

Abstract
MYCN, a member of the MYC family, is correlated with tumorigenesis, metastasis and therapy in many malignancies; however, its role in small-cell lung cancer (SCLC) remains unclear. In this study, we sought to identify the function of MYCN in SCLC chemoresistance and found that MYCN is overexpressed in chemoresistant SCLC cells. We used MYCN gain- and loss-of- function experiments to demonstrate that MYCN promotes in vitro and in vivo chemoresistance in SCLC by inhibiting apoptosis. Mechanistic investigations showed that MYCN binds to the HES1 promoter and exhibits transcriptional activity. Furthermore, MYCN mediated SCLC chemoresistance through HES1. Accordingly, the NOTCH inhibitor FLI-60 derepressed HES1 and diminished MYCN-induced chemoresistance in SCLC. Finally, the positive correlation between HES1 and MYCN was confirmed in SCLC patients. Chemoresistant SCLC patients had higher expression levels of MYCN and HES1 than patients without chemoresistant SCLC. MYCN overexpression was related to advanced clinical stage and shorter survival in SCLC. In conclusion, our study revealed that MYCN and HES1 may be potential therapeutic targets and promising predictors for SCLC.
Keywords: MYCN, small-cell lung cancer, chemoresistance, HES1, NOTCH pathway
Introduction
Lung carcinoma is the leading cause of cancer-related death worldwide [1,2]. Small-cell lung cancer (SCLC) is an aggressive malignancy and accounts for approximately 13-15% of all lung cancers [3]. Etoposide (VP16)- and cisplatin (CDDP)-based chemotherapies are the first-line treatment for SCLC. Although SCLC is initially sensitive to these treatments, most patients rapidly develop drug resistance, leading to chemotherapy failure. Owing to the lack of an effective second-line treatment strategy for these individuals, the prognosis of SCLC is very poor, with a discouraging 5-year overall survival [4]. Therefore, it is vital to fully elucidate the molecular mechanisms of chemoresistance and discover novel effective, diagnostic and therapeutic markers for SCLC.
The MYC oncogene family is a class of transcription factors with a basic-helix-loop-helix/leucine-zipper domain and includes the following three members: MYC, MYCL, and MYCN [5,6]. MYCN is located on human chromosome 2p24.3. In contrast to MYC and MYCL, MYCN is mainly amplified in embryonic and neuroendocrine tumors [7]. Research on MYCN is mainly focused on neuroblastoma [8] and has indicated that MYCN amplification is strongly associated with proliferation [9,10], migration [11], invasion, apoptosis [12,13], drug resistance [14,15], and poor clinical outcomes for aggressive neuroblastoma. SCLC is also a neuroendocrine tumor, and the frequent and significant amplification of MYCN was found in SCLC through whole-genome and transcriptome sequencing analyses [16-19]. However, little is known about the role of MYCN in SCLC. Our previous cDNA microarray analysis revealed that MYCN is differentially expressed between chemosensitive and chemoresistant SCLC cells. Therefore, our research focused on the relationship and mechanism between MYCN and drug resistance.
The Notch pathway is a fundamental signaling system that mediates critically important functions by direct cell-cell contact. Notch proteins are surface transmembrane receptors. Upon binding with their ligands (Delta and Serrate/Jagged), the Notch receptors drive the expression of various target genes, including the HES/HEY gene families among others [20,21]. Previous studies have shown that the Notch pathway is involved in the occurrence, development, stemness and drug resistance of many tumors, such as leukemia [22], prostate cancer [23], breast cancer [24,25], and lung cancer [26-28]. NOTCH1 has been confirmed to be related to the chemoresistance of SCLC [29]. HES1 is a substrate of the Notch pathway and plays a key role in the multidrug resistance of various tumors [30-32], but the functions of HES1 in SCLC chemoresistance have not been reported.
In this study, we found that MYCN was significantly upregulated in SCLC chemoresistant cell lines. Functionally, MYCN promoted the chemoresistance of SCLC cells in vitro and in vivo by inhibiting drug-induced apoptosis; mechanistically, MYCN increased HES1 transcription and expression by binding to the HES1 promoter and activating the NOTCH pathway. Furthermore, MYCN was associated with chemotherapy response, clinical stage and poor prognosis in SCLC patients. Taken together, these findings suggest that MYCN and HES1 may be useful candidates for SCLC diagnosis and therapy.
Materials and methods
Patients and tissue samples
A total of 42 SCLC patient tissues were collected from the First Affiliated Hospital of University of South China (Heyang, China) between January 2014 and January 2017. Clinicopathological data, including age, gender, stage, chemotherapy response and survival time, were collected. Chemotherapy response was categorized as ‘chemosensitive’ (PR or CR) and ‘chemoresistant’ (PD or SD) based on the Response Evaluation Criteria in Solid Tumors (RECIST Edition 1.1). The follow-up time was calculated from the date of diagnosis to the date of either death or the last known follow-up. Written informed consent was obtained from all patients, and the protocol was approved by the First Affiliated Hospital of University of South China.
Cell culture
The human SCLC cell lines NCI-H69, NCI-H69AR and NCI-H446 were purchased from American Type Culture Collection (ATCC, USA), whereas the human SCLC cell lines NCI-H526, NCI-H82, NCI-H209, NCI-146, and NCI-345 were generous gifts from Dr. Ji Lin of MD Anderson Cancer Center. All cell lines were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum and antibiotics (100 mg/mL penicillin and 100 mg/mL streptomycin).
Reagents and antibodies
The chemotherapeutic drugs cisplatin (DDP; Shandong, China), etoposide (VP-16; Jiangsu, China) and adriamycin (ADM; Jiangsu, China) were obtained from the First Affiliated Hospital of University of South China (Heyang, China) and reconstituted according to the manufacturer’s instructions. The Notch inhibitor FLI-60 was purchased from Selleck (USA).
Primary antibodies included antibodies against MYCN (#51705), JAG2 (#2210), HES1 (#11988; CST, USA); HES1 (sc-166410; Santa Cruz, USA); BCL2 (#12789-1-AP), BAX (50599-2-Ig), NOTCH1 (#20687-1-AP; Proteintech, USA); and GAPDH (AP0066; Bioworld, USA). Secondary antibodies included HRP-goat anti-mouse IgG (E030110-02), HRP-goat anti-rat IgG (E030120-02; Earthox, USA); goat anti-rabbit IgG (H+L) highly cross-adsorbed Alexa Fluor Plus 488 (# A32731), and goat anti-mouse IgG (H+L) highly cross-adsorbed, Alexa Fluor Plus555 (# A32727; Invitrogen, USA).
Cell transfection
Small interfering RNAs (siRNAs) targeting MYCN (siMYCN) and HES1 (siHES1) as well as a negative control siRNA (siNC) were purchased from GenePharma (Shanghai, China). An overexpression plasmid (pcDNA3.1-MYCN) and negative control plasmid (pcDNA3.1-NC) were also synthesized by GenePharma (Shanghai, China). The siRNA and overexpression plasmids were transfected using Lipofectamine 3000 and Opti-MEM I (Invitrogen, USA) according to the manufacturer’s instructions.
The lentiviral expression system containing shRNAs or overexpression plasmids for MYCN (LV-shMYCN or LV-MYCN) and their corresponding negative controls (LV-shNC or LV-NC) were constructed by GenePharma (Shanghai, China). All lentiviral vectors expressed enhanced green fluorescent protein (GFP) and puromycin resistance genes. After target cells were infected for 48 h, they were selected with 2.0 μg/ml puromycin (Solarbio, China).
The siRNA and shRNA sequences are as follows: siMYCN#1, sense (5’-3’) CACCAAGGCUGUCACCACATT, antisense (5’-3’) UGUGGUGACAGCCUUGGUGTT; siMYCN#2, sense (5’-3’) GCCACUGAGUAUGUCCACUTT, antisense (5’-3’) AGUGGACAUACUCAGUGGCTT; siHES1#1, sense (5’-3’) UCUCCGAUUUCUUCGCCAATT, antisense (5’-3’) UUGGCGAAGAAAUCGGAGATT; siHES1#2, sense (5’-3’) GCCUCAAGAUUCGCAUUCATT, antisense (5’-3’) UGAAUGCGAAUCUUGAGGCT; siNC, sense (5’-3’) UCCUCCGAACGUGUCACGUTT, antisense (5’-3’) ACGUGACACGUUCGGAGAATT; LV-shMYCN (5’-3’) GCCACTGAGTATGTCCACT; and LV-shNC (5’-3’) TTCTCCGAACGTGTCACGT. The transfection efficiency was validated by qRT-PCR and Western blotting.
