

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer mortality among men and women in the United States. Its incidence has been on the rise, with a projected two-fold increase by 2030. PDAC carries a poor prognosis due to a lack of effective screening tools, limited understanding of pathophysiology, and ineffective treatment modalities. Recently, there has been a revolution in the world of oncology with the advent of novel treatments to combat this disease. However, the 5-year survival of PDAC remains unchanged at a dismal 8%. The aim of this review is to bring together several studies and identify various recent modalities that have been promising in treating PDAC.
Keywords: Pancreatic cancer, immunotherapy, cancer vaccine, BRCA, check point inhibitor, targeted therapy, hypoxia induced resistance
Introduction
Cancer is the second-leading cause of death in the United States following cardiovascular disease. Pancreatic cancer is the fourth leading cause of death due to cancer in the US in 2019. In fact, it is projected to become the second-leading cause by 2030 [1]. This disease has a 5-year survival rate of 8% [2]. The most common forms of pancreatic cancers are exocrine cancers, which comprise of 95% of all pancreatic cancers [3,4]. Of these exocrine cancers, the most common and aggressive form is pancreatic ductal adenocarcinoma (PDAC). PDAC accounts for approximately 85% of all pancreatic tumors. Other histological variants of pancreatic cancer include adeno-squamous carcinoma, colloid carcinoma, hepatoid carcinoma, medullary carcinoma, signet-ring cell carcinoma, undifferentiated carcinoma, and undifferentiated carcinoma with osteoclast-like giant cells [5]. All of these have different pathogeneses and carry different prognoses.
Here, we will focus on different treatments that are available for PDAC and note possible areas for future treatment development.
Targeted therapy
Targeted therapies mainly focus on trans- receptor membrane proteins (TRMPs). Cell membranes express surface molecules that serve as targets for clinical intervention. Various targets include epidermal growth factor (EGFR/Erb1), vascular endothelial growth factor (VEGF 1, 2, and 3), human epidermal growth factor receptor-2/human erythroblastic oncogene B-2 (HER2/ERBB2), fibroblastic growth factor receptor (FGFR), and insulin-like growth factor-1 (IGF-1).
Recently, the Southwest Oncology Group conducted a phase III trial testing a combination of cetuximab (EGFR inhibitors) and gemcitabine compared to gemcitabine alone [6]. Results showed that there were no differences in survival rates between the two arms of the study (6.3 vs 5.9 months, respectively; P = 0.19). A preclinical study demonstrated the overexpression of EGFR during the formation of a complex between NFATc1 and C-JUN in de-differentiated mouse acinar cells [7]. In a clinical setting, this overexpression led to the activation of Sox9 transcription and induction of acinar ductal metaplasia in patients with chronic pancreatitis [7]. Targeting this pathway may be valuable in preventing PDAC in patients with chronic pancreatitis [7].
VEGF receptor is another member in the surface tyrosine kinase family. VEGF allows tumors to gain ample blood supply and thus continue cell proliferation. Overexpression of VEGF is commonly associated with poor prognosis [6]. A study by Korc has shown a correlation between blood vessel density, tumor VEGF-A levels, and disease progression [8]. The phase 3 CALGB trial observed patients treated with either gemcitabine and a placebo or gemcitabine and bevacizumab (VEGF inhibitor) [9]. This trial did not observe any difference in overall survival [OS] (5.9 vs 5.8 months, respectively; p = 0.95). Another phase II trial exploring maintenance with sunitinib (a multi-receptor tyrosine kinase inhibitor) after first-line chemotherapy versus solely observation showed an improvement in two-year OS in treating metastatic PDAC in the observation versus sunitinib arms [OS 7.1% (95% CI 0-16.8%) vs 22.9% (95% CI 5.8-40.0% P = 0.11, respectively)] [10]. However, a phase III trial is needed to confirm this outcome. Another ongoing phase I trial [NCT02902484] is studying nintedanib (a multi-receptor inhibitor) combined with standard chemotherapy (gemcitabine and nab-paclitaxel) in metastatic PDAC.
Insulin-like growth factor (IGF) is a signaling protein that is overexpressed in PDAC. High levels of IGF-1 are associated with highly-aggressive tumors and poor prognosis [11]. IGF-1 has been proposed to confer resistance against EGFR inhibitors [6]. Preclinical studies suggest that targeting both EGFR and IGFR pathways potentiates growth inhibition and apoptosis [6]. An ongoing phase II trial [NCT02399137] is studying istiratumab (MM-141), an engineered bispecific monoclonal antibody that blocks the IGF-1R and ErbB3 pathways by binding to HER3 and IGF-1 receptors [12]. Another preclinical study in mouse models showed that small IGF-1 receptors and insulin receptor reversible inhibitors of IGF-1R/IR signaling (BMS-754807) reduce relative PDAC volume when used in tandem with nab-paclitaxel [13].
Another preclinical study has demonstrated that IGF-1 and heregulin (HRG) are the most potent out of all protein kinase B (AKT) activators. Therefore, these growth factors may play a role in reducing pancreatic cancer cell response to gemcitabine or nab-paclitaxel. Istiratumab (MM-141) has been shown to intensify gemcitabine and paclitaxel sensitivity through the inhibition of AKT phosphorylation in vivo [14].
Intracytoplasmic signal transduction
RAS is the first molecule in the MAP kinase pathway [15,16]. This cascade involves phosphorylation at every step in the RAS-RAF-MEK-ERK pathway with nuclear transcription as the final outcome (Figure 1). The vast majority of PDAC patients have KRAS mutations [17,18]. In theory, targeting this pathway may play a role in cell proliferation and tumor growth; however, a previous study showed that the presence of KRAS mutations has no impact on OS [19]. Mutant KRAS has no effect on downstream signaling pathways. This may explain why targeting this pathway has failed to improve patient outcomes [19-22]. Despite all challenges, preclinical and clinical efforts are still being made. Salirasib, a Ras farnesylcysteine mimetic, has been studied in combination with gemcitabine [23]. An early phase trial has determined safe dosages of salirasib when used in combination with gemcitabine. Larger studies are needed to determine the effectiveness of this combination [6]. Another effort has been made to target the degradation of KRAS oncoproteins through the ornithine decarboxylase/antizyme (ODC/AZ) pathway. This has been shown both in vitro and in vivo to decrease KRAS levels and suppress PANC-1 cell proliferation in addition to downregulating the phosphorylation of ERK1/2. Targeting this pathway may be effective in future treatments of PDAC [24].
Figure 1.
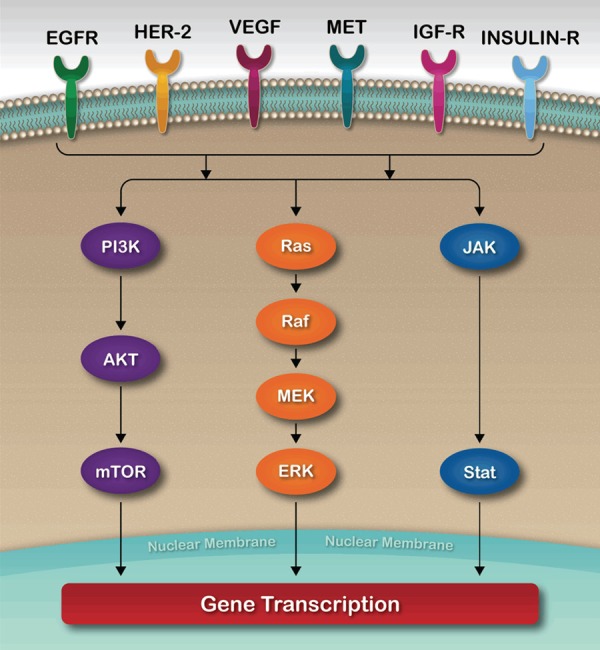
Cell Signal Transduction.
Blocking of RAS-RAF-MEK-ERK, a MAP kinase signaling pathway (Figure 1), usually fails through several escape mechanisms (19). In fact, a recent study found an inverse correlation between STAT3 and MEK signaling and resistance to RAS pathway inhibition in PDAC [25]. This study found that MEKi leads to immediate activation of STAT3, while STAT3i leads to AREG-dependent activation of the RAS pathway [25]. This combination has changed tumor growth in PDX mice through the use of patient-derived xenografts. It has also improved the survival of PKT mice while reducing serum AREG levels [25]. Moreover, MEKi and STAT3i change the pancreatic cancer microenvironment by inhibiting tumor fibrosis, preserving pancreatic integrity, and downregulating CD44+ and CD133+ cancer stem cells (CSCs) [25]. In addition, a study suggests that AREG levels may serve as key circulating prognostic biomarkers of PDAC and potential biomarkers of therapeutic resistance and response to EGFR, MEK and STAT3 inhibition [25].
To date, all other clinical studies have shown little or no valuable results in treating PDAC with traditional mTOR inhibitors [26,27]. However, the rapamycin-insensitive companion of mTOR (RICTOR) has been shown to play a critical role in human cancer initiation and progression. Targeting this component disrupts the activation of AGC kinases such as AKT and SGK. This, in turn, decreases expression of hypoxia-induced factor HIF-1α and the secretion of cancer-promoting factors in pancreatic cancer cell lines [28]. Indeed, AKT and HIF-1α expression have been associated with poor prognosis and early recurrence of PDAC [29]. High RICTOR expression in patients with resected PDAC is associated with poor survival; a recent study showed a large difference median survival (MS) between high and low RICTOR expression groups (MS 11.1 vs 24 months, respectively; P < 0.0001) [28]. Targeting RICTOR would hence be a novel therapeutic option in treating PDAC.
Hypoxia-induced resistance
The hypoxic environment is a result of poor tumor vascularity. This environment is strongly associated with increased radio-resistance, chemo-resistance and tumor metastasis. Hypoxia-induced prodrug monotherapy is generally inefficacious and must be combined with other treatments. One such application of hypoxia-induced resistance is PI3K pathway inhibition through the activation of AKT [30,31]. Preclinical evaluation using a dual-regimen therapy of an mTORC1/2 inhibitor (AZD2014) and a hypoxia-activated pro-drug (HAP) TH-302 has been shown to decrease the hypoxic fractions of HIF1α and carbonic anhydrase IX (CAIX) expression. This has helped overcome resistance to PI3K pathway targeting and inhibition of tumor growth in vivo. It may explain the reduction in AKT activity after the use of such combination therapy [32].
