

Abstract
Background and Purpose
Extracellular vesicles (EVs) are constitutively shed from cells and released by various stimuli. Their protein and RNA cargo are modified by the stimulus, and in disease conditions can carry pathological cargo involved in disease progression. Neutral sphingomyelinase 2 (nSMase2) is a major regulator in at least one of several independent routes of EV biogenesis, and its inhibition is a promising new therapeutic approach for neurological disorders. Unfortunately, known inhibitors exhibit μM potency, poor physicochemical properties, and/or limited brain penetration. Here, we sought to identify a drug‐like inhibitor of nSMase2.
Experimental Approach
We conducted a human nSMase2 high throughput screen (>365,000 compounds). Selected hits were optimized focusing on potency, selectivity, metabolic stability, pharmacokinetics, and ability to inhibit EV release in vitro and in vivo.
Key Results
We identified phenyl(R)‐(1‐(3‐(3,4‐dimethoxyphenyl)‐2,6‐dimethylimidazo[1,2‐b]pyridazin‐8‐yl)pyrrolidin‐3‐yl)‐carbamate (PDDC), a potent (pIC50 = 6.57) and selective non‐competitive inhibitor of nSMase2. PDDC was metabolically stable, with excellent oral bioavailability (%F = 88) and brain penetration (AUCbrain/AUCplasma = 0.60). PDDC dose‐dependently (pEC50 = 5.5) inhibited release of astrocyte‐derived extracellular vesicles (ADEV). In an in vivo inflammatory brain injury model, PDDC robustly inhibited ADEV release and the associated peripheral immunological response. A closely related inactive PDDC analogue was ineffective.
Conclusion and Implications
PDDC is a structurally novel, potent, orally available, and brain penetrant inhibitor of nSMase2. PDDC inhibited release of ADEVs in tissue culture and in vivo. PDDC is actively being tested in animal models of neurological disease and, along with closely related analogues, is being considered for clinical translation.
Abbreviations
- ADEV
astrocyte‐derived extracellular vesicles
- EVs
extracellular vesicles
- GFAP
glial fibrillary acidic protein
- nSMase2
neutral sphingomyelinase 2
- PDDC
phenyl(R)‐(1‐(3‐(3,4‐dimethoxyphenyl)‐2,6‐dimethylimidazo[1,2‐b]pyridazin‐8‐yl)pyrrolidin‐3‐yl)carbamate
- SM
sphingomyelin
What is already known
Inhibition of extracellular vesicle (EV) release is a promising therapeutic approach to treat neurodegenerative diseases.
Currently available neutral sphingomyelinase 2 (nSMase2) inhibitors have not been suitable for clinical development.
What this study adds
We identified PDDC, a potent, selective, orally bioavailable, and brain penetrant nSMase2 inhibitor.
PDDC dose‐dependently inhibited astrocyte‐derived extracellular vesicles both in vitro and in vivo.
What is the clinical significance
PDDC can be utilized to probe the role of nSMase2 in models of neurological disease.
PDDC and closely related analogues are being considered for clinical translation.
1. INTRODUCTION
Extracellular vesicles (EVs) are small vesicular carriers (~30–1,000 nm in diameter) composed of select lipids, proteins, RNA, and possibly DNA (De Toro, Herschlik, Waldner, & Mongini, 2015; Kourembanas, 2015; Thery, 2011; Thery, Zitvogel, & Amigorena, 2002). In addition to removing unwanted materials from cells, EVs regulate a large variety of intercellular communication underlying physiological and pathological processes (Ibrahim & Marban, 2016; Raposo & Stoorvogel, 2013; Weidle, Birzele, Kollmorgen, & Ruger, 2017). The hydrolysis of sphingomyelin (SM) to ceramide by neutral sphingomyelinase 2 (nSMase2) is a major regulator in at least one of several independent routes of biogenesis and release of EVs (Trajkovic et al., 2008). While transient increases in nSMase2 activity are part of normal physiological function, chronically activated nSMase2 and subsequent EV secretion have been implicated in modulating the immune response to brain inflammation (Dickens et al., 2017), cancer metastasis (Kosaka et al., 2013), amyloid deposition (Dinkins et al., 2016; Dinkins, Dasgupta, Wang, Zhu, & Bieberich, 2014; Sardar Sinha et al., 2018), Tau protein propagation (Asai et al., 2015), and HIV infection (Barclay et al., 2017; Dalvi, Sun, Tang, & Pulliam, 2017; Hu et al., 2012; Sun, Dalvi, Abadjian, Tang, & Pulliam, 2017). Moreover, genetic and pharmacological inhibition of nSMase2 has been demonstrated to reduce EV secretion (Asai et al., 2015; Dickens et al., 2017; Dinkins et al., 2014; Dinkins et al., 2016). These data suggest that reducing EV secretion through inhibition of nSMase2 may be a useful avenue for the treatment of diseases involving intercellular communication mediated by EVs. Unfortunately, currently available nSMase2 inhibitors exhibit low potency (pIC50 6 or lower), poor aqueous solubility, and limited brain penetration, preventing the progress of nSMase2 inhibitors to the clinic. GW4869 (Luberto et al., 2002), the most widely used inhibitor, has low inhibitory potency (pIC50 6), it is practically insoluble in water and has very poor solubility in organic solvents (e.g., solubility in DMSO = 0.2 mg·ml−1) thus limiting its clinical potential. Cambinol, an nSMase2 inhibitor we identified from a screen of commercially available small chemical libraries (Figuera‐Losada et al., 2015), exhibited better solubility but slightly lower potency (pIC50 5.3) than GW4869. However, cambinol was metabolically unstable and optimization efforts to improve its stability were unsuccessful. Here, we report the findings from a high throughput screening (HTS) campaign of >365,000 compounds and chemistry optimization efforts that identified phenyl(R)‐(1‐(3‐(3,4‐dimethoxyphenyl)‐2,6‐dimethylimidazo[1,2‐b]pyridazin‐8‐yl)pyrrolidin‐3‐yl)carbamate (PDDC) as a potent, selective inhibitor of nSMase2 with an excellent oral bioavailability and brain penetration. In proof of concept studies, we show that PDDC inhibits the release of astrocyte‐derived extracellular vesicles (ADEV) from primary cultures of astrocytes and, when administered systemically, blocks the release of ADEVs in response to an inflammatory brain lesion.
2. METHODS
2.1. Animal studies
All animal care and experimental procedures complied with the National Institutes of Health guidelines on animal care and were approved by the Johns Hopkins University Institutional Animal Care and Use Committee. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010) and with the recommendations made by the British Journal of Pharmacology. Five mice (including the glial fibrillary acidic protein [GFAP]–GFP mice) or three rats per cage were housed in a temperature and humidity‐controlled room under a 12‐hr light cycle with food and water available ad libitum and were allowed to acclimate to the colony room for at least 7 days after arrival, before any experimentation.
2.2. Screening assay
Preparation of human recombinant nSMase2 and the fluorescence‐based assay to monitor its activity in the presence or absence of potential inhibitors has been described recently (Figuera‐Losada et al., 2015). Briefly, lysates of HEK293 cells that have been transfected with full human length nSMase2 were used to catalyse the hydrolysis of SM to ceramide and phosphorylcholine. Phosphorylcholine undergoes dephosphorylation in a reaction catalysed by alkaline phosphatase (AP; 4 U·ml−1) to produce choline which in turn is oxidized by choline oxidase (0.1 U·ml−1) to betaine and hydrogen peroxide. Hydrogen peroxide is made to react with Amplex Red (50 μM) in the presence of HRP (1 U·ml−1) to generate the fluorescent molecule resorufin. Generation of fluorescence was monitored by measuring relative florescence units with excitation at 530 nm and emission at 590 nm. The intensity of fluorescence is directly proportional to the extent of SM hydrolysis. Substrate stock solution was prepared in 2% Triton X‐100 and sonicated for 1 min. Reactions were carried out for 1 hr at 37°C in 100‐mM Tris–HCl pH 7.4, 10‐mM MgCl2, and 0.2% Triton X‐100. The assay showed high reliability (Z′ = 0.8–0.9). A counter screen was concomitantly carried out to identify false positives resulting from inhibition of the coupling enzymes. For the counter screen, the AP, choline oxidase, and HRP reactions were carried out in the absence of human nSMase2 and initiated by the addition of phosphorylcholine (2 μM), the AP substrate. Compounds that showed inhibition of the coupling enzymes were considered false positives and were not used further. HTS of >365,000 compounds was performed in 1,536‐well format (0.1‐μg protein per ml human nSMase2 cell lysate 20‐μM SM in a total volume of 4 μl and 2‐hr incubation at 37°C) using a customized screening robot (Kalypsys) and a Viewflux fluorimeter. Confirmation of hits and evaluation of analogues during hit optimization was carried out in 384‐well format (total volume 50 μl) using a Molecular Devices fluorescence spectrophotometer.
2.3. pIC50 determinations and mode of inhibition studies
pIC50 determinations were made from eight‐point dose–response curves (n = 2) using the same Amplex Red fluorescence‐based assay. Data analysis and non‐linear least squares curve fitting were carried out with GraphPad Prism 7 (RRID:SCR_002798). Confidence intervals of pIC50 values were computed using the method that produces asymmetrical intervals (Prism 7). Mode of inhibition was determined using the fluorescence‐based assay detailed above. Lysate of cells expressing recombinant nSMase2 (1.9‐μg protein/50 μl) was incubated with different SM concentrations in the presence of different concentrations of PDDC for 3 hr. Rate of change of fluorescence in the presence of increasing concentrations of PDDC was plotted against sphingomyelin concentration, using GraphPad Prism, and V max and K M were obtained by a least squares fit to the Michaelis–Menten equation using non‐linear regression.