RNA isolation and real-time qRT-PCR
Total RNA was extracted from cells using TRIzol Reagent (Invitrogen, USA), and the RNA concentration was measured with a NanoDrop 2000 (Thermo, USA). cDNA was synthesized using a FastKing RT Kit (With gDNase) (Tiangen, China) according to the manufacturer’s protocol. Then, qRT-PCR was performed using Talent qPCR Premix (SYBR Green) (Tiangen, China). The relative mRNA expression levels were calculated by the 2-ΔΔCT method. All primers were as follows: MYCN, forward (5’-3’) CCACAAGGCCCTCAGTACC, reverse (5’-3’) TCCTCTTCATCATCTTCATCATCT; HES1, forward (5’-3’) GGAAATGACAGTGAAGCACCTCC, reverse (5’-3’) GAAGCGGGTCACCTCGTTCATG; JAG2, forward (5’-3’) GCTGCTACGACCTGGTCAATGA, reverse (5’-3’) AGGTGTAGGCATCGCACTGGAA; and GAPDH, forward (5’-3’) GTCTCCTCTGACTTCAACAGCG, reverse (5’-3’) ACCACCCTGTTGCTGTAGCCAA.
Western blot analysis
Total protein was extracted from cells using RIPA lysis buffer (Beyotime, China) with a protease inhibitor cocktail (Cwbiotech, China) and quantified using a BCA Protein Assay Kit (Cwbiotech, China). Protein lysates were separated by SDS-PAGE on 10% gels before they were transferred to polyvinylidene difluoride membranes (Millipore, USA). After the membranes were blocked with 5% bovine serum albumin (BSA), they were incubated with primary antibodies at 4°C overnight. Next, they were washed with a Tris-buffered saline solution containing 0.1% Tween 20 (TBST) and incubated with a secondary antibody for 1 h at room temperature. After the membranes were washed again with TBST, the protein bands were detected by chemiluminescence. All antibodies used are listed in the Reagents and antibodies section.
Cell counting kit-8 (CCK-8) assays
Cells were plated in 96-well plates at a density of 1 × 104 cells per well. Chemotherapy drugs (CDDP, VP-16, and ADM) were diluted to obtain different concentration gradients. Then, the cells were treated with the above drugs for 24 h. The absorbance at 450 nm was detected after incubation with the RPMI1640 (90 μL) and CCK-8 (10 μL) reagents (Dojindo, Japan) for 4 h. Cells incubated without drugs were set at 100% survival and were used to calculate the IC50 concentration of each chemotherapeutic drug.
In vivo tumor xenograft model
Fifty-six BALB/c nude mice (female, 4-5 weeks old, 12-16 g) were purchased from the Experimental Animal Center of Southern Medical University (Guangzhou, China). The experiment was approved by the Institutional Guidelines and Use Committee for Animal Care of Guangdong Province.
Twenty mice were randomly divided into four groups involving H69AR cells (LV-shMYCN or LV-shNC) and treatments with chemotherapy drugs or PBS, while another twenty mice were divided into four groups involving H69 cells (LV-MYCN or LV-NC) and treatments with chemotherapy drugs or PBS. The mice were subcutaneously injected in the flanks with one of the above four SCLC cells (1 × 107 cells/100 μL PBS). After one week, the mice were intraperitoneally injected with PBS or drugs (3 mg/kg CDDP and 2 mg/kg VP16) every 4 days [33].
Sixteen mice were randomly grouped into the FLI-60, drugs, FLI-60 plus drugs, and PBS groups. All groups received a subcutaneous injection of H69AR cells (1 × 107 cells/100 μL PBS) in the flanks. Mice in the drug treatment groups received an intraperitoneal injection of 3 mg/kg CDDP and 2 mg/kg VP16 every 4 days, while the FLI-06 groups received FLI-06 (1 mg/kg) via tail vein injection [34] on the same day; mice in the PBS group were administered PBS via tail vein injection.
The sizes of the tumors were measured and recorded every 4 days, and the volume was calculated with the following equation: V = 1/2 (width2 × length). The mice were euthanized 4 weeks after cell injection.
Flow cytometry analysis
Transfected cells were treated with drugs for 24 h and then collected for apoptosis analysis. The dosages of the different cell lines were as follows: H69AR (5 μg/mL DDP, 200 μg/mL VP-16, and 10 μg/mL ADM), H69 (2 μg/mL DDP, 40 μg/mL VP-16, and 0.8 μg/mL ADM), H526 (2 μg/mL DDP, 40 μg/mL VP-16, and 1.5 μg/mL ADM), and H446(5 μg/mL DDP, 20 μg/mL VP-16, and 2 μg/mL ADM). The apoptosis assay was performed with Annexin V450 and APC-Cy7 785 labels (eBioscience, USA) according to the manufacturer’s protocol. All samples were analyzed by a BD FACSVerse flow cytometer.
cDNA microarray and mRNA sequencing
cDNA microarray assays were performed as previously described [35], and mRNA-sequencing assays were performed using a BGISEQ-500 platform (BGI Genomics, Wuhan, China).
Chromatin immunoprecipitation quantitative PCR (ChIP-qPCR) assay
ChIP experiments were performed using a Pierce Agarose ChIP Kit (26156, Thermo, USA). Briefly, H69AR cells were cross-linked with formaldehyde for 10 min at 37°C and then washed twice with ice-cold PBS. The cells were lysed in 100 μL of 1% SDS lysis buffer and sheared by sonication. Proteins and DNA were pulled down with an anti-MYCN antibody (#51705, CST, USA). An anti-RNA polymerase II antibody was used as a positive control, and a normal rabbit IgG antibody was used as a negative control. DNA association was quantified by RT-qPCR (2-ΔΔCt method) using primers specific for the HES1 promoter and the JAG2 promoter and was normalized to the input. The CHIP-qPCR primers used are as follows: HES1-P1, forward (5’-3’) CCCAGAGGGAGAGTAGCAAA, reverse (5’-3’) CCCAAACTTTCTTTCCCACA; HES1-P2, forward (5’-3’) CGCAGAACCTAAAGCCTACG, reverse (5’-3’) TTCAGAAATTCCTCGTTTGGA; HES1-P3, forward (5’-3’) GCCGCTTTAACCGCAGTC, reverse (5’-3’) GCCTCCAAGTTTGCTCCTC; JAG2-P1, forward (5’-3’) GAGTAGGAGGCGGCATCTC, reverse (5’-3’) CACACCTCCGCGTGAGTC; and JAG2-P2, forward (5’-3’) CTCTTGACATGGTCCACTATCC, reverse (5’-3’) GGCCATCGCTACATTCTCTTAT.
Luciferase assays
The pGL3-basic vector (Promega) and pRL-TK vector (GenePharma) were purchased. The pGL3-basic-promoter (pGL3-HES1-P1) and negative control (pGL3-NC) plasmids were constructed by inserting the HES1 promoter region or negative control region into the pGL3-basic plasmid; the plasmids were constructed by GenePharma (Shanghai, China). Plasmid transfection was performed with Lipofectamine 3000 and Opti-MEM I (Invitrogen, USA) according to the manufacturer’s protocol. Forty-eight hours after transfection, cells were harvested. Luciferase activity was determined by the Dual-luciferase Reporter Assay System (Promega, E1910). The luminescence readings of firefly and Renilla luciferase were measured by a multimode microplate reader from BioTek.
Immunofluorescence
Cells were seeded onto glass-bottom cell culture dishes (NEST, China) 24 h before they were fixed with 4% paraformaldehyde and then permeabilized with 0.3% Triton X-100. Subsequently, the cells were blocked with 5% BSA for 1 h and then treated with the primary antibody overnight at 4°C. After three washes with PBS, the cells were incubated with a fluorescence-conjugated secondary antibody (Invitrogen, USA) in the dark for 1 h. Finally, the cell nuclei were stained with DAPI. Images were obtained by fluorescence microscopy (Leica, Germany). The antibodies used are summarized in the Reagents and antibodies section.
Immunohistochemical analyses
Tissue samples were fixed in 4% paraformaldehyde and embedded in paraffin blocks. Then, they were sectioned and analyzed for MYCN, HES1 and KI67 protein expression. Briefly, after being dewaxed and hydrated, the tissues were subjected to antigen retrieval by microwaving them in citrate buffer (10 mM citric acid, pH 6.0), blocked in 5% goat serum, and incubated with the appropriate diluted primary antibody (MYCN 1:600, HES1 1:600, or KI67 1:300) overnight at 4°C. Next, the samples were incubated with secondary antibodies for 2 h at room temperature. Subsequently, specimens were developed by DAB, and the nuclei were counterstained with hematoxylin. The sections were photographed under a microscope with an EnVision Peroxidase System and analyzed with Image-Pro Plus software.
Statistical analysis
The results are given as the mean ± SD of at least three independent experiments. The data were analyzed by SPSS 20.0 or GraphPad Prism 6.0. Comparisons of two groups were analyzed by independent samples t-test, and ANOVA was performed to analyze significant differences among more than two groups. Spearman’s rank correlation test was used to analyze the association between MYCN and HES1 expression, while the association between MYCN expression and the clinicopathological characteristics was explored by χ2 test. Survival curves were estimated using the Kaplan-Meier analysis and log-rank tests. Prognostic factors were evaluated by univariate and multivariate analyses (Cox proportional hazards model). P < 0.05 was considered statistically significant.