A preclinical study has evaluated targeting the ERK pathway with ulixertinib, a drug which has been shown to enhance the cytotoxic effect of gemcitabine [33]. Ulixertinib has an upregulation effect on the PI3K-AKT pathway through the activation of HER/ERB2 [33]. Concurrent use of gemcitabine and ulixertinib has been shown both in vivo and in vitro to create a synergistic effect in suppressing PDAC through the inhibition of PI3K and HER [33]. A phase I clinical trial [NCT02608229] is currently testing the ERK inhibitor BVD-523 in combination with nab-paclitaxel plus gemcitabine in patients with newly diagnosed metastatic PDAC.
PI3Kγ plays a critical role in immunosuppression by inhibiting adaptive immune responses through promoting immune suppressive polarization in macrophages (Figure 2). This leads to immune suppression, tumor invasion, metastasis, and desmoplasia in PDAC [34]. Therefore, targeting PI3Kγ in PDAC-bearing mice may promote CD8+ T cell-mediated tumor suppression [34]. In addition, PI3Kγ inhibition is theoretically the most potent type of inhibition possible because it lacks the downstream feedback activation of mTOR inhibitors [35]. This data demonstrates that inhibiting PI3Kγ is a promising therapeutic pathway for treating PDAC [34].
Figure 2.
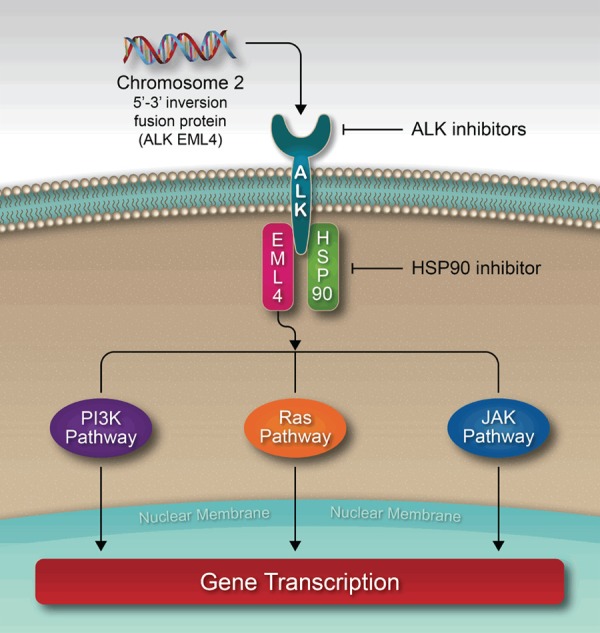
Alk-EML4/HSP90 interplay.
A study evaluating the anticancer effects of crizotinib (an ALK inhibitor) on pancreatic cancer cells found that crizotinib induces apoptosis and inhibits tumor progression and angiogenesis in pancreatic cancer cell lines by downregulating the ALK pathway [36,37]. Surprisingly, crizotinib did not suppress pancreatic cell c-MET, a cell surface receptor tyrosine kinase which induces intracytoplasmic signal transduction using various pathways including MAP kinase, PI3K, STAT, Notch and beta-catenin. This may explain why crizotinib lacks effectiveness when treating overexpressed c-MET signals in pancreatic cancer [38,39].
Targeting unfolding protein response (UPR)
Increasing protein synthesis and the protein folding capacity of the endoplasmic reticulum (ER) allows cancer cells to survive. This happens through the upregulation of UPR signaling pathways, such as the activation of the inositol-requiring enzyme 1α/X boxbinding protein (IRE1α-XBP) pathway and the overexpression of the glucose-regulated protein 78-binding immunoglobulin protein (GRP78/BIP) [40-43]. Therefore, targeting UPR pathways can alter the balance of UPR components and affect cancer cell survival [44]. Several studies indicate that the fluorinated-ONC201 analog of the imipridone family (ONC201) induces cellular stress [45-48]. ONC212, which is similar to ONC201, induces the expression of C/EBP homologous protein-10 (CHOP/GADD153) [49]. This, in turn, may also induce cellular stress [44]. A study by Lev et al evaluated the combination of either ONC201 or ONC212 with the IGF1-R inhibitor AG1024 in vitro. Results demonstrated that PANC-1 cells undergo apoptosis only when they receive a combination either of ONC201 or ONC212 with AG1024 [44]. Furthermore, ONC212 was more efficacious than ONC201 in treating different in vitro and in vivo models of human PDAC [44]. Therefore, ONC212 may be a promising drug when it is combined with chemotherapy or selected targeted therapies such as IGF1-R [44].
Heat-shock protein (HSP) inhibitors
HSP90 and ubiquitin proteasome play critical roles in the homeostasis of human pancreatic cancer cells (Figure 2) [50]. Disruption of these pathways leads to endoplasmic reticulum (ER) stress and the breakdown of pancreatic homeostasis [50]. One study evaluated the combination of ganetespib, a HSP90 inhibitor, and carfilzomib, a proteasome inhibitor, in both in vitro and in vivo pancreatic cancer cell lines. Use of these inhibitors resulted in interference of cell viability and ultimately lead to cell death. Many other studies have demonstrated reduced cell viability after use of ganetespib to induce ER stress [51-56]. In these studies, ganetespib led to the suppression of PI3K, AKT, mTOR pathways and MAP kinase pathways. The HSP90 inhibitor Y306zh prevents ATP from binding to HSP90 and thus leads to an impedance of HSP90-p23 association. Targeting HSP90 and therefore inducing ER stress could potentially be useful in treating refractory cases of PDAC [57]. Another heat shock protein called HS-345 inhibitor prevents ATP from binding to TrkA. HS-345 inhibits the TrkA/Akt signaling pathway in pancreatic cancer cells. This leads to the inhibition of cell growth and proliferation in a dose-dependent manner. It also prevents angiogenesis through the inhibition of NGF (nerve growth factor) to stop micro-vessel growth and thus induces apoptosis. A recent study demonstrated the strong anti-cancer effects of HS-345 inhibitors in three pancreatic cancer cell lines (PANC-1, MIA PaCa-2, and BxPC-3) [58].
DNA damage and homologous recombination (HR)
DNA damage in noncancerous cells is detected during the G1 phase through the G1 checkpoint of the cell cycle. In contrast, cancer cells depend on the G2 checkpoint for cellular repair and survival. DNA damage detected at the G2 checkpoint results in a reaction cascade which activates WEE1. This, in turn, stops the cell at the G2 phase and allows it to undergo repair before proceeding to the mitosis (M) phase. Most chemotherapy drugs work by damaging cancer cell DNA. Blocking off this repair mechanism may help prevent drug resistance [59].
Recently, many studies have demonstrated that inhibition of homologous repair in cancer cells by WEE1 inhibitors (AZD1775) is a possible mechanism of chemo-radio sensitization [60,61]. A pre-clinical study has analyzed the ability of AZD1775 to sensitize and inhibit HR repair in vivo on patient-derived pancreatic tumor xenografts [62]. Findings showed significant sensitization to gemcitabine in selected HR-proficient locally advanced pancreatic cancers [62]. In addition, previous studies have suggested that abrogation of the AZD1775-mediated G2 checkpoint is the primary mechanism of radio-sensitization through WEE1 inhibition [63-65]. Results from these studies showed that AZD1775 produced significant G2 checkpoint abrogation in response to gemcitabine-radiation in both BRCA2 wild type and BRCA2 null cells. This provides a foundation for clinical trial NCT02037230, which is currently testing the combination of AZD1775 with gemcitabine radiation in locally advanced pancreatic cancer patients [62]. Another study has addressed the combination of AZD1775, olaparib (a PARP inhibitor), and radiation in human pancreatic tumor models [61]. Results from this combination showed significant tumor regression and slower tumor re-growth rates. This is especially in contrast to the results of other combinations such as AZD1775 and radiation, which resulted in stable disease, and olaparib and radiation, which resulted in growth during treatment [61]. The combination of AZD1775, olaparib, and radiation is well-tolerated without obvious systemic toxicity [61].
Some pancreatic cancers are associated with BRCA mutations. BRCA is a family of breast cancer tumor suppressor genes that plays a role in DNA repair. A mutation in this gene renders cells susceptible to cancer through insufficient homologous repair. This thus makes cells sensitive to PARP1 inhibitors [66-71].
The goal of investigating olaparib together with gemcitabine is to ensure the optimal concentration of both these agents in the targeted cells [72]. The subgroup of pancreatic cancers with BRCA mutations may be treated by olaparib because poly-ADP-ribose polymerase (PARP) plays a critical role in single-strand DNA break repair [73]. Investigators have engineered a nanomedicine called GE11 peptide self-assembly amphiphilic peptide nanoparticle gemcitabine olaparib (GENP-Gem-Ola) that enhances delivery of gemcitabine and olaparib to pancreatic cancer cells with BRCA2 mutations [74]. Here, gemcitabine and PARPi combine synergistically to suppress BRCA2 mutant capan-1 cells. This study considered a potential approach in treating pancreatic cancers with mutations in DNA repair pathways through the use of GENP-Gem-Ola.
