

Abstract
Cancer-associated fibroblasts (CAFs) play critical roles in cancer progression and treatment failure. CAFs display extreme phenotypic heterogeneity and functional diversity. Some subpopulations of CAFs have the ability to reconstitute cancer stemness by promoting the expansion of cancer stem cells (CSCs) or by inducing the generation of CSCs from differentiated cancer cells. CAFs regulate cancer stemness in different types of solid tumors by activating a wide array of CSC-related signaling by secreting proteins and exosomes. As feedback, the CSCs can also induce the proliferation and further activation of CAFs to promote their CSC-supporting activities, thus completing the loop of CAF-CSC crosstalk. Current research on targeting CAF-CSC crosstalk could be classified into (i) specific depletion of CAF subpopulations that have CSC-supporting activities and (ii) targeting molecular signaling in CAF-CSC crosstalk, such as the IL6/STAT3, TGF-β/SDF-1/PI3K, WNT/β-catenin, HGF/cMET and SHH/Hh pathways. Strategies targeting CAF-CSC crosstalk may open new avenues for overcoming cancer progression and therapeutic resistance.
Keywords: CAFs, CSCs, cancer stemness, therapeutic resistance, cancer therapy
Introduction
The tumor microenvironment (TME) is one of the key unsolved issues that hampers effective cancer management in clinical practice [1] and is composed of extracellular matrix (ECM), cytokines interspersed in the ECM, and several different types of stromal cells, including fibroblasts, endothelial cells and immune cells [2]. Among the components of the TME, cancer-associated fibroblasts (CAFs) are key members of stromal cells and have been suggested to play essential roles in the complex process of tumor-stroma coevolution and tumorigenesis [3,4]. CAFs have a strong ability to produce various factors that are related to ECM synthesis or remodeling, such as collagens, fibronectins and ECM-degrading proteases. CAFs can also secrete growth factors and proinflammatory cytokines and chemokines, such as transforming growth factor-β (TGF-β), interleukin-6 (IL-6), CC-chemokine ligand 2 (CCL2), and stromal cell-derived factor 1 (SDF-1; also named CXCL-12) [3,4]. Specific cell-surface proteins on CAFs have been considered ideal markers for CAFs, and these markers include alpha smooth muscle actin (α-SMA), fibroblast activation protein (FAP), S100 calcium binding protein A4 (S100A4), platelet-derived growth factor receptor-β (PDGFRβ) and fibroblast growth factor receptor 2 (FGFR2); however, none of these markers are exclusively expressed by CAFs [1,5]. Increasing evidence suggests that CAFs are a complex and heterogeneous population of cells [4] with different cellular sources in cancers [1,4]. Once activated by a broad range of stimuli from cancer cells or the TME, CAF progenitors acquire the phenotypes of CAFs, such as the expression of α-SMA, FAP or S100A4 and the secretion of TGF-β, IL-6 or CCL2 [1]. The diversity of CAF origins and stimuli may contribute to the phenotypic heterogeneity and functional diversity of CAFs. Most studies have suggested that CAFs have tumor-promoting effects, while some reports have indicated that CAFs have antitumor functions [1,4]. The protumorigenic type of CAFs can enhance tumorigenesis, metastasis, angiogenesis and immune evasion of cancer cells by either the secretion of proteins (including chemokines, cytokines and growth factors) and exosomes or the direct cell-cell contact with tumor cells [4]. In addition to regulating cancer progression, CAFs also play critical roles in hampering the effect of cancer therapies, especially immunotherapy [6] and chemotherapy [1,4]. Although most CAFs have potent protumorigenic roles, some CAF subsets actually have tumor-suppressive functions in specific cancers or at specific cancer stages. The antitumor functions of CAFs are mediated by their regulation of antitumor immunity [7]. The more recent studies showed that CAFs may induce the generation and expansion of cancer stem cells (CSCs) [8], which are involved in multiple steps of cancer progression and drug resistance development [8,9].
CSCs are a small, self-renewing cell population within the bulk tumor mass and are capable of generating new tumors [9]. Compared with differentiated cancer cells, which have limited regenerative and tumorigenic capacities, CSCs have extensive proliferative potential to generate primary tumors and form disseminated metastatic tumors [10]. During tumor pathogenesis, genetic and epigenetic changes cause the transformation of normal stem cells, or even differentiated cells, into CSCs [11]. These mutated CSCs have extensive abilities to promote tumor growth, immune evasion, metastasis, chemoresistance and radioresistance, thereby leading to cancer progression and therapeutic failure [10]. Moreover, TME factors, including CAFs and their derived stimuli, can induce CSC clonal expansion and tumorigenic activities [8]. The amount and activity of CSCs within the tumor determine cancer stemness and correlate with disease progression and therapy efficacy. Notably, CSCs might undergo further mutations and thus increase their heterogeneity and generate diverse phenotypes of cancer cells [11]. The identification of CSCs is important to CSC studies and the development of CSC-targeting intervention strategies. CSC identification is generally based on cell surface markers. CD24, CD26, CD29 (also called β1-integrin), CD34, CD44, CD133, CD166, aldehyde dehydrogenase (ALDH) and Ep-CAM (also called CD326) have been identified as CSC-specific surface markers in different cancers [8]. In addition, activated pathways, including the WNT/β-catenin, MEK, Notch, Hedgehog (Hh), phosphoinositide 3 kinase (PI3K)/AKT and TGF-β pathways, are utilized by CSCs for their survival, proliferation and stemness maintenance [8].
To date, very few reviews focusing on CAF-regulated cancer stemness have been reported. It is essential to provide an overview of CAF-regulated cancer stemness for developing more efficient cancer therapeutic approaches. In this review, we will focus on the current knowledge in identifying CAF subsets that regulate cancer stemness and discuss the crosstalk between CAFs and tumor cells for regulating cancer stemness in different cancer models. This review intends to open a new window for devising novel strategies to overcome cancer progression and therapeutic resistance.
CAFs support cancer stemness in different cancers
It has been reported that cancer stemness can be enhanced by CAFs in several cancer types (Table 1). A cell culture model in which tumor cells are cultured with conditioned media (CM) from CAFs or a coculture system with tumor cells and CAFs is often used to study the influence of CAFs on different CSCs in vitro. CAFs are able to induce the expression of stemness markers (e.g., Sox2, Bmi-1 and CD44), enhance sphere formation, and promote the self-renewal and expansion of CSCs in breast [12-14], prostate [15,16], lung [14], colorectal [17,18], gastric [19], and liver cancers [20]. Although the conditioned media from CAFs can regulate CSC properties in most in vivo models [1-9], it is noteworthy that direct cell-cell interactions are required for the regulation of cancer stemness by CAFs in some cases [17]. Additionally, in vivo experiments have also indicated the roles of CAF-regulated cancer stemness in enhancing the in vivo tumorigenicity in breast [13,14], prostate [16], colorectal [21], gastric [19] and liver cancers [20]. Cancer stemness is correlated with chemoresistance and cancer metastasis. Both in vitro and in vivo experiments have demonstrated that CAFs can promote drug resistance [14,17,18,20] and cancer metastasis [14,16,21] through the regulation of cancer stemness in different cancers. CAF-regulated cancer stemness may also affect cancer relapse [22].
Table 1.
CAFs regulate cancer stemness in different cancers
| Cancer types | CAF-regulated signatures | References | |
|---|---|---|---|
|
| |||
| Stemness marker | Induced/Expanded CSCs | ||
| Breast | Sox2, Bmi-1, Nanog and CD44 | CD24-CD44+ cells; | [12-14] |
| PKH67high cells; | |||
| ALDH+ cells | |||
| Prostate | Oct4, Sox2, and Nanog | CD24-CD44+ cells; CD133+ cells | [15,16] |
| Lin-Sca1highCD49fhigh cells | |||
| Lung | ABCG2 | ALDH+ cells | [14] |
| Colon | Not reported | ALDH+ cells | [17,18,21] |
| Gastric | ABCG2, CD44, Nanog and ALDH1 etc. | ★SP (side population) cells | [19] |
| Liver | Oct4, and Nanog | CD44+ cells; CD47+ cells; | [20] |
| CD90 cells; CD24+ cells | |||
SP cells were defined as the subset of cells that exhibited a low Hoechst33342 staining pattern and disappeared with the use of verapamil.
Abbreviations: Sox2, SRY-box transcription factor 2; Oct4, Octamer-binding transcription factor 4; ABCG2, ATP binding cassette subfamily G member 2.
Key components and pathways involved in CAF-regulated cancer stemness
As summarized in Table 2, the CAF-derived stimuli that regulate CSC properties may be different among various types of cancer. These stimuli include CAF-secreted chemokines, cytokines, growth factors and exosomes, and direct CAF-tumor cell interactions.
Table 2.