Results
Reduced expression of MYCN sensitizes small-cell lung cancer (SCLC) cells to chemotherapy in vitro
Our previous cDNA microarray analysis showed a 2.3-fold upregulation of MYCN expression in H69AR cells compared with the expression in the parental H69 cells (Figure 1A); these results were confirmed by RT-qPCR and Western blotting (Figures 1B, S1A). Therefore, we hypothesized that MYCN may play an important role in the chemoresistance of SCLC cells. First, we selected nine SCLC cell lines to detect their MYCN expression levels. Only three cell lines, H69AR, H69 and H526, had amplified MYCN expression (Figures 1C, S1B). At the same time, we confirmed by immunofluorescence that MYCN is mainly localized in the nucleus (Figure 1D). We chose the above 3 cell lines, as well as H446 cells that do not express MYCN, for subsequent studies.
Figure 1.
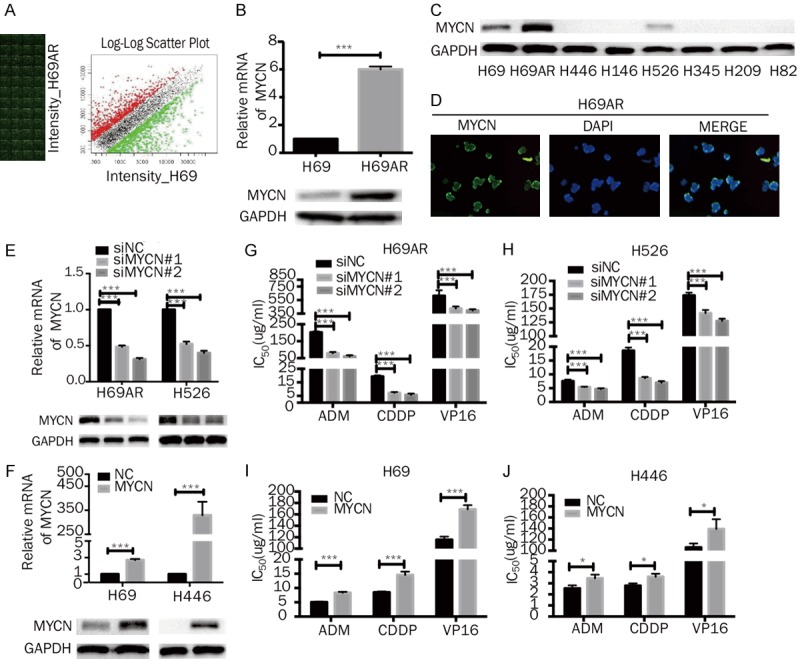
Effects of MYCN on the chemoresistance of SCLC in vitro. A. cDNA expression profile showed that MYCN is differentially expressed between H69AR cells and H69 cells. B. RT-qPCR and Western blot analysis of MYCN expression in H69 and H69AR cells. C. Western blot analysis of MYCN expression in eight SCLC cell lines (H69, H69AR, H446, H146, H526, H345, H209, and H82). D. The cellular localization of MYCN was confirmed by immunofluorescence staining of H69AR cells. E, F. RT-qPCR and Western blot analyses of MYCN expression in H69AR and H526 cells transfected with siRNA targeting MYCN or NC siRNA and in H69 and H446 cells transfected with pcDNA3.1-MYCN or NC plasmids. G-J. CCK-8 assays showed that MYCN knockdown decreased the IC50 values of the chemotherapeutic agents (ADM, CDDP, and VP-16) in H69AR and H526 cells, whereas MYCN overexpression increased the IC50 values of these compounds in H69 and H446 cells. Error bars indicate the mean ± SD from three independent experiments. *, P < 0.05; ***, P < 0.001.
We first knocked down MYCN expression with two independent MYCN siRNAs (siMYCN#1 and siMYCN#2) in the H69AR and H526 cell lines (Figures 1E, S1C). Meanwhile, we developed MYCN-overexpressing sublines, H69MYCN and H446MYCN, by transfecting H69 and H446 cells with pcDNA3.1-MYCN (Figures 1F, S1D). CCK-8 assays were performed to evaluate the effect of chemotherapeutic drugs (ADM, CDDP and VP16) on the viability of the four SCLC cell lines and their sensitivity to the drugs 24 h after the treatment. The two siMYCN clones (H69AR-siMYCN and H526-siMYCN) displayed more sensitivity to ADM, CDDP and VP16 than the siNC clone, as indicated by the lower IC50 values (Figure 1G, 1H). In addition, the overexpressing sublines (H69MYCN and H446MYCN) showed less sensitivity to ADM, CDDP and VP16 than the NC clone, as exhibited by the higher IC50 values (Figure 1I, 1J). Collectively, these results indicate that MYCN upregulation or downregulation could significantly affect the sensitivity of SCLC cells to chemotherapeutic drugs, suggesting that MYCN expression may be associated with chemoresistance in SCLC.
MYCN enhances tumor growth and chemoresistance in vivo
The effect of MYCN on chemoresistance was further investigated in an in vivo tumor model. First, we developed H69 and H69AR cell lines with stable upregulation and downregulation of MYCN, respectively, via lentivirus (Figures 2A, 2D, S2A, S2B). Compared with the LV-NC cell-based tumors, tumors derived from H69 cells with MYCN overexpression were increased in size and showed accelerated growth in mice as well as exhibited reduced sensitization to CDDP and VP16 (Figure 2B, 2C). The proliferative indicator Ki-67 was highly expressed in MYCN-overexpressing cells (Figure 2G, 2H). Conversely, we observed that compared with the LV-shNC clones, the H69AR cells with MYCN knockdown had smaller mean volumes and a slower rate of subcutaneous tumor growth in mice and showed significant sensitivity to CDDP and VP16 (Figure 2E, 2F). Furthermore, Ki-67 was expressed at lower levels in tumors derived from MYCN knockdown cells than in the negative control cells (Figure 2G, 2H). These results provide evidence that MYCN promotes tumor growth and enhances drug resistance to VP16- and CDDP- based chemotherapy.
Figure 2.
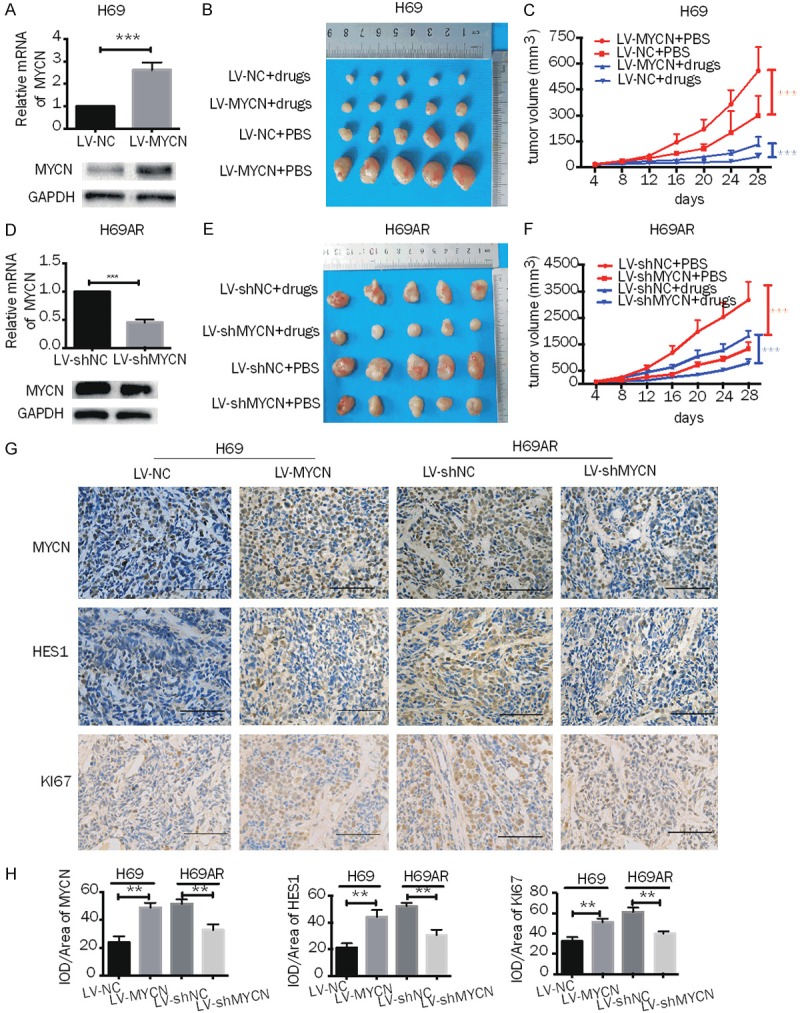
Effects of MYCN on chemoresistance and growth of SCLC in vivo. A, D. RT-qPCR and Western blot analysis of MYCN expression in H69 cells transfected with lentiviral-based MYCN overexpression plasmids and in H69AR cells transfected with lentiviral-based MYCN knockdown plasmids and their corresponding control vectors. B, E. Subcutaneous tumor formation experiments were performed in nude mice injected with H69 cells containing vectors with stably upregulated MYCN or control (LV-MYCN or LV-NC, respectively) or with H69AR cells containing vectors with stably downregulated MYCN or control (LV-shNC or LV-shMYCN, respectively); drugs (CDDP+VP-16) or PBS were intraperitoneally injected (n = 5 mice for each group). C, F, The growth curve of the tumor volumes of mouse groups injected with H69 cells with stable MYCN overexpression or with H69AR cells with stably MYCN knockdown. G, H. Histopathological features and representative IHC staining of MYCN, HES1 and Ki67 in tumor tissues from mice injected with H69 cells with stable overexpression of MYCN or with H69AR cells with stable knockdown of MYCN (magnification 400x). Scale bars, 100 μm. Error bars indicate the mean ± SD from three independent experiments. **, P < 0.01. ***, P < 0.001.
