

Abstract
DNA metabarcoding is a sophisticated molecular tool that can enhance biological surveys of freshwater plankton communities by providing broader taxonomic coverage and, for certain groups, higher taxonomic resolution compared to morphological methods. We conducted 18S rRNA gene metabarcoding analyses on 214 water samples collected over a four-month period from multiple sites within a freshwater reservoir. We detected 1,314 unique operational taxonomic units that included various metazoans, protists, chlorophytes, and fungi. Alpha diversity differed among sites, suggesting local habitat variation linked to differing species responses. Strong temporal variation was detected at both daily and monthly scales. Diversity and relative abundance patterns for several protist groups (including dinoflagellates, ciliates, and cryptophytes) differed from arthropods (e.g., cladocerans and copepods), a traditional focus of plankton surveys. This suggests that the protists respond to different environmental dimensions and may therefore provide additional information regarding ecosystem status. Comparison of the sequence-based population survey data to conventional-based data revealed similar trends for taxa that were ranked among the most abundant in both approaches, although some groups were missing in each data set. These results highlight the potential benefit of supplementing conventional biological survey approaches with metabarcoding to obtain a more comprehensive understanding of freshwater plankton community structure and dynamics.
Keywords: Aquatic arthropods, Biological survey, Community dynamics, Protists
Introduction
Society is dependent on natural and man-made lakes to reliably provide a wide variety of ecosystem services, including drinking water, irrigation, fish stocks, hydroelectric energy storage, recreation sites/resources, and flood control (McMahon & Farmer, 2004). These services, in turn, depend on lake ecosystems being able to support diverse, balanced, and integrated aquatic communities (i.e., possessing biological integrity, sensu Karr, 1981). Perturbations that impact the ecology of freshwater lakes can lead to fish-killing hypoxia (Diaz, 2001), harmful algal blooms (Smayda, 2008), outbreaks of disease among humans and wildlife (Patz et al., 2004; Walker & Winton, 2010; Wilde et al., 2014), and obstruction of human resource use (Lynch et al., 2016). Some key priorities of lake management are to anticipate and mitigate these adverse outcomes, evaluate the effectiveness of management practices, and develop a better understanding of the mechanisms that drive lake dynamics (Holdren et al., 2001; USEPA, 2017). These goals rest on the ability to assess the status and trends of lake ecosystems, by monitoring potentially subtle changes in lake plankton community structure and dynamics indicative of environmental stressors such as pollution (Carpenter et al., 1998; Schulz, 2004), nuisance aquatic invasive species (Leung et al., 2002), and climate change (Wagner & Adrian, 2011). The impacts of pollution in particular, especially in the form of organic nitrogen and phosphorus and legacy toxics such as polychlorinated biphenyls, may be exacerbated in lake systems, as residence times are often much longer than in other aquatic habitats such as rivers and streams (Chang et al. 2012; Hayes et al. 2017).
Standard procedures for the biological survey of inland lakes typically focus on a common set of ecological indicators, such as important individual taxa (e.g., arthropods, diatoms, and green algae—Kentucky DEP, 2016; Indiana DEM, 2016) or measurements/indices of biodiversity (Jørgensen et al., 2016). Historically, biological monitoring has relied on identification of taxa via morphological traits. Morphological taxonomy can be effective for plankton monitoring but has limitations. For example, lack of diagnostic morphological characters can result in overly coarse or erroneous assessments of taxonomic diversity, especially among certain closely related organisms (Ko et al., 2013; Chan et al., 2014). In addition, for most taxa, the approach requires careful preparation of specimens to protect and showcase the characters, increasingly scarce taxonomic expertise (Hopkins & Freckleton, 2002; Tautz et al., 2003), and time-consuming inspection (Blaise & Férard, 2005). Many of the organisms amenable to morphological analysis are visible to the naked eye, such as fish (Naigaga et al., 2011), mayflies (Edsall et al., 2005), and cladocerans (Siciliano et al., 2015). Diatoms and other phytoplankton are also important ecological indicators in lake surveys but are not often tallied with high taxonomic resolution (Blinn & Herbst 2003; Edlund et al., 2011).
Molecular genetic techniques such as DNA metabarcoding can enable researchers to obtain more comprehensive profiles of aquatic communities quickly, economically, and with less dependence on taxonomic expertise (Rees et al., 2014; Trebitz et al., 2017). Such profiles would include groups of organisms that are difficult to monitor using morphological approaches yet potentially improve the discriminatory power of plankton community assessments. For example, microscopic heterotrophic protists and phytoplankton offer potential advantages over larger, slower-reproducing organisms in terms of their sensitivity to environmental change, spatial distribution, and response times (Xu et al., 2001; Sherr & Sherr, 2002; Fenchel, 2008). Moreover, molecular genetic techniques are amenable to automation, which may provide efficiencies even for organisms that can be readily identified via traditional microscopy (Emilson et al., 2017).
DNA sequence-based assessment of aquatic biodiversity has been available for more than three decades (Pace et al., 1986; Schmidt et al., 1991) and is considered routine within the field of microbiology (Yarza et al., 2014). 16S rRNA gene sequencing, in particular, has been useful in elucidating ecologically relevant patterns of spatial and temporal biodiversity among various prokaryotic groups (Techtmann et al., 2017). The 16S rRNA gene is useful for studies requiring deep taxonomic resolution across multiple lineages, being universally present in prokaryotes and showing high sequence homology among closely related phylogenetic groups (Woese & Fox, 1977). For eukaryotes, the analogous 18S rRNA gene has similar taxonomic attributes and has been used in a number of studies to census various eukaryotic groups including fish (Hänfling et al., 2016), chironomids (Bista et al., 2017), amphibians (Valentini et al., 2016), and diatoms (Vasselon et al., 2017). However, studies utilizing metabarcoding for the purpose of monitoring changes in the structure of diverse eukaryotic plankton communities over space and time are rare.
We utilized a DNA metabarcoding approach that allowed us to measure assemblage patterns among a wide diversity of eukaryotic groups, including arthropods, diatoms, green algae, heterotrophic protists, and fungi (Creer et al., 2010; Hadziavdic et al., 2014). Specifically, we performed 18S rRNA gene sequence analyses on water samples collected on multiple dates from multiple sites within Lake Harsha (Batavia, Ohio, USA), a eutrophic freshwater reservoir used for drinking water, flood control, and recreation. Our goal was to assess the utility of the metabarcoding approach for monitoring spatial and temporal dynamics of freshwater eukaryotic plankton in lake monitoring programs. Our data provide insights regarding the community dynamics of the system but also highlight some of the limitations of the metabarcoding approach that must be considered.