Immunotherapy, vaccination, and checkpoint blockade
Immune system responses against cancer cells have been studied for many years. It has been widely recognized that immune responses actively protect the body from suspicious invasion by cancer cells [75]. For instance, cancer cells are attacked by immune system cells such as natural killer (NK) cells and cytotoxic T-cells [76]. However, cancer cells always try to prevent themselves from being attacked by these cells by making themselves invisible to the immune system. Furthermore, cancer cells alter their tumor microenvironment metabolism in order to avoid being attacked by the immune system and continue growing with impunity [77]. Moreover, cancer cells downregulate the expression of antigen presenting molecules such as major histocompatibility antigen class I (MHC I) [78]. PDAC cells induce immune system tolerance by interacting with activated tumor antigen-specific T-cells. This process is called immune privilege [77]. For instance, PDAC cells downregulate Fas receptor signaling and augment Fas ligand expression, which in turn induce apoptosis of activated antitumor cytotoxic T cells [79-81]. FoxP3 (forkhead box P3), a transcription regulator, is highly expressed on both T-regulatory (Treg) and PDAC cells. The mechanism controlled by FoxP3 plays a crucial role in suppressing the proliferation of cytotoxic T cells [82]. In addition, PDAC cells secrete granulocyte-macrophage colony-stimulating factor (GM-CSF), which promotes the infiltration of myeloid derived cells into the tumor microenvironment. This, in turn, creates a safe environment for tumor cells and allows for aggressive tumor behavior to continue [83]. T-regs also invade the PDAC microenvironment and suppress cancer immunity [84].
A study has evaluated the targeting of mesothelin in animal models of PDAC [85]. Mesothelin is a peptide that is overexpressed in assorted cancers such as ovarian cancer and mesothelioma [86]. In this study, targeting mesothelin antigen activated cytotoxic T cells induced substantial tumor suppression.
Mucin-1 (MUC1) is a cell surface gene that is associated with large membrane glycoproteins expressed in PDAC cells. A phase I/II study evaluated the role of MUC1 in PDAC. Here, 12 patients underwent surgical resection and received MUC1-pulsed autologous dendritic cells as adjuvant treatment. Four out of the 12 patients were able to survive 4 years postsurgery [87]. MUC1 has been engineered to express antigenic epitopes, prevent the development of self-tolerance, and enhance immune activity. Studies of this engineered MUC1 gene have shown promising outcomes in murine models; however, treatment through this approach has yet to be investigated in clinical studies [88].
Telomerase is commonly overexpressed in cancer cells. This overexpression allows it to become a possible target for immunotherapy [77]. A combination of telomerase and GM-CSF has been shown to provide immunity against tumors through early signaling [89]. A phase I trial (NCT02960594) has identified human telomerase reverse transcriptase (hTERT), a subunit of the telomerase enzyme, as a single agent that can be combined with IL-12 for treating solid tumors such as PDAC. However, similar results have not been replicated; for example, a phase III trial studying a chemotherapy and telomerase peptide combination has not demonstrated any improvement in survival [90].
Anti-cancer vaccinations
Cancer vaccines strategies have been investigated for treating pancreatic cancer. Here, the purpose of vaccination is to enhance endogenous anti-tumor immune responses [91]. Examples of vaccines that have been developed include whole cell vaccines (e.g. Algenpantucel-L), peptide vaccines, dendritic cell (DC) vaccines, and recombinant virus-based vaccines [91].
Algenpantucel-L vaccine
Algenpantucel-L (HyperAcute ™ Pancreas) is a whole cell vaccine composed of two irradiated cancer cell lines (HAPa-1, HAPa-2) that have been genetically engineered to express murine enzyme α-GT (alpha-1,3-galactosyltransferase) [92]. Another treatment called α-Gal (alpha-1,3 galactosyl epitopes) mediated vaccine immunotherapy has been investigated for treatment and prevention of melanoma, pancreatic, and prostate cancers [92-94]. Algenpantucel-L works by evoking an innate immune reaction against cancer cells. This begins with hyperacute rejection and continues with phagocytosis of tumor cells [95-97]. Hyperacute rejection of cancer cells produces anti-αGal antibodies that cause complement-mediated destruction of xenografts [96]. Limited data has shown that mounting humoral immunity to algenpantucel-L is associated with enriched survival outcome. More randomized trials such as the Immunotherapy for Pancreatic Resectable Cancer Study (IMPRESS) are needed to confirm these results [91].
IMPRESS (NCT01072981), a Phase III randomized control trial, investigated the use of algenpantucel-L at 300 million cells per dose. Results showed a 1-year disease-free survival (DFS) of 81% and 1-year overall survival (OS) of 96% [95]. According to a 2016 press release, overall survival in the control and experimental groups were 30.4 and 27.3 months, respectively [77]. Another study investigating the use of algenpantucel-L is PILLAR (NCT01836432), a Phase III randomized control study. Here, algenpantucel-L given at 300 million cells per dose was combined with chemotherapy regimens (e.g. FOLFIRINOX or gemcitabine/nab-paclitaxel) and chemoradiation (e.g. capecitabine or 5-FU based). PILLAR was recently terminated in February 2019. All of these regimens will be tested in patients with locally advanced and borderline resectable PDAC [95].
GVAX/CRS-207 vaccine
GM-CSF (granulocyte-macrophage colony-stimulating factor) is a cytokine that promotes growth and differentiation of dendritic cells (DCs). Dendritic cells play a critical role in immune responses because they are the most efficient antigen-presenting cells (APCs) [91].
During a phase I study, investigators created GVAX, a line of engineered pancreatic tumor cells that secrete GM-CSF. Early results have shown a favorable safety profile and enhanced antitumor immunity [98]. Clinical studies are starting to evaluate the potential advantages of GVAX use in treating PDAC [77]. A phase I safety study combined the use of GVAX with cyclophosphamide (Cy) at a low dose of 250 mg/m2. Here, Cy was used to decrease T-reg cellular activity [99]. Results showed that survival outcomes were superior with GVAX and Cy compared to GVAX alone [100]. In another study, adding Cy to GVAX was associated with improved OS and enhanced mesothelin-specific T-cell responses compared to use of GVAX alone [91]. A phase II trial [101] enrolled patients who were previously treated for advanced PDAC using a variety of regimens. Here, patients were divided into two arms of treatment. Arm (A) received 2 doses of Cy/GVAX followed by four doses of CRS-207, a live-attenuated Listeria strain that induces tumor-associated antigens. Arm (B) received six doses of Cy/GVAX alone. Overall survival was superior in arm A (6.1 versus 3.9 months, P = 0.02). In addition, the response of mesothelin-specific CD8+ T-cell was associated with an improved course in both groups [77].
Currently, using cyclophosphamide (Cy) and GVAX with or without CSR-207 combined with chemotherapy and checkpoint inhibitors is being tested in neoadjuvant settings (trials NCT00727441 and NCT02451982). Use of Cy/GVAX and CRS-207 with or without nivolumab (a PD-1 inhibitor) is currently being examined in neoadjuvant and adjuvant settings in the treatment of metastatic PDAC (trials NCT02243371 and NCT02451982) [77]. A randomized phase IIB study titled the “Safety and Efficacy of Combination Listeria/GVAX Pancreas Vaccine in the Pancreatic Cancer Setting” (NCT02004262) examined these treatments in metastatic PDAC patients who were previously managed with other treatments. Patients were randomly assigned to receive 1 of 3 treatments: (A) Cy/GVAX plus CRS-207, (B) CRS-207 alone, or (C) single chemotherapy alone [91]. Unfortunately, results were unsatisfactory. Use of Cy/GVAX in combination with CRS-207 did not show increased efficacy compared to use of chemotherapy alone. However, there was improved survival in group B compared to group C (5.4 vs 4.6 months, respectively) [77].
Although cancer vaccines are able to activate antitumor immunity, their sole use has not proven to significantly improve patient outcomes in a clinical setting. Thus, scientists have tried to use vaccines along with immune modulatory agents [102] to see if outcomes can be improved through their combination. A small phase I study investigated the combination of ipilimumab (a CTLA inhibitor) and GVAX in treating advanced PDAC [102]. Overall survival outcomes were better in patients treated with GVAX and ipilimumab compared to patients treated with ipilimumab alone (5.7 vs 3.6 months, respectively; HR 0.51, P = 0.072). Another ongoing study is currently investigating the use of vaccines and immune modulatory agents in treating locally advanced PDAC [77]. In this phase II trial, (NCT02648282), the combination investigated is Cy/GVAX, SBRT (stereotactic body radiation therapy), and pembrolizumab (a PD-1 inhibitor).
Survivin vaccine
Survivin, an inhibitor from the apoptosis family, is a well-known tumor-related antigen that functions to suppress caspase [77,91]. Survivin has been studied in cancer vaccines because of its ability to negatively regulate apoptosis [77,91]. Survivin is expressed in the majority of PDAC cells but not in normal tissue cells [103]. Survivin vaccines have been shown to be efficacious in a few case studies. In one case, a patient with gemcitabine-resistant PDAC went into complete remission after use of a survivin vaccine [104]. This remission did not last though; PDAC disease progressed after the vaccine was discontinued. Kameshima et al conducted a similar study of a HLA-A2 restricted survivin-peptide based vaccine in a series of 6 patients [105]. These 6 patients were had stage III or IV PDAC and were either treatment-naïve or had previously been treated with other regimens. Results showed that more than 50% of patients had a immunologic response associated with clinical advantage in combatting PDAC [105].
Wobser et al cited a patient with refractory stage IV pancreatic cancer who was also treated with a HLA-A2 restricted survivin-based peptide vaccine [91,104,105]. This patient achieved 8 months of complete remission because of immune-reactivity against survivin antigens [104]. Despite the presence of these promising preliminary studies, survivin-based vaccines still have not been tested in pancreatic cancer clinical trials [91].
Wilms tumor 1 and dendritic cell vaccines
Wilms tumor 1 (WT1) is a mutated peptide that is expressed in various cancers, including PDAC. It has been used to sensitize effector T-cells in treating pancreatic cancer [106]. WT1 was rated as the best target antigen for cancer vaccines among 75 tumor-associated antigens (TAAs) selected by a 2009 National Cancer Institute (NCI) prioritization project [107]. DCs are considered the most efficient APCs capable of presenting TAAs to CD8+ and CD4+ T-cells; in addition, they can also prime naïve T-cells [91,108]. In a recent study, DCs were created to present WT1 via either MHC class I, II, or I/II models [77]. The best clinical response was detected through the MHC class I/II combined model. This response was associated with an increased delayed hypersensitivity reaction.