The signatures of CSC-supporting CAFs
| Cancer types | CAF markers | Activated signaling in CAFs | CAF-derived mediators for CSC regulation | References |
|---|---|---|---|---|
| Breast | α-SMA, CD10, GPR77 | NF-kB pathway; STAT3 pathway | IL-6, IL-8, SDF-1, CCL2 HMGB1 | [12-14,22] |
| Hedgehog signaling; | ||||
| Autophagic pathway | ||||
| Prostate | α-SMA-FAP+ | IL-6/IL-6R signaling | Metalloproteases | [16,36] |
| Lung | α-SMA, CD10, GPR77 | NF-kB pathway | IL-6, IL-8 | [14] |
| Colon | α-SMA, CD44, FAP and S100A4 etc. | Hypoxic inducible pathway | CD44, Exosomal lncRNA H19, Wnts | [17,18,42] |
| Gastric | α-SMA | Not reported | TGFβ | [19] |
| Liver | α-SMA, FAP | Not reported | HGF, IL6 | [20,25] |
Abbreviations: α-SMA, α smooth muscle actin; GPR77, G protein-coupled receptor 77; SDF-1, stromal cell-derived factor 1; HMGB1, high mobility group box 1; FAP, fibroblast activation protein alpha; S100A4, S100 calcium-binding protein A4; HGF, hepatocyte growth factor.
Chemokines
SDF-1 secreted by CAFs has been found to promote the generation of CSCs and sustain their tumorigenesis and metastatic activity in breast cancer [12] and colorectal cancer [21]. CAF-secreted SDF-1 regulates the CSC phenotype of cancer cells by interacting with the cancer cell-expressed receptor CXCR4 [12] and then activating the Wnt/β-catenin [12,21] and/or PI3K/AKT pathways [21] in cancer cells. The SDF-1/CXCR4 chemokine axis also activates other CSC-related signaling pathways, such as NOTCH signaling in breast cancer cells [12,23]. In cancers with SDF-1 secretion, CXCR4+ cancer cells can obtain enhanced stemness compared with CXCR4- cancer cells since CAF-secreted SDF1 can stimulate the CSC phenotype and induce the expansion of them. In line with this finding, CXCR4 has been considered a biomarker for radioresistant CSCs [24]. CCL2 is another chemokine released by CAFs and has the ability to regulate CSC properties. Tsuyada et al. showed that breast CAF-secreted CCL2 can induce self-renewal and expansion of CSCs in breast cancer. Consistently, a tumor model generated by cotransplantation of primary breast cancer cells and CAFs into the mammary fat pads of NOD/SCID/IL-2Rg-null mice suggested that CCL2 is critical for CAF regulation of tumorigenesis in vivo. CAF-secreted CCL2 has also been found to regulate the CSC phenotype by activating NOTCH1 signaling in breast cancer cells [13].
Cytokines and growth factors
It has been shown that IL-6 secreted by CAFs promotes the stemness of CD24+ liver cells [25]. Similarly, IL-8 from CAFs can regulate the cancer stemness and chemoresistance of cancer cells [26]. In most cases, CAFs regulate the epithelial-mesenchymal transition (EMT) and CSC properties of cancer cells via the combined production of IL-6 and IL-8. Su et al. showed that IL-6 and IL8 can be secreted by CD10+GPR77+ CAFs and can promote tumorigenicity and chemoresistance by supporting cancer stemness in breast and lung cancers [14]. In line with this finding, simultaneous blocking of IL-6 and IL-8 completely inhibits CAF-induced human melanoma cell invasiveness [27]. CAF-derived IL-6 supports the CSC phenotype by activating the STAT3 pathway and/or TGF-β/Smad signaling [28,29]. IL-8 released by CAFs may sustain the stemness of cancer cells via interactions with its receptors, CXCR1 and CXCR2, which are expressed by cancer cells [30,31].
CAF-secreted growth factors, such as TGF-β and hepatocyte growth factor (HGF), are involved in the regulation of tumorigenicity, metastasis and drug resistance [1,32]. Recent studies demonstrated that TGF-β released by CAFs can sustain the stemness of scirrhous gastric cancer cells [19] and promote the EMT program by activating TGF-β/Smad signaling in breast cancer [33]. Additionally, autocrine TGF-β can promote SDF-1 secretion via the activation of HSF1 in breast CAFs and thus regulate the CSC properties of breast cancer cells through the SDF-1/CXCR4 axis [12,34]. Therefore, TGF-β released by CAFs may regulate cancer stemness either in a direct manner or in an indirect manner. Similarly, CAF-secreted HGF can also regulate CSC phenotypes through different pathways. CAF-secreted HGF is able to activate cMET-dependent signaling in liver cancer cells [20] and colorectal cancer cells [35]. The cMET/FRA1/HEY1 cascade induced by the CAF-secreted HGF plays critical roles in the regulation of liver cancer stemness [20]. It remains unclear whether the cMET-dependent signaling activated by CAF-secreted HGF can regulate the CSC properties of colorectal cancer cells. However, it has been shown that colorectal CAF-secreted HGF can support the stemness of colorectal cancer cells by activating the Wnt/β-catenin and PI3K signaling pathways [21].
Other secreted proteins
Except for chemokines, cytokines and growth factors, other CAF-secreted proteins, such as metalloproteases and high-mobility group box 1 (HMGB1), may also regulate the CSC phenotype. Giannoni et al. showed that metalloproteases secreted by prostate CAFs elicit a Rac1b/COX-2-mediated release of reactive oxygen species (ROS) in cancer cells, which is mandatory for supporting the stemness of metastatic prostate cancer cells [16,36]. In these studies, the EMT signaling and proinflammatory signature induced by the CAF-secreted metalloproteases can increase the expression of stem cell markers, sphere-forming capacity and self-renewal activity of prostate cancer cells. HMGB1, a nuclear protein, can also induce proinflammatory signaling when released by apoptotic cells or autophagic cells [37]. It has been demonstrated that HMGB1 released by autophagic CAFs can enhance the stemness and tumorigenicity of luminal breast cancer cells by activating its receptor, toll-like receptor 4 (TLR4), expressed by cancer cells [22].
Exosomes
Exosomes, which are closely associated with cancer progression and treatment failure [38], are critical mediators of tumor-CAF crosstalk. Exosomes released by CAFs can contribute to cancer progression and drug resistance by regulating the proliferation, metastasis and CSC properties of cancer cells [39,40]. Donnarumma et al. showed that exosomal miRNAs, including miRs-21, -378e, and -143, are essential for CAF-regulated stemness and the EMT phenotype of breast cancer cells [41]. Ren et al. demonstrated that CAFs promote the stemness and oxaliplatin resistance of colorectal cancer via exosomal lncRNA H19 [18]. To regulate the CSC properties of cancer cells, miRNAs [41] or lncRNAs [18] should be shuttled by CAF-derived exosomes into cancer cells. Additionally, after being transferred to tumor cells, lncRNA H19 can activate the β-catenin pathway, acting as a competing endogenous RNA sponge for miR-141 [18]. In addition to noncoding RNAs, exosomal proteins such as Wnt ligands can also promote cancer stemness. Hu et al. de-monstrated that CAF-secreted exosomal Wnts can support CSC characteristics and drug resistance by inducing colorectal cancer cell dedifferentiation [42]. In pancreatic ductal adenocarcinoma (PDAC), CAFs can provide a stem cell niche for PDAC cells without the ability to produce Wnts and support their in vivo tumorigenesis by secreting Wnts [43]. Moreover, Wnts secreted by esophageal CAFs can induce the EMT and invasiveness of cancer cells, which are considered hallmarks of CSCs [5,44]. Taken together, CAFs induce the activation of the WNT/β-catenin pathway in cancer cells and thus regulate their CSC phenotypes by secreting different factors, such as soluble SDF-1 and HGF, exosomal lncRNA H19 and exosomal Wnt ligands.
Membrane proteins
Except for secreted factors, direct cell-cell contact is also required for CAFs in regulating cancer stemness [17], suggesting the critical role of membrane proteins in CAF regulation of the CSC phenotype. Kinugasa et al. demonstrated that CD44 expressed on CAFs acts as a functional cell-surface molecule that is essential for supporting the stemness and drug resistance of colorectal cancer cells [17]. CD44 expressed on CAFs is implicated in the regulation of CAF-secreted SDF-1, which has been reported to stimulate the CSC properties of cancer cells. It remains unclear whether CD44 is involved in the direct cell-cell interaction of CAFs and cancer cells. More membrane molecules on CAFs have been reported to play indirect roles in the regulation of cancer stemness. As described above, CD10+GPR77+ CAFs act as a protumorigenic CAF subpopulation that can sustain the stemness and enhance the chemoresistance of breast and lung cancer cells [14]. GPR77 is a functional CAF membrane molecule that can be activated by cancer-derived stimuli and then induces CAF intracellular NF-kB signaling, which is involved in cancer stemness regulation via the production of IL-6 and IL-8 [14]. CD10 serves as a marker for the CAF subpopulation that supports the CSC phenotype and tumorigenesis of tumor cells in breast, lung and colorectal cancers [14,45]; however, the function and mechanism of CD10 on stemness regulation remain unknown.