MYCN affects chemoresistance in SCLC by regulating drug-induced apoptosis
To determine the possible mechanisms of MYCN on chemoresistance, an apoptosis assay was carried out by flow cytometry analysis in cells with up- or downregulation of MYCN after they were exposed to chemotherapeutic drugs (ADM, CDDP and VP16) for 24 h. The results showed that the early apoptotic fractions of MYCN-knockdown H69AR and H526 cells were significantly higher than those of siNC cells (Figure 3A, 3E). In contrast, the early apoptotic fractions of MYCN-overexpressing H69 and H446 cells were significantly lower than those of NC cells (Figure 3B, 3F). In addition, BCL-2 expression was decreased in MYCN-downregulated H69AR and H526 cells after treatment with ADM, DDP or VP16, while BAX expression was increased (Figures 3C, 3G, S3A, S3C), while MYCN overexpression in H69 and H446 cells produced the opposite result (Figures 3D, 3H, S3B, S3D). These results suggest that MYCN influenced the chemoresistance of SCLC by regulating drug-induced apoptosis.
Figure 3.

MYCN induces chemoresistance mainly by decreasing drug-induced apoptosis. A, E. Representative dot plots of flow cytometry analyzing the effect of MYCN on early apoptosis and the summary of the cumulative data showing the percentage of early apoptotic cells in MYCN-downregulated SCLC cells (H69AR and H526) after exposure to cytotoxic drugs (ADM, CDDP, and VP16) for 24 h. B, F. Flow cytometry analysis of early apoptosis in MYCN-overexpressing SCLC cells (H69 and H446) induced by exposure to cytotoxic drugs (ADM, CDDP, and VP16) for 24 h. C, D, G, H. Apoptosis-related proteins (BCL2 and BAX) were measured by Western blot in MYCN-downregulated (H69AR and H526) or -upregulated (H69 and H446) SCLC cells after exposure to anticancer drugs for 24 h. Error bars indicate the mean ± SD from three independent experiments. ***, P < 0.001.
MYCN regulates the NOTCH pathway, and HES1 is a direct transcriptional target of MYCN
To identify the targets of MYCN, we performed RNA sequencing (RNA-seq) analysis in H69AR-siNC cells and H69AR-siMYCN#2 cells, the latter of which had a higher suppression efficiency than H69AR-siMYCN#1. A total of 1067 differentially expressed genes were identified (|Fold Change| ≥ h.00 and FDR ≤ 0.001) (Figure 4A). Then, we conducted KEGG pathway enrichment analysis (Figure 4B). The Notch signaling pathway attracted our attention because this pathway is associated with tumor resistance, stemness, and proliferation. There are six differentially expressed genes in this pathway (Figure 4C), all of which are downregulated in MYCN-depleted cells. According to our previous cDNA microarray (Figure 1A), the gene expression of HES1 and JAG2 was higher in H69AR cells than in the parental H69 cells, so we hypothesized that these two genes were related to drug resistance. Considering that the other four genes (HES7, HES5, LFNG, and MFNG) were only secondary changes caused by MYCN and had no relationship to drug resistance, we chose to verify HES1 and JAG2. RT-qPCR showed that HES1 and JAG2 expression had indeed decreased with the down regulation of MYCN in H69AR cells (Figure 4D). Subsequently, Western blotting analysis confirmed that JAG2 and HES1 were significantly downregulated in MYCN-depleted cells and upregulated in MYCN-overexpressing cells (Figures 4E, 4F, S4A, S4B). These findings of HES1 were also confirmed by immunohistochemistry (IHC) analysis of xenografts (Figure 2G, 2H). NOTCH1 was a differentially expressed gene in the H69 and H69AR cDNA microarray (Figure 1A) but not in the MYCN-related RNA-seq analysis (Figure 4A). Considering that NOTCH1 is the hallmark protein of this pathway and had been proven as a chemoresistance gene in SCLC [29], we detected NOTCH1 expression by Western blot. Intere-stingly, NOTCH1 was positively correlated with MYCN expression at the protein level (Figures 4E, 4F, S4A, S4B). All these data show that MYCN could regulate the NOTCH pathway.
Figure 4.
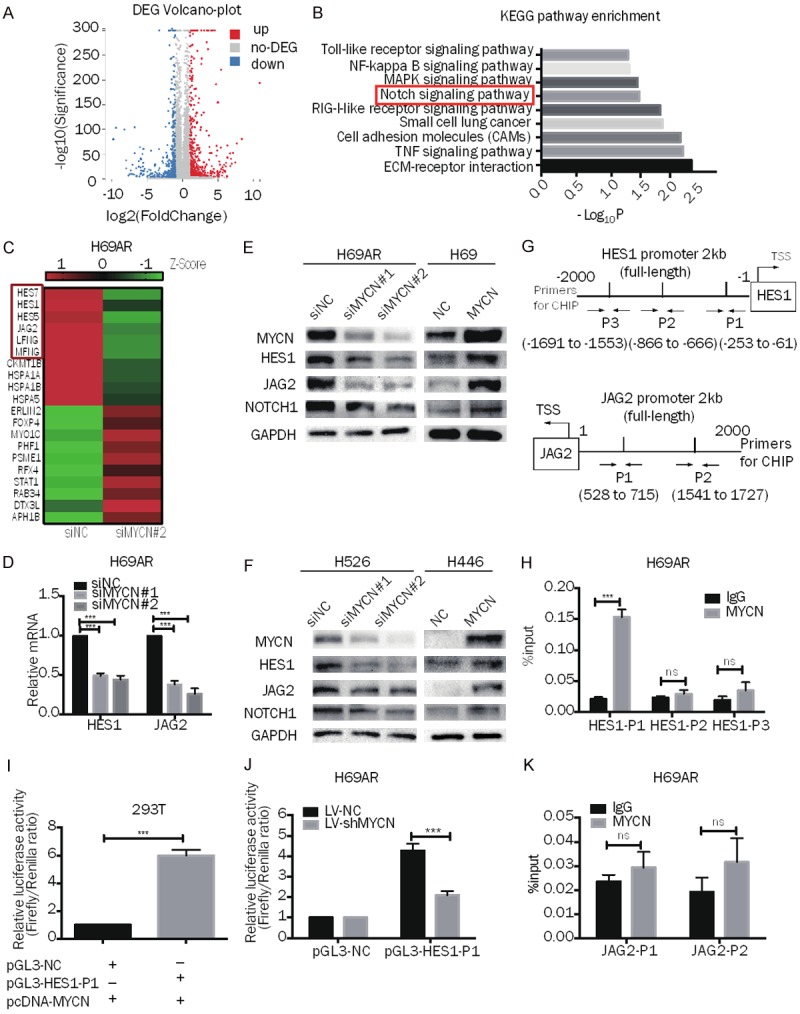
MYCN regulates the NOTCH pathway and targets HES1 directly in SCLC. A. Volcano plot showing the differentially expressed genes between H69AR-siNC cells and H69AR-siMYCN#2 cells. B. -log10 transformations of the P values of 9 representative and significantly enriched KEGG pathways; the NOTCH pathway is in the red frame. C. Representative heatmap showing 10 downregulated and 10 upregulated genes (6 genes from the NOTCH pathway are shown in the red frame). D. RT-qPCR analysis of HES1 and JAG2 expression in MYCN downregulated H69AR cells and the corresponding control cells. E, F. Western blot analysis of MYCN, HES1, JAG2, and NOTCH1 expression in MYCN-downregulated (H69AR and H526) or MYCN-upregulated (H69 and H446) SCLC cells. G. Prediction of the MYCN binding site and location of the ChIP-qPCR primers in the HES1 promoter region (HES1-P1, HES1-P2, HES1-P3) and the JAG2 promoter region (JAG2-P1, JAG2-P2). H. ChIP-qPCR for MYCN or immunoglobulin G (IgG) at the HES1-P1, HES1-P2 and HES1-P3 regions in H69AR cells. I. Dual-luciferase assay indicating that the activity of the cotransfected HES1-P1 promoter reporter and the pcDNA3.1-MYCN plasmid is stronger than that of the NC promoter reporter and pcDNA3.1-MYCN plasmid group in 293T cells. J. Dual-luciferase assay showing that the activity of the HES1-P1 promoter reporter is weaker in H69AR-LV-shMYCN cells than in LV-shNC cells. K. ChIP-qPCR for MYCN or immunoglobulin G (IgG) at the JAG2-P1 and JAG2-P2 regions in H69AR cells. Error bars indicate the mean ± SD from three independent experiments; ***P < 0.05.