Use of a biweekly MHC-restricted WT1 vaccine in tandem with gemcitabine also appears to be a safe approach in treating patients with advanced PDAC [109]. Therefore, numerous studies have concentrated on using DC-based cancer vaccines to initiate and spread TAA-specific antitumor immune responses and augment cell lymphocytes (CTLs) [110]. Moreover, cancer peptides, a type of personalized peptide with the capability to activate pre-existing host immunity in an HLA-specific manner, have been investigated to overcome progressive self-tolerance to cancer-related antigens. Early phase studies of these peptide approaches have demonstrated both tolerable safety profiles and significant clinical benefits in both chemotherapy-responsive and chemotherapy-resistant patients with advanced-stage PDAC [111,112].
All pancreatic cancer cells express WT1 in the cytoplasm and nucleus [113]. Therefore, reactivating the immune systems of patients with pancreatic cancer by targeting WT1 may be a potentially therapeutic target. WT1-specific CTLs target not only PDAC cells but also tumor vascular endothelial cells and myeloid-derived suppressor cells (MDSCs). Therefore, targeting WT1 may result in good clinical outcomes [114-116]. A multimodal therapy strategy, comprised of DC/WT1-I vaccines, chemotherapy, radiation, and/or surgery, may be promising in treating advanced PDAC [117].
Checkpoint blockade
T-cells play a critical role in protecting the body against various diseases, including cancers [118]. T-cells eliminate cancer cells by identifying the tumor-associated antigens on their surfaces. CD8+ effector T-cells, also called cytotoxic T lymphocytes (CTLs), orchestrate diverse immune responses with CD4+ helper T-cells [119]. Multiple mechanisms of immune suppression, such as poor dendritic cell (DC) activation, poor tumor-associated antigen presentation, and overexpression of inhibitory ligands, suppress the activity of T-cells and thus allow tumor growth to continue [120,121].
A pivotal mechanism underlying immune resistance is immune inhibition, which is also called an immune checkpoint. Immune checkpoints play key roles in mediating immune tolerance and protecting tissues from collateral damage [122]. Examples of immune checkpoints include cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), programmed cell death protein 1 and its ligand (PD-1/PD-L1). These immune checkpoints can negatively regulate the tumor specific T-cells [123,124]. Immune checkpoint blockades, such as anti-PD-1/PD-L1 and anti-CTLA4 antibodies, have been approved by the U.S. Food and Drug Administration (FDA) to treat various types of cancers [125-128]. However, use of single checkpoint inhibitors produces insufficient immune reactions. Thus, studies are trying combinations of checkpoint inhibitors. One such recently completed study, a phase II trial (NCT02558894), studied the combination of durvalumab with tremelimumab (anti-PD-L1 and anti-CTLA4 antibodies) [77].
CD40 upregulates T-cell function and PD-L1 expression [129,130]. Therefore, treatment through CD40 agonists may be promising in treating PDAC. The combination of CD40 agonists and gemcitabine in 28 chemotherapy-naive patients with advanced PDAC yielded decreased FDG uptake in the hepatic lesions of 4 patients [129]. Furthermore, depletion of tumor-associated fibroblasts may enhance tumor specific T-cell infiltration by targeting the CXCl12-CXCR4 axis when combined with an anti-PD-L1 antibody [131] This activates signaling pathways to promote cellular proliferation and subsequent survival [132].
Immune modulation
Indoleamine 2,3-dioxygenase (IDO) is a cytosolic, heme-containing enzyme that catalyzes the first and rate-limiting step in the metabolism of L-tryptophan to kynurenine. This thus leads to tryptophan depletion. IDO utilizes immunomodulatory effects by suppressing T-cell proliferation, preventing memory T-cell formation, and inducing regulatory T-cell differentiation [91]. Expressed by APCs, IDO is induced by interferon-γ and other pro-inflammatory cytokines. In vivo models have shown that the main activity of IDO is to inhibit T-cell responses to autoantigens and fetal alloantigens [133,134]. The immunosuppressive effects of myeloid derived suppressor cells (MDSCs) may depend on IDO activity [135]. Therefore, inhibition of IDO activity increases tumor specific T-cell responses and decreases conversion to T-reg-like cells [136].
In a pancreatic cancer tumor model, Manuel et al revealed notable antitumor activity when using a combination of (A) a Salmonella-based therapy targeting IDO and PEGPH20, (B) an enzyme capable of depleting tumor hyaluronic acid, and (C) potentially enhancing immune cells infiltration in PC tumor stroma [137].
Most recently, a completed phase I/II clinical trial [91] evaluated the combination of indoximod, an IDO inhibitor, with gemcitabine and nab-paclitaxel in the first-line treatment of 80 patients with metastatic PDAC (NLG2104, NCT02077881). Results are still pending. Another IDO1 enzyme inhibitor, GDC-0919, is being evaluated in phase Ib clinical trials to target solid tumors in combination with PDL1 inhibition (MPDL3280A) (NCT02471846).
Anti-OX40 agonist therapies are planned to begin shortly [91]. OX40 is also called “T cell co-stimulation”. Immune co-stimulators work by providing the signal to expand and proliferate CD8 and CD4 helper T-cells [138]. Preclinical studies have demonstrated that using anti-OX40 mAbs and OX40L-Fc fusion proteins can enhance antitumor immunity and improve tumor-free survival [139,140].
Discussion
Pancreatic adenocarcinoma continues to be a disease with a grim prognosis. Results from the PRODIGE [141] and JASPAC-1 trials [142] are promising; however, in general, the long-term PDAC outcomes are dismal. There is a growing need to actively look for a either a curative drug, regimen or technique. Chemotherapeutic drugs are toxic; and pancreatic surgery and radiation are associated with morbidity. This review looks into many recent studies that investigate the underlying disease process as well as possible cures. Because EGFR is overexpressed in chronic pancreatitis, drug treatments that block EGFR may prove promising. IGF-1 overexpression confers resistance to EGFR; hence, blocking these two receptors together may be even more beneficial. Targeting the MAP kinase pathway alone has not proved to be fruitful due to availability of several escape pathways. Inhibiting both MEK, a part of the MAP kinase pathway, and STAT results in depleting tumor fibrosis. This allows tumors to become more susceptible to chemotherapy and radiation therapy responses. Hypoxia-induced resistance acts via PI3K pathway. Targeting PI3K stimulates T-cells to attack the tumor cells; thus, combining PI3K inhibitors and immunotherapy may be a promising combination. Targeting tumor cell protein synthesis and unfolding along with IGF-1 inhibition is also a promising combination. HSPs maintain tumor cell homeostasis. Thus, targeting them in combination with proteasome inhibitors such as carfilzomib results in cell death.
The growth phase in which a cancer cell repairs itself is different from that of normal cells. This knowledge is useful in developing chemotherapy regimens in combination with DNA repair blockers such as PARP inhibitors. MUC1 and mesothelin are surface glycoproteins that are overexpressed in tumor cells. Using dendritic cells to target MUC1 and CAR-T or NK cells to target mesothelin are options for future immunotherapy trials. Whole cell vaccines using irradiated pancreatic cells (such as algenpantucel-L) promote cancer cell attack by the innate immune system.
Engineered cells that make GM-CSF promote dendritic cell differentiation. Dendritic cells have the ability to attack cancer cells in combination with cyclophosphamide to reduce T-regs. This results in improved OS. A great example of this mechanism involves the use of CRS207, a live-attenuated vaccine engineered through double-deleted Listeria (LADD). Here, CRS207 expresses tumor-associated antigens (TAAs) and induces an immune response that specifically targets mesothelin, a TAA that is especially overproduced by PDAC cells but not by non-cancerous cells. Phagocytes such as dendritic cells engulf the Listeria-encased vaccine and induces the immune system to target such mesothelin-producing cells. As a result, PDAC cells are specifically targeted [143].
Survivin is a cell surface antigen that is expressed only by PDAC cells. Continuous use of survivin vaccines has yielded excellent results.
WT1 is a mutated peptide expressed in the cytoplasm and nuclei of all PDAC cells. Targeting WT1 cells through the use of dendritic cell vaccines has been shown to be an effective treatment. Inhibiting IDO in tumor cells increases their tumor specific T-cell response. OX 40, a T-cell co-stimulator, is being studied along with checkpoint inhibitors. These have been shown to enhance the body’s immunity against PDAC.
Overall, all of these developing medications and vaccines are promising treatments in combatting PDAC. As the incidence of PDAC rises, it is of utmost importance to treat this disease effectively in the coming years. While we continue with chemotherapy regimens, radiation therapy, and surgical resection, we await the development of novel drugs to treat and cure this disease.
Acknowledgements
We would like to thank Matthew Landry for the figures in this paper.
Disclosure of conflict of interest
None.