CAF-CSC interaction loop
The bi-directional activation between cancer cells and stromal cells is critical to cancer cell phenotypes and functions and influences cancer progression and treatment resistance [1,4]. Only when specific signaling pathways are activated by corresponding stimuli from cancer cells or the TME can CAFs acquire their phenotypes for sustaining cancer stemness. For example, CM from the breast cancer cell lines BT474 and MDA361 but not from the noncancerous mammary epithelial cell line MCF10A can activate STAT3 signaling in CAFs and then induce CCL2 production, which is essential to regulating the stemness of cancer cells [13]. The NF-kB pathway in CAFs is also implicated in the regulation of CAF phenotype-stimulating cancer stemness. Persistent activation of the NF-kB pathway in CD10+GPR77+ CAFs from breast or lung cancer is required for the production and secretion of IL-6 and IL-8, exerting the ability to promote cancer stemness [5]. NF-kB signaling in CD10+GPR77+ CAFs can be activated by autocrine or tumor-derived C5a, one of the complement mediators [14]. Additionally, TGF-β and IL-6 are two well-known factors that regulate the crosstalk between cancer cells and CAFs. In lung cancer, tumor-derived TGF-β can drive α-SMA+ CAFs to produce IL-6, which supports the stemness of cancer cells and in turn increases TGF-β secretion by cancer cells [29]. Furthermore, tumor-derived IL-6 can promote the differentiation and activation of α-SMA-FAP+ CAFs and regulate their CSC-stimulating abilities by inducing metalloprotease production [16,36]. After being induced by CAFs or other stimuli, CSCs will in turn release specific factors to maintain the CAF phenotype with CSC-stimulating functions. Valenti et al. showed that CSCs from mammary gland tumors secrete the Hedgehog ligand SHH, which activates the CAF Hedgehog signaling pathway and thus induces CAFs to secrete factors promoting the expansion and self-renewal of CSCs [46].
The origin of CAFs and the stimuli they receive influences CAF phenotype with specific secretomes, whereas the cancer cells featured with specific membrane receptors and intracellular signaling pathways reciprocally have impacts on the response of cancer cells to CAF-derived stimuli. In breast cancer, CAF-secreted CCL-2 failed to induce CSCs in MCF-7 cells because of their lack of CXCR-2 receptor; however, CAF-secreted CCL-2 strongly induces CSCs in BT474 and MDA361 cells that have high expression of CXCR-2 [13]. Therefore, the regulation of cancer stemness by CAFs is an effect of complicated cancer-stroma crosstalk.
Targeted strategies for disrupting CAF-CSC crosstalk
CAFs have emerged as one of the key factors that regulate the CSC phenotype of cancer cells, so targeting cancer-CAF crosstalk for cancer-stemness regulation should be a feasible strategy to suppress cancer progression or reduce treatment failure. Strategies targeting CAF-CSC crosstalk can be mainly classified into two categories: direct depletion of CAF subpopulations that have CSC-stimulating abilities and interference of the activation of signaling or downstream effectors (Figure 1).
Figure 1.
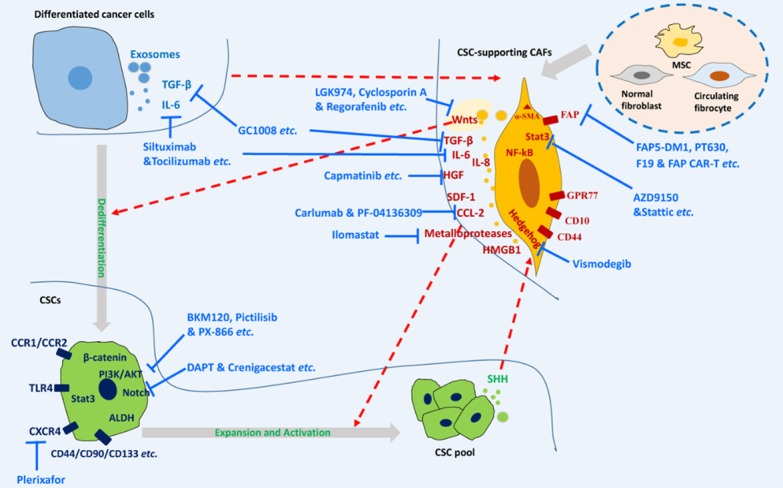
Targeting CAF-CSC crosstalk for cancer therapy. CSC-supporting CAFs can be activated by surrounding stimuli, including cancer cell-derived factors, and then regulate cancer stemness by inducing the dedifferentiation of differentiated cancer cells or inducing the expansion and activation of CSCs via their secreted proteins and exosomes. CAF-CSC crosstalk can be blocked by direct depletion of CSC-supporting CAFs or various drug candidates targeting molecular signaling pathways between CAFs and CSCs. CSCs: cancer stem cells; CAFs: cancer-associated fibroblasts; MSC: mesenchymal stem cell; FAP: fibroblast activation protein.
Direct depletion of CSC-stimulating CAFs
Recently, researchers have put much effort into identifying specific CAF cell-surface markers for in vitro sorting of CAFs and in vivo depletion of CAFs. α-SMA or FAP is often expressed on the CSC-stimulating CAFs (Table 2). Since selective depletion of the α-SMA+ CAFs in a mouse model of spontaneous PDAC not only enhances tumor hypoxia, the EMT, and the CSC phenotype but also increases the tumor infiltration of Treg cells that suppress antitumor immunity and promote tumor progression, targeting α-SMA+ myofibroblasts may not be a proper strategy for cancer treatment [7]. However, as shown in Table 3, depletion of FAP+ CAFs via different strategies demonstrated promising antitumor efficacy in various preclinical studies [47-50]. More importantly, inhibition of FAP+ CAFs with the small-molecule dipeptidyl peptidase inhibitor PT100 enhanced the efficacy of cancer therapy with oxaliplatin in a mouse colon cancer model [51], suggesting that direct depletion of FAP+ CAFs could lead to a reduction in the CSCs that are associated with chemoresistance. However, clinical investigations for such treatment paradigms did not show significant antitumor efficacy [52,53]. In addition, FAP can also serve as a target for chimeric antigen receptor T (CAR-T) cell strategies in cancer treatment. In a preclinical study, FAP-specific CAR-T cells arrested the growth of desmoplastic human lung cancer xenografts and syngeneic murine pancreatic cancers [54]. In contrast, another study demonstrated that CAR-T cells had limited antitumor efficacy and even induced lethal toxicity since they also recognized and killed multipotent bone marrow stromal cells (BMSCs) that expressed FAP [55].
Table 3.
Strategies for direct depletion of FAP+ CAFs
| Strategy types | Method or drug candidate | Cancer types | Clinical trial | References |
|---|---|---|---|---|
| Genetic depletion | LacZ knock-in | Lung, Colon | Not reported | [47] |
| Conditional depletion | Diphtheria toxin | Pancreatic, Lung | Not reported | [48] |
| FAP-targeting killing | FAP5-DM1: maytansinoid-conjugated antibody | Pancreatic, Lung, Colon | Not reported | [49] |
| αFAP-PE38: toxin-conjugated antibody | Breast, Melanoma | Not reported | [50] | |
| Pharmacological inhibition: Inhibitors | PT630, PT100 | Lung, Colon | Not reported | [47,51] |
| Pharmacological inhibition: Antibodies | F19 (sibrotuzumab) | Lung, Colon | (1) Clinically safe; (2) Showed tumor inhibiting potential in a phase I trial of advanced cancers with FAP-expressing stroma; (3) Failed in another early phase II trial in patients with metastatic colorectal cancer | [52,53] |
| CAR-T strategy | FAP-specific CAR-T | Lung, Pancreatic | No | [54,55] |
Collectively, the direct targeting of CAFs still faces numerous obstacles and challenges. Neither FAP nor α-SMA is exclusively expressed by CAFs. Moreover, some subsets of CAFs may suppress the CSC phenotype, and thus their depletion may enhance the EMT and stemness of cancer cells [56]. Further studies are needed to develop novel strategies targeting more specific cell-surface markers of CSC-stimulating CAF subpopulations. Previous studies showed that the cell-surface molecule CD44 is associated with the CSC-stimulating activity of α-SMA+ CAFs in colorectal cancer [17], and CD10 or GPR77 can serve as a cell-surface marker for CSC-stimulating CAFs in breast, lung or colorectal cancer [14,45]. Thus, the recognition of combined markers, including membrane molecules associated with CSC-stimulating properties and canonical CAF cell-surface markers, such as FAP, should be able to enhance the specificity of targeting CSC-stimulating CAFs. Roybal et al. designed a novel CAR-T cell, the dual-receptor AND-gate T cell, which is only armed and activated in the presence of dual antigen tumor cells [57]. Novel dual-receptor AND-gate T cells can also be developed to only recognize CAFs with dual surface markers and thus deplete CAFs more specifically.