Materials and methods
Sampling of Lake Harsha
A total of 214 water samples were collected from May to September 2015, at five separate sampling sites within Lake Harsha (See Fig. 1). At three sites (LH1, LH2, and LH3), surface water grab samples were collected by boat using sterile 1-L sample bottles (Nalgene, Rochester, NY). At one location, equivalent volumes were collected using the same type of containers but siphoned from intake pipes for a drinking water plant that pump water from the surface (LH4-S) and from a fixed elevation that is generally ~ 6.1 m (20 feet) below the surface (LH4-D). The samples were collected on a weekly basis, except during a period of peak algal blooms (June 4–July 1, 2016), when sampling frequency was increased to multiple days a week. All samples were transported on ice to the U.S. Environmental Protection Agency research facility in Cincinnati, OH, within 2 h of collection. On five of the sampling dates, inclement weather prevented sampling of all sites except for those at the intake (LH4-S and LH4-D). On two other sampling dates, LH4-S samples could not be obtained due to a malfunction in the surface-water intake pipe.
Fig. 1.
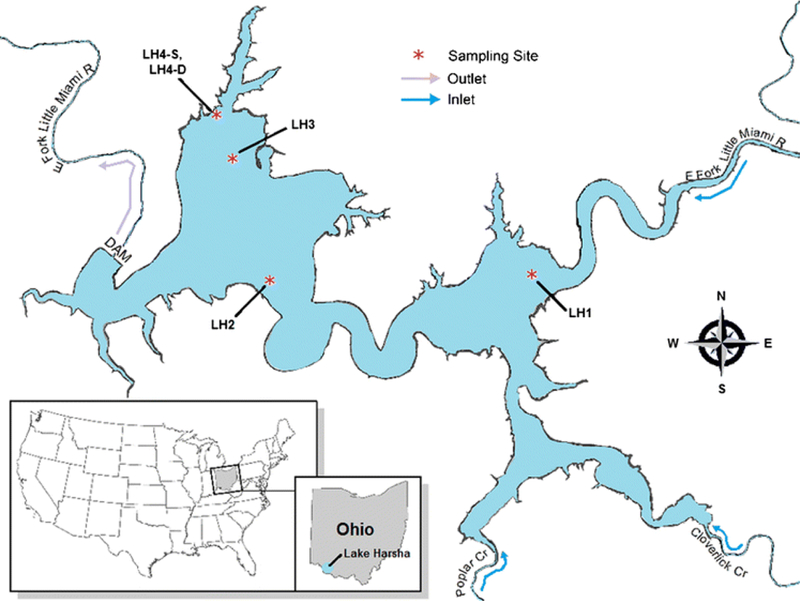
Map of Lake Harsha, Batavia, OH, showing the locations of the sampling sites. LH1 (lat. 39.022506, long. − 84.094618) is in the inlet basin of the lake. LH2 (lat. 39.02, long. − 84.1311) is at the entrance to the outlet basin of the lake, near the lake’s main beach area. LH3 (lat. 39.032506, long. − 84.137661) is in the outlet basin of the lake, near the dam and the intake tower for the Bob McEwen drinking water treatment plant. LH4-S and LH4-D (lat. 39.0367, long. − 84.1381) are at the intake tower, where samples were collected from the surface (LH4-S) and from a depth of ~ 7 m below the surface (LH4-D)
Sample processing and molecular analyses
From each 1-L water sample, a single 100-mL subsample was filtered onto a 0.4-μm-pore-size, 47-mm-diameter polycarbonate membrane (GE Osmonics). Membranes were transferred to sterile microcentrifuge tubes and stored at − 80 °C until subsequent manipulations were performed. Nucleic acid extractions and development of sequencing libraries were conducted as described elsewhere (Kapoor et al., 2016) with minor modifications. Briefly, nucleic acid was extracted directly from the filtered biomass (i.e., membranes) using AllPrep DNA/RNA Mini Kit (Qiagen GmbH, Hilden, Germany) following the manufacturer’s instructions. The concentration and purity of DNA were determined using Qubit dsDNA HS assay kits and the Qubit 2.0 Fluorometer (Life Technologies).
DNA extracts were used as PCR templates, and barcoded primers targeting the V4 region of the 18S rRNA gene (SSU_F04 GCTTGTCTCAAAGATTAAGCC and SSU_R22 GCCTGCTGCCTTCCTTGGA) were used to construct sequencing libraries for each sample tested. These primers were originally developed to target meiofaunal biodiversity based on NCBI sequence analysis but have been shown to have broad phylogenetic coverage (Creer et al., 2010). The reactions used to generate the sequencing libraries (i.e., one library per sample) were performed in 25-μL volumes using the Ex Taq kit (Takara) with 200 nM of forward and reverse primers and ~ 10 ng of DNA template. Cycling conditions consisted of an initial 5-min denaturing step at 94 °C, followed by 30 cycles of 45 s at 94 °C, 60 s at 50 °C, and 90 s at 72 °C and a final elongation step of 10 min at 72 °C. Agarose gel electrophoresis was used to confirm the size of the amplification products. PCR products for each of the samples were then pooled and size-selected (i.e., based on the range of expected size product) prior to multiplex sequencing on an Illumina Miseq benchtop sequencer using pair-end 250 bp kits at the Cincinnati Children’s Hospital DNA Core facility.
Read filtering and taxonomical assignments
Paired-end sequence reads were merged with USEARCH v9.2 (Edgar, 2010), and primer sequences were removed with Cutadapt v 1.14 (Martin, 2011). USEARCH was then used to filter low quality reads that contained one or more expected errors per sequence based on cumulative Phred scores. Unique sequences were identified, and those with < 4 observations in the total data set were removed. This threshold helped ensure that sequence errors did not lead to spurious results but likely removed some infrequent but valid biological sequences. Remaining sequences were then screened to remove chimeras and clustered into operational taxonomic units (OTUs) of 97% similarity or more, after which all quality-filtered reads were mapped to these OTUs.