References
- 1.Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, Maitra A, DePinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016;30:355–385. doi: 10.1101/gad.275776.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 3.Zhou J, Enewold L, Stojadinovic A, Clifton GT, Potter JF, Peoples GE, Zhu K. Incidence rates of exocrine and endocrine pancreatic cancers in the united states. Cancer Causes Control. 2010;21:853–861. doi: 10.1007/s10552-010-9512-y. [DOI] [PubMed] [Google Scholar]
- 4.Fesinmeyer MD, Austin MA, Li CI, De Roos AJ, Bowen DJ. Differences in survival by histologic type of pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:1766–1773. doi: 10.1158/1055-9965.EPI-05-0120. [DOI] [PubMed] [Google Scholar]
- 5.Hruban RH, Takaori K, Klimstra DS, Adsay NV, Albores-Saavedra J, Biankin AV, Biankin SA, Compton C, Fukushima N, Furukawa T, Goggins M, Kato Y, Kloppel G, Longnecker DS, Luttges J, Maitra A, Offerhaus GJ, Shimizu M, Yonezawa S. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004;28:977–987. doi: 10.1097/01.pas.0000126675.59108.80. [DOI] [PubMed] [Google Scholar]
- 6.Amanam I, Chung V. Targeted therapies for pancreatic cancer. Cancers (Basel) 2018;10 doi: 10.3390/cancers10020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen NM, Singh G, Koenig A, Liou GY, Storz P, Zhang JS, Regul L, Nagarajan S, Kuhnemuth B, Johnsen SA, Hebrok M, Siveke J, Billadeau DD, Ellenrieder V, Hessmann E. NFATc1 Links EGFR signaling to induction of Sox9 transcription and acinar-ductal transdifferentiation in the pancreas. Gastroenterology. 2015;148:1024–1034. e1029. doi: 10.1053/j.gastro.2015.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korc M. Pathways for aberrant angiogenesis in pancreatic cancer. Mol Cancer. 2003;2:8. doi: 10.1186/1476-4598-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kindler HL, Niedzwiecki D, Hollis D, Sutherland S, Schrag D, Hurwitz H, Innocenti F, Mulcahy MF, O'Reilly E, Wozniak TF, Picus J, Bhargava P, Mayer RJ, Schilsky RL, Goldberg RM. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the cancer and leukemia group B (CALGB 80303) J. Clin. Oncol. 2010;28:3617–3622. doi: 10.1200/JCO.2010.28.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reni M, Cereda S, Milella M, Novarino A, Passardi A, Mambrini A, Di Lucca G, Aprile G, Belli C, Danova M, Bergamo F, Franceschi E, Fugazza C, Ceraulo D, Villa E. Maintenance sunitinib or observation in metastatic pancreatic adenocarcinoma: a phase II randomised trial. Eur J Cancer. 2013;49:3609–3615. doi: 10.1016/j.ejca.2013.06.041. [DOI] [PubMed] [Google Scholar]
- 11.Giordano G, Pancione M, Olivieri N, Parcesepe P, Velocci M, Di Raimo T, Coppola L, Toffoli G, D’Andrea MR. Nano albumin bound-paclitaxel in pancreatic cancer: current evidences and future directions. World J Gastroenterol. 2017;23:5875–5886. doi: 10.3748/wjg.v23.i32.5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lugovskoy AA, Curley M, Baum J, Adams S, Iadevaia S, Rimkunas V, Camblin A, Nie L, Tan G, Johnson B, Mathews S, Horgan K, Louis CU, Czibere AG, Arnedos M, Soria J-C, Bahleda R, Shields A, LoRusso PM, Saleh M, Isakoff SJ. Abstract CT237: preclinical characterization and first-in-human study of MM-141, a dual antibody inhibitor of IGF-1R and ErbB3. Cancer Res. 2015;75:CT237. [Google Scholar]
- 13.Awasthi N, Scire E, Monahan S, Grojean M, Zhang E, Schwarz MA, Schwarz RE. Augmentation of response to nab-paclitaxel by inhibition of insulin-like growth factor (IGF) signaling in preclinical pancreatic cancer models. Oncotarget. 2016;7:46988–47001. doi: 10.18632/oncotarget.9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Camblin AJ, Pace EA, Adams S, Curley MD, Rimkunas V, Nie L, Tan G, Bloom T, Iadevaia S, Baum J, Minx C, Czibere A, Louis CU, Drummond DC, Nielsen UB, Schoeberl B, Pipas JM, Straubinger RM, Askoxylakis V, Lugovskoy AA. Dual inhibition of IGF-1R and ErbB3 enhances the activity of gemcitabine and Nab-paclitaxel in preclinical models of pancreatic cancer. Clin Cancer Res. 2018;24:2873–2885. doi: 10.1158/1078-0432.CCR-17-2262. [DOI] [PubMed] [Google Scholar]
- 15.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401–1409. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 16.Salama AK, Flaherty KT. BRAF in melanoma: current strategies and future directions. Clin Cancer Res. 2013;19:4326–4334. doi: 10.1158/1078-0432.CCR-13-0779. [DOI] [PubMed] [Google Scholar]
- 17.Bournet B, Buscail C, Muscari F, Cordelier P, Buscail L. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: hopes and realities. Eur J Cancer. 2016;54:75–83. doi: 10.1016/j.ejca.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 18.Caldas C, Hahn SA, Hruban RH, Redston MS, Yeo CJ, Kern SE. Detection of K-ras mutations in the stool of patients with pancreatic adenocarcinoma and pancreatic ductal hyperplasia. Cancer Res. 1994;54:3568–3573. [PubMed] [Google Scholar]
- 19.Lemstrova R, Brynychova V, Hughes DJ, Hlavac V, Dvorak P, Doherty JE, Murray HA, Crockard M, Oliverius M, Hlavsa J, Honsova E, Mazanec J, Kala Z, Lovecek M, Havlik R, Ehrmann J, Strouhal O, Soucek P, Melichar B, Mohelnikova-Duchonova B. Dysregulation of KRAS signaling in pancreatic cancer is not associated with KRAS mutations and outcome. Oncol Lett. 2017;14:5980–5988. doi: 10.3892/ol.2017.6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou L, Baba Y, Kitano Y, Miyake K, Zhang X, Yamamura K, Kosumi K, Kaida T, Arima K, Taki K, Higashi T, Imai K, Hashimoto D, Yamashita Y, Chikamoto A, Beppu T, Tan X, Baba H. KRAS, BRAF, and PIK3CA mutations, and patient prognosis in 126 pancreatic cancers: pyrosequencing technology and literature review. Med Oncol. 2016;33:32. doi: 10.1007/s12032-016-0745-9. [DOI] [PubMed] [Google Scholar]
- 21.Oliveira-Cunha M, Hadfield KD, Siriwardena AK, Newman W. EGFR and KRAS mutational analysis and their correlation to survival in pancreatic and periampullary cancer. Pancreas. 2012;41:428–434. doi: 10.1097/MPA.0b013e3182327a03. [DOI] [PubMed] [Google Scholar]
- 22.Schultz NA, Roslind A, Christensen IJ, Horn T, Hogdall E, Pedersen LN, Kruhoffer M, Burcharth F, Wojdemann M, Johansen JS. Frequencies and prognostic role of KRAS and BRAF mutations in patients with localized pancreatic and ampullary adenocarcinomas. Pancreas. 2012;41:759–766. doi: 10.1097/MPA.0b013e31823cd9df. [DOI] [PubMed] [Google Scholar]
- 23.Bustinza-Linares E, Kurzrock R, Tsimberidou AM. Salirasib in the treatment of pancreatic cancer. Future Oncol. 2010;6:885–891. doi: 10.2217/fon.10.71. [DOI] [PubMed] [Google Scholar]
- 24.Ma Y, Xu J, Huang P, Bai X, Gao H. Ubiquitin-independent, proteasome-mediated targeted degradation of KRAS in pancreatic adenocarcinoma cells using an engineered ornithine decarboxylase/antizyme system. IUBMB Life. 2019;71:57–65. doi: 10.1002/iub.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagathihalli NS, Castellanos JA, Lamichhane P, Messaggio F, Shi C, Dai X, Rai P, Chen X, VanSaun MN, Merchant NB. Inverse correlation of STAT3 and MEK signaling mediates resistance to RAS pathway inhibition in pancreatic cancer. Cancer Res. 2018;78:6235–6246. doi: 10.1158/0008-5472.CAN-18-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kordes S, Klumpen HJ, Weterman MJ, Schellens JH, Richel DJ, Wilmink JW. Phase II study of capecitabine and the oral mTOR inhibitor everolimus in patients with advanced pancreatic cancer. Cancer Chemother Pharmacol. 2015;75:1135–1141. doi: 10.1007/s00280-015-2730-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Javle MM, Shroff RT, Xiong H, Varadhachary GA, Fogelman D, Reddy SA, Davis D, Zhang Y, Wolff RA, Abbruzzese JL. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC Cancer. 2010;10:368. doi: 10.1186/1471-2407-10-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt KM, Hellerbrand C, Ruemmele P, Michalski CW, Kong B, Kroemer A, Hackl C, Schlitt HJ, Geissler EK, Lang SA. Inhibition of mTORC2 component RICTOR impairs tumor growth in pancreatic cancer models. Oncotarget. 2017;8:24491–24505. doi: 10.18632/oncotarget.15524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye LY, Zhang Q, Bai XL, Pankaj P, Hu QD, Liang TB. Hypoxia-inducible factor 1alpha expression and its clinical significance in pancreatic cancer: a meta-analysis. Pancreatology. 2014;14:391–397. doi: 10.1016/j.pan.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 30.Chae YC, Vaira V, Caino MC, Tang HY, Seo JH, Kossenkov AV, Ottobrini L, Martelli C, Lucignani G, Bertolini I, Locatelli M, Bryant KG, Ghosh JC, Lisanti S, Ku B, Bosari S, Languino LR, Speicher DW, Altieri DC. Mitochondrial Akt regulation of hypoxic tumor reprogramming. Cancer Cell. 2016;30:257–272. doi: 10.1016/j.ccell.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo J, Chakraborty AA, Liu P, Gan W, Zheng X, Inuzuka H, Wang B, Zhang J, Zhang L, Yuan M, Novak J, Cheng JQ, Toker A, Signoretti S, Zhang Q, Asara JM, Kaelin WG Jr, Wei W. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science. 2016;353:929–932. doi: 10.1126/science.aad5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conway JRW, Warren SC, Herrmann D, Murphy KJ, Cazet AS, Vennin C, Shearer RF, Killen MJ, Magenau A, Mélénec P, Pinese M, Nobis M, Zaratzian A, Boulghourjian A, Da Silva AM, Del Monte-Nieto G, Adam ASA, Harvey RP, Haigh JJ, Wang Y, Croucher DR, Sansom OJ, Pajic M, Caldon CE, Morton JP, Timpson P. Intravital imaging to monitor therapeutic response in moving hypoxic regions resistant to PI3K pathway targeting in pancreatic cancer. Cell Rep. 2018;23:3312–3326. doi: 10.1016/j.celrep.2018.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang H, Xu M, Li L, Grierson P, Dodhiawala P, Highkin M, Zhang D, Li Q, Wang-Gillam A, Lim KH. Concurrent HER or PI3K inhibition potentiates the antitumor effect of the ERK inhibitor ulixertinib in preclinical pancreatic cancer models. Mol Cancer Ther. 2018;17:2144–2155. doi: 10.1158/1535-7163.MCT-17-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, Schmid MC, Sun P, Mose E, Bouvet M, Lowy AM, Valasek MA, Sasik R, Novelli F, Hirsch E, Varner JA. Macrophage PI3Kgamma drives pancreatic ductal adenocarcinoma progression. Cancer Discov. 2016;6:870–885. doi: 10.1158/2159-8290.CD-15-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bilsland JG, Wheeldon A, Mead A, Znamenskiy P, Almond S, Waters KA, Thakur M, Beaumont V, Bonnert TP, Heavens R, Whiting P, McAllister G, Munoz-Sanjuan I. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology. 2008;33:685–700. doi: 10.1038/sj.npp.1301446. [DOI] [PubMed] [Google Scholar]