To substantiate a putative interaction between the HES1 promoter and MYCN, we performed ChIP followed by qPCR (ChIP-qPCR) to probe for genomic occupancy of MYCN at the HES1 promoter sequence. Three qPCR probe sets (HES1-P1, HES1-P2, and HES1-P3) were designed based on the NCBI and UCSC databases and previous studies [36] (Figure 4G). The qPCR analysis of the chromatin pulled down by anti-MYCN antibodies implicated a preferential enrichment of MYCN occupancy at the HES1-P1 sequence (Figure 4H), supporting the notion that MYCN directly targets HES1. To verify whether MYCN binds to the promoter region of JAG2, we also designed two primers [36,37] (Figure 4G) and attempted ChIP-qPCR detection, but no evidence of direct binding was found (Figure 4K).
To further confirm that MYCN binds to the HES1 promoter and affects transcriptional activity, we cloned the HES1-P1 sequence into a luciferase reporter plasmid. The MYCN plasmid (pcDNA 3.1 MYCN) was then cotransfected with a luciferase reporter (pGL3-HES1-P1) and a Renilla reporter plasmid into 293T cells, and the relative luciferase activity demonstrated that a positive response was induced by MYCN (Figure 4I). We transfected the luciferase and Renilla reporter plasmids into H69AR-LV-shNC and H69AR-LV-shMYCN cells and the relative luciferase activity in H69AR-LV-shMYCN cells was significantly lower than that in H69AR-LV-shNC cells (Figure 4J). Taken together, these data confirm that MYCN positively regulates HES1 transcription.
HES1 contributes to MYCN-induced chemoresistance in SCLC
Next, we further investigated whether HES1 contributes to MYCN-mediated chemoresistance in SCLC and found that the HES1 mRNA and protein levels were significantly higher in chemoresistant H69AR cells than in H69 cells (Figures 5A, S5A). We also detected HES1 expression levels in nine SCLC cell lines (Figures 5B, S5A). All cell lines expressed HES1 to varying degrees, and the cell lines with relatively high MYCN expression, for example, H69 and H69AR cells, also had higher HES1 expression. Cells with relatively low or no expression of MYCN, such as H526 and H446 cells, had lower expression of HES1. It was also found by immunofluorescence that HES1 is mainly localized in the nucleus, followed by localization at the cell membrane (Figure 5C). We knocked down HES1 expression in H69AR and H526 cells by siRNA (Figures 5D, S5B), and HES1 downregulation resulted in markedly increased IC50 values (Figure 5E, 5F), indicating that HES1 positively regulates SCLC chemoresistance.
Figure 5.
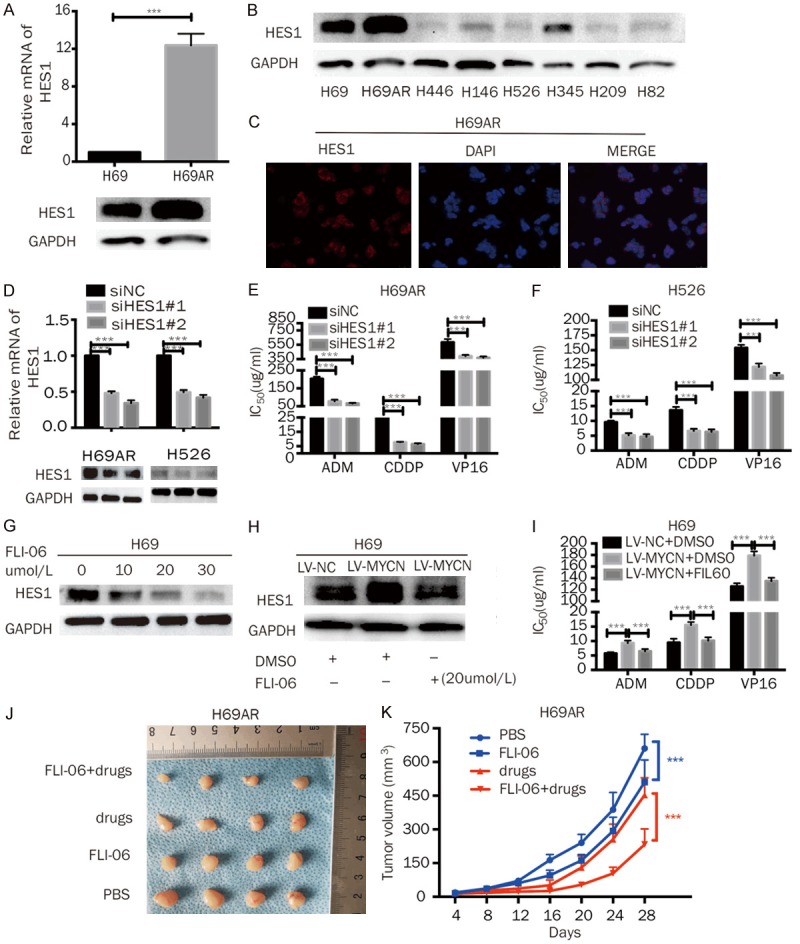
MYCN promotes the chemoresistance of SCLC through HES1. A. RT-qPCR and Western blot analysis of HES1 expression in H69 and H69AR cells. B. Western blot analysis of HES1 expression in eight SCLC cell lines (H69, H69AR, H446, H146, H526, H345, H209, and H82). C. The cellular localization of HES1 was confirmed by immunofluorescence staining of H69AR cells. D. RT-qPCR and Western blot analysis of MYCN in H69AR and H526 cells transfected with siRNA against HES1. E, F. CCK-8 assays showed that HES1 knockdown decreased the IC50 values of chemotherapeutic agents (ADM, CDDP, and VP-16) in H69AR and H526 cells. G. Western blotting analysis revealed that 24 h of FLI-06 treatment inhibited HES1 expression in a dose-dependent manner. H. Western blotting showed that MYCN overexpression increased HES1 expression; however, the increased HES1 expression was diminished by FLI-06. I. A CCK-8 assay showed that the IC50 values were significantly increased in H69-LV-MYCN cells compared with those in the control cells, and the inhibition of HES1 by FLI-06 in MYCN-overexpressing cells could abate the increase in the IC50 values mediated by MYCN upregulation. J. Effect of FLI-06 with or without chemotherapy drugs (CDDP+VP16) on subcutaneous tumor growth injected with H69AR cells (n = 4 per group). K. Growth curve of tumor volumes of H69AR cells using FLI-06 with or without drugs (CDDP+VP16). Error bars indicate the mean ± SD from three independent experiments; ***P < 0.05.
FLI-06 is an inhibitior of the Notch pathway [38]. We added FLI-06 to H69 cells at doses ranging from 10 μmol/L to 30 μmol/L for 24 h. Western blotting analysis revealed that FLI-06 inhibited HES1 expression in a dose-dependent manner (Figures 5G, S5C). Based on these data, we chose 20 μmol/L for subsequent rescue experiments. The Western blotting results demonstrated that MYCN overexpression increased HES1; however, the increased HES1 expression was diminished by FLI-06 (Figures 5H, S5D). The CCK-8 assay was conducted by adding FLI-06 to MYCN-overexpressing H69-LV-MYCN cells to inhibit HES1. The results showed that the IC50 values significantly increased in the H69-LV-MYCN cells compared with those in the empty-vector control cells, and the inhibition of HES1 by FLI-06 in MYCN-overexpressing cells abated the increase in the IC50 values mediated by MYCN upregulation (Figure 5I). In addition, to determine whether FLI-06 modulates chemoresistance in vivo, we used subcutaneous xenograft models of H69AR cells. The results showed that the tumor growth and volumes could be significantly inhibited by the combination of FLI-06 and chemotherapy (Figure 5J, 5K). Additionally, a smaller tumor volume and slower growth was found in mice administered either treatment (FLI-06 or chemotherapy) alone compared with the PBS mice (Figure 5J, 5K). These results indicate that MYCN mediates the chemoresistance of SCLC through HES1.