2.4. Selectivity measurements
The first two selectivity measurements were against the related enzymes AP (a phosphomonoesterase) and acid sphingomyelinase (a PDE). Evaluation for inhibition of AP is the same as the counter screen as this is one of the coupling enzymes in the screening assay. A negative counter screen is an indication that the compound is not an AP inhibitor. Acid sphingomyelinase activity was measured using a commercially available kit (http://www.echelon‐inc.com/index.php?module=Products&func=detail&id=598) that is already routinely used by us and has been described by Li et al. (2010). The assay uses a substrate specific for acid sphingomyelinase that when hydrolyzed makes a fluorescent product (ex/em 360/460 nm) that is proportional to acid sphingomyelinase activity. PDDC was also evaluated in Eurofins SafetyScreen44, a panel of 44 selected targets recommended by major pharmaceutical companies to identify undesirable off‐target activities (Bowes et al., 2012).
2.5. Metabolic stability
Phase I metabolic stability assays were conducted in mouse (Xenotech LLC, USA) or human (Corning Gentest, USA) liver microsomes, as described previously with minor modifications (Rais et al., 2016). Briefly, for Phase I stability, the reaction was carried out using potassium phosphate buffer (100 mM, pH 7.4), in the presence of an NADPH regenerating system (compound final concentration was 1 μM; 0.2 mg·ml−1 microsomes). Compound disappearance was monitored over time using an LC and tandem MS method. Chromatographic analysis was performed using an Accela ultra high‐performance system consisting of an analytical pump and an autosampler coupled with a TSQ Vantage mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA). Separation of analyte was achieved at ambient temperature using Agilent Eclipse Plus column (100 × 2.1 mm i.d.) packed with a 1.8‐μm C18 stationary phase. The mobile phase consisted of 0.1% formic acid in acetonitrile and 0.1% formic acid in water with gradient elution. The [M + H]+ ion transitions for PDDC were m/z 488.050 → 394.249, 378.18 and for losartan (IS) were m/z 423.130 → 207.090, 180.071.
2.6. In vivo pharmacokinetics
Male CD1 mice (25–30 g) were obtained from Harlan and maintained on a 12‐hr light–dark cycle with ad libitum access to food and water. A total of 63 mice were used in the pharmacokinetic experiments. PDDC was dosed at 10 mg·kg−1 i.v., p.o., or i.p. at a dosing volume of 10 ml·kg−1. PDDC was administered using a formulation consisting of 5% DMSO/5% Tween‐80/90% saline (v/v/v) to obtain a solubility of 1 mg·mL−1 and dosed at a volume of 10 ml·kg−1. Blood and brain tissue were collected at 0.183, 0.25, 0.5, 1, 3, 6, and 8 hr after dosing (n = 3 per time point). Mice were killed by pentobarbital injection at 0.17, 0.33, 0.5, 1, 3, 6, and 8 hr after drug administration. Blood was collected via cardiac puncture and placed into iced EDTA‐coated BD microtainers; plasma was harvested from blood by centrifugation at 3,000×g for 15 min and stored at −80°C. Brain tissues were harvested following blood collection and immediately snap frozen in liquid nitrogen and stored at −80°C until LC–MS analysis. Calibration standards were prepared using naïve mouse plasma or brain with additions of PDDC. PDDC standards and samples were extracted from plasma and brain by a one‐step protein precipitation using acetonitrile (100% v/v) containing internal standard (losartan 500 nM). The samples were vortexed (30 sec) and centrifuged at 10,000 x g (10 min at 4 °C). An aliquot of the supernatant (50 μl) was diluted with water (50 μl) and transferred to a 250‐μl polypropylene vial sealed with a Teflon cap and analysed via LC/MS/MS as described above. Plasma concentrations (pmol·ml−1) as well as tissue concentrations (pmol·g−1) were determined and plots of mean plasma concentration versus time were constructed for PK analysis.
Non‐compartmental analysis modules in Phoenix WinNonlin version 7.0 (Certara USA, Inc., Princeton, NJ) were used to assess clearance (Cl), volume of distribution (Vd), and exposures measured by AUC0–t. The maximum plasma concentration (Cmax) and the time to Cmax (Tmax) were determined directly from the individual plasma concentration‐time profiles. The absolute bioavailability (F) of the oral doses was calculated by using the following equation: F = (AUC0–t PO/AUCI0–t IV) × (DoseIV/DosePO).
2.7. PDDC brain protein binding
Brain protein binding for PDDC was determined using the rapid equilibrium dialysis method (8K MWCO; Thermo Scientific, USA) as previously described (Waters, Jones, Williams, & Sohal, 2008). Briefly, naïve mouse brain tissue was homogenized in PBS, pH 7.4 to obtain a homogenate 33.33% w/v. PDDC was added to the brain homogenate, to a final concentration of 10 μM. The homogenate (300 μl) was subjected to equilibrium dialysis against 550 μl of PBS, pH 7.4 at 37°C with gentle shaking at 450 r.p.m. After equilibration (5 hr), aliquots of dialysed brain homogenate (50 μl) and buffer (50 μl) were transferred to tubes and matrices were matched. Samples were extracted by single‐step protein precipitation with 300‐μl acetonitrile containing 0.5‐μM losartan as internal standard, vortex mixed for 30 s, and centrifuged at 10,000×g for 10 min at 4°C. The supernatants were analysed by LC–MS/MS. Peak area ratios were calculated for each side by taking ratio of area of analyte to internal standard. % Protein bound was calculated using the formula: [100 − (Peak area ratio in buffer/Peak area ratio in brain homogenate ×) 100]. Free concentrations were obtained by multiplying the free fraction by total concentrations at Cmax and Cmin in the PK profile.
2.8. Isolation of rat primary astrocytes
Rat primary astrocyte cell cultures were established and maintained as previously described (Chaudhuri et al., 2018). Briefly, Sprague–Dawley rats (RRID:RGD_70508; Charles River, Wilmington, MA) were killed by decapitation at postnatal Day 1. Cerebral cortices were mechanically dissociated in HBSS and plated on poly‐d‐lysine (1 mg·ml−1, Sigma) coated T175 culture flasks (Corning) containing DMEM/F‐12 media (Gibco BRL) supplemented with 10% FBS (Gibco BRL), D‐glucose (final concentration 25 mM; Sigma), and 1% antibiotic/antimitotic solution (104 unit of penicillin G per ml, 10‐mg streptomycin per ml, and 25‐μg amphotericin B per ml; Sigma). After 7 days, culture flasks were secured to a rotary shaker (Thermo Scientific) and rotated at 200 r.p.m. in a tissue culture incubator (Thermo Scientific) for 18 hr at 37°C in a 5% CO2 environment. Less adherent cells were removed from the media and fresh media added to the cells. Cells were split when close to confluent and at Passage 2, cultures were 98% astrocytes with Type I morphology. Astrocytes were used for experiments between Passages 3 and 10.
2.9. Measurement of EV secretion in vitro
Inhibition of EV release from primary astrocytes by test compounds was carried out as previously described (Dickens et al., 2017). Briefly, rat primary astrocytes were seeded onto 6‐well plates at a density of 20,000 cells per well. Astrocytes were washed with PBS and media without FBS 24 hr after seeding. Absence of FBS mimics a trophic factor withdrawal stimulus causing EVs to be released from astrocytes via an nSMase2‐dependent pathway. Astrocytes were then treated with test compounds at different concentrations: 0.03, 0.1, 0.3, 1, 3, and 10 μM. A stock solution of the test compound (including PDDC) was made in DMSO (30 mM) and diluted in culture medium to arrive to the final concentrations in 0.02% DMSO. DMSO (0.02%) was used as control. Two hours after treatment, media was collected and centrifuged at 2,700×g for 15 min at 4°C. The supernatant was collected and the number of EVs quantified using ZetaView Nanoparticle Tracker (Particle Metrix GmBH, Meerbusch, Germany) and the corresponding ZetaVeiw software (8.03.04.01). Nanosphere size standard 100 nm (Thermo Scientific) was used to calibrate the instrument prior to sample readings. Instrument pre‐acquisition parameters were set to 23°C, a sensitivity of 65, a frame rate of 30 frames per second, a shutter speed of 100, and laser pulse duration equal to that of shutter duration. Post‐acquisition parameters were set to a minimum brightness of 25, a maximum size of 200 pixels, and a minimum size of 10 pixels. For each sample, 1 ml of the supernatant was injected into the sample‐carrier cell and the particle count measured at five positions, with two cycles of reading per position. The cell was washed with PBS after every sampling. Concentration of EVs per ml (±SEM) was calculated from three independent experiments.