- 37.Yan HH, Jung KH, Son MK, Fang Z, Kim SJ, Ryu YL, Kim J, Kim MH, Hong SS. Crizotinib exhibits antitumor activity by targeting ALK signaling not c-MET in pancreatic cancer. Oncotarget. 2014;5:9150–9168. doi: 10.18632/oncotarget.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong SW, Jung KH, Lee HS, Son MK, Yan HH, Kang NS, Lee J, Hong SS. SB365, pulsatilla saponin D, targets c-Met and exerts antiangiogenic and antitumor activities. Carcinogenesis. 2013;34:2156–2169. doi: 10.1093/carcin/bgt159. [DOI] [PubMed] [Google Scholar]
- 39.Di Renzo MF, Poulsom R, Olivero M, Comoglio PM, Lemoine NR. Expression of the met/hepatocyte growth factor receptor in human pancreatic cancer. Cancer Res. 1995;55:1129–1138. [PubMed] [Google Scholar]
- 40.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14:581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 41.Hendershot LM. The ER function BiP is a master regulator of ER function. Mt Sinai J Med. 2004;71:289–297. [PubMed] [Google Scholar]
- 42.Lee AS. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods. 2005;35:373–381. doi: 10.1016/j.ymeth.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 43.Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6:45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 44.Lev A, Lulla AR, Wagner J, Ralff MD, Kiehl JB, Zhou Y, Benes CH, Prabhu VV, Oster W, Astsaturov I, Dicker DT, El-Deiry WS. Anti-pancreatic cancer activity of ONC212 involves the unfolded protein response (UPR) and is reduced by IGF1-R and GRP78/BIP. Oncotarget. 2017;8:81776–81793. doi: 10.18632/oncotarget.20819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ishizawa J, Kojima K, Chachad D, Ruvolo P, Ruvolo V, Jacamo RO, Borthakur G, Mu H, Zeng Z, Tabe Y, Allen JE, Wang Z, Ma W, Lee HC, Orlowski R, Sarbassov dos D, Lorenzi PL, Huang X, Neelapu SS, McDonnell T, Miranda RN, Wang M, Kantarjian H, Konopleva M, Davis RE, Andreeff M. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci Signal. 2016;9:ra17. doi: 10.1126/scisignal.aac4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kline CL, Van den Heuvel AP, Allen JE, Prabhu VV, Dicker DT, El-Deiry WS. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2alpha kinases. Sci Signal. 2016;9:ra18. doi: 10.1126/scisignal.aac4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan G, Wu L, Li B, An J. Primary malignant melanoma of the cervix: report of 14 cases and review of literature. Oncotarget. 2017;8:73162–73167. doi: 10.18632/oncotarget.17183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Endo Greer Y, Lipkowitz S. ONC201: stressing tumors to death. Sci Signal. 2016;9:fs1. doi: 10.1126/scisignal.aad7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fornace AJ Jr, Alamo I Jr, Hollander MC. DNA damage-inducible transcripts in mammalian cells. Proc Natl Acad Sci U S A. 1988;85:8800–8804. doi: 10.1073/pnas.85.23.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Belalcazar A, Shaib WL, Farren MR, Zhang C, Chen Z, Yang L, Lesinski GB, El-Rayes BF, Nagaraju GP. Inhibiting heat shock protein 90 and the ubiquitin-proteasome pathway impairs metabolic homeostasis and leads to cell death in human pancreatic cancer cells. Cancer. 2017;123:4924–4933. doi: 10.1002/cncr.30944. [DOI] [PubMed] [Google Scholar]
- 51.Bull EE, Dote H, Brady KJ, Burgan WE, Carter DJ, Cerra MA, Oswald KA, Hollingshead MG, Camphausen K, Tofilon PJ. Enhanced tumor cell radiosensitivity and abrogation of G2 and S phase arrest by the Hsp90 inhibitor 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin. Clin Cancer Res. 2004;10:8077–8084. doi: 10.1158/1078-0432.CCR-04-1212. [DOI] [PubMed] [Google Scholar]
- 52.Wang B, Chen Z, Yu F, Chen Q, Tian Y, Ma S, Wang T, Liu X. Hsp90 regulates autophagy and plays a role in cancer therapy. Tumour Biol. 2016;37:1–6. doi: 10.1007/s13277-015-4142-3. [DOI] [PubMed] [Google Scholar]
- 53.He W, Ye X, Huang X, Lel W, You L, Wang L, Chen X, Qian W. Hsp90 inhibitor, BIIB021, induces apoptosis and autophagy by regulating mTOR-Ulk1 pathway in imatinib-sensitive and -resistant chronic myeloid leukemia cells. Int J Oncol. 2016;48:1710–1720. doi: 10.3892/ijo.2016.3382. [DOI] [PubMed] [Google Scholar]
- 54.Mori M, Hitora T, Nakamura O, Yamagami Y, Horie R, Nishimura H, Yamamoto T. Hsp90 inhibitor induces autophagy and apoptosis in osteosarcoma cells. Int J Oncol. 2015;46:47–54. doi: 10.3892/ijo.2014.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qing G, Yan P, Xiao G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IkappaB kinase (IKK) Cell Res. 2006;16:895–901. doi: 10.1038/sj.cr.7310109. [DOI] [PubMed] [Google Scholar]
- 56.Lang SA, Moser C, Gaumann A, Klein D, Glockzin G, Popp FC, Dahlke MH, Piso P, Schlitt HJ, Geissler EK, Stoeltzing O. Targeting heat shock protein 90 in pancreatic cancer impairs insulin-like growth factor-I receptor signaling, disrupts an interleukin-6/signal-transducer and activator of transcription 3/hypoxia-inducible factor-1alpha autocrine loop, and reduces orthotopic tumor growth. Clin Cancer Res. 2007;13:6459–6468. doi: 10.1158/1078-0432.CCR-07-1104. [DOI] [PubMed] [Google Scholar]
- 57.Xue N, Jin J, Liu D, Yan R, Zhang S, Yu X, Chen X. Antiproliferative effect of HSP90 inhibitor Y306zh against pancreatic cancer is mediated by interruption of AKT and MAPK signaling pathways. Curr Cancer Drug Targets. 2014;14:671–683. doi: 10.2174/1568009614666140908101523. [DOI] [PubMed] [Google Scholar]
- 58.Seo JH, Jung KH, Son MK, Yan HH, Ryu YL, Kim J, Lee JK, Hong S, Hong SS. Anti-cancer effect of HS-345, a new tropomyosin-related kinase A inhibitor, on human pancreatic cancer. Cancer Lett. 2013;338:271–281. doi: 10.1016/j.canlet.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 59.Iwata T, Uchino T, Koyama A, Johmura Y, Koyama K, Saito T, Ishiguro S, Arikawa T, Komatsu S, Miyachi M, Sano T, Nakanishi M, Shimada M. The G2 checkpoint inhibitor CBP-93872 increases the sensitivity of colorectal and pancreatic cancer cells to chemotherapy. PLoS One. 2017;12:e0178221. doi: 10.1371/journal.pone.0178221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mueller S, Hashizume R, Yang X, Kolkowitz I, Olow AK, Phillips J, Smirnov I, Tom MW, Prados MD, James CD, Berger MS, Gupta N, Haas-Kogan DA. Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro Oncol. 2014;16:352–360. doi: 10.1093/neuonc/not220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karnak D, Engelke CG, Parsels LA, Kausar T, Wei D, Robertson JR, Marsh KB, Davis MA, Zhao L, Maybaum J, Lawrence TS, Morgan MA. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin Cancer Res. 2014;20:5085–5096. doi: 10.1158/1078-0432.CCR-14-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kausar T, Schreiber JS, Karnak D, Parsels LA, Parsels JD, Davis MA, Zhao L, Maybaum J, Lawrence TS, Morgan MA. Sensitization of pancreatic cancers to gemcitabine chemoradiation by WEE1 kinase inhibition depends on homologous recombination repair. Neoplasia. 2015;17:757–766. doi: 10.1016/j.neo.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caretti V, Hiddingh L, Lagerweij T, Schellen P, Koken PW, Hulleman E, van Vuurden DG, Vandertop WP, Kaspers GJ, Noske DP, Wurdinger T. WEE1 kinase inhibition enhances the radiation response of diffuse intrinsic pontine gliomas. Mol Cancer Ther. 2013;12:141–150. doi: 10.1158/1535-7163.MCT-12-0735. [DOI] [PubMed] [Google Scholar]