Elevated MYCN expression correlates with poor survival and chemotherapy response in SCLC patients
To further evaluate the clinical significance of MYCN expression in SCLC and the correlation between MYCN and HES1, we analyzed the GSE60052 dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE60052) and 42 SCLC samples from patients at our hospital by IHC. GSE60052 contained the RNA sequencing data of 79 SCLC specimens and 7 paracancerous specimens. The mRNA expression of MYCN was higher in SCLC tissues than in paracancerous lung tissues, and MYCN levels were positively correlated with HES1 expression in the dataset (Figure 6A, 6B). The IHC results of the 42 SCLC patient samples showed that the samples from chemorefractory patients had a higher frequency of MYCN expression (55%) than the samples from chemosensitive patients (10%) (Figure 6C, 6D; Table 1). Similarly, the rate of HES1 positivity was much higher in the SCLC specimens from chemorefractory patients (66.7%) than those from drug-sensitive patients (39.1%) (Figure 6C, 6D). Moreover, we found that the MYCN levels were positively correlated with HES1 expression in the SCLC patient samples (Figure 6E). Kaplan-Meier analysis revealed that high MYCN levels were associated with poor overall survival (P = 0.000) (Figure 6F). Finally, as shown in Table 1, IHC analysis indicated that MYCN protein expression was higher in extensive-stage patients than in limited-stage patients (P = 0.003). The univariate and multivariate analyses showed that the stage, drug sensitivity, and MYCN expression were independent prognostic factors (P = 0.000, P = 0.000, and P = 0.000, respectively; Table 2) in SCLC. Collectively, these results indicate that high MYCN levels were correlated with a decreased response to chemotherapy, poor survival, and advanced clinical stages in SCLC patients.
Figure 6.

MYCN was correlated with poor chemotherapy response and prognosis in SCLC patients. A. Detection of the mRNA expression of MYCN in SCLC tissues and paracancerous lung tissues from the GSE60052 dataset. B. Correlation analysis of the mRNA expression of MYCN and HES1 in the GSE60052 dataset. C. Representative IHC staining of MYCN or HES1 in samples from normal alveolar epithelium and from patients with chemosensitive and refractory SCLC (magnification 400x). D. The MYCN- or HES1-positive expression rate was frequently increased in chemoresistant SCLC tissues compared to chemosensitive SCLC tissues. E. Correlation analysis of MYCN and HES1 by IHC in 42 SCLC patient samples. F. Kaplan-Meier analysis of the overall survival of 42 SCLC patients based on MYCN expression. -, negative; +, positive. Scale bars, 100 μm; Error bars indicate the mean ± SD from three independent experiments. ***P < 0.05.
Table 1.
MYCN expression and its relationship with the clinicopathological characteristics of 42 SCLC patients
| Clinic pathological features | n | Expression of MYCN | ||
|---|---|---|---|---|
|
| ||||
| + | - | P | ||
| Gender | 0.395 | |||
| Male | 33 | 8 | 25 | |
| Female | 9 | 1 | 8 | |
| Age | 0.862 | |||
| < 56 | 13 | 3 | 10 | |
| ≥ 56 | 29 | 6 | 23 | |
| Clinical stage | 0.003 | |||
| Limited disease | 27 | 2 | 25 | |
| Extensive disease | 15 | 7 | 8 | |
| Chemotherapy response | 0.002 | |||
| Sensitive group | 31 | 3 | 28 | |
| Refractory group | 11 | 6 | 5 | |
-, negative; +, positive for MYCN. The P-value was calculated by Pearson χ2-test.
Table 2.
Univariate and multivariate analyses of potential prognostic factors associated with the overall survival of 42 SCLC patients
| Univariate analysis | Multivariate analysis | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Variables | Hazard ratio | 95% CI | P value | Hazard ratio | 95% CI | P value |
| Gender | 0.730 | 0.258-2.059 | 0.551 | 1.807 | 0.521-6.275 | 0.351 |
| Age, y | 1.154 | 0.379-3.517 | 0.800 | 1.656 | 0.485-5.656 | 0.421 |
| Disease stage | 19.574 | 4.318-88.731 | 0.000 | 29.344 | 3.897-220.936 | 0.001 |
| Drug-sensitivity | 6.858 | 2.658-17.700 | 0.000 | 4.240 | 1.079-16.657 | 0.038 |
| MYCN expression | 0.104 | 0.038-0.284 | 0.000 | 0.062 | 0.013-0.282 | 0.000 |
CI, confidence interval; The P-value was calculated by Cox proportional hazards model.
Discussion
SCLC is an aggressive neuroendocrine carcinoma with a high mutational burden [39]. The amplification of MYC family members is one of the common molecular genetic alterations in this malignancy, occurring in 18-31% of SCLCs [17]. Thus, determining the mechanism how MYCN amplification alters responses to chemotherapy in SCLC may have significant therapeutic implications. Emerging evidence has demonstrated that MYCN is involved in chemoresistance in neuroblastoma [14,15] and in tongue cancer [40]. However, the role of MYCN in the chemoresistance of SCLC remains unclear. In our study, we initially identified that MYCN was significantly upregulated in SCLC chemoresistant cells (Figures 1B, S1A) and clinical samples (Figure 6C, 6D). Downregulation or overexpression of MYCN could potentiate or weaken cell chemosensitivity in vitro and in vivo (Figures 1G-J, 2B, 2C, 2E, 2F), respectively, by regulating apoptosis (Figures 3A-H, S3A-D). To the best of our knowledge, this is the first study to investigate the function of MYCN in SCLC chemoresistance.
MYCN has been identified as a key transcriptional regulator involved in tumors biology. Hossain MS et al. found that NLRR1 is a direct transcriptional target of oncogenic MYCN and is important in the regulation of cell proliferation and apoptosis in neuroblastoma cells [10]. Chen L et al. reported that p53 is a direct transcriptional target of MYCN in neuroblastoma and is likely to be involved in MYCN induced p53-dependent apoptosis [13]. Inspired by the study showing that MYCN is involved in chemoresistance by directly regulating the MRP1 promoter in neuroblastoma [14], we hypothesized that the same regulatory relationship may exist in SCLC. Unfortunately, we were unable to confirm our hypothesis in this study (the data was not shown). So we performed RNA sequencing (RNA-seq) analysis (Figure 4A). The preliminary results revealed that HES1 is a candidate target gene of MYCN in SCLC (Figure 4C). Through up- and downregulation of MYCN, HES1 expression was increased and decreased, respectively (Figures 4D-F, S4A, S4B). Furthermore, using MYCN ChIP-qPCR and a dual-luciferase reporter assay, we confirmed that MYCN binds to the HES1 promoter and affects transcriptional activity (Figure 4G-K). Recently, our study revealed that the MYCN opposite strand MYCNOS is a super-enhancer involved in SCLC chemoresistance [41]. O’Brien EM et al. confirmed that MYCNOS regulates MYCN protein levels and affects growth of MYCN-amplified rhabdomyosarcoma and neuroblastoma cells [42]. We will further confirm whether this regulatory relationship exists in SCLC and clarify the role of MYCN as a super enhancer.
HES1 is a downstream target gene of Notch pathway. Previous studies have shown that HES1 mainly acts as an important regulator of cell proliferation, differentiation, invasion, cancer stem cell properties and tumorigenicity [43-45], while other reports revealed that HES1 plays an important role in drug resistance in non- small-cell lung cancer [31,44], prostate cancer [46], and colorectal cancer [32]. However, whether HES1 regulates chemoresistance in SCLC remains poorly understood. In this study, we confirmed that repressing HES1 could overcome chemoresistance in SCLC (Figure 5E, 5F). Down-regulation of HES1 can reverse the increase in drug resistance caused by MYCN overexpression (Figure 5I). We also verified the positive correlation between HES1 and MYCN in both clinical samples (Figure 6B, 6E) and animal experiments (Figure 2G, 2H). In the future, we aim to develop cell lines with stable upregulation and downregulation of HES1, and to validate their drug resistance in animal experiments. Goto N etc. reported that HES1 invovled in the fuction of tumor stem-like cells of the Intestine [45], so we hypothesize that HES1 may enhance the SCLC chemoresistance by modulating cancer stem cells. More research is required to investigate this hypothesis.