Taxonomy was assigned to OTUs by comparing two reference databases using three different classification approaches. This was done primarily to enhance taxonomic resolution of protists, which appeared poorly represented in both databases. In the first approach, OTU sequences were classified using the SILVA reference database (SSU Ref NR 128, accessed April 7, 2017) with SINA Online (Pruesse et al., 2012) based on an identity threshold of 0.8. The remaining two approaches utilized the NCBI Genbank reference database (downloaded January 17, 2017), which was queried to subset nucleotide sequences with 80% or better identity to OTUs over the full sequence length using BLAST+ (Camacho et al., 2009) and Entrez Direct (Kans, 2017) tools. Matches to taxonomically uninformative environmental samples were filtered out using keyword searches (e.g., “uncultured eukaryote”). Remaining sequences were trimmed to the aligned length of the OTU and then taxonomy information was downloaded using Entrez efetch. USEARCH’s utax classifier (-utax option) was used on this database as the second taxonomy classification approach. For the third classification approach, reference sequences from this filtered NCBI Genbank database that had at least 98% full-length BLAST identity to OTUs were considered matches and tallied at each taxonomic rank (kingdom to species); the taxonomic string with the highest tally was assigned to the OTU. The fraction of matches to the assigned taxonomy versus other taxa at each taxonomic rank was used as an indicator of the strength of match.
To create a consensus taxonomy, the SINA classification was treated as the default classification since it employs the manually curated and routinely updated SILVA reference database (Quast et al., 2013; Yilmaz et al., 2014). In cases where either or both of the NCBI classification approaches were in agreement with the SILVA classification but provided more taxonomic precision, the NCBI classification was nominally accepted pending confirmation by phylogenetic analysis; in cases where the NCBI and SILVA classifications were in disagreement, the SILVA classification was used. Comparison of putative taxonomies to phylogenetic trees provided a check against overclassification and, in a few cases, provided further taxonomic resolution. Phylogenetic relationships among OTUs were estimated using Neighbor Joining in MEGA (Kumar et al., 2016) using the Kimura 2-parameter substitution model and compared to nominal taxonomic classifications. Taxonomic classifications that were outliers within phylogenetic groupings were reclassified at a higher taxonomic rank (reduced precision), if appropriate, or marked unclassified. Unclassified OTUs that were highly divergent (branch lengths > 0.5) from taxonomically classified OTUs were removed from the analysis as likely chimeric sequences.
Analyses of spatial and temporal biodiversity patterns
Alpha diversity was calculated using the Shannon–Wiener index. Differences among sites in terms of alpha diversity were evaluated using a Friedman rank sum test, controlling for temporal variation. Post hoc tests were constructed with the PMCMR package of R, version 3.4.0 (R Core Team, 2013), using Conover’s method, controlling for family wise error rate with the Holm–Bonferroni method. Beta diversity was measured using the Bray–Curtis dissimilarity statistic after converting OTU abundances to frequencies within samples. Patterns of beta diversity were assessed via non-metric multidimensional scaling ordination (NMDS), conducted using the software package PC-ORD v7.02 (McCune & Mefford, 2016). Additional NMDS ordinations were performed on subsets of the data corresponding to arthropods (classical indicator taxa) and several major protist clades to investigate differences in the distributions and population dynamics of these taxa. Strength and significance of spatial and temporal differences among samples were evaluated using permutational multivariate analysis of variance (PERMANOVA) of Bray–Curtis distances, using the command “adonis” in the R package “vegan” (Anderson 2001; Oksanen et al., 2017).
Results
Taxa detected
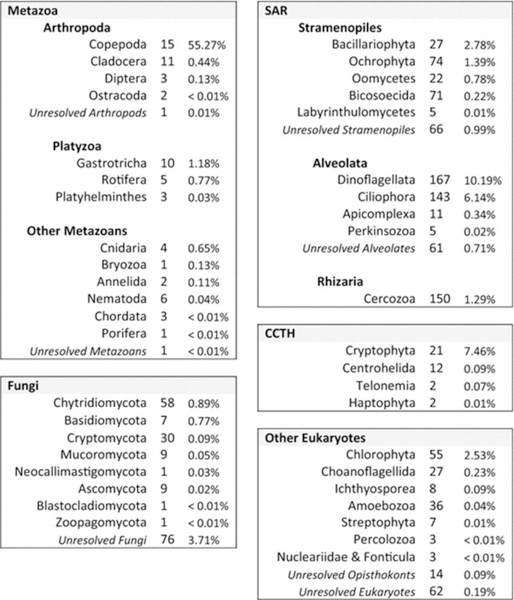
18S rRNA gene metabarcoding yielded 10,698,543 sequences (total across all samples), which were partitioned among 1,341 unique OTUs. Rarefaction curves estimated with USEARCH indicated that OTU detection saturated at subsample sizes of < 60% of the sample totals in all cases (Supplemental Material, Fig. S1; note that the rarefaction curves were influenced by the removal of OTUs with n < 4 prior to analysis). A total of 1,080 OTUs were taxonomically resolved using the SILVA reference database while 69 were further resolved by the USEARCH UTAX approach and 165 were further resolved by the BLAST identity approach. The remaining 27 unclassified OTUs were highly divergent from other OTUs and from each other in the Neighbor Joining tree and were excluded from analysis as likely chimeric sequences. Following their removal, the remaining 1314 OTUs were minimally classified with certainty to Eukaryota, although 62 OTUs could not be resolved beyond this level (Table 1). Further analysis of phylogenetic relationships in the tree revealed 33 inconsistent taxonomic classifications that were reclassified at a higher (less precise) taxonomic level. In addition, 50 OTUs that were poorly resolved by the classification algorithms but tightly grouped within specific phylogenetic clades were assigned the consensus taxonomy of OTUs in those clades.
Table 1.
Taxonomic groups detected via 18S rRNA gene metabarcoding. Listed beside each group are the number of OTUs found within the group (out of the total 1,314 OTUs) and the percent of DNA sequences that the group contributed to the grand total (10,698,543 sequences)
 |
Spatial and temporal biodiversity patterns
Alpha diversity measured by the Shannon–Wiener index differed significantly among sample sites within the lake (Friedman χ2 = 27.549, P < 0.001). Post hoc pairwise comparisons revealed that LH1 and LH4-D differed from each other and from all the other sites (Fig. 2A). Alpha diversity estimates for LH2, LH3, and LH4-S were not significantly different. Alpha diversity also varied significantly over the study period (Friedman χ2 = 63.643, P = 0.006; Fig. S2). Species richness (number of OTUs) showed patterns similar to Shannon–Wiener and varied significantly among sites and over time (P < 0.001).