2.10. Measurement of EV secretion in vivo
Striatal injections and EV measurements were performed as previously described by our group (Dickens et al., 2017; McCluskey, Campbell, Anthony, & Allan, 2008). Briefly, male (2–3 month), GFAP–GFP transgenic mice that allow for the evaluation of fluorescent EVs in plasma that are generated in the brain (Jackson Laboratories, n = 69) were anaesthetized with 3% isoflurane (Baxter) in oxygen (Airgas) and placed in a stereotaxic frame (Stoelting Co). We chose male mice to avoid effect of female specific hormones (i.e., oestrogen) in inflammation that is known to be anti‐inflammatory (Zhang et al., 2014). A small burr hole was drilled in the skull over the left striatum using a dental drill (Fine Scientific Tools). IL‐1β (0.1 ng/3 μl) was injected (total volume of 3 μl) at the rate 0.5 μl·min−1 via a pulled glass capillary (tip diameter < 50 μm; McCluskey et al., 2008). The stereotaxic coordinates based on bregma as reference point were A/P + 1; M/L −2; and −3 D/V (Dickens et al., 2017; Paxinos & Franklin, 2001). Saline was used as a control. When PDDC or the inactive analogue 5 were used, they were given i.p. (10 mg·kg−1, 5% DMSO, 5% Tween‐80 in saline) 30 min before IL‐1β injection. Mice subjected to intrastriatal injection of IL‐1β were also injected with the NSAID carprofen (rimadyl, 5 mg·kg−1, i.p.) and closely monitored during recovery; no adverse reactions were observed. Following infusion, the capillary was held in place for 5 min to allow for the solution to diffuse into the tissue. Thirty‐six mice were utilized to perform EVs counts and liver cytokine analysis. Thirty‐three mice were used for histology. Mice were killed at 2 hr (EV release and liver cytokines) or at 24 hr (histology) post‐IL‐1β treatment with overdose of anaesthetic (isoflurane). Blood samples were taken at death by cardiac puncture with heparin (Sigma) coated syringes and EDTA tubes (BD). Blood was immediately centrifuged at 2,700×g for 15 min (20°C) to obtain plasma. Plasma was further centrifuged at 10,000 g for 15 min (4°C) to generate platelet‐free plasma. This procedure removes large particles such as apoptotic bodies. Quantitation of Plasma EVs: Dynabeads M‐450 Epoxy (Invitrogen) were coupled with anti‐GFP antibody (Thermo Fisher) at a ratio of 200‐μg antibody per 4 × 108 beads. Plasma from GFAP–GFP mouse (50 μl) was incubated with 2 × 107 anti‐GFP antibody‐coupled Dynabeads at 4°C overnight. The beads were washed and placed on a magnet to separate EVs bound to anti‐GFP antibody‐coupled Dynabeads. The precipitated EVs were eluted using 0.1‐M glycine (pH 3.0). The concentration of immunoprecipitated GFP + EVs was quantified using ZetaView nanoparticle tracking analysis (Particle Metrix) as described above. The data were collected by an investigator blinded to experimental conditions.
2.11. Western blot analysis
All antibody ‐based procedures used in this study comply with the recommendations made by the British Journal of Pharmacology. Proteins were resolved by 10% SDS–PAGE and transferred to PVDF membranes (Bio‐Rad). Nonspecific binding sites were blocked with 5% (w/v) milk in TBS containing 0.1% Tween 20 (TBS‐T). After blocking, blots were incubated overnight with the primary polyclonal antibodies to GFP (1:1,000; Thermo Fisher Scientific), CD63 (1:200; Santa Cruz Biotechnology), flotillin1 (1:1,000; Abcam), TSG101 (1:1,000; BD Biosciences), mitofilin (1:5,000; Thermo Fisher Scientific), and α‐actinin (1:1,000; GeneTex). After washes with TBS‐T, blots were incubated for 2 hr with the corresponding IgG HRP‐linked secondary antibody (1:1,000; Cell Signaling Technology) and developed by enhanced chemiluminescence. Image analysis was performed using a G: BOX imaging system (Syngene).
2.12. Cytokine measurements
RNA was isolated from fresh frozen tissues (10 to 50 mg) using the RNeasy Mini Kit (Qiagen). Total RNA was reverse transcribed and quantified using previously published methods (Westberry, Trout, & Wilson, 2010). For quantitative real‐time PCR, each reaction contained SYBR Green Master Mix (12.5 ml; Life Technologies), diethyl pyrocarbonate H2O (10.5 ml), forward and reverse primers to CCL2, TNF‐α, IL‐6, IL‐1β, IL‐17A, IL‐10, IGFR1, and CXCL1 (0.5 ml each; Sigma‐Aldrich), and cDNA (1 ml). Each 96‐well plate included a nontemplate control, and samples were analysed in triplicate on an Applied Biosystems 7300 (Life Technologies). Cycling parameters were as follows: one cycle for 2 min at 50°C, one cycle for 10 min at 95°C, and 40 cycles for 15 s at 95°C and for 1 min at 60°C. The change in threshold cycle (ΔCt) for each sample was normalized to β‐actin, and ΔΔCt was calculated by comparing ΔCt for the treatment group to the average ΔCt of the control group (Livak & Schmittgen, 2001).
2.13. Immunohistochemistry
Coronal brain sections (20 to 40 mm) were prepared using a cryostat microtome (Leica). Endogenous peroxidase activity was quenched using a 1% solution of H2O2 in methanol, and primary antibody Ly6b (1:1,000, AbD Serotec) was incubated at 4°C overnight. Sections were washed (3× PBS), and biotinylated secondary antibody (1:100, Vector Laboratories) was added at room temperature for 2 hr. Staining was visualized using an avadin‐biotin complex (1:100 of A and B, Vector Laboratories) and DAB‐HCl using a microscope to monitor staining progression. Stereological quantitation was performed using a one‐in‐five series (200‐mm spacing), from the rostral point of bregma −1.22 mm to the caudal point of bregma −2.80 mm as previously described (Chen, Hui, Geiger, Haughey, & Geiger, 2013).
2.14. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). pIC50 determinations were made from eight point dose response curves run in duplicate. Data analysis and non‐linear least squares curve fitting were carried out with GraphPad Prism 7 [RRID: SCR_002798]. Confidence intervals of pIC50 values were computed using the method that produces asymmetrical intervals (Prism 7). Rate of change of fluorescence vs. sphingomyelin concentration in the presence of increasing concentrations of PDDC in the mode of inhibition experiments were plotted in GraphPad Prism and Vmax and Km were obtained by a least‐squares fit to the Michaelis‐Menten equation using non‐linear regression. Extra cellular vesicles were quantified using ZetaView Nanoparticle tracker (Particle Metrix GmBH, Meerbusch, Germany) and the corresponding Zetaview software (8.03.04.01). pEC50 values and corresponding 95% confidence asymmetrical intervals, CI (95%), were obtained from a non‐linear fit of log [inhibitor] vs. response using GraphPad Prism 7. Results are the average of three or four independent experiments. Time points in pharmacokinetic profiles correspond to the average of three independent determinations; non‐compartmental‐analysis modules in Phoenix WinNonlin version 7.0 (Certara USA, Inc., Princeton, NJ) were used to assess clearance (Cl), volume of distribution (Vd) and exposures measured by area under the curve (AUC0‐t). The maximum plasma concentration (Cmax) and the time to Cmax (Tmax) were determined directly from the individual plasma concentration‐time profiles. Absolute bioavailability (F) of oral doses was calculated by using the equation F = (AUC0‐t PO/AUCI0‐t IV) x (DoseIV/DosePO). None of the in vitro biochemical experiments were performed or analysed in a blinded manner. Image analysis of Western blots was performed using a G: BOX imaging system (Syngene). Liver cytokine levels under different treatments were measured by qRT‐PCR of RNA isolated from fresh frozen liver tissue. Results are n = 5 per treatment, except for the 30 mg kg‐1 dose where n = 3 due to limited number of mice (2 h cohort). Finally, leukocyte influx was quantified in coronal brain sections using primary antibodies to leukocytes (Ly6b). Results are from 10 mice per treatment (24 h cohort); exceptions here, also due to limited mouse availability were saline control and the inactive compound 5 (n=5‐8); quantitation of leukocyte influx was carried out using one‐way ANOVA with Tukey's post hoc test, when F achieved statistical significance (P <.05).
2.15. Materials
Details of the syntheses of PDDC, compound 5, and other analogues are shown in Figure 1 and their corresponding authentication is provided in the Supporting Information. The pCMV6 entry vector containing C‐terminal Flag‐tagged SMPD3 gene used to transfect into HEK293 cells (RRID:CVCL_0045) was obtained from Origene (Rockville, MD, USA). SM and Amplex Red reagent were purchased from Life Technologies Corporation (Grand Island, NY, USA) and HRP from Worthington Biochemical Corporation (Lakewood, NJ, USA). All other enzymes, reagents, and buffers were obtained from Millipore Sigma (St. Louis, MO, USA). Adult (2–3 month) male GFAP–GFP mice were obtained from Jackson Laboratories (USA).
Figure 1.
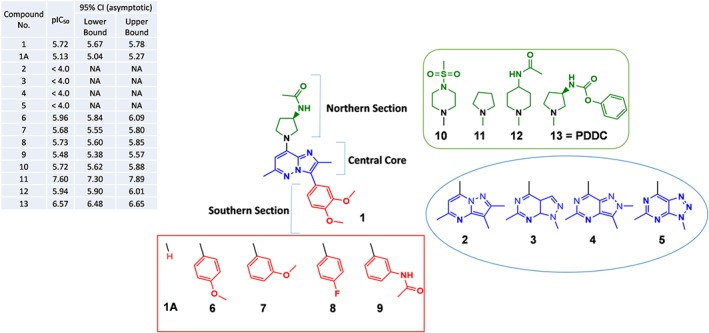
Representative structure–activity relationship (SAR) studies of enantiomer hit 1. Substitutions focused on changes in the central core (blue moiety), in the southern aromatic ring (red moiety), and in the northern acetyl amide substitution on the pyrrolidine ring (green moiety). Inhibition of nSMase2 by each compound is listed on the table at the upper left corner; inhibition values are given as average pIC50 of at least two independent determinations with corresponding 95% confidence asymmetrical intervals: CI (95%). Changes in the central core—The imidazo[1,2‐b]pyridazine ring was substituted with other fused bicyclic heterocycles including three other pyrazolo pyrimidines (2, 3, and 4) and a triazolo pyrimidine (5). Changes in the southern section—Substituents in this position (6, 7, 8, and 9) were phenyl‐derived moieties. Changes in the northern section—Pyrrolidine acetamide group moiety in 1 was replaced by various heterocycles like methyl sulfonyl piperazine or piperidine acetamide moieties that gave 10 and 12 respectively. The unsubstituted pyrrolidine derivative resulting from removal of the acetamide substitution on the pyrrolidine ring gave 11. Replacement of the acetamide group on the pyrrolidine ring with a phenyl carbamoyl group resulted in compound 13 (PDDC)
2.16. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
3. RESULTS
3.1. HTS assays against human nSMase2 identified a new inhibitor scaffold with low μM potency
We adapted the Amplex Red fluorescence assay to screen for inhibitors of human nSMase2 as we have reported previously (Figuera‐Losada et al., 2015). We screened for inhibitors of nSMase2 using the Molecular Libraries Small Molecule Repository and the Institute of Organic Chemistry internal library (>365,000 compounds). After counter screening for false positives (i.e., compounds that inhibit coupling enzymes in the screening assay), considerations of drug‐like properties, confirmatory dose–responses, and potency, the hit of most interest was (R)‐N‐(1‐(3‐(3,4‐dimethoxyphenyl)‐2,6‐dimethylimidazo[1,2‐b]pyridazin‐8‐yl)pyrrolidin‐3‐yl)acetamide with pIC50 = 5.72, CI (95% [5.67, 5.78]; 1 in Figure 1). Compound 1 underwent significant structural optimization to identify potent, selective nM level inhibitors as detailed below.