Our recent study revealed that the Notch signaling pathway is enriched in drug-resistant cell line H69AR [41]. Interestingly, in this study we found that the Notch signaling pathway is also enriched in downstream genes regulated by MYCN (Figure 4B). There was a JAG2/NOTCH1/HES1 axis mediated by MYCN in SCLC (Figures 4E, 4F, S4A, S4B). Many novel targeted gene therapies and therapies that target signal pathways are now being actively tested in clinical trials for SCLC. For example, PARP inhibitors, EZH2 inhibitors, the inhibitory Notch ligand DLL3, and Aurora kinase have all undergone clinical trial testing [3]. In this study, we used FLI-06, a novel secretion inhibitor that blocks the secretion of Notch pre-receptors from the endoplasmic reticulum (ER), thereby suppressing the Notch signaling pathway. Lu Z et al. showed that FLI-06 suppresses proliferation, and induces apoptosis and cell cycle arrest in esophageal squamous cell carcinoma [47]. In our study, we found that FLI-06 could inhibit the expression of HES1 and diminished MYCN-induced chemoresistance in SCLC cell lines (Figures 5G-I, S5C, S5D). In a nude mouse model, the combination of FLI-06 and anticancer drugs significantly reduced the tumor volume derived from the MYCN amplified cell line H69AR, which suggests a synergistic effect of FLI-06 with chemotherapy in MYCN amplified SCLC cells (Figure 5J, 5K). Our findings could have potential clinical implications in which patients with refractory SCLC and high MYCN expression may benefit from combination chemotherapy with a notch inhibitor. Notably, additional mechanistic studies and clinical trials will be required to pursue this hypothesis.
In summary, our results demonstrate that MYCN promotes the chemoresistance of SCLC by regulating the HES1 promoter. Our study implies that FLI-60 treatment combined with chemotherapy can partially overcome MYCN-mediated drug resistance in SCLC cells. Therefore, we conclude that MYCN and HES1 may be potential targets that block chemoresistance in SCLC.
Acknowledgements
The authors thank Dr. Peng Luo of Zhujiang Hospital for Biological information analysis and Dr. Xiaoxing You of University of South China for critically reading for the manuscript. This work was supported by grants from the National Natural Science Foundation of China (No.81572244, No.81772458, No.81702255); the Natural Science Foundation of Guangdong Province (key) (No.2015A030311028); the Scientific Program of Health Commission of Hunan Province (No.C2019116).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 3.Sabari JK, Lok BH, Laird JH, Poirier JT, Rudin CM. Unravelling the biology of SCLC: implications for therapy. Nat Rev Clin Oncol. 2017;14:549–561. doi: 10.1038/nrclinonc.2017.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicholson AG, Chansky K, Crowley J, Beyruti R, Kubota K, Turrisi A, Eberhardt WE, van Meerbeeck J, Rami-Porta R Staging and Prognostic Factors Committee, Advisory Boards, and Participating Institutions; Staging and Prognostic Factors Committee Advisory Boards and Participating Institutions. The international association for the study of lung cancer lung cancer staging project: proposals for the revision of the clinical and pathologic staging of small cell lung cancer in the forthcoming eighth edition of the tnm classification for lung cancer. J Thorac Oncol. 2016;11:300–11. doi: 10.1016/j.jtho.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 5.Sos ML, Dietlein F, Peifer M, Schöttle J, Balke-Want H, Müller C, Koker M, Richters A, Heynck S, Malchers F, Heuckmann JM, Seidel D, Eyers PA, Ullrich RT, Antonchick AP, Vintonyak VV, Schneider PM, Ninomiya T, Waldmann H, Büttner R, Rauh D, Heukamp LC, Thomas RK. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc Natl Acad Sci U S A. 2012;109:17034–9. doi: 10.1073/pnas.1207310109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richards MW, Burgess SG, Poon E, Carstensen A, Eilers M, Chesler L, Bayliss R. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc Natl Acad Sci U S A. 2016;113:13726–13731. doi: 10.1073/pnas.1610626113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boon K, Caron HN, van Asperen R, Valentijn L, Hermus MC, van Sluis P, Roobeek I, Weis I, Voûte PA, Schwab M, Versteeg R. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 2001;20:1383–93. doi: 10.1093/emboj/20.6.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whittle SB, Smith V, Doherty E, Zhao S, McCarty S, Zage PE. Overview and recent advances in the treatment of neuroblastoma. Expert Rev Anticancer Ther. 2017;17:369–386. doi: 10.1080/14737140.2017.1285230. [DOI] [PubMed] [Google Scholar]
- 9.Lasorella A, Noseda M, Beyna M, Yokota Y, Iavarone A. Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature. 2000;407:592–598. doi: 10.1038/35036504. [DOI] [PubMed] [Google Scholar]
- 10.Hossain MS, Ozaki T, Wang H, Nakagawa A, Takenobu H, Ohira M, Kamijo T, Nakagawara A. N-MYC promotes cell proliferation through a direct transactivation of neuronal leucine-rich repeat protein-1 (NLRR1) gene in neuroblastoma. Oncogene. 2008;27:6075–6082. doi: 10.1038/onc.2008.200. [DOI] [PubMed] [Google Scholar]
- 11.Zhu S, Zhang X, Weichert-Leahey N, Dong Z, Zhang C, Lopez G, Tao T, He S, Wood AC, Oldridge D, Ung CY, van Ree JH, Khan A, Salazar BM, Lummertz da Rocha E, Zimmerman MW, Guo F, Cao H, Hou X, Weroha SJ, Perez-Atayde AR, Neuberg DS, Meves A, McNiven MA, van Deursen JM, Li H, Maris JM, Look AT. LMO1 synergizes with MYCN to promote neuroblastoma initiation and metastasis. Cancer Cell. 2017;32:310–323. doi: 10.1016/j.ccell.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He J, Gu L, Zhang H, Zhou M. Crosstalk between MYCN and MDM2-p53 signal pathways regulates tumor cell growth and apoptosis in neuroblastoma. Cell Cycle. 2014;10:2994–3002. doi: 10.4161/cc.10.17.17118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen L, Iraci N, Gherardi S, Gamble LD, Wood KM, Perini G, Lunec J, Tweddle DA. p53 is a direct transcriptional target of MYCN in neuroblastoma. Cancer Res. 2010;70:1377–1388. doi: 10.1158/0008-5472.CAN-09-2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manohar CF, Bray JA, Salwen HR, Madafiglio J, Cheng A, Flemming C, Marshall GM, Norris MD, Haber M, Cohn SL. MYCN-mediated regulation of the MRP1 promoter in human neuroblastoma. Oncogene. 2004;23:753–762. doi: 10.1038/sj.onc.1207151. [DOI] [PubMed] [Google Scholar]
- 15.Agarwal S, Milazzo G, Rajapakshe K, Bernardi R, Chen Z, Barbieri E, Koster J, Perini G, Coarfa C, Shohet JM. MYCN acts as a direct co-regulator of p53 in MYCN amplified neuroblastoma. Oncotarget. 2018;9:20323–20338. doi: 10.18632/oncotarget.24859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwakawa R, Takenaka M, Kohno T, Shimada Y, Totoki Y, Shibata T, Tsuta K, Nishikawa R, Noguchi M, Sato-Otsubo A, Ogawa S, Yokota J. Genome-wide identification of genes with amplification and/or fusion in small cell lung cancer. Genes Chromosomes Cancer. 2013;52:802–16. doi: 10.1002/gcc.22076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim YH, Girard L, Giacomini CP, Wang P, Hernandez-Boussard T, Tibshirani R, Minna JD, Pollack JR. Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene. 2005;25:130–138. doi: 10.1038/sj.onc.1208997. [DOI] [PubMed] [Google Scholar]
