE3): structure, localization and function. Cardiovasc Hematol Agents Med Chem. 2009;7:206–211. doi: 10.2174/187152509789105453. [DOI] [PubMed] [Google Scholar]
- 31.Degerman E, Ahmad F, Chung YW, Guirguis E, Omar B, Stenson L, Manganiello V. From PDE3B to the regulation of energy homeostasis. Curr Opin Pharmacol. 2011;11:676–682. doi: 10.1016/j.coph.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davari AS, Abnous K, Mehri S, Ghandadi M, Hadizadeh F. Synthesis and biological evaluation of novel pyridine derivatives as potential anticancer agents and phosphodiesterase-3 inhibitors. Bioorg Chem. 2014;57:83–89. doi: 10.1016/j.bioorg.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 33.Shimizu K, Murata T, Okumura K, Manganiello VC, Tagawa T. Expression and role of phosphodiesterase 3 in human squamous cell carcinoma KB cells. Anticancer Drugs. 2002;13:875–880. doi: 10.1097/00001813-200209000-00014. [DOI] [PubMed] [Google Scholar]
- 34.Nazir M, Senkowski W, Nyberg F, Blom K, Edqvist PH, Jarvius M, Andersson C, Gustafsson MG, Nygren P, Larsson R, Fryknas M. Targeting tumor cells based on phosphodiesterase 3A expression. Exp Cell Res. 2017;361:308–315. doi: 10.1016/j.yexcr.2017.10.032. [DOI] [PubMed] [Google Scholar]
- 35.Schwarz DA, Katayama CD, Hedrick SM. Schlafen, a new family of growth regulatory genes that affect thymocyte development. Immunity. 1998;9:657–668. doi: 10.1016/s1074-7613(00)80663-9. [DOI] [PubMed] [Google Scholar]
- 36.Puck A, Aigner R, Modak M, Cejka P, Blaas D, Stockl J. Expression and regulation of Schlafen (SLFN) family members in primary human monocytes, monocyte-derived dendritic cells and T cells. Results Immunol. 2015;5:23–32. doi: 10.1016/j.rinim.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neumann B, Zhao L, Murphy K, Gonda TJ. Subcellular localization of the Schlafen protein family. Biochem Biophys Res Commun. 2008;370:62–6. doi: 10.1016/j.bbrc.2008.03.032. [DOI] [PubMed] [Google Scholar]
- 38.Bustos O, Naik S, Ayers G, Casola C, Perez-Lamigueiro MA, Chippindale PT, Pritham EJ, de la Casa-Esperón E. Evolution of the Schlafen genes, a gene family associated with embryonic lethality, meiotic drive, immune processes and orthopoxvirus virulence. Gene. 2009;447:1–11. doi: 10.1016/j.gene.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sassano A, Mavrommatis E, Arslan AD, Kroczynska B, Beauchamp EM, Khuon S, Chew TL, Green KJ, Munshi HG, Verma AK, Plataniasa LC. Human Schlafen 5 (SLFN5) is a regulator of motility and invasiveness of renal cell carcinoma cells. Mol Cell Biol. 2015;35:2684–98. doi: 10.1128/MCB.00019-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zoppoli G, Regairaz M, Leo E, Reinhold WC, Varma S, Ballestrero A, Doroshow JH, Pommier Y. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc Natl Acad Sci U S A. 2012;109:15030–15035. doi: 10.1073/pnas.1205943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He T, Zhang M, Zheng R, Zheng S, Linghu E, Herman JG, Guo M. Methylation of SLFN11 is a marker of poor prognosis and cisplatin resistance in colorectal cancer. Epigenomics. 2017;9:849–862. doi: 10.2217/epi-2017-0019. [DOI] [PubMed] [Google Scholar]
- 42.Li M, Kao E, Gao X, Sandig H, Limmer K, Pavon-Eternod M, Jones TE, Landry S, Pan T, Weitzman MD, David M. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature. 2012;491:125–128. doi: 10.1038/nature11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nogales V, Reinhold WC, Varma S, Martinez-Cardus A, Moutinho C, Moran S, Heyn H, Sebio A, Barnadas A, Pommier Y, Esteller M. Epigenetic inactivation of the putative DNA/RNA helicase SLFN11 in human cancer confers resistance to platinum drugs. Oncotarget. 2016;7:3084–3097. doi: 10.18632/oncotarget.6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang SW, Thomas A, Murai J, Trepel JB, Bates SE, Rajapakse VN, Pommier Y. Overcoming resistance to DNA-targeted agents by epigenetic activation of schlafen 11 (SLFN11) expression with class i histone deacetylase inhibitors. Clin Cancer Res. 2018;24:1944–1953. doi: 10.1158/1078-0432.CCR-17-0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kovalenko PL, Basson MD. Schlafen 12 expression modulates prostate cancer cell differentiation. J Surg Res. 2014;190:177–184. doi: 10.1016/j.jss.2014.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pulkka OP, Gebreyohannes YK, Wozniak A, Mpindi JP, Tynninen O, Icay K, Cervera A, Keskitalo S, Murumagi A, Kulesskiy E, Laaksonen M, Wennerberg K, Varjosalo M, Laakkonen P, Lehtonen R, Hautaniemi S, Kallioniemi O, Schoffski P, Sihto H, Joensuu H. Anagrelide for gastrointestinal stromal tumor. Clin Cancer Res. 2019;25:1676–1687. doi: 10.1158/1078-0432.CCR-18-0815. [DOI] [PubMed] [Google Scholar]
- 47.de Waal L, Lewis TA, Rees MG, Tsherniak A, Wu X, Choi PS, Gechijian L, Hartigan C, Faloon PW, Hickey MJ, Tolliday N, Carr SA, Clemons PA, Munoz B, Wagner BK, Shamji AF, Koehler AN, Schenone M, Burgin AB, Schreiber SL, Greulich H, Meyerson M. Identification of cancer-cytotoxic modulators of PDE3A by predictive chemogenomics. Nat Chem Biol. 2016;12:102–108. doi: 10.1038/nchembio.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mavrommatis E, Fish EN, Platanias LC. The schlafen family of proteins and their regulation by interferons. J Interferon Cytokine Res. 2013;33:206–10. doi: 10.1089/jir.2012.0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma YT, Yang Y, Cai P, Sun DY, Sanchez-Murcia PA, Zhang XY, Jia WQ, Lei L, Guo M, Gago F, Wang H, Fang WS. A series of enthalpically optimized docetaxel analogues exhibiting enhanced antitumor activity and water solubility. J Nat Prod. 2018;81:524–533. doi: 10.1021/acs.jnatprod.7b00857. [DOI] [PubMed] [Google Scholar]
- 50.Lv G, Sun D, Zhang J, Xie X, Wu X, Fang W, Tian J, Yan C, Wang H, Fu F. Lx2-32c, a novel semi-synthetic taxane, exerts antitumor activity against prostate cancer cells in vitro and in vivo. Acta Pharm Sin B. 2017;7:52–58. doi: 10.1016/j.apsb.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.