Fig. 2.
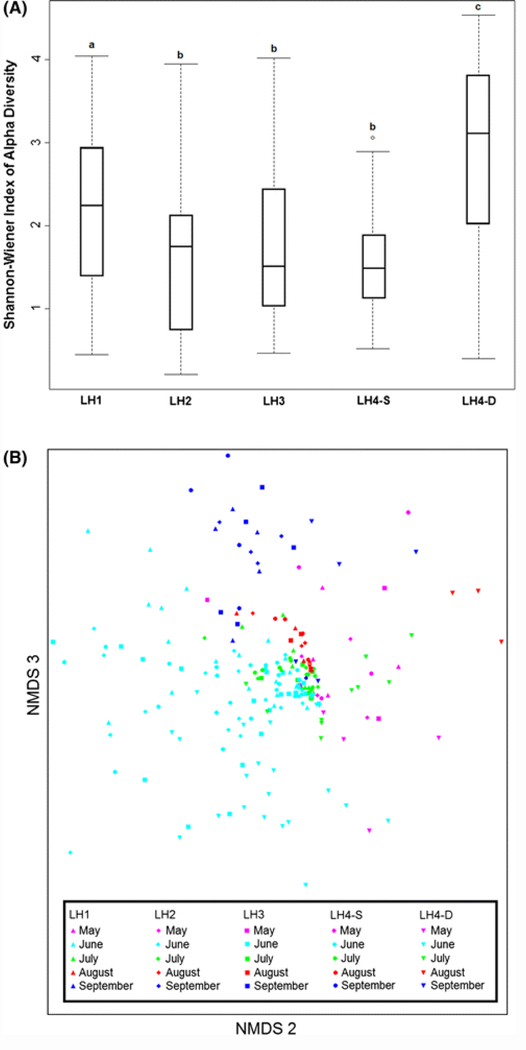
Alpha and beta diversity among lake samples. A Box-and-whisker plot comparing the Shannon–Wiener index values at each of the 5 sampling sites. B Reduced-space NMDS plot showing spatio-temporal variation in the planktonic community’s taxonomic composition based on Bray–Curtis dissimilarity (The best solution was a three-dimensional ordination; mean stress in relation to dimensionality = 11.823. Only dimensions 2 and 3 of the ordination are presented for illustrative purposes)
NMDS ordinations used to evaluate beta diversity patterns among all OTUs suggested that the overall taxonomic composition of LH4-D differed from other sites. In contrast to alpha diversity patterns, no clear differences between LH1 and other sites were apparent. In general, temporal variation (differences among sample months) had a much stronger impact on beta diversity patterns than site-to-site variation (Fig. 2B). Quantification of beta diversity patterns using PERMANOVA revealed no significant site-to-site variation. A slight (R2 = 0.07) but highly significant effect of month-to-month variation was observed; however, more variation was attributed to short-term (day-to-day) differences affecting all sites (R2 = 0.41).
Comparing beta diversity for different taxonomic groups, we found no discernible changes in community structure for arthropods over space and time, other than a few samples from LH4-D in August being distinct from all others (Fig. 3A). On the other hand, conspicuous changes in community structure among SAR groups such as stramenopiles were apparent (Fig. 3B). Figure 4 depicts changes over time in the relative abundances of the dominant arthropods, phytoplankton, and protozoa at each site. Copepods (Arthropoda) were the dominant planktonic eukaryote in all surface water samples (copepod sequences comprised > 20% of all sequences in each sample on most days and comprised > 90% of all sequences on multiple occasions). However, in the deep-water samples (LH4-D), copepod sequences generally comprised < 20% and often < 1% of the total. Declines in copepod relative abundance within the surface water samples appeared, in some cases, to coincide with increases in copepod relative abundance within the LH4-D samples. Cladocerans, the second most abundant arthropod group, only contributed < 10% (usually < 1%) of sequences at all sites throughout the study period. Several weeks into the study, the relative abundance of dinoflagellate sequences increased sharply in all surface water samples (from < 10% to ≥ 30% to > 80% in all but LH2), yet exhibited no such pattern at LH4-D. These spikes coincided with declines in copepod relative abundance (e.g., from ≥ 70% before May 28 to < 30% until June 8 at LH3). Also of note were the greater relative abundances of groups such as the ciliates (Ciliophora) at the deep site (LH4-D) as compared to the surface water sites and the peak relative abundances (40–50%) of phytoplankton such as cryptophytes and diatoms toward the start and end of the study period (Fig. 4).
Fig. 3.
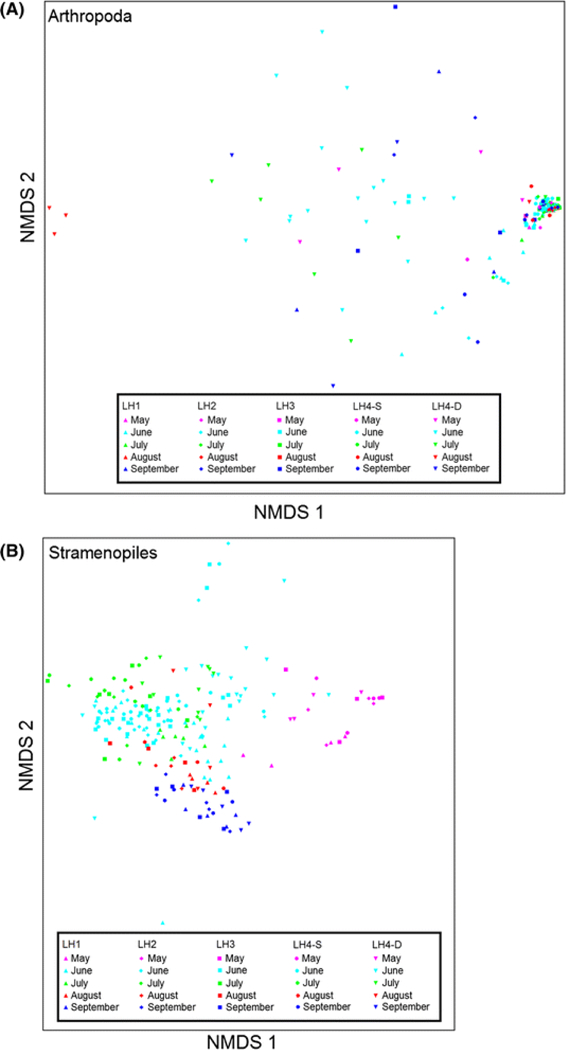
Reduced-space NMDS plots illustrating spatio-temporal relationships for A arthropods (classical indicators) and B stramenopiles (microbial eukaryotes) based on Bray–Curtis dissimilarity (The best solutions were two dimensional ordinations in both cases; mean stress in relation to dimensionality = 10.507 and 20.206, respectively)
Fig. 4.