Targeting CAF-CSC crosstalk molecular signaling
Targeting the IL-6/STAT3 pathway and its downstream effectors
Tumor-derived IL-6 has the ability to activate the STAT3 pathway in CAFs [58], thus sustaining the CSC-stimulating phenotype of CAFs by inducing their metalloprotease secretion [36]. Additionally, the activation of STAT3 signaling by autocrine or CAF-derived IL-6 in cancer cells is also implicated in the mediation of cancer stemness [14,25,28]. Blocking IL-6/IL-6R/STAT3 signaling with the STAT3 inhibitor Stattic has been shown to reduce the stemness of breast cancer cells in preclinical studies [2]. Other agents for inhibition of IL-6/IL-6R/STAT3 signaling have also exhibited antitumor efficacy in preclinical studies, and the early phase clinical trials for some of these inhibitors in cancer patients are ongoing [59] (Table 4). Among these agents used in current clinical trials, AZD9150, a second-generation STAT3 antisense oligonucleotide, has shown antitumor activity with a maximum-tolerated dose of 3 mg/kg in a clinical trial of treatment-refractory lymphoma and NSCLC [60] (Table 5). Most of the clinical trials for blocking IL-6/IL-6R/STAT3 did not show effective antitumor activity in patients with solid tumors [59]. The failure of these clinical trials may be partially caused by the absence of reliable markers that are predictive of treatment response. In the crosstalk between cancer cells and CAFs, the CCL2/CXCR2/NOTCH pathway can be activated in response to IL-6/IL-6R/STAT3 signaling. Treatment with CCL2 neutralizing antibodies or inhibitors of the NOTCH activating α- and γ-secretases effectively attenuated the stemness of breast cancer cells or glioblastoma cells and suppressed their metastasis in preclinical studies [13,61]. However, clinical investigations on CCL2/CXCR2/NOTCH blocking show limited clinical activity in patients with solid tumors [62,63]. Matrix metalloprotease (MMP) secretion serves as a downstream effector of IL-6/IL-6R/STAT3 signaling in prostate cancer, and the MMP inhibitor Ilomastat is sufficient to suppress the CAF-regulated CSC phenotype of prostate cancer cells [36]. However, to the best of our knowledge, no clinical trials investigating the safety, tolerance, and bioactivity of MMP inhibitors in cancer patients have been reported.
Table 4.
Drug candidates targeting molecular signaling for CAF-CSC crosstalk
| Molecular Pathways or key effectors | Cancers types | Targets | Drug candidates★ | References |
|---|---|---|---|---|
| IL-6/IL-6R/STAT3 signaling and their effectors | Breast, Prostate, Lung, Liver, Glioblastoma | IL-6/IL-6R | Siltuximab, Tocilizumab, Olamkicept | [2,13,14,25,28,36,58-63] |
| STAT3 | Stattic, C188-9, OPB-31121, OPB-51602, AZD9150, STAT3 decoy oligonucleotide | |||
| CCL2/CCR2 | Carlumab, PF-04136309 | |||
| NOTCH | DAPT, INCB3619, GSI II, Crenigacestat | |||
| MMP | Ilomastat | |||
| TGF-β/SDF-1/CXCR4/PI3K | Gastric, Breast, Colon, Prostate, Renal | TGF-β/TGF-βR | GC1008, TβM, Ki26894, LY2109761, LY3022859, PF-03446962 | [12,19,34,64-76] |
| SDF-1/CXCR4 | Plerixafor | |||
| PI3K/AKT | BKM120, Ly294002, Pictilisib, Buparlisib, Ipatasertib, Alpelisib, PQR309, PX-866 | |||
| HGF/cMET | Colon, Liver | HGF/cMET | CpdA, Volitinib, Capmatinib, Crizotinib, SU11274, PHA665752, TC-N19, LY2875358, LY2801653, JNJ-61186372 | [20,35,77-87] |
| WNT/β-catenin | Colon, Pancreatic, Esophageal | WNT | LGK974, Cyclosporin A, Wnt-C59 | [42-44,88-92] |
| β-catenin | ||||
| SHH/Hedgehog | Breast, Colon, Prostate | SHH/Hedgehog | Vismodegib, Sonidegib | [46,93-96] |
(1) All the drug candidates listed here have shown antitumor activities in preclinical studies; (2) The underlined drug candidates listed here have been tested for their CAF-CSC blocking abilities; (3) The drug candidates listed here in bold have been used in clinical investigations.
Abbreviations: SDF-1, stromal cell-derived factor 1; HGF, hepatocyte growth factor; cMET, tyrosine-protein kinase Met; SHH, sonic hedgehog signaling molecule.
Table 5.
Clinical trials showing bioactivity of CAF-CSC blocking strategies
| Drug | Description | Indication | Study phase | Trial results | References |
|---|---|---|---|---|---|
| AZD9150 | STAT3 antisense oligonucleotide | Advanced-stage pancreatic cancer, NSCLC, and CRC; Metastatic HNSCC; Advanced-stage and/or metastatic hepatocellular carcinoma | I, II | Showed antitumor activity with a maximum-tolerated dose of 3 mg/kg in patients with treatment-refractory lymphoma and NSCLC | [60] |
| PX-866 | Pan-isoform inhibitor of Class I PI3K | Recurrent or metastatic castration-resistant prostate cancer | II | At 12 weeks, 33% patients were progression-free with 5% of partial objective responses when treated with single-agent PX-866 | [73] |
| Alpelisib | Alpha-specific PI3K inhibitor | Japanese patients with advanced solid tumors; Estrogen receptor-positive advanced breast cancer; HER2-positive metastatic breast cancer | I, Ib | Single treatment with alpelisib, or combined therapies with alpelisib and fulvestrant, or T-DM1 all exhibited clinical activity | [74] |
| PQR309 | Dual PI3K and mTORC1/2 inhibitor | Advanced solid tumors | I | Partial response in a patient with metastatic thymic cancer, 24% disease volume reduction in a patient with sinonasal cancer, and stable disease for more than 16 weeks in a patient with clear cell Bartholin’s gland cancer | [75] |
| Pictilisib | Pan-PI3K inhibitor | Advanced breast cancer | Ib | Two (3.4%) patients experienced complete responses, and 17 (29.3%) patients had partial responses when treated with pictilisib and paclitaxel together | [76] |
| Capmatinib | Multitarget cMET inhibitor | Japanese patients with advanced solid tumors | I | Exhibited antitumor activity in 8/44 patients | [87] |
| Vismodegib | Hedgehog inhibitor | Basal cell carcinoma | FDA approved | N/A | [95] |
| Sonidegib | Hedgehog inhibitor | Triple negative breast cancer (TNBC) | I | In total, 25% of patients with metastatic TNBC derived clinical benefit from combination therapy with sonidegib and docetaxel | [96] |
Abbreviations: NSCLC, non-small cell lung cancer; CRC, colorectal cancer; HNSCC, head and neck squamous cell carcinoma; PI3K, phosphatidylinositol 3-kinase.
Targeting the TGF-β/SDF-1/PI3K pathway
Autocrine signaling loops mediated by TGF-β and SDF1 in CAFs have been found to promote cancer-stemness-supporting CAF phenotypes in breast and gastric cancers [12,19,34]. CAF-derived SDF1 plays roles in cancer progression through its interaction with its receptor CXCR4 [12]. As reported in preclinical studies, the gastric CSC population that was upregulated by CM from gastric CAFs was significantly decreased by anti-TGFβ1 neutralizing antibodies or by a TGF-βR inhibitor (Ki26894) [19], and blocking SDF-1/CXCR4 signaling with the CXCR4 antagonist AMD3100 (plerixafor) significantly inhibited CSC populations in breast [12], colon [64] or renal cancer cells [65]. Unfortunately, none of the clinical trials in patients with solid tumors have shown effective antitumor activity for these strategies targeting TGFβ/TGF-βR or SDF-1/CXCR4 [66-69]. PI3K/AKT activation in cancer cells induced by CAF-derived factors, such as SDF-1, TGF-β and HEF, can mediate CSC phenotypes [21]. Blocking PI3K/AKT signaling with the PI3K inhibitor BKM120 or Ly294002 is capable of killing CSCs in different cancers, including colon [21,70], prostate [71] and breast cancers [72], by single-drug or combination therapy in preclinical studies. Interestingly, PI3K inhibitors, including PX-866 [73], alpelisib [74], PQR309 [75] and pictilisib [76], have shown antitumor activity in clinical trials for patients with solid tumors (Table 5).
Targeting the HGF/cMET or WNT/β-catenin pathway
HGF regulates CSC phenotypes in liver or colorectal cancer cells by acting on their cMET signaling [20,35] or β-catenin signaling [21]; CAF-secreted WNT ligands induce CSCs or metastasis in colorectal, pancreatic and esophageal cancers through the WNT/β-catenin pathway [42-44]. Numerous preclinical studies have demonstrated that specific cMET inhibitors [77-84] or multitargeted inhibitors [79,85,86], which inhibit HGF/cMET signaling and other signaling simultaneously, have the ability to suppress cancer cell growth, enhance cancer cell sensitivity to radiotherapy and medical therapy, and reduce cancer recurrence in different cancer models (Table 4). Recently, a phase I study in Japanese patients with advanced solid tumors showed that capmatinib (INC280), a multitargeted cMET inhibitor, exhibited antitumor activity in 8/44 patients [87], thereby supporting further drug development of capmatinib. WNT/β-catenin inhibitors, including LGK974 [88], Wnt-C59 [89] and cyclosporin A [90], have been found to suppress the survival or proliferation of CSCs in different cancers in preclinical studies. Phase I clinical trials using LGK974 or cyclosporin A in patients with advanced solid malignancies that are dependent on WNT ligands are ongoing [91,92].