3.2. Chemical optimization of compound 1 led to the identification nSMase2 inhibitors with nM potency
Due to the lack of structural information on any nSMase2‐inhibitor complex, we approached potency optimization of the initial hit through ligand‐based optimization. Preliminary structure–activity relationship studies on compound 1 were focused on (a) changes in the central core (blue moiety), (b) changes in the southern aromatic ring (red moiety), and (c) changes in the northern acetyl amide substitution on the pyrrolidine ring (green moiety; Figure 1). Changes in the central core including substitution of the imidazo[1,2‐b]pyridazine ring with other pyrazolo pyrimidines (2, 3, and 4) and a triazolo pyrimidine (5) resulted in loss of inhibitory activity (pIC50 < 4.0; Figure 1). We concluded that the imidazo[1,2‐b]pyridazine central core is an essential part of the pharmacophore possibly due to a unique electron distribution within the bicyclic heterocycle. Changes in the southern section including removal of the southern moiety (1A) resulted in significantly less potency (pIC50 = 5.13, CI [95%; 5.04, 5.27]). We examined numerous substituents in this position (e.g., 6, 7, 8, and 9) with most phenyl‐derived moieties exhibiting inhibitory potency, comparable to that of the original hit (1) (pIC50s in the range 5.48–5.96; Figure 1). Changes in the northern section including replacement of the acetamide group in 1 with various heterocycles like methyl sulfonyl piperazine (10) (pIC50 = 5.72, CI [95%; 5.62, 5.88]) or piperidine acetamide (12) (pIC50 = 5.94, CI [95%; 5.90, 6.01]) did not show improvement in potency. There was a breakthrough when we removed the acetamide substitution to produce 11 with pIC50 = 7.60, CI (95%; [5.90, 6.01]). Compound 11, however, exhibited poor metabolic stability. Replacement of the acetamide group on the pyrrolidine ring with a phenyl carbamoyl group resulted in PDDC (13) with pIC50 = 6.57, CI (95%; [6.48, 6.65]; Figure 1). PDDC exhibited good metabolic stability and was chosen for further pharmacological characterization as described below.
3.3. PDDC is a noncompetitive inhibitor of nSMase2
The mechanism for nSMase2 inhibition of PDDC was determined using the Amplex Red fluorescence‐based nSMase2 activity assay. Lysates from cells expressing recombinant nSMase2 (1.9‐μg protein/50 μl) were incubated with an escalating dose range of both SM and PDDC for 3 hr. PDDC exhibited noncompetitive inhibition with respect to SM. The maximal rate of SM hydrolysis (V max) decreased with increasing concentrations of inhibitor, but the binding constant (K M) remained the same (Figure 2a). A Lineweaver–Burk linear transformation of the data illustrates the noncompetitive mode of inhibition (Figure 2b). A secondary plot of K M apparent/V max versus [PDDC] gave a Ki value of 0.3 μM (pKi = 6.52; Figure 2c), in close agreement with pIC50 6.57.
Figure 2.
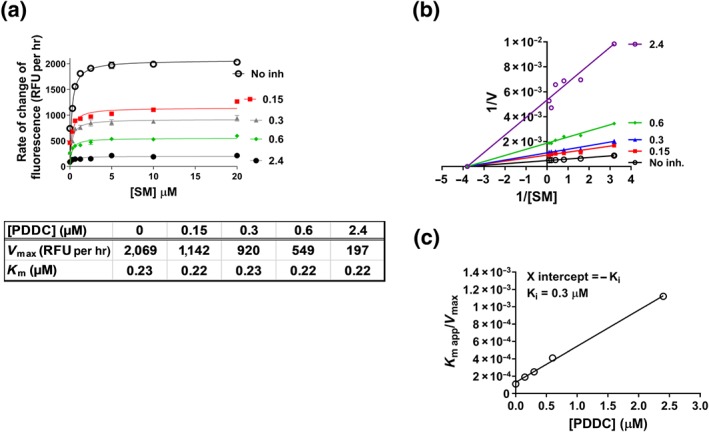
Mechanism of inhibition of nSMAse2 by PDDC. (a) Rate of reaction plotted against substrate (SM) concentration, in the presence of several concentrations of PDDC. Human nSMase2 cell lysate (0.1 μg·μl−1) was incubated with increasing concentrations of SM and coupling reagents for 2 hr at 37°C before measuring fluorescence. V max and K M values were obtained from non‐linear regression fits to Michaelis–Menten kinetics using prism. (b) Lineweaver–Burk plot to illustrate the non‐competitive inhibition by PDDC. (c) Secondary plot (K M app/V max vs. [PDDC]) to obtain the binding constant (Ki = −X intercept) of PDDC
3.4. PDDC showed selectivity for nSMase2 versus related enzymes and in a broad selectivity screen
Selectivity of PDDC for nSMase2 was first evaluated against the related PDEs AP and acid sphingomyelinase. The pIC50 for inhibition of AP was <4.0 (highest concentration tested was 100 μM; Figure S1). Similar results were observed for acid sphingomyelinase (data not shown). We then evaluated PDDC in the Eurofins SafetyScreen44, a panel of 44 selected targets recommended by major pharmaceutical companies to establish undesirable off‐target activity profiles (Bowes et al., 2012). There were 4/44 positive hits at 10 μM that included the α1A adrenoceptor, Ca2+ and Na+ channels, and dopamine transporter (Table S1). However, all four positive hits were in the 47–84% inhibition range, suggesting at least a 20‐fold segregation between inhibition of these off targets and inhibition of nSMase2. Of note, PDDC did not inhibit hERG or two additional PDEs (3A and 4D2) in the panel of selected targets. PDDC selectivity with respect to enzymes from related families is consistent with a non‐competitive mode of inhibition, as PDDC is likely to be acting at a site other than the catalytic site.
3.5. PDDC showed metabolic stability in mouse and human microsomes
We evaluated PDDC for Phase I (NADPH) metabolic stability using human and mouse liver microsomes as previously described (Rais et al., 2016). The fraction of PDDC remaining over time was determined by LC–MS/MS. Incubation of PDDC with mouse microsomes in the presence of NADPH resulted in slight time‐dependent loss of PDDC with >60% remaining after a 1‐hr incubation. In human liver microsomes, PDDC remained intact with 100% remaining after a 1‐hr incubation (Figure 3). These results indicate that PDDC does not have major liver metabolic liabilities that would preclude its use as an in vivo probe.
Figure 3.
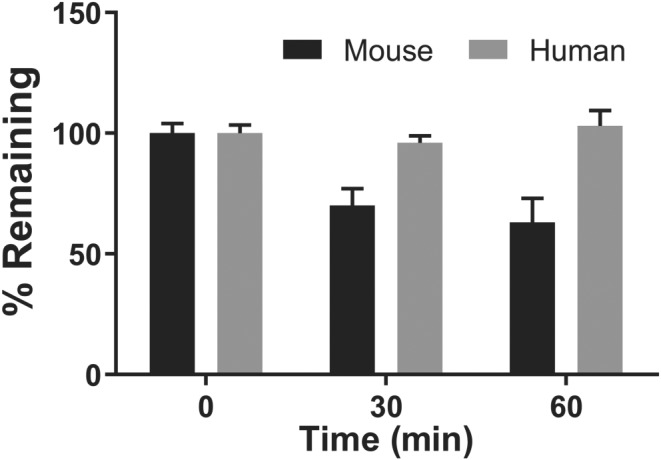
Phase I metabolic stability of PDDC in mouse and human liver microsomes. PDDC was evaluated for Phase I metabolic stability using human and mouse liver microsomes (fortified with NADPH). PDDC was completely stable to Phase I metabolism in human and moderately stable in mouse (100% and 63% remaining at 1 hr, in human and mouse respectively). Data shown are means ± SEM from n = 3 experiments
3.6. PDDC exhibited excellent plasma and brain exposures following i.v., p.o., and i.p. administrations
We next evaluated the in vivo pharmacokinetics of PDDC in mice. Mice were administered 10 mg·kg−1 PDDC through i.v., p.o., and i.p. routes, and plasma as well as brain levels of PDDC were measured at predetermined time points. Following i.v. administration, PDDC exhibited first‐order kinetics with a Cmax of 22.10 ± 1.8 nmol·ml−1, low clearance 9.40 ± 0.42 ml·min−1·kg−1, moderate volume of distribution 1.05 ± 0.15 L·kg−1, and exposures (AUC0–t) of 32.89 ± 2.16 nmol*hr·ml−1. The brain exposure (AUC0–t) was 19.24 ± 1.20 nmol*hr·g−1 giving an excellent brain to plasma ratio of 60%. Absorption was rapid following both p.o. and i.p. administration with a peak plasma Cmax of 5.45 ± 0.48 and 9.34 ± 0.12 nmol·ml−1, achieved at 1 and 0.25 hr after dosing respectively. The Cmax values in brain were 2.61 ± 0.43 and 7.80 ± 0.72 nmol·g−1 after p.o. and i.p. administrations respectively. Corresponding brain AUC0–t were 13.09 ± 0.93 and 27.17 ± 2.21 nmol*hr·g−1. These values resulted in AUCbrain/AUCplasma of 0.5 and 0.6 after p.o. and i.p. administrations respectively (Figure 4a,b). True elimination t 1/2 following i.v. administration in mice was t 1/2 = 1.30 ± 0.25 hr. The pharmacokinetic profiles and corresponding calculated parameters underline four major features: first, PDDC showed low clearance from plasma (Figure 4b). Second, PDDC exhibited high p.o. bioavailability as calculated from plasma AUC values (Figure 4b): %F = 88. Third, PDDC exhibited excellent brain penetration: brain/plasma 0.5–0.6 regardless of route of administration. And fourth, PDDC exhibited high exposures in both plasma and brain as indicated by the AUC measurements. As a result, PDDC levels were two‐fold to three‐fold its IC50 (0.270 μM) for inhibition of nSMase2 8 hr following 10 mg·kg−1 i.p. or p.o. dosing (Figure 4a).