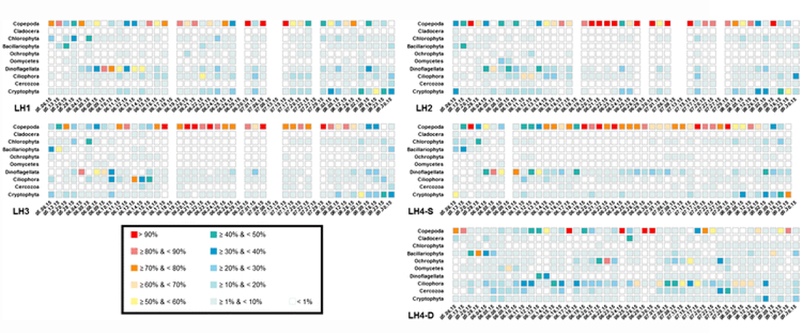
Time series of taxa that attained relative abundances ≥ 10% on at least one occasion at least one of the five sampling sites. These consisted of arthropods, phytoplankton, and heterotrophic protists, the two most dominant taxa being the copepods (Arthropoda) and dinoflagellates (SAR; see also Table 1)
Discussion
In this study, we used DNA metabarcoding to describe the spatial and temporal dynamics of planktonic eukaryotic assemblages in a freshwater reservoir over a four-month period. We were able to characterize diversity patterns among arthropods, fungi, green algae, and protist groups. While we observed that the planktonic community composition (as measured by the different phyla identified) was similar in different study sites, distributional patterns varied both over time and among various locations within the lake, suggesting that the groups identified responded to different environmental cues. Our results are based on analysis of relative abundance of 18S rRNA gene sequences for each taxon identified. Theoretically, relative abundances of DNA sequences may provide a reasonable measure of the relative biomass for many taxa (Gihring et al., 2012; Lovell et al., 2015). Indeed, good correlations between metabarcoding results and traditional approaches to measuring biomass have been observed (Kelly et al., 2014; Pochon et al., 2015). However, differences in 18S rRNA gene copies per genome, number of cells and organism biovolume during different developmental stages can introduce some important artifacts that may impact relative density estimates (Leray and Knowlton, 2016). While relative abundances of gene sequences are imprecise measures of population sizes, we assumed that these biases do not greatly affect intra-taxon comparisons among samples and that it is then possible to use these data to examine comparative differences in identified taxa among different sites and sampling dates.
Effective monitoring relies on detecting and accurately identifying the different organisms and their abundance in a sample. The number of taxa identified in our study is higher than other surveys conducted in this lake using microscopy-based methods (Chen et al., 2017, for phytoplankton). This is despite reducing the number of OTUs by excluding sequences that were present less than four times in our sequencing library. The latter is necessary to eliminate artifacts introduced by sequencing errors (Pawlowski et al., 2016). It should be noted that metabarcoding data are often dominated by low-abundance OTUs (Simon et al., 2015), and reducing artifacts due to sequencing errors comes at the expense of eliminating bona fide taxa that form part of the “rare biosphere” (Sogin et al., 2006; Bachy et al., 2013). However, we expected to see greater diversity with the metabarcoding approach as some of the taxa identified cannot be easily discriminated by morphological analysis (Pawlowski et al., 2016).
Metabarcoding-based detection relies on efficient amplification of the different eukaryotic groups within a sample. To determine potential biases of the primer set used in our study, we compared the population dynamics of taxa that were ranked most abundant in our metabarcoding study with results obtained from other independent studies using morphological analyses. For the zooplankton, the comparisons were made to data collected in May, June, and August of 2016 using plankton nets (6-m vertical draw, 150-μm mesh) for three different sites (LH1, LH2, LH4; US Army Corps of Engineers unpublished data). For the phytoplankton comparison, we used data reported by Chen et al. (2017) for surface water samples collected from the same Lake Harsha sites during the same sampling period. Overall, the comparisons revealed similar population abundance trends among some taxa. In comparison to the plankton net draws, both methods showed that calanoid copepods were the most dominant taxa for each of the three months (calanoid copepods contributing 83–98% of the DNA sequences and 18–57% of the total number of individual specimens identified via morphology) and that ostracods and rotifers were relatively rare across all sites throughout time (< 1–7%). While both methods identified cyclopoids and cladocerans, their relative abundance was several times higher when estimated via morphological analyses. This may reflect the vertical distribution of these organisms during collection time (i.e., lower in the water column during the day), the integrated depth profile because of using the plankton nets, the exclusion of other eukaryotic taxa due to the plankton net pore size, and differences in the number of cells per organism and 18S rRNA gene copy number per genome (White & McLaren, 2000). Interannual variation in plankton communities may have also played a role. While the focus of the above comparison was on larger-size zooplankton, several studies have also observed similar diversity of freshwater microbial eukaryotes using morphological and metabarcoding analyses (Kammerlander et al., 2015; Debroas et al., 2017; Piredda et al., 2017),
In comparison to phytoplankton data (Chen et al., 2017), dinoflagellates (Dinophyta/Dinoflagellata) and diatoms (Bacillariophyta) were observed in similar relative abundance (cyanobacteria were excluded in the analysis as we used eukaryote-specific 18S rRNA gene primers). Specifically, dinoflagellates and diatoms comprised approximately 44 and 12%, respectively, of the total number of phytoplankton sequences while these groups were estimated to comprise approximately 59 and 9% of the total phytoplankton biomass based on morphological analysis. Temporal changes in abundance in each of these two groups were also qualitatively similar (e.g., both methods detected a sharp increase in dinoflagellate abundance in June and maximum diatom abundance in May). Moreover, monthly trends were different for green algae (Chlorophyta) and cryptophytes (Cryptophyta), with metabarcoding consistently reporting lower relative abundances of green algae and higher relative abundances of cryptophytes. Potential amplification biases due to primer mismatches and differences in rRNA gene copy numbers may also explain some of these differences.