Targeting the SHH/Hedgehog pathway
It has been demonstrated that the Hedgehog inhibitor vismodegib is effective in reducing CAF and CSC expansion and thus inhibiting proliferation and triggering apoptosis of cancer cells in different cancers, including breast [46], colon [93] and prostate cancers [94]. Moreover, vismodegib (GDC-0449/Erivedge) has been approved by the FDA for the treatment of basal cell carcinoma [95]. Another Hedgehog inhibitor, sonidegib, also exhibited the ability to suppress CAF activation and reduce CSCs in triple negative breast cancer (TNBC) [96]. More importantly, a phase I clinical trial has shown that 3 of 12 patients with metastatic TNBC derived a meaningful clinical benefit from combination therapy with sonidegib and docetaxel [96]. Therefore, vismodegib and sonidegib treatment could be considered new strategies for overcoming cancer stemness and increasing the sensitivity of cancer cells to chemotherapy. However, smoothened (Smo) mutations that confer constitutive activity and drug resistance might emerge during vismodegib treatment [95]. For this reason, new effective Hh inhibitors are being developed, and their CSC-inhibiting bioactivities are being studied [95].
Conclusions and perspectives
Here, we want to emphasize the importance of CAFs in supporting the CSC properties of human cancers. CAFs have heterogeneous phenotypes and functions and can be classified as protumorigenic and antitumorigenic CAFs [1,4]. The CSC-promoting CAF subpopulations have different phenotypes compared with those without CSC-supporting activity. Additionally, the phenotypes of CSC-promoting CAF subpopulations may vary among different cancer types and cancer niches. As shown in Table 2, the CSC-promoting abilities of CAFs in different cancers are related to their specific membrane markers, signaling activation and secretomes. These molecular characteristics of CAFs facilitate the phenotype identification of CAF subpopulations with CSC-supporting activity in a specific cancer type; however, further studies are required to improve these identifications. Additionally, specific membrane markers, such as CD10, GPR77 and CD44, could be used for the in vivo depletion of CSC-supporting CAFs. Since CAFs also contain antitumorigenic subpopulations, strategies targeting CSC-supporting CAFs may be more appropriate for cancer intervention than depleting all CAF populations [1,4]. However, more studies are required to identify specific membrane markers for differentiating CSC-supporting CAF subpopulations in human cancers.
CSCs can transform between states in response to signals from CAFs, exhibiting distinct features and abilities to disseminate and give rise to metastatic lesions, which further influence cancer progression, cancer relapse and treatment response [10,11]. Thus, beneficial therapies could be derived from blocking the CAF-CSC crosstalk in human cancers (Figure 1). However, it seems that most of the drug candidates targeting a single pathway in CAF-CSC crosstalk fail to significantly improve the antitumor efficacy in cancer patients [59,62,63,66-69]. Only a few candidates, such as the PI3K inhibitor, have shown gratifying antitumor effects when used singly in broad clinical studies [73-76,87,95] (Table 5). One possible explanation for the single drug treatment failure is that the CAF subsets with CSC-supporting activities in one cancer can sustain the CSC phenotype of cancer cells through different pathways. Thus, combination therapy targeting multiple pathways of CAF-CSC crosstalk is required to enhance the antitumor activities. Moreover, identification of reliable biomarkers that are predictive of treatment response would also be indispensable for the clinical application of CAF-CSC crosstalk blockade. The failure in some clinical trials of CAF-CSC targeting strategies was due to the lack of reliable biomarkers for predicting treatment response [59,62,63,66-69]. As CSCs have the ability to drive cancer chemoresistance or radioresistance [1,4], proper strategies blocking CAF-CSC crosstalk could also be designed to enhance the sensitivity to these therapies, eventually leading to the suppression of cancer progression and relapse. Moreover, some signal transductions involved in the CAF-CSC crosstalk can regulate the suppression of antitumor immunity. For example, TGF-β/SDF-1 signaling [97], STAT3/CCL2 signaling [98], and WNT/β-catenin signaling [99] have been reported to regulate both CAF-CSC crosstalk and antitumor immunity. Thus, strategies blocking these pathways could be used to enhance the antitumor efficacy of immunotherapy, including PD-1/PDL-1 blockade. In addition, CAF-CSC crosstalk-targeted strategies could also enhance the efficacy of antiangiogenesis therapeutics, as it has been reported that cancer intervention with VEGF antibodies can induce CAF-regulated CSCs and reduce their antitumor effect [17]. Targeting CAF-CSC crosstalk is still a crucial challenge in cancer interventions. Further studies on CAF-regulated cancer stemness will open a new window for designing effective strategies to overcome cancer progression and therapeutic resistance.
Acknowledgements
We appreciate Mr. Yangzhi Wang from University of California at Berkeley for proof-reading. The work of the authors has been funded by grants from the National Key R&D Program of China (2017YFA0503900), the National Natural Science Foundation of China (81372583 and 81772957), the Science and Technology Program of Guangdong Province in China (2017B030301016), the RGC Collaborative Research Fund (C7065-18GF) and the Industry and Information Technology Foundation of Shenzhen (20180309100135860).
Disclosure of conflict of interest
None.
References
- 1.Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. 2019;18:99–115. doi: 10.1038/s41573-018-0004-1. [DOI] [PubMed] [Google Scholar]
- 2.Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 4.LeBleu VS, Kalluri R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis Model Mech. 2018;11 doi: 10.1242/dmm.029447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang C, Fu L, Fu J, Hu L, Yang H, Rong TH, Li Y, Liu H, Fu SB, Zeng YX, Guan XY. Fibroblast growth factor receptor 2-positive fibroblasts provide a suitable microenvironment for tumor development and progression in esophageal carcinoma. Clin Cancer Res. 2009;15:4017–4027. doi: 10.1158/1078-0432.CCR-08-2824. [DOI] [PubMed] [Google Scholar]
- 6.Ziani L, Chouaib S, Thiery J. Alteration of the antitumor immune response by cancer-associated fibroblasts. Front Immunol. 2018;9:414. doi: 10.3389/fimmu.2018.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, De Jesus-Acosta A, Sharma P, Heidari P, Mahmood U, Chin L, Moses HL, Weaver VM, Maitra A, Allison JP, LeBleu VS, Kalluri R. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2015;28:831–833. doi: 10.1016/j.ccell.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Najafi M, Farhood B, Mortezaee K. Cancer stem cells (CSCs) in cancer progression and therapy. J Cell Physiol. 2019;234:8381–8395. doi: 10.1002/jcp.27740. [DOI] [PubMed] [Google Scholar]
- 9.Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124–1134. doi: 10.1038/nm.4409. [DOI] [PubMed] [Google Scholar]
- 10.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 11.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 12.Huang M, Li Y, Zhang H, Nan F. Breast cancer stromal fibroblasts promote the generation of CD44+CD24- cells through SDF-1/CXCR4 interaction. J Exp Clin Cancer Res. 2010;29:80. doi: 10.1186/1756-9966-29-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, Luu T, Li AX, Wu X, Ye W, Chen S, Zhou W, Yu Y, Wang YZ, Ren X, Li H, Scherle P, Kuroki Y, Wang SE. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012;72:2768–2779. doi: 10.1158/0008-5472.CAN-11-3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su S, Chen J, Yao H, Liu J, Yu S, Lao L, Wang M, Luo M, Xing Y, Chen F, Huang D, Zhao J, Yang L, Liao D, Su F, Li M, Liu Q, Song E. CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. 2018;172:841–856. e816. doi: 10.1016/j.cell.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 15.Liao CP, Chen LY, Luethy A, Kim Y, Kani K, MacLeod AR, Gross ME. Androgen receptor in cancer-associated fibroblasts influences stemness in cancer cells. Endocr Relat Cancer. 2017;24:157–170. doi: 10.1530/ERC-16-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giannoni E, Bianchini F, Calorini L, Chiarugi P. Cancer associated fibroblasts exploit reactive oxygen species through a proinflammatory signature leading to epithelial mesenchymal transition and stemness. Antioxid Redox Signal. 2011;14:2361–2371. doi: 10.1089/ars.2010.3727. [DOI] [PubMed] [Google Scholar]