Figure 4.
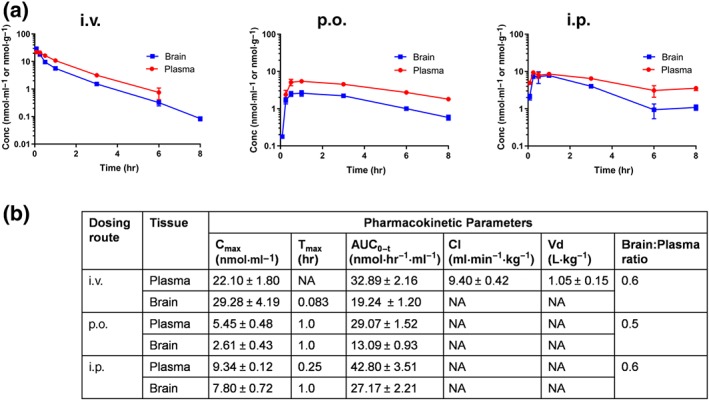
Pharmacokinetics of PDDC after i.v., p.o., and i.p. administrations. Following i.v., i.p., or p.o. administration (10 mg·kg−1) in CD1 mice, plasma and brain levels of PDDC were measured at 0.183, 0.25, 0.50, 1, 3, 6, and 8 hr after dosing (n = 3 per time point) via LC/MS/MS. (b) Pharmacokinetic parameters corresponding to PK profiles in (a). Non‐compartmental analysis modules in WinNonlin® (version 5.3) were used to assess pharmacokinetic parameters. Relative bioavailabilities (%F) for the p.o. and i.p. routes were 85% and 130% respectively
3.7. PDDC inhibited leukocyte transmigration in response to a focal inflammatory brain lesion
Several lines of evidence have shown that nSMase2 is a critical regulator of EV secretion (Asai et al., 2015; Dickens et al., 2017; Dinkins et al., 2014; Kosaka et al., 2013; Trajkovic et al., 2008). We first evaluated PDDC for its ability to inhibit ADEV release from primary cultured astrocytes as a consequence of inhibition of nSMase2. Mouse primary astrocytes were activated by FBS withdrawal as previously described (Dickens et al., 2017) and treated with PDDC or a closely related inactive structural analogue (5, pIC50 < 4.0 Figure 1) over a range of concentrations (0.03 ‐ 30 μM). Two hours after treatment, ADEVs were isolated from the media and characterized (quantity and size) by nanoparticle tracking analysis. PDDC produced a dose‐dependent reduction in ADEV release with pEC50 5.5, CI (95%; [5.3, 5.6]; Figure 5). Inactive analogue 5 had no effect on EV release (Figure 5). As positive control, we used cambinol, a known inhibitor of nSMase2 (Figuera‐Losada et al., 2015); cambinol exhibited a dose‐dependent reduction of ADEV release with pEC50 4.7, CI (95%; [4.5, 4.8]). The 12‐fold decrease in potency in going from PDDC to cambinol in this functional assay is in close agreement with the corresponding potencies in the cell‐free assay: pIC50s for PDDC and cambinol are 6.57 and 5.3 (Figuera‐Losada et al., 2015) respectively. In order to rule out the possibility that inhibition of EV release by PDDC was due to cell death, we measured astrocyte viability as monitored by the LDH assay under the same conditions we carried out the EV release measurements (2‐hr incubation with PDDC up to 30 μM). No deleterious effects were observed upon incubation with PDDC (Table S2) indicating inhibition of EV release by PDDC was not due to cell death. We have also determined that PDDC blocks the release of EVs from H9 cells (a human T‐cells line), U1 cells (a macrophage cell line), and primary CD4 cells with a minimal effective dose of 0.3 μM but is not toxic to these cells with a 10‐μM dose, the highest dose tested (data not shown) which is 30‐fold higher than its EC50 in these immune cells.
Figure 5.
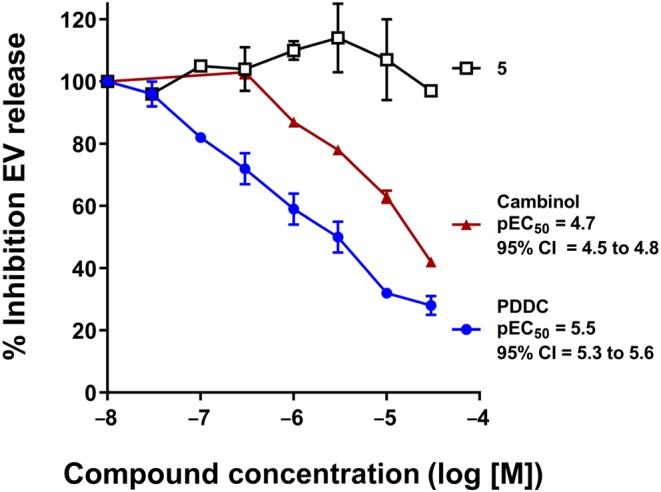
Inhibition of EV secretion by PDDC and 5 in glial cells. Primary astrocytes were treated in parallel incubations with PDDC, 5, or cambinol in the 0.03–30 μM range as indicated; DMSO (0.02%) was used as vehicle control (set as 100% on y‐axis). Media was collected after 2‐hr incubation and centrifuged at 2,700 g for 15 min at 4°C. Supernatant was collected and the number of extracellular vesicles (EVs) was quantified using ZetaView Nanoparticle Tracker. The data points correspond to per cent inhibition of EV release (±SEM) over a concentration range for each compound; 100% release corresponds to 4.87 × 107 ± 0.098 EVs. Results are the average of three or four independent assays. Each independent assay was the average of three replicates. pEC50 values and corresponding 95% confidence asymmetrical intervals, CI (95%), were obtained from a non‐linear fit of log [inhibitor] against response, using GraphPad Prism 7
We have recently shown that nSMase2 regulates the secretion of ADEVs in response to a focal inflammatory brain lesion. ADEVs shed in response to IL‐1β, enter into blood, and target multiple organ systems. Protein and miRNA cargo of these ADEVs regulate an acute cytokine response in liver that primes leukocytes which transmigrate to the site of inflammatory brain lesion (Dickens et al., 2017). We used this model to evaluate the ability of PDDC to inhibit ADEVs release in vivo. Two groups of GFAP/GFP mice were administered an intrastriatal injection of IL‐1β and dosed i.p. with 1, 10,or 30 mg·kg−1 PDDC or the inactive analogue (10 mg·kg−1) 0.5 hr before IL‐1β injection (Figure 6a). GFP+ ADEVs were isolated from plasma 2 hr following IL‐1β administration and characterized for size and quantity by nanoparticle tracking analysis. ADEV size remained the same within experimental error for the different treatments (Figure S2). EVs isolated from vehicle‐treated mice as measured by western blot analysis exhibited low levels of tetraspan protein CD63, the lipid raft protein flotilin‐1, and the ESCRT protein TSG101. Intensity of signals of these proteins increased in EVs isolated from mice treated with IL‐1β remained high in EVs isolated from mice treated with compound 5 (10 mg·kg−1) and returned to low levels in EVs isolated from mice treated with PDDC (10 mg·kg−1 i.p.; Figure 6b). Intrastriatal administration of IL‐1β increased the number of GFP+ ADEVs in plasma more than two‐fold (Figure 6c). The pharmacokinetic results (Figure 4) showed that total PDDC concentration in brain at 2 hr after 10 mg·kg−1 i.p. administration was approximately 5 μM, which exceeded the nSMase2 pIC50 6.57 (0.27 μM). In order to show a dose–response and given the limited solubility of PDDC, we used 1, 10, and 30 mg·kg−1 doses. We observed a dose–response effect on inhibition of ADEV release from 0 to 10 mg·kg−1 where a maximal inhibition of ADEV release (83 ± 13%) was achieved at 10 mg·kg−1. When PDDC was used at 30 mg·kg−1, it was not significantly different from inhibition at 10 mg·kg−1 although a trend towards less inhibition was observed (Figure 6c). This latter result may have been due to the limited solubility and precipitation of PDDC in the 30 mg·kg−1 dosing solution. Administration of the inactive structural analogue had no effect on IL‐1β‐induced ADEV release into plasma, further establishing the relevance of nSMAse2 inhibition (Figure 6c). The free brain fraction (fubrain) of PDDC was determined to be 2.2 ± 0.02 using rapid equilibrium dialysis. The calculated free concentrations following a 10 mg·kg−1 i.p. dose ranged from 171.5 to 21 nM (Cmax − Cmin). To more directly evaluate target engagement in vivo, we also quantified brain nSMase2 activity ex vivo from mice used in the dose–response efficacy study. Inhibition of nSMase2 activity (±SEM) was found to be 3 ± 1, 20 ± 2, and 14 ± 3 at 1, 10, and 30 mg·kg−1 doses respectively. These results are in congruence with the calculated free drug concentrations (e.g., at 10 mg·kg−1 i.p., the maximal free fraction of 172 nM approximates to half the IC50 of PDDC [270 nM] and provides 20% enzyme inhibition).