We also noted differences in taxonomic coverage between metabarcoding- and microscopy-based approaches. While we detected several genera within the eukaryotic phytoplankton phyla reported by Chen et al. (2017), specifically, Cyclotella (Bacillariophyta), Skeletonema (previously classified as Melosira, Bacillariophyta), Cryptomonas (Cryptophyta), Ceratium (Dinophyta/Dinoflagellata), and Peridinium (Dinophyta/Dinoflagellata), we did not detect genera such as Euglena (Euglenophyta) and Chlamydomonas (Chlorophyta). The lack of detection of Euglena can be explained by the poor sequence match of the sequencing primers used (particularly the forward primer). The case of Chlamydomonas is more intriguing as both primers have perfect matches with publicly available Chlamydomonas sequences. This organism is unicellular and contains a low number of rRNA gene copies per nuclear genome (Merchant et al., 2007), which in great part explains its absence from our sequencing libraries. On the other hand, metabarcoding detected several eukaryotic phytoplankton phyla that were not reported by Chen et al. (i.e., Colponemidia, Eustigmatophyta, Haptophyta, Ochrophyta, Silicoflagellata, and Streptophyta), including 14 phytoplankton that we were able to assign genus-level identification: Thalassiosira (Bacillariophyta); Tetraselmis and Nephroselmis (Chlorophyta); Goniomonas (Cryptophyta); Azadinium, Gymnodinium, Oxyrrhis, Peridiniopsis, Scrippsiella, and Spiniferodinium (Dinoflagellata); Chrysochromulina (Haptophyta); Paraphysomonas and Mallomonas (Ochrophyta); Closterium (Streptophyta). Some of these taxa are relatively rare and morphologically cryptic (e.g., Goniomonas, Paraphysomonas) or endosymbiotic in nature and therefore simply easy to overlook amidst other organisms and debris within samples. Overall, these findings demonstrate that metabarcoding can yield comparable results to conventional methods for several abundant eukaryotic taxa, but that each method has different limitations as far as accurately describing the eukaryotic composition in this lake.
The 18S rRNA gene metabarcoding approach revealed extensive within-clade diversity and was able to detect distinct seasonal and spatial community patterns for taxa not typically amenable to such analysis using traditional methods. Among these different phylogenetic groups, we found distinct distributional patterns that suggest they responded to different environmental cues in the lake environment. Biodiversity patterns were strongly structured temporally, as reflected both in alpha and beta diversity measures. Temporal differences explained about half of beta diversity in the PERMANOVA analysis. Surprisingly, long-term (monthly) trends explained only a small percentage of the differences among samples. Instead, short-term (day-to-day) variation explained most temporal variation in beta diversity. A similar temporal pattern was observed for alpha diversity, with large changes in diversity occurring over relatively short time intervals but no clear long-term trend. Nonetheless, typical seasonal changes such as spikes in the abundances of green algae and diatoms at the beginning and end of the study period were apparent (Fig. 4). It is not clear what factors drove the rapid changes in community composition, as they could be linked to many pulse disturbances in the environment (e.g., storms, cloud cover, wind, or changes in the community composition and activity of prokaryotes).
Consistent differences in biodiversity patterns likely reflect ecologically relevant differences between sites. It should be noted that consistent differences in biodiversity patterns between LH4-D and other sites are not surprising, and likely reflect niche differences related to effects of depth-related factors such as light penetration, dissolved oxygen, temperature (LH4-D often being below the thermocline), food resources, and (for certain taxa) availability of competitor or predator-free space. The larger alpha diversity of LH1 compared to other surface sites is, however, perhaps more interesting. In relation to other sites, LH1 is near a more forested area and a primary influent stream to the lake. LH1 also is reported to have higher concentrations of phosphorus during the study period than other sites (Chen et al. 2017). Subsequently, the alpha diversity differences we observed could easily reflect a species filtering effect as organisms disperse further from lake influents. That neither the NMDS ordinations nor the PERMANOVA detected a strong spatial beta diversity signal suggests that spatial differences in eukaryotic plankton alpha diversity was more the result of selection by local environmental conditions (filtering) than dispersal (spatial reconfiguration of plankton communities; see Havel & Shurin, 2004 for a review). This in turn suggests that alpha diversity of eukaryotic plankton based on 18S rRNA gene metabarcoding could be a useful indicator for discriminating ecological condition.
Regarding group-specific patterns, arthropods (predominantly copepods) were highly abundant throughout most of the sampling period, whereas green algae (Chlorophyta), cryptophytes, and diatoms (Bacillariophyta) were most abundant near the start and end of the study period. The latter three taxa are obligate phototrophs and thus may be responding to environmental conditions that promote photosynthesis (e.g., increased availability of photosynthetically active radiation) or to herbivory by heterotrophs such as copepods and ciliates (Ciliophora). However, dinoflagellates, which are predominantly mixotrophic (capable of engaging in photosynthesis and heterotrophy simultaneously), did not exhibit the same pattern. Dinoflagellate peak abundance lagged that of the phototrophs. Dinoflagellates are known to feed on diatoms and may mediate competition among diatoms and other phytoplankton (Buskey, 1997; Jeong et al., 2004; Sherr & Sherr, 2007). Ciliates and cercozoans often comprised 20–40% of the LH4-D samples, which is surprising at first glance, given low availability of light and oxygen at this location, but they and other protozoa have been found to have remarkable tolerance to these conditions due to their ability to feed on bacteria and smaller protists (Finlay, 1981). Various fungi and other eukaryotes with generally low relative abundances likely also contributed to differences over time and among sites.