- 64.Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, Buchholz TA, Molkentine JM, Mason KA, Meyn RE. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res. 2011;17:5638–5648. doi: 10.1158/1078-0432.CCR-11-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sarcar B, Kahali S, Prabhu AH, Shumway SD, Xu Y, Demuth T, Chinnaiyan P. Targeting radiation-induced G(2) checkpoint activation with the Wee-1 inhibitor MK-1775 in glioblastoma cell lines. Mol Cancer Ther. 2011;10:2405–2414. doi: 10.1158/1535-7163.MCT-11-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science. 2013;339:700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, Haffty BG, Tommiska J, Blomqvist C, Drapkin R, Adams DJ, Nevanlinna H, Bartek J, Tarsounas M, Ganesan S, Jonkers J. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, Xu X, Deng CX, Finkel T, Nussenzweig M, Stark JM, Nussenzweig A. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boersma V, Moatti N, Segura-Bayona S, Peuscher MH, van der Torre J, Wevers BA, Orthwein A, Durocher D, Jacobs JJL. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5’ end resection. Nature. 2015;521:537–540. doi: 10.1038/nature14216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49:858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, Jaspers JE, Watanabe K, Pieterse M, Kersbergen A, Sol W, Celie PHN, Schouten PC, van den Broek B, Salman A, Nieuwland M, de Rink I, de Ronde J, Jalink K, Boulton SJ, Chen J, van Gent DC, Bartek J, Jonkers J, Borst P, Rottenberg S. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521:541–544. doi: 10.1038/nature14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Drean A, Lord CJ, Ashworth A. PARP inhibitor combination therapy. Crit Rev Oncol Hematol. 2016;108:73–85. doi: 10.1016/j.critrevonc.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 73.Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science. 2017;355:1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Du C, Qi Y, Zhang Y, Wang Y, Zhao X, Min H, Han X, Lang J, Qin H, Shi Q, Zhang Z, Tian X, Anderson GJ, Zhao Y, Nie G, Yang Y. Epidermal growth factor receptor-targeting peptide nanoparticles simultaneously deliver gemcitabine and olaparib to treat pancreatic cancer with breast cancer 2 (BRCA2) mutation. ACS Nano. 2018;12:10785–10796. doi: 10.1021/acsnano.8b01573. [DOI] [PubMed] [Google Scholar]
- 75.Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007;117:1137–1146. doi: 10.1172/JCI31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 77.Sahin IH, Askan G, Hu ZI, O’Reilly EM. Immunotherapy in pancreatic ductal adenocarcinoma: an emerging entity? Ann Oncol. 2017;28:2950–2961. doi: 10.1093/annonc/mdx503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pandha H, Rigg A, John J, Lemoine N. Loss of expression of antigen-presenting molecules in human pancreatic cancer and pancreatic cancer cell lines. Clin Exp Immunol. 2007;148:127–35. doi: 10.1111/j.1365-2249.2006.03289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- 80.von Bernstorff W, Spanjaard RA, Chan AK, Lockhart DC, Sadanaga N, Wood I, Peiper M, Goedegebuure PS, Eberlein TJ. Pancreatic cancer cells can evade immune surveillance via nonfunctional Fas (APO-1/CD95) receptors and aberrant expression of functional fas ligand. Surgery. 1999;125:73–84. doi: 10.1067/msy.2099.93570. [DOI] [PubMed] [Google Scholar]
- 81.Whiteside TL. Tumor-induced death of immune cells: its mechanisms and consequences. Semin Cancer Biol. 2002;12:43–50. doi: 10.1006/scbi.2001.0402. [DOI] [PubMed] [Google Scholar]
- 82.Hinz S, Pagerols-Raluy L, Oberg HH, Ammerpohl O, Grussel S, Sipos B, Grutzmann R, Pilarsky C, Ungefroren H, Saeger HD, Kloppel G, Kabelitz D, Kalthoff H. Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Cancer Res. 2007;67:8344–8350. doi: 10.1158/0008-5472.CAN-06-3304. [DOI] [PubMed] [Google Scholar]
- 83.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chellappa S, Hugenschmidt H, Hagness M, Line PD, Labori KJ, Wiedswang G, Tasken K, Aandahl EM. Regulatory T cells that co-express RORgammat and FOXP3 are pro-inflammatory and immunosuppressive and expand in human pancreatic cancer. Oncoimmunology. 2016;5:e1102828. doi: 10.1080/2162402X.2015.1102828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li M, Bharadwaj U, Zhang R, Zhang S, Mu H, Fisher WE, Brunicardi FC, Chen C, Yao Q. Mesothelin is a malignant factor and therapeutic vaccine target for pancreatic cancer. Mol Cancer Ther. 2008;7:286–296. doi: 10.1158/1535-7163.MCT-07-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hassan R, Bera T, Pastan I. Mesothelin: a new target for immunotherapy. Clin Cancer Res. 2004;10:3937–3942. doi: 10.1158/1078-0432.CCR-03-0801. [DOI] [PubMed] [Google Scholar]
- 87.Lepisto AJ, Moser AJ, Zeh H, Lee K, Bartlett D, McKolanis JR, Geller BA, Schmotzer A, Potter DP, Whiteside T, Finn OJ, Ramanathan RK. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther. 2008;6:955–964. [PMC free article] [PubMed] [Google Scholar]
- 88.Deguchi T, Tanemura M, Miyoshi E, Nagano H, Machida T, Ohmura Y, Kobayashi S, Marubashi S, Eguchi H, Takeda Y, Ito T, Mori M, Doki Y, Sawa Y. Increased immunogenicity of tumor-associated antigen, mucin 1, engineered to express alpha-gal epitopes: a novel approach to immunotherapy in pancreatic cancer. Cancer Res. 2010;70:5259–5269. doi: 10.1158/0008-5472.CAN-09-4313. [DOI] [PubMed] [Google Scholar]
- 89.Bernhardt SL, Gjertsen MK, Trachsel S, Moller M, Eriksen JA, Meo M, Buanes T, Gaudernack G. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br J Cancer. 2006;95:1474–1482. doi: 10.1038/sj.bjc.6603437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Middleton G, Silcocks P, Cox T, Valle J, Wadsley J, Propper D, Coxon F, Ross P, Madhusudan S, Roques T, Cunningham D, Falk S, Wadd N, Harrison M, Corrie P, Iveson T, Robinson A, McAdam K, Eatock M, Evans J, Archer C, Hickish T, Garcia-Alonso A, Nicolson M, Steward W, Anthoney A, Greenhalf W, Shaw V, Costello E, Naisbitt D, Rawcliffe C, Nanson G, Neoptolemos J. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol. 2014;15:829–840. doi: 10.1016/S1470-2045(14)70236-0. [DOI] [PubMed] [Google Scholar]
- 91.McCormick KA, Coveler AL, Rossi GR, Vahanian NN, Link C, Chiorean EG. Pancreatic cancer: Update on immunotherapies and algenpantucel-L. Hum Vaccin Immunother. 2016;12:563–575. doi: 10.1080/21645515.2015.1093264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hardacre JM, Mulcahy M, Small W, Talamonti M, Obel J, Krishnamurthi S, Rocha-Lima CS, Safran H, Lenz HJ, Chiorean EG. Addition of algenpantucel-L immunotherapy to standard adjuvant therapy for pancreatic cancer: a phase 2 study. J Gastrointest Surg. 2013;17:94–100. doi: 10.1007/s11605-012-2064-6. [DOI] [PubMed] [Google Scholar]
- 93.Hemstreet GP 3rd, Rossi GR, Pisarev VM, Enke CA, Helfner L, Hauke RJ, Tennant L, Ramsey WJ, Vahanian NN, Link CJ. Cellular immunotherapy study of prostate cancer patients and resulting IgG responses to peptide epitopes predicted from prostate tumor-associated autoantigens. J Immunother. 2013;36:57–65. doi: 10.1097/CJI.0b013e3182780abc. [DOI] [PubMed] [Google Scholar]
- 94.Rossi GR, Unfer RC, Seregina T, Link CJ. Complete protection against melanoma in absence of autoimmune depigmentation after rejection of melanoma cells expressing alpha(1,3) galactosyl epitopes. Cancer Immunol Immunother. 2005;54:999–1009. doi: 10.1007/s00262-005-0667-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Coveler AL, Rossi GR, Vahanian NN, Link C, Chiorean EG. Algenpantucel-L immunotherapy in pancreatic adenocarcinoma. Immunotherapy. 2016;8:117–125. doi: 10.2217/imt.15.113. [DOI] [PubMed] [Google Scholar]
- 96.Link CJ Jr, Seregina T, Atchison R, Hall A, Muldoon R, Levy JP. Eliciting hyperacute xenograft response to treat human cancer: alpha(1,3) galactosyltransferase gene therapy. Anticancer Res. 1998;18:2301–2308. [PubMed] [Google Scholar]
- 97.Galili U, Anaraki F, Thall A, Hill-Black C, Radic M. One percent of human circulating B lymphocytes are capable of producing the natural anti-Gal antibody. Blood. 1993;82:2485–2493. [PubMed] [Google Scholar]
- 98.Jaffee EM, Hruban RH, Biedrzycki B, Laheru D, Schepers K, Sauter PR, Goemann M, Coleman J, Grochow L, Donehower RC, Lillemoe KD, O’Reilly S, Abrams RA, Pardoll DM, Cameron JL, Yeo CJ. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J. Clin. Oncol. 2001;19:145–156. doi: 10.1200/JCO.2001.19.1.145. [DOI] [PubMed] [Google Scholar]
- 99.Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, Eberlein TJ, Hsieh CS, Linehan DC. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182:1746–1755. doi: 10.4049/jimmunol.182.3.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Laheru D, Lutz E, Burke J, Biedrzycki B, Solt S, Onners B, Tartakovsky I, Nemunaitis J, Le D, Sugar E, Hege K, Jaffee E. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: a pilot study of safety, feasibility, and immune activation. Clin Cancer Res. 2008;14:1455–1463. doi: 10.1158/1078-0432.CCR-07-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, Springett G, Morse M, Zeh H, Cohen D, Fine RL, Onners B, Uram JN, Laheru DA, Lutz ER, Solt S, Murphy AL, Skoble J, Lemmens E, Grous J, Dubensky T Jr, Brockstedt DG, Jaffee EM. Safety and survival with GVAX pancreas prime and listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015;33:1325–1333. doi: 10.1200/JCO.2014.57.4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Le DT, Lutz E, Uram JN, Sugar EA, Onners B, Solt S, Zheng L, Diaz LA Jr, Donehower RC, Jaffee EM, Laheru DA. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother. 2013;36:382–389. doi: 10.1097/CJI.0b013e31829fb7a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Satoh K, Kaneko K, Hirota M, Masamune A, Satoh A, Shimosegawa T. Expression of survivin is correlated with cancer cell apoptosis and is involved in the development of human pancreatic duct cell tumors. Cancer. 