Figure 6.
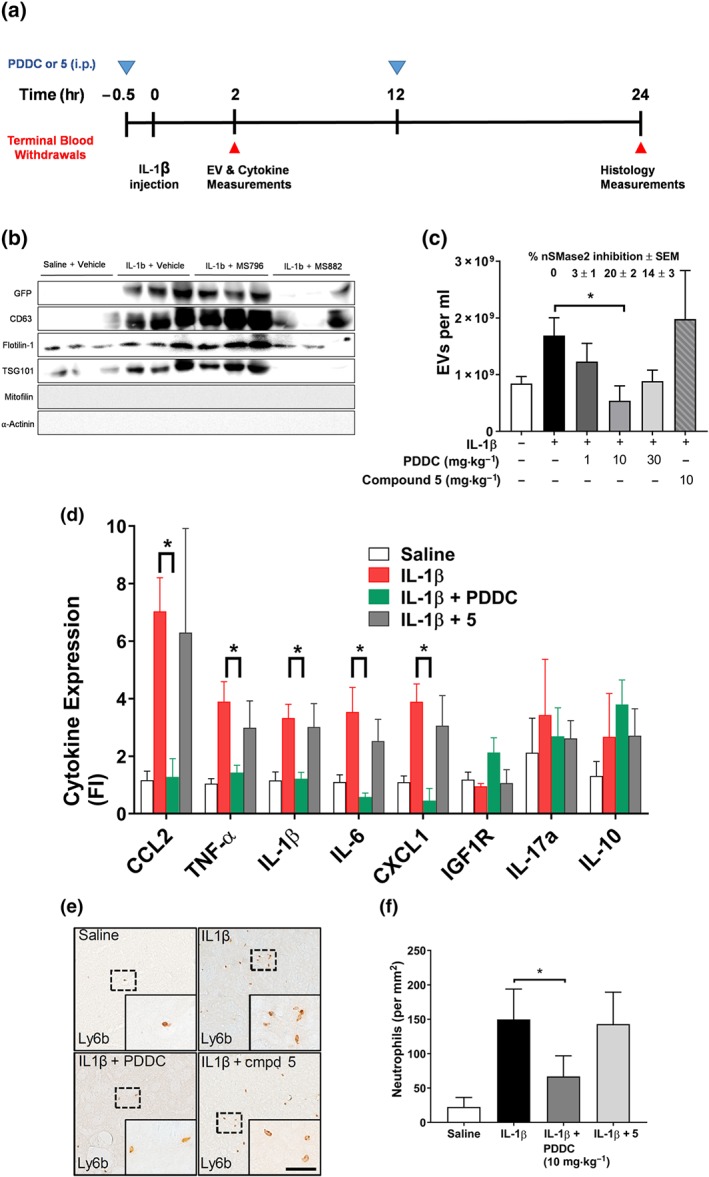
Inhibition of EV and leukocyte migration in an in vivo model of brain injury. (a) Schedule of procedures. Mice were given PDDC or compound 5 (10 mg·kg−1 i.p.) 0.5 hr before striatal injections of saline (control) or IL‐1β (0 hr). Dosing (blue triangles) and terminal blood collections (red triangles) were carried out at the times indicated. There were two animal cohorts: the first, killed at 2 hr, was used for plasma EVs and liver cytokine measurements; the second, killed at 24 hr, was used for histology measurements. (b) Western analyses of EV preparation from different treatments to monitor exosome biomarkers. (c) GFP‐exosome vesicle secretion—plasma levels of GFP–EVs isolated from mice under different treatments, including PDDC (dose–response) and compound 5 (10 mg·kg−1), at 2.5 hr after dosing . Data are mean ± SD from n = 5 per treatment, except for 30 mg·kg−1 PDDC, when n = 3. *P < .05, significant effect of PDDC (10 mg·kg−1 i.p.); one‐way ANOVA with Tukey's post hoc test. There was a significant increase in EVs released after IL‐1β alone, compared with release in control animals. There was no significant difference observed between IL‐1β and IL‐1β plus compound 5 groups. Average per cent inhibition of nSMase2 activity in striatum of mice treated with different doses of PDDC compared to that of IL‐1β‐treated mice is shown above the bars. (d) Liver cytokine levels after 10 mg kg‐1 dose as measured by qRT‐PCR of RNA isolated from fresh frozen liver tissue. Samples were analysed in triplicate. Data shown are means ± SEM from n=5 mice. *P < .05, significantly different as indicated; one‐way ANOVA with Tukey's post hoc test. (e) Representative photomicrographs of Ly6b + cells in the striatum of mice 24 hr after injection of saline (top, left panel), IL‐1β (top, right panel), IL‐1β + PDDC (10 mg·kg−1, bottom, left panel), and IL‐1β + 5 (bottom right panel). Insets: Magnification of the indicated regions. Leukocyte influx was quantified in coronal brain sections using primary antibodies to leukocytes (Ly6b); scale bar, 200 μm. The results for the 24‐hr cohort was 10 mice per treatment. The exceptions here, also due to limited mouse availability, were saline control and the inactive compound 5 (n = 5–8). (f) Quantitation of data in (e). *P < .05, significant effect of PDDC, as indicated; one‐way ANOVA with Tukey's post hoc test. There was no significant difference observed between IL‐1β and IL‐1β plus compound 5 groups
Consistent with our previous report, striatal injection of IL‐1β increased liver expression of the leukocyte chemokine CCL2, acute phase cytokines TNF‐α, IL‐1β, IL‐6, and CXCL1. This acute liver cytokine response was blocked when PDDC was administered before the striatal injection of IL‐1β (Figure 6d). Histology measurements were carried out on a second cohort of mice that were given an additional dose at 12 hr of both PDDC and compound 5 (10 mg·kg−1 i.p.). These mice were killed at 24 hr a time where given the pharmacokinetics and pharmacological findings mentioned above we would expect to see downstream effects of nSMase2 inhibition. Immunohistochemical analysis of coronal brain sections showed a prominent influx of leukocytes in mice 24 hr following the intrastriatal administration of IL‐1β, compared to mice administered intrastriatal saline (Figure 6e). PDDC reduced leukocyte influx induced following intrastriatal IL‐1β by 65 ± 24%. The inactive structural analogue did not reduce leukocyte influx (Figure 6e,f). The results show that peripheral administration of PDDC blocks IL‐1β‐triggered release of ADEVs into blood and prevents the associated influx of leukocytes to the site of inflammatory brain lesion.
4. DISCUSSION AND CONCLUSION
While nSMase2 has emerged as an important player in the aetiology of diseases associated with altered EV release (Asai et al., 2015; Dickens et al., 2017; Dinkins et al., 2014; Kosaka et al., 2013), currently, available inhibitors have limitations including low potency (pIC50 6 or lower), poor solubility, and/or limited brain penetration (Figuera‐Losada et al., 2015). Moreover, their chemical structures have not been amenable to optimization into drug‐like molecules. In search for novel scaffolds of nSMase2 inhibitors, we carried out an HTS campaign in collaboration with the National Center for Advancing Translational Sciences and the Institute of Chemistry and Biochemistry in the Czech Republic (>365,000 compounds). Systematic optimization of one of the screening hits led to PDDC, a potent (pIC50 6.57, CI [95%; 6.48, 6.65]; Figure 1), non‐competitive (Figure 2), and selective inhibitor (Supporting Information). PDDC was metabolically stable (Figure 3) and exhibited substantial brain penetration (AUCbrain/AUCplasma = 0.5–0.6) and excellent exposure with concentrations in the brain that were well above its IC50 by 8 hr after dosing, using either i.p. or p.o. administration of a 10 mg·kg−1 dose (Figure 4).
The drug‐like features of this nSMase2 inhibitor allowed us to explore the relevance of the role of nSMase2 as modulator of ceramide synthesis during EV manufacturing. PDDC inhibited ADEV release in a dose‐dependent manner with pEC50 5.5, CI (95%; [5.3, 5.6]). This effect was consistent with mediation through nSMase2 inhibition as a structurally related inactive analogue was without effect. Moreover, cambinol, a known nSMase2 inhibitor, also exhibited a dose‐dependent inhibition of ADEV release with weaker potency (pEC50 4.7, CI [4.5, 4.8]; Figure 5) in correlation with cambinol's lower inhibitory potency in the cell‐free assay (pIC50 5.3, compared with. 6.57 for PDDC). Next, we used a model of brain inflammation to evaluate the ability of PDDC to inhibit ADEVs release in vivo. Using GFAP–GFP mice, we could follow fluorescent‐labelled EVs originating from astrocytes. PDDC inhibited ADEV release into plasma in close correlation with ex vivo measurements of nSMase2 inhibition demonstrating target engagement. The finding that it is not necessary to fully inhibit nSMase2 activity to see a pharmacological effect bodes well for studies in humans where there could be a concern about fully inhibiting the enzyme. Importantly, we show that inhibition of ADEV release results in inhibition of cytokine up‐regulation and subsequent neutrophil migration from the periphery into the brain an effect associated with lingering damage from brain injury (Smith et al., 1997; Wilcockson, Campbell, Anthony, & Perry, 2002). In contrast, 5, a structurally related but inactive analogue of PDDC, did not have any effect in this in vivo model (Figure 6). The results point to the centrality of the role of nSMase2 during inflammatory brain injury (Figure 7) and support the use of inhibitors of this enzyme for the treatment of diseases associated with EV dysfunction (Figure 7). PDDC is actively being used as a tool in animal models of CNS disease and its structural scaffold is currently being optimized for clinical translation.