- 18.Cristea S, Sage J. Is the canonical RAF/MEK/ERK signaling pathway a therapeutic target in SCLC? J Thorac Oncol. 2016;11:1233–1241. doi: 10.1016/j.jtho.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peifer M, Fernández-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, Menon R, Koker M, Dahmen I, Müller C, Di Cerbo V, Schildhaus HU, Altmüller J, Baessmann I, Becker C, de Wilde B, Vandesompele J, Böhm D, Ansén S, Gabler F, Wilkening I, Heynck S, Heuckmann JM, Lu X, Carter SL, Cibulskis K, Banerji S, Getz G, Park KS, Rauh D, Grütter C, Fischer M, Pasqualucci L, Wright G, Wainer Z, Russell P, Petersen I, Chen Y, Stoelben E, Ludwig C, Schnabel P, Hoffmann H, Muley T, Brockmann M, Engel-Riedel W, Muscarella LA, Fazio VM, Groen H, Timens W, Sietsma H, Thunnissen E, Smit E, Heideman DA, Snijders PJ, Cappuzzo F, Ligorio C, Damiani S, Field J, Solberg S, Brustugun OT, Lund-Iversen M, Sänger J, Clement JH, Soltermann A, Moch H, Weder W, Solomon B, Soria JC, Validire P, Besse B, Brambilla E, Brambilla C, Lantuejoul S, Lorimier P, Schneider PM, Hallek M, Pao W, Meyerson M, Sage J, Shendure J, Schneider R, Büttner R, Wolf J, Nürnberg P, Perner S, Heukamp LC, Brindle PK, Haas S, Thomas RK. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–10. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Previs RA, Coleman RL, Harris AL, Sood AK. Molecular pathways: translational and therapeutic implications of the notch signaling pathway in cancer. Clin Cancer Res. 2015;21:955–961. doi: 10.1158/1078-0432.CCR-14-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lobry C, Oh P, Aifantis I. Oncogenic and tumor suppressor functions of Notch in cancer: it’s NOTCH what you think. J Exp Med. 2011;208:1931–5. doi: 10.1084/jem.20111855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez-Martin M, Ferrando A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood. 2017;129:1124–1133. doi: 10.1182/blood-2016-09-692582. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Sha J, Yang G, Huang X, Bo J, Huang Y. Activation of Notch pathway is linked with epithelial-mesenchymal transition in prostate cancer cells. Cell Cycle. 2017;16:999–1007. doi: 10.1080/15384101.2017.1312237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kontomanolis EN, Kalagasidou S, Pouliliou S, Anthoulaki X, Georgiou N, Papamanolis V, Fasoulakis ZN. The notch pathway in breast cancer progression. ScientificWorldJournal. 2018;2018:2415489. doi: 10.1155/2018/2415489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barnawi R, Al-Khaldi S, Majed Sleiman G, Sarkar A, Al-Dhfyan A, Al-Mohanna F, Ghebeh H, Al-Alwan M. Fascin is critical for the maintenance of breast cancer stem cell pool predominantly via the activation of the notch self-renewal pathway. Stem Cells. 2016;34:2799–2813. doi: 10.1002/stem.2473. [DOI] [PubMed] [Google Scholar]
- 26.Yuan X, Wu H, Han N, Xu H, Chu Q, Yu S, Chen Y, Wu K. Notch signaling and EMT in non-small cell lung cancer: biological significance and therapeutic application. J Hematol Oncol. 2014;7:87. doi: 10.1186/s13045-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikezawa Y, Sakakibara-Konishi J, Mizugaki H, Oizumi S, Nishimura M. Inhibition of Notch and HIF enhances the antitumor effect of radiation in Notch expressing lung cancer. Int J Clin Oncol. 2017;22:59–69. doi: 10.1007/s10147-016-1031-8. [DOI] [PubMed] [Google Scholar]
- 28.Lim JS, Ibaseta A, Fischer MM, Cancilla B, O’Young G, Cristea S, Luca VC, Yang D, Jahchan NS, Hamard C, Antoine M, Wislez M, Kong C, Cain J, Liu YW, Kapoun AM, Garcia KC, Hoey T, Murriel CL, Sage J. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature. 2017;545:360–364. doi: 10.1038/nature22323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hassan WA, Yoshida R, Kudoh S, Kameyama H, Hasegawa K, Niimori-Kita K, Ito T. Notch1 controls cell chemoresistance in small cell lung carcinoma cells. Thoracic Cancer. 2016;7:123–128. doi: 10.1111/1759-7714.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu ZH, Dai XM, Du B. Hes1: a key role in stemness, metastasis and multidrug resistance. Cancer Biol Ther. 2015;16:353–9. doi: 10.1080/15384047.2015.1016662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Meng Q, Qiao W, Ma R, Ju W, Hu J, Lu H, Cui J, Jin Z, Zhao Y, Wang Y. miR-181b/Notch2 overcome chemoresistance by regulating cancer stem cell-like properties in NSCLC. Stem Cell Res Ther. 2018;9:327. doi: 10.1186/s13287-018-1072-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun L, Ke J, He Z, Chen Z, Huang Q, Ai W, Wang G, Wei Y, Zou X, Zhang S, Lan P, Hong C. HES1 promotes colorectal cancer cell resistance to 5-Fu by inducing Of EMT and ABC transporter proteins. J Cancer. 2017;8:2802–2808. doi: 10.7150/jca.19142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Q, Zeng F, Sun Y, Qiu Q, Zhang J, Huang W, Huang J, Huang X, Guo L. Etk interaction with PFKFB4 modulates chemoresistance of small-cell lung cancer by regulating autophagy. Clin Cancer Res. 2018;24:950–962. doi: 10.1158/1078-0432.CCR-17-1475. [DOI] [PubMed] [Google Scholar]
- 34.Jiang S, Han S, Chen J, Li X, Che H. Inhibition effect of blunting Notch signaling on food allergy through improving TH1/TH2 balance in mice. Ann Allergy Asthma Immunol. 2017;118:94–102. doi: 10.1016/j.anai.2016.10.024. [DOI] [PubMed] [Google Scholar]
- 35.Guo L, Liu Y, Bai Y, Sun Y, Xiao F, Guo Y. Gene expression profiling of drug-resistant small cell lung cancer cells by combining microRNA and cDNA expression analysis. Eur J Cancer. 2010;46:1692–702. doi: 10.1016/j.ejca.2010.02.043. [DOI] [PubMed] [Google Scholar]
- 36.Jin L, Vu T, Yuan G, Datta PK. STRAP promotes stemness of human colorectal cancer via epigenetic regulation of the NOTCH pathway. Cancer Res. 2017;77:5464–5478. doi: 10.1158/0008-5472.CAN-17-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ, Alam H, Lv J, Yun K, Gopalakrishnan V, Flores ER, Northcott PA, Rajaram V, Li W, Shilatifard A, Sillitoe RV, Chen K, Lee MG. MLL4 is required to maintain broad H3K4me3 peaks and super-enhancers at tumor suppressor genes. Mol Cell. 2018;70:825–841. doi: 10.1016/j.molcel.2018.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu DP, Zhou Y. Astrocyte elevated gene 1 (AEG-1) acts as a promoter gene in clear cell renal cell carcinoma cell growth and metastasis. Med Sci Monit. 2018;24:8213–8223. doi: 10.12659/MSM.911010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sundaresan V, Lin VT, Liang F, Kaye FJ, Kawabata-Iwakawa R, Shiraishi K, Kohno T, Yokota J, Zhou L. Significantly mutated genes and regulatory pathways in SCLC-a meta-analysis. Cancer Genet. 2017;216-217:20–28. doi: 10.1016/j.cancergen.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng G, Li N, Jia X, Peng C, Luo L, Deng Y, Yin J, Song Y, Liu H, Lu M, Zhang Z, Gu Y, He Z. MYCN-mediated miR-21 overexpression enhances chemo-resistance via targeting CADM1 in tongue cancer. J Mol Med. 2016;94:1129–1141. doi: 10.1007/s00109-016-1417-0. [DOI] [PubMed] [Google Scholar]
- 41.Bao J, Li M, Liang S, Yang Y, Wu J, Zou Q, Fang S, Chen S, Guo L. Integrated high-throughput analysis identifies super enhancers associated with chemoresistance in SCLC. BMC Med Genomics. 2019;12:67. doi: 10.1186/s12920-019-0520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Brien EM, Selfe JL, Martins AS, Walters ZS, Shipley JM. The long non-coding RNA MYCNOS-01 regulates MYCN protein levels and affects growth of MYCN-amplified rhabdomyosarcoma and neuroblastoma cells. BMC Cancer. 2018;18:217. doi: 10.1186/s12885-018-4129-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spitschak A, Meier C, Kowtharapu B, Engelmann D, Pützer BM. MiR-182 promotes cancer invasion by linking RET oncogene activated NF-κB to loss of the HES1/Notch1 regulatory circuit. Mol Cancer. 2017;16:24. doi: 10.1186/s12943-016-0563-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan Q, Chen X, Han Y, Lei T, Wu Q, Yu X, Wang L, Fan Z, Wang S. Modification of α2,6-sialylation mediates the invasiveness and tumorigenicity of non-small cell lung cancer cells in vitro and in vivo via Notch1/Hes1/MMPs pathway. Int J Cancer. 2018;143:2319–2330. doi: 10.1002/ijc.31737. [DOI] [PubMed] [Google Scholar]
- 45.Goto N, Ueo T, Fukuda A, Kawada K, Sakai Y, Miyoshi H, Taketo MM, Chiba T, Seno H. Distinct roles of HES1 in normal stem cells and tumor stem-like cells of the intestine. Cancer Res. 2017;77:3442–3454. doi: 10.1158/0008-5472.CAN-16-3192. [DOI] [PubMed] [Google Scholar]
- 46.Farah E, Li C, Cheng L, Kong Y, Lanman NA, Pascuzzi P, Lorenz GR, Zhang Y, Ahmad N, Li L, Ratliff T, Liu X. NOTCH signaling is activated in and contributes to resistance in enzalutamide-resistant prostate cancer cells. J Biol Chem. 2019;294:8543–8554. doi: 10.1074/jbc.RA118.006983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu Z, Ren Y, Zhang M, Fan T, Wang Y, Zhao Q, Liu HM, Zhao W, Hou G. FLI-06 suppresses proliferation, induces apoptosis and cell cycle arrest by targeting LSD1 and Notch pathway in esophageal squamous cell carcinoma cells. Biomed Pharmacother. 2018;107:1370–1376. doi: 10.1016/j.biopha.2018.08.140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.