2001;92:271–278. doi: 10.1002/1097-0142(20010715)92:2<271::aid-cncr1319>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 104.Wobser M, Keikavoussi P, Kunzmann V, Weininger M, Andersen MH, Becker JC. Complete remission of liver metastasis of pancreatic cancer under vaccination with a HLA-A2 restricted peptide derived from the universal tumor antigen survivin. Cancer Immunol Immunother. 2006;55:1294–1298. doi: 10.1007/s00262-005-0102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kameshima H, Tsuruma T, Kutomi G, Shima H, Iwayama Y, Kimura Y, Imamura M, Torigoe T, Takahashi A, Hirohashi Y, Tamura Y, Tsukahara T, Kanaseki T, Sato N, Hirata K. Immunotherapeutic benefit of alpha-interferon (IFNalpha) in survivin2B-derived peptide vaccination for advanced pancreatic cancer patients. Cancer Sci. 2013;104:124–129. doi: 10.1111/cas.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Koido S, Homma S, Okamoto M, Takakura K, Mori M, Yoshizaki S, Tsukinaga S, Odahara S, Koyama S, Imazu H, Uchiyama K, Kajihara M, Arakawa H, Misawa T, Toyama Y, Yanagisawa S, Ikegami M, Kan S, Hayashi K, Komita H, Kamata Y, Ito M, Ishidao T, Yusa S, Shimodaira S, Gong J, Sugiyama H, Ohkusa T, Tajiri H. Treatment with chemotherapy and dendritic cells pulsed with multiple wilms’ tumor 1 (WT1)-specific MHC class I/II-restricted epitopes for pancreatic cancer. Clin Cancer Res. 2014;20:4228–4239. doi: 10.1158/1078-0432.CCR-14-0314. [DOI] [PubMed] [Google Scholar]
- 107.Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, Matrisian LM. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 109.Nishida S, Koido S, Takeda Y, Homma S, Komita H, Takahara A, Morita S, Ito T, Morimoto S, Hara K, Tsuboi A, Oka Y, Yanagisawa S, Toyama Y, Ikegami M, Kitagawa T, Eguchi H, Wada H, Nagano H, Nakata J, Nakae Y, Hosen N, Oji Y, Tanaka T, Kawase I, Kumanogoh A, Sakamoto J, Doki Y, Mori M, Ohkusa T, Tajiri H, Sugiyama H. Wilms tumor gene (WT1) peptide-based cancer vaccine combined with gemcitabine for patients with advanced pancreatic cancer. J Immunother. 2014;37:105–114. doi: 10.1097/CJI.0000000000000020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Garg AD, Vara Perez M, Schaaf M, Agostinis P, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: dendritic cell-based anticancer immunotherapy. Oncoimmunology. 2017;6:e1328341. doi: 10.1080/2162402X.2017.1328341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yanagimoto H, Shiomi H, Satoi S, Mine T, Toyokawa H, Yamamoto T, Tani T, Yamada A, Kwon AH, Komatsu N, Itoh K, Noguchi M. A phase II study of personalized peptide vaccination combined with gemcitabine for non-resectable pancreatic cancer patients. Oncol Rep. 2010;24:795–801. doi: 10.3892/or_00000923. [DOI] [PubMed] [Google Scholar]
- 112.Yutani S, Komatsu N, Yoshitomi M, Matsueda S, Yonemoto K, Mine T, Noguchi M, Ishihara Y, Yamada A, Itoh K, Sasada T. A phase II study of a personalized peptide vaccination for chemotherapy-resistant advanced pancreatic cancer patients. Oncol Rep. 2013;30:1094–1100. doi: 10.3892/or.2013.2556. [DOI] [PubMed] [Google Scholar]
- 113.Kanai T, Ito Z, Oji Y, Suka M, Nishida S, Takakura K, Kajihara M, Saruta M, Fujioka S, Misawa T, Akiba T, Yanagisawa H, Shimodaira S, Okamoto M, Sugiyama H, Koido S. Prognostic significance of Wilms’ tumor 1 expression in patients with pancreatic ductal adenocarcinoma. Oncol Lett. 2018;16:2682–2692. doi: 10.3892/ol.2018.8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Koido S, Okamoto M, Kobayashi M, Shimodaira S, Sugiyama H. Significance of wilms’ tumor 1 antigen as a cancer vaccine for pancreatic cancer. Discov Med. 2017;24:41–49. [PubMed] [Google Scholar]
- 115.Wagner KD, Cherfils-Vicini J, Hosen N, Hohenstein P, Gilson E, Hastie ND, Michiels JF, Wagner N. The Wilms’ tumour suppressor Wt1 is a major regulator of tumour angiogenesis and progression. Nat Commun. 2014;5:5852. doi: 10.1038/ncomms6852. [DOI] [PubMed] [Google Scholar]
- 116.Koido S, Okamoto M, Shimodaira S, Sugiyama H. Wilms’ tumor 1 (WT1)-targeted cancer vaccines to extend survival for patients with pancreatic cancer. Immunotherapy. 2016;8:1309–1320. doi: 10.2217/imt-2016-0031. [DOI] [PubMed] [Google Scholar]
- 117.Hanada S, Tsuruta T, Haraguchi K, Okamoto M, Sugiyama H, Koido S. Long-term survival of pancreatic cancer patients treated with multimodal therapy combined with WT1-targeted dendritic cell vaccines. Hum Vaccin Immunother. 2019;15:397–406. doi: 10.1080/21645515.2018.1524238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 119.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Speiser DE, Ho PC, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol. 2016;16:599–611. doi: 10.1038/nri.2016.80. [DOI] [PubMed] [Google Scholar]
- 121.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–182. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- 124.Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013;73:3591–3603. doi: 10.1158/0008-5472.CAN-12-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, Vanderford TH, Chennareddi L, Silvestri G, Freeman GJ, Ahmed R, Amara RR. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458:206–210. doi: 10.1038/nature07662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv324. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384–3391. doi: 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, Troxel AB, Sun W, Teitelbaum UR, Vonderheide RH, O’Dwyer PJ. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19:6286–6295. doi: 10.1158/1078-0432.CCR-13-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zippelius A, Schreiner J, Herzig P, Muller P. Induced PD-L1 expression mediates acquired resistance to agonistic anti-CD40 treatment. Cancer Immunol Res. 2015;3:236–244. doi: 10.1158/2326-6066.CIR-14-0226. [DOI] [PubMed] [Google Scholar]
- 131.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, Teichmann SA, Janowitz T, Jodrell DI, Tuveson DA, Fearon DT. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110:20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dewan MZ, Ahmed S, Iwasaki Y, Ohba K, Toi M, Yamamoto N. Stromal cell-derived factor-1 and CXCR4 receptor interaction in tumor growth and metastasis of breast cancer. Biomed Pharmacother. 2006;60:273–276. doi: 10.1016/j.biopha.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 133.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 134.Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, Marshall B, Chandler P, Antonia SJ, Burgess R, Slingluff CL Jr, Mellor AL. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. 2002;297:1867–1870. doi: 10.1126/science.1073514. [DOI] [PubMed] [Google Scholar]
- 135.Meisel R, Zibert A, Laryea M, Gobel U, Daubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;103:4619–4621. doi: 10.1182/blood-2003-11-3909. [DOI] [PubMed] [Google Scholar]
- 136.Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, Leffet L, Hansbury MJ, Thomas B, Rupar M, Waeltz P, Bowman KJ, Polam P, Sparks RB, Yue EW, Li Y, Wynn R, Fridman JS, Burn TC, Combs AP, Newton RC, Scherle PA. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010;115:3520–3530. doi: 10.1182/blood-2009-09-246124. [DOI] [PubMed] [Google Scholar]
- 137.Manuel ER, Chen J, D’Apuzzo M, Lampa MG, Kaltcheva TI, Thompson CB, Ludwig T, Chung V, Diamond DJ. Salmonella-based therapy targeting indoleamine 2,3-dioxygenase coupled with enzymatic depletion of tumor hyaluronan induces complete regression of aggressive pancreatic tumors. Cancer Immunol Res. 2015;3:1096–1107. doi: 10.1158/2326-6066.CIR-14-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Patients CCST. OX40 Agonists: Boosting Cancer Immunotherapy. 2017, August 29 [Available from: https://blog. crownbio.com/ox40-ox40l-immune-co-stimulators]
- 139.Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34. doi: 10.3389/fonc.2015.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jensen SM, Maston LD, Gough MJ, Ruby CE, Redmond WL, Crittenden M, Li Y, Puri S, Poehlein CH, Morris N, Kovacsovics-Bankowski M, Moudgil T, Twitty C, Walker EB, Hu HM, Urba WJ, Weinberg AD, Curti B, Fox BA. Signaling through OX40 enhances antitumor immunity. Semin Oncol. 2010;37:524–532. doi: 10.1053/j.seminoncol.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, Chone L, Francois E, Artru P, Biagi JJ, Lecomte T, Assenat E, Faroux R, Ychou M, Volet J, Sauvanet A, Breysacher G, Di Fiore F, Cripps C, Kavan P, Texereau P, Bouhier-Leporrier K, Khemissa-Akouz F, Legoux JL, Juzyna B, Gourgou S, O’Callaghan CJ, Jouffroy-Zeller C, Rat P, Malka D, Castan F, Bachet JB. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med. 2018;379:2395–2406. doi: 10.1056/NEJMoa1809775. [DOI] [PubMed] [Google Scholar]
- 142.Uesaka K, Boku N, Fukutomi A, Okamura Y, Konishi M, Matsumoto I, Kaneoka Y, Shimizu Y, Nakamori S, Sakamoto H, Morinaga S, Kainuma O, Imai K, Sata N, Hishinuma S, Ojima H, Yamaguchi R, Hirano S, Sudo T, Ohashi Y. Adjuvant chemotherapy of S-1 versus gemcitabine for resected pancreatic cancer: a phase 3, open-label, randomised, non-inferiority trial (JASPAC 01) Lancet. 2016;388:248–257. doi: 10.1016/S0140-6736(16)30583-9. [DOI] [PubMed] [Google Scholar]
- 143.News IO. CRS-207 [Available from: https://immuno-oncologynews. com/crs-207/]