Our data were generated using a primer set that targets the V4 region of the 18S rRNA gene, a region that has been determined to be adequate for biodiversity assessments (Hadziavdic et al., 2014). Targeting a different region of the 18S rRNA gene is an option, but studies have shown that each region might introduce some biases and as a result the use of multiple primers is recommended (Bradley et al., 2016). As 18S rRNA gene sequence databases are expanded with taxonomically curated voucher specimens, particularly from taxonomic groups that are currently poorly represented, the utility of the overall approach herein described will significantly increase. Similar arguments have been made for other genes (i.e., CO1, rcbL), suggesting that additional sequencing data is needed to understand the different limitations of each gene locus (Yang et al., 2017). Despite these limitations, we identified several groups that were not described when morphological analysis was used (Chen et al., 2017), which is in agreement with comparative studies assessing the diversity of microbial eukaryotes (Debroas et al., 2017; Rimet et al., 2018).
Notwithstanding current sequencing data gaps, the capability to examine thousands of samples in a matter of days to weeks is presently viable via the use of dual-indexing primers and high throughput sequencing technologies (i.e., Illumina MiSeq and HiSeq). Moreover, as future studies provide additional data on the differences in 18S rRNA gene copy number per genome (information that is unknown for the vast majority of aquatic eukaryotic species), the value and use of sequencing-based analysis for estimating the relative abundance of taxonomic groups in aquatic systems will likely increase (Stoddard et al., 2014; Roller et al., 2016). From an environmental management standpoint, metabarcoding provides the opportunity to integrate traditional and emerging approaches to develop molecular biotic indices that can more rapidly, frequently, and robustly be used in water quality assessments (Pawlowski et al., 2016; Keck et al., 2017) and in the detection of non-indigenous species (Bucklin et al., 2016) across a larger number of geographic locations. From an ecological standpoint, molecular biodiversity surveys can also improve our understanding of the biological interactions at different trophic levels (Wurzbacher et al., 2017), and help us identify physiologically active organisms (i.e., when coupling both RNA and DNA pools as PCR templates—Wurzbacher et al., 2017; Lepère et al., 2016) and novel evolutionary radiations (Yi et al., 2017).
In summary, the results from our study further highlight the value of incorporating metabarcoding data into lake biological monitoring efforts to encompass a broader diversity of organisms. We believe that this added taxonomic breadth is valuable for lake management as different taxa may react to environmental conditions in very different ways. As processing times are shortened by metabarcoding, sampling frequency and spatial coverage could be increased, providing an ability to more comprehensively assess lake biological processes and ecological trends that are relevant to ecological health/vulnerability and enhance our understanding of lake processes. Using multiple genetic markers should enhance detection and taxonomic resolution of a wider diversity of planktonic microbial eukaryotes. The combined use of morphology and phylogenetically informative genetic markers will make plankton monitoring data more broadly useful in answering questions such as what level of genetic diversity is relevant to the overall functional stability of a system, how to discriminate between rare species and potential sequencing artifacts, how abundant and rare taxa are dispersed at different geographic scales, and how different populations of closely related taxa react to different perturbations.
Supplementary Material
Acknowledgements
We gratefully acknowledge Joel Allen, Mia Varner, Dana Macke, Kit Daniels, and Armah de la Cruz for their role in collecting, transporting and processing the lake water samples used in this study and Chris Nietch and Jade Young for their roles in providing the USACE zooplankton data. The U.S. Environmental Protection Agency, through its Office of Research and Development, partially funded and participated in the research described herein. Any opinions expressed in this paper are those of the authors and do not necessarily reflect the views of the agency; therefore, no official endorsement should be inferred. Any mention of trade names or commercial products does not constitute endorsement or recommendation for use.
References
- 1.Anderson MJ, 2001. A new method for non-parametric multivariate analysis of variance. Austral Ecology 26: 32–46. [Google Scholar]
- 2.Bachy C,Dolan JR, López-García P,Deschamps P & David Moreira, 2013. Accuracy of protist diversity assessments: morphology compared with cloning and direct pyrosequencing of 18S rRNA genes and ITS regions using the conspicuous tintinnid ciliates as a case study. The ISME Journal 7: 244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bista I,Carvalho GR, Walsh K,Seymour M,Hajibabae M,Lallias D,Christmas M & Creer S,2017. Annual time-series analysis of aqueous eDNA reveals ecologically relevant dynamics of lake ecosystem biodiversity. Nature Communications 8: 14087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blaise C & Férard JF, 2005. Small-Scale Freshwater Toxicity Investigations, Vol. 1 Springer, Berlin: 1–68. [Google Scholar]
- 5.Blinn, D. W. & D. B. Herbst, 2003. Use of diatoms and soft algae as indicators of environmental determinants in the Lahontan Basin, USA. Annual Report for California State Water Resources Board Contract Agreement 704558.01.CT766.