Figure 7.
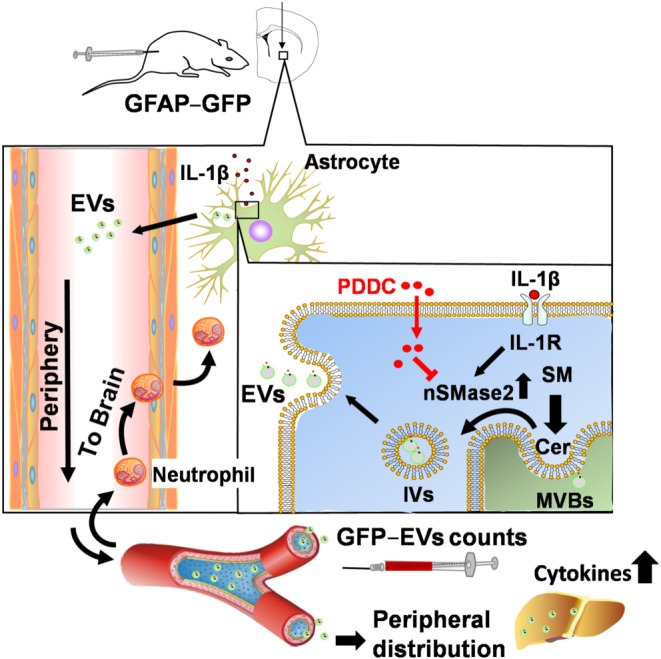
Role of nSMase2 during inflammatory brain injury and effect of nSMase2 inhibition by PDDC. Intracerebral injection of IL‐1β activates the IL‐1β receptor which turn activates nSMase2. nSMase2‐catalysed hydrolysis of sphingomyelin (SM) produces long‐chain ceramides (Cer). Increase in ceramide production at multivesicular bodies (MVBs) leads to the formation of intraluminal vesicles (IVs) and budding of extracellular vesicles (EVs) that are then shed from astrocytes and released into the periphery. ADEVs can be identified in plasma because they are GFP labelled. Astrocytic EVs promote crossing of leukocytes into the brain as a result of cytokine production in the liver. PDDC injection inhibits nSMase2 in astrocytes, prevents exosome biosynthesis, and results in inhibition of migration of leukocytes from periphery to brain and prevention of damage associated with infiltration of these immune cells into the brain
AUTHOR CONTRIBUTIONS
C.R., R.R., N.J.H., R.N., and B.S.S. were responsible for the experimental design and writing of the manuscript. M.S. performed the chemical syntheses and analyses. A.G.T. carried out the IC50 determinations and mechanism of inhibition studies. R.D. performed the metabolic stability and PK studies. A.D.C. and Z.L. carried out in vitro inhibition of EV secretion in glial cells and analyses of EVs in samples generated from GFAP–GFP in vivo studies. S.‐W.Y. was responsible for in vivo studies using GFAP–GFP mice and for the illustration in Figure 7.
CONFLICT OF INTEREST
Authors B.S.S., C.R., A.G.T., N.J.H., R.N. and M.S. are listed inventors in a non‐provisional patent application filed by Johns Hopkins Technology Ventures (PCT/US2018/012699) covering the compounds included in this manuscript.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design and Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1. Selectivity of PDDC for nSMase2 vs. coupling enzymes in fluorescence Amplex Red assay ‐ pIC50 curves were generated in duplicate twelve‐point dose response curves using the Amplex red Fluorescence‐based assay. Lysate of cells expressing human recombinant nSMase2 (1.9 μg protein/50 μL) was incubated with sphingomyelin (20 μM) and detecting reagents (alkaline phosphatase 4 U/mL, choline oxidase 0.1 U/mL, horse radish peroxidase 0.1 U/mL, and Amplex Red 50 μM) in the presence of different concentrations of PDDC for 3 h (total reaction volume 50 μL). Percent inhibition vs. inhibitor concentration was plotted in GraphPad Prism. Data analysis and non‐linear least squares curve fitting were carried out with GraphPad Prism 7. Reagents in the counter screen to establish selectivity against coupling enzymes (alkaline phosphatase, choline oxidase, and horse radish peroxidase) were the same as for the pIC50 determination as for inhibition of nSMase2 except that nSMase2 was not present and phosphorylcholine (2 μM) was used as substrate for alkaline phosphatase. CI (95%): 95% asymptotic confidence interval; lower bound – upper bound
Table S1. Evaluation of PDDC at 10 μM in Eurofin's SafetyScreen 44
Table S2. Effect of PDDC on astrocyte viability as measured by lactate dehydrogenase (LDH) activity released into cell media – Primary astrocytes were treated in parallel incubations with PDDC using the same conditions as those used for the measurement of EV release from astrocytes (2h incubation up to 30 μM). Media was collected and used as LDH source to follow disappearance of NADH (1 mM) absorbance at 340 nM in the presence of sodium pyruvate (20 mM). Maximal LDH activity was obtained by using mammalian protein extraction reagent (M‐PER, ThermoScientific) and normalized to 100%
Figure S2. Extracellular Vesicle (EV) size in an in vivo model of brain injury – EVs were isolated from plasma 2 h following treatments with vehicle, IL‐1β, IL‐1β + PDDC (10 mg/kg IP) and IL‐1β + compound 5 (10 mg/kg IP). Plasma was collected and centrifuged at 2700 g for 15 min at 4°C. Supernatant was collected and size of EVs was measured using a Zetaview nanoparticle Tracker.
ACKNOWLEDGEMENTS
This research was supported by National Institutes of Health Grants RO1 MH107659 (C.R.) and P30 MH075673 (B.S.S. and N.J.H.), TEDCO Maryland Innovation Initiative Grant award (C.R.), the Institute of Organic Chemistry and Biochemistry of the Academy of Sciences of the Czech Republic (Grant RVO 61388963) and by the European Regional Development Fund; OP RDE; Project: “Chemical biology for drugging undruggable targets (ChemBioDrug)” (No.CZ.02.1.01/0.0/0.0/16_019/0000729) (R.N. and M.S.).
Rojas C, Sala M, Thomas AG, et al. A novel and potent brain penetrant inhibitor of extracellular vesicle release. Br J Pharmacol. 2019;176:3857–3870. 10.1111/bph.14789
Contributor Information
Camilo Rojas, Email: crojas2@jhmi.edu.
Norman J. Haughey, Email: nhaughey@jhmi.edu.
Radim Nencka, Email: radim.nencka@uochb.cas.cz.
Barbara Slusher, Email: bslusher@jhmi.edu.