- 17.Kinugasa Y, Matsui T, Takakura N. CD44 expressed on cancer-associated fibroblasts is a functional molecule supporting the stemness and drug resistance of malignant cancer cells in the tumor microenvironment. Stem Cells. 2014;32:145–156. doi: 10.1002/stem.1556. [DOI] [PubMed] [Google Scholar]
- 18.Ren J, Ding L, Zhang D, Shi G, Xu Q, Shen S, Wang Y, Wang T, Hou Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics. 2018;8:3932–3948. doi: 10.7150/thno.25541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasegawa T, Yashiro M, Nishii T, Matsuoka J, Fuyuhiro Y, Morisaki T, Fukuoka T, Shimizu K, Shimizu T, Miwa A, Hirakawa K. Cancer-associated fibroblasts might sustain the stemness of scirrhous gastric cancer cells via transforming growth factor-beta signaling. Int J Cancer. 2014;134:1785–1795. doi: 10.1002/ijc.28520. [DOI] [PubMed] [Google Scholar]
- 20.Lau EY, Lo J, Cheng BY, Ma MK, Lee JM, Ng JK, Chai S, Lin CH, Tsang SY, Ma S, Ng IO, Lee TK. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-Met/FRA1/HEY1 signaling. Cell Rep. 2016;15:1175–1189. doi: 10.1016/j.celrep.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 21.Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M, Apuzzo T, Sperduti I, Volpe S, Cocorullo G, Gulotta G, Dieli F, De Maria R, Stassi G. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell. 2014;14:342–356. doi: 10.1016/j.stem.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 22.Zhao XL, Lin Y, Jiang J, Tang Z, Yang S, Lu L, Liang Y, Liu X, Tan J, Hu XG, Niu Q, Fu WJ, Yan ZX, Guo DY, Ping YF, Wang JM, Zhang X, Kung HF, Bian XW, Yao XH. High-mobility group box 1 released by autophagic cancer-associated fibroblasts maintains the stemness of luminal breast cancer cells. J Pathol. 2017;243:376–389. doi: 10.1002/path.4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farnie G, Clarke RB. Mammary stem cells and breast cancer--role of Notch signalling. Stem Cell Rev. 2007;3:169–175. doi: 10.1007/s12015-007-0023-5. [DOI] [PubMed] [Google Scholar]
- 24.Trautmann F, Cojoc M, Kurth I, Melin N, Bouchez LC, Dubrovska A, Peitzsch C. CXCR4 as biomarker for radioresistant cancer stem cells. Int J Radiat Biol. 2014;90:687–699. doi: 10.3109/09553002.2014.906766. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Wang R, Xiong S, Wang X, Zhao Z, Bai S, Wang Y, Zhao Y, Cheng B. Cancer-associated fibroblasts promote the stemness of CD24(+) liver cells via paracrine signaling. J Mol Med (Berl) 2019;97:243–255. doi: 10.1007/s00109-018-1731-9. [DOI] [PubMed] [Google Scholar]
- 26.Peng CY, Wang TY, Lee SS, Hsieh PL, Liao YW, Tsai LL, Fang CY, Yu CC, Hsieh CS. Let-7c restores radiosensitivity and chemosensitivity and impairs stemness in oral cancer cells through inhibiting interleukin-8. J Oral Pathol Med. 2018;47:590–597. doi: 10.1111/jop.12711. [DOI] [PubMed] [Google Scholar]
- 27.Jobe NP, Rosel D, Dvorankova B, Kodet O, Lacina L, Mateu R, Smetana K, Brabek J. Simultaneous blocking of IL-6 and IL-8 is sufficient to fully inhibit CAF-induced human melanoma cell invasiveness. Histochem Cell Biol. 2016;146:205–217. doi: 10.1007/s00418-016-1433-8. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Sun W, Shen W, Xia M, Chen C, Xiang D, Ning B, Cui X, Li H, Li X, Ding J, Wang H. Long non-coding RNA DILC regulates liver cancer stem cells via IL-6/STAT3 axis. J Hepatol. 2016;64:1283–1294. doi: 10.1016/j.jhep.2016.01.019. [DOI] [PubMed] [Google Scholar]
- 29.Shintani Y, Fujiwara A, Kimura T, Kawamura T, Funaki S, Minami M, Okumura M. IL-6 secreted from cancer-associated fibroblasts mediates chemoresistance in NSCLC by increasing epithelial-mesenchymal transition signaling. J Thorac Oncol. 2016;11:1482–1492. doi: 10.1016/j.jtho.2016.05.025. [DOI] [PubMed] [Google Scholar]
- 30.Liotti F, Collina F, Pone E, La Sala L, Franco R, Prevete N, Melillo RM. Interleukin-8, but not the related chemokine CXCL1, sustains an autocrine circuit necessary for the properties and functions of thyroid cancer stem cells. Stem Cells. 2017;35:135–146. doi: 10.1002/stem.2492. [DOI] [PubMed] [Google Scholar]
- 31.Jin F, Miao Y, Xu P, Qiu X. IL-8 regulates the stemness properties of cancer stem cells in the small-cell lung cancer cell line H446. Onco Targets Ther. 2018;11:5723–5731. doi: 10.2147/OTT.S161760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tao L, Huang G, Song H, Chen Y, Chen L. Cancer associated fibroblasts: an essential role in the tumor microenvironment. Oncol Lett. 2017;14:2611–2620. doi: 10.3892/ol.2017.6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-beta signalling. Br J Cancer. 2014;110:724–732. doi: 10.1038/bjc.2013.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scherz-Shouval R, Santagata S, Mendillo ML, Sholl LM, Ben-Aharon I, Beck AH, Dias-Santagata D, Koeva M, Stemmer SM, Whitesell L, Lindquist S. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;158:564–578. doi: 10.1016/j.cell.2014.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gibbons AV, Lin JE, Kim GW, Marszalowicz GP, Li P, Stoecker BA, Blomain ES, Rattan S, Snook AE, Schulz S, Waldman SA. Intestinal GUCY2C prevents TGF-beta secretion coordinating desmoplasia and hyperproliferation in colorectal cancer. Cancer Res. 2013;73:6654–6666. doi: 10.1158/0008-5472.CAN-13-0887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, Chiarugi P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010;70:6945–6956. doi: 10.1158/0008-5472.CAN-10-0785. [DOI] [PubMed] [Google Scholar]
- 37.Ding JW, Luo CY, Wang XA, Zhou T, Zheng XX, Zhang ZQ, Yu B, Zhang J, Tong XH. Glycyrrhizin, a high-mobility group box 1 inhibitor, improves lipid metabolism and suppresses vascular inflammation in apolipoprotein E knockout mice. J Vasc Res. 2018;55:365–377. doi: 10.1159/000495310. [DOI] [PubMed] [Google Scholar]
- 38.Yu S, Cao H, Shen B, Feng J. Tumor-derived exosomes in cancer progression and treatment failure. Oncotarget. 2015;6:37151–37168. doi: 10.18632/oncotarget.6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li YY, Tao YW, Gao S, Li P, Zheng JM, Zhang SE, Liang J, Zhang Y. Cancer-associated fibroblasts contribute to oral cancer cells proliferation and metastasis via exosome-mediated paracrine miR-34a-5p. EBioMedicine. 2018;36:209–220. doi: 10.1016/j.ebiom.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu Y, Yan C, Mu L, Huang K, Li X, Tao D, Wu Y, Qin J. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS One. 2015;10:e0125625. doi: 10.1371/journal.pone.0125625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donnarumma E, Fiore D, Nappa M, Roscigno G, Adamo A, Iaboni M, Russo V, Affinito A, Puoti I, Quintavalle C, Rienzo A, Piscuoglio S, Thomas R, Condorelli G. Cancer-associated fibroblasts release exosomal microRNAs that dictate an aggressive phenotype in breast cancer. Oncotarget. 2017;8:19592–19608. doi: 10.18632/oncotarget.14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu YB, Yan C, Mu L, Mi YL, Zhao H, Hu H, Li XL, Tao DD, Wu YQ, Gong JP, Qin JC. Exosomal Wnt-induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance. Oncogene. 2019;38:1951–1965. doi: 10.1038/s41388-018-0557-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seino T, Kawasaki S, Shimokawa M, Tamagawa H, Toshimitsu K, Fujii M, Ohta Y, Matano M, Nanki K, Kawasaki K, Takahashi S, Sugimoto S, Iwasaki E, Takagi J, Itoi T, Kitago M, Kitagawa Y, Kanai T, Sato T. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell. 2018;22:454–467. e456. doi: 10.1016/j.stem.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 44.Fu L, Zhang C, Zhang LY, Dong SS, Lu LH, Chen J, Dai Y, Li Y, Kong KL, Kwong DL, Guan XY. Wnt2 secreted by tumour fibroblasts promotes tumour progression in oesophageal cancer by activation of the Wnt/beta-catenin signalling pathway. Gut. 2011;60:1635–1643. doi: 10.1136/gut.2011.241638. [DOI] [PubMed] [Google Scholar]
- 45.Zhu Y, Zheng JJ, Yang F, Nie QQ, Zhu ZL, Deng H. Expression of CD10 in cancer-associated fibroblasts and its effect on initiation and progression of colorectal carcinoma. Zhonghua Bing Li Xue Za Zhi. 2016;45:859–865. doi: 10.3760/cma.j.issn.0529-5807.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 46.Valenti G, Quinn HM, Heynen G, Lan L, Holland JD, Vogel R, Wulf-Goldenberg A, Birchmeier W. Cancer stem cells regulate cancer-associated fibroblasts via activation of hedgehog signaling in mammary gland tumors. Cancer Res. 2017;77:2134–2147. doi: 10.1158/0008-5472.CAN-15-3490. [DOI] [PubMed] [Google Scholar]