REFERENCES
- Alexander, S. P. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The concise guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174(Suppl 1), S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai, H. , Ikezu, S. , Tsunoda, S. , Medalla, M. , Luebke, J. , Haydar, T. , … Ikezu, T. (2015). Depletion of microglia and inhibition of exosome synthesis halt τ propagation. Nature Neuroscience, 18, 1584–1593. 10.1038/nn.4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay, R. A. , Schwab, A. , DeMarino, C. , Akpamagbo, Y. , Lepene, B. , Kassaye, S. , … Kashanchi, F. (2017). Exosomes from uninfected cells activate transcription of latent HIV‐1. The Journal of Biological Chemistry, 292, 11682–11701. 10.1074/jbc.M117.793521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowes, J. , Brown, A. J. , Hamon, J. , Jarolimek, W. , Sridhar, A. , Waldron, G. , & Whitebread, S. (2012). Reducing safety‐related drug attrition: The use of in vitro pharmacological profiling. Nature Reviews. Drug Discovery, 11, 909–922. 10.1038/nrd3845 [DOI] [PubMed] [Google Scholar]
- Chaudhuri, A. D. , Dastgheyb, R. M. , Yoo, S. W. , Trout, A. , Talbot, C. C. Jr. , Hao, H. , … Haughey, N. J. (2018). TNFα and IL‐1β modify the miRNA cargo of astrocyte shed extracellular vesicles to regulate neurotrophic signaling in neurons. Cell Death & Disease, 9, 363 10.1038/s41419-018-0369-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. , Hui, L. , Geiger, N. H. , Haughey, N. J. , & Geiger, J. D. (2013). Endolysosome involvement in HIV‐1 transactivator protein‐induced neuronal amyloid β production. Neurobiology of Aging, 34, 2370–2378. 10.1016/j.neurobiolaging.2013.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: updated andsimplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvi, P. , Sun, B. , Tang, N. , & Pulliam, L. (2017). Immune activated monocyte exosomes alter microRNAs in brain endothelial cells and initiate an inflammatory response through the TLR4/MyD88 pathway. Scientific Reports, 7, 9954 10.1038/s41598-017-10449-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Toro, J. , Herschlik, L. , Waldner, C. , & Mongini, C. (2015). Emerging roles of exosomes in normal and pathological conditions: New insights for diagnosis and therapeutic applications. Frontiers in Immunology, 6, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickens, A. M. , Tovar‐y‐Romo, L. B. , Yoo, S. W. , Trout, A. L. , Bae, M. , Kanmogne, M. , … Haughey, N. J. (2017). Astrocyte‐shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Science Signaling, 10(473), pii: eaai7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkins, M. B. , Dasgupta, S. , Wang, G. , Zhu, G. , & Bieberich, E. (2014). Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer's disease. Neurobiology of Aging, 35, 1792–1800. 10.1016/j.neurobiolaging.2014.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkins, M. B. , Enasko, J. , Hernandez, C. , Wang, G. , Kong, J. , Helwa, I. , … Bieberich, E. (2016). Neutral sphingomyelinase‐2 deficiency ameliorates Alzheimer's disease pathology and improves cognition in the 5XFAD mouse. The Journal of Neuroscience, 36, 8653–8667. 10.1523/JNEUROSCI.1429-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figuera‐Losada, M. , Stathis, M. , Dorskind, J. M. , Thomas, A. G. , Bandaru, V. V. , Yoo, S. W. , et al. (2015). Cambinol, a novel inhibitor of neutral sphingomyelinase 2 shows neuroprotective properties. PLoS ONE, 10, e0124481 10.1371/journal.pone.0124481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, G. , Yao, H. , Chaudhuri, A. D. , Duan, M. , Yelamanchili, S. V. , Wen, H. , … Buch, S. (2012). Exosome‐mediated shuttling of microRNA‐29 regulates HIV Tat and morphine‐mediated neuronal dysfunction. Cell Death & Disease, 3, e381 10.1038/cddis.2012.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim, A. , & Marban, E. (2016). Exosomes: Fundamental biology and roles in cardiovascular physiology. Annual Review of Physiology, 78, 67–83. 10.1146/annurev-physiol-021115-104929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , Altman, D. G. , & Group NCRRGW (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka, N. , Iguchi, H. , Hagiwara, K. , Yoshioka, Y. , Takeshita, F. , & Ochiya, T. (2013). Neutral sphingomyelinase 2 (nSMase2)‐dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. The Journal of Biological Chemistry, 288, 10849–10859. 10.1074/jbc.M112.446831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourembanas, S. (2015). Exosomes: Vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annual Review of Physiology, 77, 13–27. 10.1146/annurev-physiol-021014-071641 [DOI] [PubMed] [Google Scholar]
- Li, J. , Yu, W. , Tiwary, R. , Park, S. K. , Xiong, A. , Sanders, B. G. , & Kline, K. (2010). α‐TEA‐induced death receptor dependent apoptosis involves activation of acid sphingomyelinase and elevated ceramide‐enriched cell surface membranes. Cancer Cell International, 10, 40 10.1186/1475-2867-10-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, K. J. , & Schmittgen, T. D. (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2(−ΔΔC(T)) method. Methods, 25, 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Luberto, C. , Hassler, D. F. , Signorelli, P. , Okamoto, Y. , Sawai, H. , Boros, E. , … Smith, G. K. (2002). Inhibition of tumor necrosis factor‐induced cell death in MCF7 by a novel inhibitor of neutral sphingomyelinase. The Journal of Biological Chemistry, 277, 41128–41139. 10.1074/jbc.M206747200 [DOI] [PubMed] [Google Scholar]
- McCluskey, L. , Campbell, S. , Anthony, D. , & Allan, S. M. (2008). Inflammatory responses in the rat brain in response to different methods of intra‐cerebral administration. Journal of Neuroimmunology, 194, 27–33. 10.1016/j.jneuroim.2007.11.009 [DOI] [PubMed] [Google Scholar]
- Paxinos, G. , & Franklin, K. B. J. (2001). The mouse brain in stereotaxic coordinates (Second ed.). Academic Press. [Google Scholar]
- Rais, R. , Jancarik, A. , Tenora, L. , Nedelcovych, M. , Alt, J. , Englert, J. , … Slusher, B. S. (2016). Discovery of 6‐diazo‐5‐oxo‐l‐norleucine (DON) prodrugs with enhanced CSF delivery in monkeys: A potential treatment for glioblastoma. Journal of Medicinal Chemistry, 59, 8621–8633. 10.1021/acs.jmedchem.6b01069 [DOI] [PubMed] [Google Scholar]
- Raposo, G. , & Stoorvogel, W. (2013). Extracellular vesicles: Exosomes, microvesicles, and friends. The Journal of Cell Biology, 200, 373–383. 10.1083/jcb.201211138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardar Sinha, M. , Ansell‐Schultz, A. , Civitelli, L. , Hildesjo, C. , Larsson, M. , Lannfelt, L. , … Hallbeck, M. (2018). Alzheimer's disease pathology propagation by exosomes containing toxic amyloid‐β oligomers. Acta Neuropathologica, 136, 41–56. 10.1007/s00401-018-1868-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, D. H. , Chen, X. H. , Pierce, J. E. , Wolf, J. A. , Trojanowski, J. Q. , Graham, D. I. , & McIntosh, T. K. (1997). Progressive atrophy and neuron death for one year following brain trauma in the rat. Journal of Neurotrauma, 14, 715–727. 10.1089/neu.1997.14.715 [DOI] [PubMed] [Google Scholar]
- Sun, B. , Dalvi, P. , Abadjian, L. , Tang, N. , & Pulliam, L. (2017). Blood neuron‐derived exosomes as biomarkers of cognitive impairment in HIV. Aids, 31, F9–F17. 10.1097/QAD.0000000000001595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery, C. (2011). Exosomes: Secreted vesicles and intercellular communications. F1000 Biology Reports, 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery, C. , Zitvogel, L. , & Amigorena, S. (2002). Exosomes: Composition, biogenesis and function. Nature Reviews. Immunology, 2, 569–579. 10.1038/nri855 [DOI] [PubMed] [Google Scholar]
- Trajkovic, K. , Hsu, C. , Chiantia, S. , Rajendran, L. , Wenzel, D. , Wieland, F. , … Simons, M. (2008). Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science, 319, 1244–1247. 10.1126/science.1153124 [DOI] [PubMed] [Google Scholar]
- Waters, N. J. , Jones, R. , Williams, G. , & Sohal, B. (2008). Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. Journal of Pharmaceutical Sciences, 97, 4586–4595. 10.1002/jps.21317 [DOI] [PubMed] [Google Scholar]
- Weidle, U. H. , Birzele, F. , Kollmorgen, G. , & Ruger, R. (2017). The multiple roles of exosomes in metastasis. Cancer Genomics Proteomics, 14(1), 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westberry, J. M. , Trout, A. L. , & Wilson, M. E. (2010). Epigenetic regulation of estrogen receptor α gene expression in the mouse cortex during early postnatal development. Endocrinology, 151, 731–740. 10.1210/en.2009-0955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcockson, D. C. , Campbell, S. J. , Anthony, D. C. , & Perry, V. H. (2002). The systemic and local acute phase response following acute brain injury. Journal of Cerebral Blood Flow and Metabolism, 22, 318–326. 10.1097/00004647-200203000-00009 [DOI] [PubMed] [Google Scholar]
- Zhang, Q. G. , Wang, R. , Tang, H. , Dong, Y. , Chan, A. , Sareddy, G. R. , … Brann, D. W. (2014). Brain‐derived estrogen exerts anti‐inflammatory and neuroprotective actions in the rat hippocampus. Molecular and Cellular Endocrinology, 389, 84–91. 10.1016/j.mce.2013.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Selectivity of PDDC for nSMase2 vs. coupling enzymes in fluorescence Amplex Red assay ‐ pIC50 curves were generated in duplicate twelve‐point dose response curves using the Amplex red Fluorescence‐based assay. Lysate of cells expressing human recombinant nSMase2 (1.9 μg protein/50 μL) was incubated with sphingomyelin (20 μM) and detecting reagents (alkaline phosphatase 4 U/mL, choline oxidase 0.1 U/mL, horse radish peroxidase 0.1 U/mL, and Amplex Red 50 μM) in the presence of different concentrations of PDDC for 3 h (total reaction volume 50 μL). Percent inhibition vs. inhibitor concentration was plotted in GraphPad Prism. Data analysis and non‐linear least squares curve fitting were carried out with GraphPad Prism 7. Reagents in the counter screen to establish selectivity against coupling enzymes (alkaline phosphatase, choline oxidase, and horse radish peroxidase) were the same as for the pIC50 determination as for inhibition of nSMase2 except that nSMase2 was not present and phosphorylcholine (2 μM) was used as substrate for alkaline phosphatase. CI (95%): 95% asymptotic confidence interval; lower bound – upper bound
Table S1. Evaluation of PDDC at 10 μM in Eurofin's SafetyScreen 44
Table S2. Effect of PDDC on astrocyte viability as measured by lactate dehydrogenase (LDH) activity released into cell media – Primary astrocytes were treated in parallel incubations with PDDC using the same conditions as those used for the measurement of EV release from astrocytes (2h incubation up to 30 μM). Media was collected and used as LDH source to follow disappearance of NADH (1 mM) absorbance at 340 nM in the presence of sodium pyruvate (20 mM). Maximal LDH activity was obtained by using mammalian protein extraction reagent (M‐PER, ThermoScientific) and normalized to 100%
Figure S2. Extracellular Vesicle (EV) size in an in vivo model of brain injury – EVs were isolated from plasma 2 h following treatments with vehicle, IL‐1β, IL‐1β + PDDC (10 mg/kg IP) and IL‐1β + compound 5 (10 mg/kg IP). Plasma was collected and centrifuged at 2700 g for 15 min at 4°C. Supernatant was collected and size of EVs was measured using a Zetaview nanoparticle Tracker.