- 47.Santos AM, Jung J, Aziz N, Kissil JL, Pure E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest. 2009;119:3613–3625. doi: 10.1172/JCI38988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA, Fearon DT. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 49.Ostermann E, Garin-Chesa P, Heider KH, Kalat M, Lamche H, Puri C, Kerjaschki D, Rettig WJ, Adolf GR. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin Cancer Res. 2008;14:4584–4592. doi: 10.1158/1078-0432.CCR-07-5211. [DOI] [PubMed] [Google Scholar]
- 50.Fang J, Hu B, Li S, Zhang C, Liu Y, Wang P. A multi-antigen vaccine in combination with an immunotoxin targeting tumor-associated fibroblast for treating murine melanoma. Mol Ther Oncolytics. 2016;3:16007. doi: 10.1038/mto.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li M, Li M, Yin T, Shi H, Wen Y, Zhang B, Chen M, Xu G, Ren K, Wei Y. Targeting of cancer associated fibroblasts enhances the efficacy of cancer chemotherapy by regulating the tumor microenvironment. Mol Med Rep. 2016;13:2476–2484. doi: 10.3892/mmr.2016.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hofheinz RD, al-Batran SE, Hartmann F, Hartung G, Jager D, Renner C, Tanswell P, Kunz U, Amelsberg A, Kuthan H, Stehle G. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie. 2003;26:44–48. doi: 10.1159/000069863. [DOI] [PubMed] [Google Scholar]
- 53.Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W, Divgi CR, Hanson LH, Mitchell P, Gansen DN, Larson SM, Ingle JN, Hoffman EW, Tanswell P, Ritter G, Cohen LS, Bette P, Arvay L, Amelsberg A, Vlock D, Rettig WJ, Old LJ. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. 2003;9:1639–1647. [PubMed] [Google Scholar]
- 54.Lo A, Wang LS, Scholler J, Monslow J, Avery D, Newick K, O’Brien S, Evans RA, Bajor DJ, Clendenin C, Durham AC, Buza EL, Vonderheide RH, June CH, Albelda SM, Pure E. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. 2015;75:2800–2810. doi: 10.1158/0008-5472.CAN-14-3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, Espeli M, Kraman M, McKenna B, Wells RJ, Zhao Q, Caballero OL, Larder R, Coll AP, O’Rahilly S, Brindle KM, Teichmann SA, Tuveson DA, Fearon DT. Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med. 2013;210:1137–1151. doi: 10.1084/jem.20122344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Fernandez-Barrena MG, Fernandez-Zapico ME, Iacobuzio-Donahue C, Olive KP, Stanger BZ. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim WA. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell. 2016;164:770–779. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karakasheva TA, Lin EW, Tang Q, Qiao E, Waldron TJ, Soni M, Klein-Szanto AJ, Sahu V, Basu D, Ohashi S, Baba K, Giaccone ZT, Walker SR, Frank DA, Wileyto EP, Long Q, Dunagin MC, Raj A, Diehl JA, Wong KK, Bass AJ, Rustgi AK. IL-6 mediates cross-talk between tumor cells and activated fibroblasts in the tumor microenvironment. Cancer Res. 2018;78:4957–4970. doi: 10.1158/0008-5472.CAN-17-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15:234–248. doi: 10.1038/nrclinonc.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, Fowler N, Zhou T, Schmidt J, Jo M, Lee SJ, Yamashita M, Hughes SG, Fayad L, Piha-Paul S, Nadella MV, Mohseni M, Lawson D, Reimer C, Blakey DC, Xiao X, Hsu J, Revenko A, Monia BP, MacLeod AR. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med. 2015;7:314ra185. doi: 10.1126/scitranslmed.aac5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ding X, Ding C, Wang F, Deng W, Yu M, Meng Q, Sun P. Effects of NOTCH1 signaling inhibitor gamma-secretase inhibitor II on growth of cancer stem cells. Oncol Lett. 2018;16:6095–6099. doi: 10.3892/ol.2018.9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, Li S, Seetharam S, Puchalski TA, Takimoto C, Elsayed Y, Dawkins F, de Bono JS. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs. 2013;31:760–768. doi: 10.1007/s10637-012-9869-8. [DOI] [PubMed] [Google Scholar]
- 63.Even C, Lassen U, Merchan J, Le Tourneau C, Soria JC, Ferte C, Ricci F, Diener JT, Yuen E, Smith C, Oakley GJ 3rd, Benhadji KA, Massard C. Safety and clinical activity of the Notch inhibitor, crenigacestat (LY3039478), in an open-label phase I trial expansion cohort of advanced or metastatic adenoid cystic carcinoma. Invest New Drugs. 2019 doi: 10.1007/s10637-019-00739-x. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang SS, Han ZP, Jing YY, Tao SF, Li TJ, Wang H, Wang Y, Li R, Yang Y, Zhao X, Xu XD, Yu ED, Rui YC, Liu HJ, Zhang L, Wei LX. CD133(+)CXCR4(+) colon cancer cells exhibit metastatic potential and predict poor prognosis of patients. BMC Med. 2012;10:85. doi: 10.1186/1741-7015-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiao W, Gao Z, Duan Y, Yuan W, Ke Y. Notch signaling plays a crucial role in cancer stem-like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma. J Exp Clin Cancer Res. 2017;36:41. doi: 10.1186/s13046-017-0507-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.den Hollander MW, Bensch F, Glaudemans AW, Oude Munnink TH, Enting RH, den Dunnen WF, Heesters MA, Kruyt FA, Lub-de Hooge MN, Cees de Groot J, Pearlberg J, Gietema JA, de Vries EG, Walenkamp AM. TGF-beta antibody uptake in recurrent high-grade glioma imaged with 89Zr-fresolimumab PET. J Nucl Med. 2015;56:1310–1314. doi: 10.2967/jnumed.115.154401. [DOI] [PubMed] [Google Scholar]
- 67.Lacouture ME, Morris JC, Lawrence DP, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Berzofsky JA, Hsu FJ, Guitart J. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor beta by the monoclonal antibody fresolimumab (GC1008) Cancer Immunol Immunother. 2015;64:437–446. doi: 10.1007/s00262-015-1653-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cohn A, Lahn MM, Williams KE, Cleverly AL, Pitou C, Kadam SK, Farmen MW, Desaiah D, Raju R, Conkling P, Richards D. A phase I dose-escalation study to a predefined dose of a transforming growth factor-beta1 monoclonal antibody (TbetaM1) in patients with metastatic cancer. Int J Oncol. 2014;45:2221–2231. doi: 10.3892/ijo.2014.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Necchi A, Giannatempo P, Mariani L, Fare E, Raggi D, Pennati M, Zaffaroni N, Crippa F, Marchiano A, Nicolai N, Maffezzini M, Togliardi E, Daidone MG, Gianni AM, Salvioni R, De Braud F. PF-03446962, a fully-human monoclonal antibody against transforming growth-factor beta (TGFbeta) receptor ALK1, in pre-treated patients with urothelial cancer: an open label, single-group, phase 2 trial. Invest New Drugs. 2014;32:555–560. doi: 10.1007/s10637-014-0074-9. [DOI] [PubMed] [Google Scholar]
- 70.Cheng Y, Wen G, Sun Y, Shen Y, Zeng Y, Du M, Zhu G, Wang G, Meng X. Osteopontin promotes colorectal cancer cell invasion and the stem cell-like properties through the PI3K-AKT-GSK/3beta-beta/catenin pathway. Med Sci Monit. 2019;25:3014–3025. doi: 10.12659/MSM.913185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang L, Ning J, Wakimoto H, Wu S, Wu CL, Humphrey MR, Rabkin SD, Martuza RL. Oncolytic herpes simplex virus and PI3K inhibitor BKM120 synergize to promote killing of prostate cancer stem-like cells. Mol Ther Oncolytics. 2019;13:58–66. doi: 10.1016/j.omto.2019.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu Y, Zhang XB, Liu JJ, Zhang S, Zhang J. NVP-BKM120 in combination with letrozole inhibit human breast cancer stem cells via PI3K/mTOR pathway. Zhonghua Yi Xue Za Zhi. 2019;99:1075–1080. doi: 10.3760/cma.j.issn.0376-2491.2019.14.008. [DOI] [PubMed] [Google Scholar]
- 73.Hotte SJ, Chi KN, Joshua AM, Tu D, Macfarlane RJ, Gregg RW, Ruether JD, Basappa NS, Finch D, Salim M, Winquist EW, Torri V, North S, Kollmannsberger C, Ellard SL, Eigl BJ, Tinker A, Allan AL, Beja K, Annala M, Powers J, Wyatt AW, Seymour L Canadian Cancer Trials Group (formerly NCIC Clinical Trials Group) A phase II study of PX-866 in patients with recurrent or metastatic castration-resistant prostate cancer: canadian cancer trials group study IND205. Clin Genitourin Cancer. 2019;17:201–208. e1. doi: 10.1016/j.clgc.2019.03.005. [DOI] [PubMed] [Google Scholar]
- 74.Ando Y, Iwasa S, Takahashi S, Saka H, Kakizume T, Natsume K, Suenaga N, Quadt C, Yamada Y. Phase I study of alpelisib (BYL719), an alpha-specific PI3K inhibitor, in Japanese patients with advanced solid tumors. Cancer Sci. 2019;110:1021–1031. doi: 10.1111/cas.13923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wicki A, Brown N, Xyrafas A, Bize V, Hawle H, Berardi S, Cmiljanovic N, Cmiljanovic V, Stumm M, Dimitrijevic S, Herrmann R, Pretre V, Ritschard R, Tzankov A, Hess V, Childs A, Hierro C, Rodon J, Hess D, Joerger M, von Moos R, Sessa C, Kristeleit R. First-in human, phase 1, dose-escalation pharmacokinetic and pharmacodynamic study of the oral dual PI3K and mTORC1/2 inhibitor PQR309 in patients with advanced solid tumors (SAKK 67/13) Eur J Cancer. 2018;96:6–16. doi: 10.1016/j.ejca.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 76.Schoffski P, Cresta S, Mayer IA, Wildiers H, Damian S, Gendreau S, Rooney I, Morrissey KM, Spoerke JM, Ng VW, Singel SM, Winer E. A phase Ib study of pictilisib (GDC-0941) in combination with paclitaxel, with and without bevacizumab or trastuzumab, and with letrozole in advanced breast cancer. Breast Cancer Res. 2018;20:109. doi: 10.1186/s13058-018-1015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gaule P, Mukherjee N, Corkery B, Eustace AJ, Gately K, Roche S, O’Connor R, O’Byrne KJ, Walsh N, Duffy MJ, Crown J, O’Donovan N. Dasatinib treatment increases sensitivity to c-met inhibition in triple-negative breast cancer cells. Cancers (Basel) 2019;11 doi: 10.3390/cancers11040548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen X, Guan Z, Lu J, Wang H, Zuo Z, Ye F, Huang J, Teng L. Synergistic antitumor effects of cMet inhibitor in combination with anti-VEGF in colorectal cancer patient-derived xenograft models. J Cancer. 2018;9:1207–1217. doi: 10.7150/jca.20964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cheng H, Chua V, Liao C, Purwin TJ, Terai M, Kageyama K, Davies MA, Sato T, Aplin AE. Co-targeting HGF/cMET signaling with MEK inhibitors in metastatic uveal melanoma. Mol Cancer Ther. 2017;16:516–528. doi: 10.1158/1535-7163.MCT-16-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cozzo AJ, Sundaram S, Zattra O, Qin Y, Freemerman AJ, Essaid L, Darr DB, Montgomery SA, McNaughton KK, Ezzell JA, Galanko JA, Troester MA, Makowski L. cMET inhibitor crizotinib impairs angiogenesis and reduces tumor burden in the C3(1)-Tag model of basal-like breast cancer. Springerplus. 2016;5:348. doi: 10.1186/s40064-016-1920-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gavine PR, Ren Y, Han L, Lv J, Fan S, Zhang W, Xu W, Liu YJ, Zhang T, Fu H, Yu Y, Wang H, Xu S, Zhou F, Su X, Yin X, Xie L, Wang L, Qing W, Jiao L, Su W, Wang QM. Volitinib, a potent and highly selective c-Met inhibitor, effectively blocks c-Met signaling and growth in c-MET amplified gastric cancer patient-derived tumor xenograft models. Mol Oncol. 2015;9:323–333. doi: 10.1016/j.molonc.2014.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Moschetta M, Basile A, Ferrucci A, Frassanito MA, Rao L, Ria R, Solimando AG, Giuliani N, Boccarelli A, Fumarola F, Coluccia M, Rossini B, Ruggieri S, Nico B, Maiorano E, Ribatti D, Roccaro AM, Vacca A. Novel targeting of phospho-cMET overcomes drug resistance and induces antitumor activity in multiple myeloma. Clin Cancer Res. 2013;19:4371–4382. doi: 10.1158/1078-0432.CCR-13-0039. [DOI] [PubMed] [Google Scholar]
- 83.Saigusa S, Toiyama Y, Tanaka K, Yokoe T, Fujikawa H, Matsushita K, Okugawa Y, Inoue Y, Uchida K, Mohri Y, Kusunoki M. Inhibition of HGF/cMET expression prevents distant recurrence of rectal cancer after preoperative chemoradiotherapy. Int J Oncol. 2012;40:583–591. doi: 10.3892/ijo.2011.1200. [DOI] [PubMed] [Google Scholar]
- 84.Crosswell HE, Dasgupta A, Alvarado CS, Watt T, Christensen JG, De P, Durden DL, Findley HW. PHA665752, a small-molecule inhibitor of c-Met, inhibits hepatocyte growth factor-stimulated migration and proliferation of c-Met-positive neuroblastoma cells. BMC Cancer. 2009;9:411. doi: 10.1186/1471-2407-9-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu DW, Chen TC, Huang HS, Lee H. TC-N19, a novel dual inhibitor of EGFR and cMET, efficiently overcomes EGFR-TKI resistance in non-small-cell lung cancer cells. Cell Death Dis. 2016;7:e2290. doi: 10.1038/cddis.2016.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moores SL, Chiu ML, Bushey BS, Chevalier K, Luistro L, Dorn K, Brezski RJ, Haytko P, Kelly T, Wu SJ, Martin PL, Neijssen J, Parren PW, Schuurman J, Attar RM, Laquerre S, Lorenzi MV, Anderson GM. A novel bispecific antibody targeting EGFR and cMet is effective against EGFR inhibitor-resistant lung tumors. Cancer Res. 2016;76:3942–3953. doi: 10.1158/0008-5472.CAN-15-2833. [DOI] [PubMed] [Google Scholar]
- 87.Esaki T, Hirai F, Makiyama A, Seto T, Bando H, Naito Y, Yoh K, Ishihara K, Kakizume T, Natsume K, Myers A, Doi T. Phase I dose-escalation study of capmatinib (INC280) in Japanese patients with advanced solid tumors. Cancer Sci. 2019;110:1340–1351. doi: 10.1111/cas.13956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kahlert UD, Suwala AK, Koch K, Natsumeda M, Orr BA, Hayashi M, Maciaczyk J, Eberhart CG. Pharmacologic wnt inhibition reduces proliferation, survival, and clonogenicity of glioblastoma cells. J Neuropathol Exp Neurol. 2015;74:889–900. doi: 10.1097/NEN.0000000000000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cheng Y, Phoon YP, Jin X, Chong SY, Ip JC, Wong BW, Lung ML. Wnt-C59 arrests stemness and suppresses growth of nasopharyngeal carcinoma in mice by inhibiting the Wnt pathway in the tumor microenvironment. Oncotarget. 2015;6:14428–14439. doi: 10.18632/oncotarget.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang G, Shen J, Sun J, Jiang Z, Fan J, Wang H, Yu S, Long Y, Liu Y, Bao H, Zhang KX, Han K, Zhu M, Zheng Y, Lin Z, Jiang C, Guo M. Cyclophilin a maintains glioma-initiating cell stemness by regulating Wnt/beta-catenin signaling. Clin Cancer Res. 2017;23:6640–6649. doi: 10.1158/1078-0432.CCR-17-0774. [DOI] [PubMed] [Google Scholar]
- 91.Krishnamurthy A, Dasari A, Noonan AM, Mehnert JM, Lockhart AC, Leong S, Capasso A, Stein MN, Sanoff HK, Lee JJ, Hansen A, Malhotra U, Rippke S, Gustafson DL, Pitts TM, Ellison K, Davis SL, Messersmith WA, Eckhardt SG, Lieu CH. Phase Ib results of the rational combination of selumetinib and cyclosporin a in advanced solid tumors with an expansion cohort in metastatic colorectal cancer. Cancer Res. 2018;78:5398–5407. doi: 10.1158/0008-5472.CAN-18-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ahmed K, Shaw HV, Koval A, Katanaev VL. A second WNT for old drugs: drug repositioning against WNT-dependent cancers. Cancers (Basel) 2016;8 doi: 10.3390/cancers8070066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu C, Hu S, Cheng J, Wang G, Tao K. Smoothened antagonist GDC-0449 (Vismodegib) inhibits proliferation and triggers apoptosis in colon cancer cell lines. Exp Ther Med. 2017;13:2529–2536. doi: 10.3892/etm.2017.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tong W, Qiu L, Qi M, Liu J, Hu K, Lin W, Huang Y, Fu J. GANT-61 and GDC-0449 induce apoptosis of prostate cancer stem cells through a GLI-dependent mechanism. J Cell Biochem. 2018;119:3641–3652. doi: 10.1002/jcb.26572. [DOI] [PubMed] [Google Scholar]
- 95.Infante P, Alfonsi R, Ingallina C, Quaglio D, Ghirga F, D’Acquarica I, Bernardi F, Di Magno L, Canettieri G, Screpanti I, Gulino A, Botta B, Mori M, Di Marcotullio L. Inhibition of Hedgehog-dependent tumors and cancer stem cells by a newly identified naturally occurring chemotype. Cell Death Dis. 2016;7:e2376. doi: 10.1038/cddis.2016.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, Skhinas JN, Collot R, Yang J, Harvey K, Johan MZ, Cooper C, Nair R, Herrmann D, McFarland A, Deng N, Ruiz-Borrego M, Rojo F, Trigo JM, Bezares S, Caballero R, Lim E, Timpson P, O’Toole S, Watkins DN, Cox TR, Samuel MS, Martin M, Swarbrick A. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9:2897. doi: 10.1038/s41467-018-05220-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pu N, Zhao G, Gao S, Cui Y, Xu Y, Lv Y, Nuerxiati A, Wu W. Neutralizing TGF-beta promotes anti-tumor immunity of dendritic cells against pancreatic cancer by regulating T lymphocytes. Cent Eur J Immunol. 2018;43:123–131. doi: 10.5114/ceji.2018.77381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, Dang Y, Chu Y, Fan J, He R. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. 2016;76:4124–4135. doi: 10.1158/0008-5472.CAN-15-2973. [DOI] [PubMed] [Google Scholar]
- 99.Holtzhausen A, Zhao F, Evans KS, Tsutsui M, Orabona C, Tyler DS, Hanks BA. Melanoma-derived Wnt5a promotes local dendritic-cell expression of IDO and immunotolerance: opportunities for pharmacologic enhancement of immunotherapy. Cancer Immunol Res. 2015;3:1082–1095. doi: 10.1158/2326-6066.CIR-14-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]