- 6.Bradley IM, Pinto AJ & Guest JS, 2016. Design and evaluation of Illumina MiSeq-compatible, 18S rRNA gene-specific primers for improved characterization of mixed phototrophic communities. Applied & Environmental Microbiology 82: 5878–5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bucklin A,Lindeque PK, Rodriguez-Ezpeleta N,Albaina A,& Lehtiniemi M,2016. Metabarcoding of marine zooplankton: prospects, progress and pitfalls. Journal of Plankton Research 38: 393–400. [Google Scholar]
- 8.Buskey EJ, 1997. Behavioral components of feeding selectivity of the heterotrophic dinoflagellate Protoperidinium pellucidum. Marine Ecology Progress Series 153: 77–89. [Google Scholar]
- 9.Camacho C,Coulouris G,Avagyan V,Ma N,Papadopoulos J,Bealer K,& Madden TL, 2009. BLAST+: architecture and applications. BMC Bioinformatics 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carpenter SR, Caraco NF, Correll DL, Howarth RW, Sharpley AN & Smith VH, 1998. Nonpoint pollution of surface waters with phosphorous and nitrogen. Ecological Applications 8: 559–568. [Google Scholar]
- 11.Chan A,Chiang L-P, Hapuarachchi HC, Tan C-H, Pang S-C, Lee R,Lee K-S, Ng L-C & Lam-Phua S-G, 2014. DNA barcoding: complementing morphological identification of mosquito species in Singapore. Parasites & Vectors 7: 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang F,Pagano JJ, Crimmins BS, Milligan MS, Xia X,Hopke PK, & Holsen TM, 2012. Temporal trends of polychlorinated biphenyls and organochlorine pesticides in Great Lakes fish, 1999–2009. Science of The Total Environment 439: 284–290. [DOI] [PubMed] [Google Scholar]
- 13.Chen K,Allen J & Lu J,2017. Community structures of phytoplankton with emphasis on toxic cyanobacteria in an Ohio inland lake during bloom season. Journal of Water Resources and Protection 9: 1299–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Creer S,Fonseca VG, Porazinska DL, Giblin-Davis RM, Sung W,Power DM, Packer M,Carvalho GR, Blaxter ML, Lambshead PJ & Thomas WK, 2010. Ultrasequencing of the meiofaunal biosphere: practice, pitfalls and promises. Molecular Ecology 19: 4–20. [DOI] [PubMed] [Google Scholar]
- 15.Debroas D,Domaizon I,Humbert JF, Jardillier L,Lepère C,Oudart A & Taib N,2017. Overview of freshwater microbial eukaryotes diversity: a first analysis of publicly available metabarcoding data. FEMS Microbiology Ecology. 10.1093/femsec/fix023. [DOI] [PubMed] [Google Scholar]
- 16.Diaz RJ, 2001. Overview of hypoxia around the world. Journal of Environmental Quality 30: 275–281. [DOI] [PubMed] [Google Scholar]
- 17.Edsall TA, Bur MT, Gorman OT & Schaeffer JS, 2005. Burrowing mayflies as indicators of ecosystem health: status of populations in western Lake Erie, Saginaw Bay and Green Bay. Aquatic Ecosystem Health & Management 8: 107–116. [Google Scholar]
- 18.Edgar RC, 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461. [DOI] [PubMed] [Google Scholar]
- 19.Edlund MB, Ramstack JM, Engstrom DR, Elias JE, & Lafrancois BM, 2011. Biomonitoring using diatoms and paleolimnology in the western Great Lakes national parks Natural Resource Technical Report NPS/GLKN/NRTR—2011/447. National Park Service, Fort Collins, Colorado. [Google Scholar]
- 20.Emilson CE, Thompson DG, Venier LA, Porter TM, Swystun T,Chartrand D,Capell S,& Hajibabaei M.. 2017. DNA metabarcoding and morphological macroinvertebrate metrics reveal the same changes in boreal watersheds across an environmental gradient. Scientific Reports 7, Article number: 12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fenchel T,2008. The microbial loop—25 years later. Journal of Experimental Marine Biology and Ecology 366: 99–103. [Google Scholar]
- 22.Finlay BJ, 1981. Oxygen availability and seasonal migrations of ciliated protozoa in a freshwater lake. Microbiology 123: 173–178. [Google Scholar]
- 23.Gihring TM, Green SJ & Schadt CW, 2012. Massively parallel rRNA gene sequencing exacerbates the potential for biased community diversity comparisons due to variable library sizes. Environmental Microbiology 14: 285–290. [DOI] [PubMed] [Google Scholar]
- 24.Hadziavdic K,Lekang K,Lanzen A,Jonassen I,Thompson EM & Troedsson C,2014. Characterization of the 18S rRNA gene for designing universal eukaryote specific primers. PLoS ONE 9: e87624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hänfling B,Lawson HL, Read DS, Hahn C,Li J,Nichols P,Blackman RC, Oliver A & Winfield IJ, 2016. Environmental DNA metabarcoding of lake fish communities reflects long-term data from established survey methods. Molecular Ecology 25: 3101–3119. [DOI] [PubMed] [Google Scholar]
- 26.Harrell FE Jr., Dupont C,& others. 2017. Hmisc: Harrell Miscellaneous. R package version 4.0–3. https://CRAN.R-project.org/package=Hmisc. [Google Scholar]
- 27.Havel JE & Shurin JB, 2004. Mechanisms, effects, and scales of dispersal in freshwater zooplankton. Limnology and Oceanography 49: 1229–1238. [Google Scholar]
- 28.Hayes NM, Deemer BR, Corman JR, Razavi NR, & Strock KE, 2017. Key differences between lakes and reservoirs modify climate signals: A case for a new conceptual model. Limnology and Oceanography Letters 2: 47–62. [Google Scholar]
- 29.Holdren C,Jones W,& Taggart J,2001. Managing lakes and reservoirs. North American Lake Management Society and Terrene Institute, in cooperation with Office of Water, Assessment and Watershed Protection Division, Madison, WI. [Google Scholar]
- 30.Hopkins GW & Freckleton RP, 2002. Declines in the numbers of amateur and professional taxonomists: implications for conservation. Animal Conservation Forum 5: 245–249. [Google Scholar]
- 31.Indiana Department of Environmental Management. 2016. Indiana’s 2016 consolidated assessment and listing methodology (CALM). http://www.in.gov/idem/nps/files/ir_2016_report_apndx_l_attch_1.pdf. Accessed 19 July 2017.
- 32.Jeong HJ, Yoo YD, Kim ST & Kang NS, 2004. Feeding by the heterotrophic dinoflagellate Protoperidinium bipes on the diatom Skeletonema costatum. Inter-Research Aquatic Microbial Ecology 36: 171–179. [Google Scholar]
- 33.Jørgensen SE, Xu F-L & Costanza R,2016. Handbook of ecological indicators for assessment of ecosystem health, 2nd ed. CRC Press Taylor & Francis Group, New York. [Google Scholar]
- 34.Kammerlander B,Breiner H-W, Filker S,Sommaruga R,Sonntag B & Stoeck T,2015. High diversity of protistan plankton communities in remote high mountain lakes in the European Alps and the Himalayan mountains. FEMS Microbiology Ecology 91(4): fiv010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kans J,2017. Entrez Direct: E-utilities on the UNIX Command Line. https://www.ncbi.nlm.nih.gov/books/NBK179288/?report=reader#!po=0.574713. Accessed 12 July 2017.
- 36.Kapoor V,Elk M,Li X,Impellitteri CA & Santo Domingo JW, 2016. Effects of Cr(III) and Cr(VI) on nitrification inhibition as determined by SOUR, function-specific gene expression and 16S rRNA sequence analysis of wastewater nitrifying enrichments. Chemosphere 147: 361–367. [DOI] [PubMed] [Google Scholar]
- 37.Karr JR, 1981. Assessment of biotic integrity using fish communities. Fisheries 6: 21–27. [Google Scholar]
- 38.Keck F,Vasselon V,Tapolczai K,Rimet F,& Bouchez A,2017. Freshwater biomonitoring in the Information Age. Frontiers in Ecology and the Environment 15: 266–274. [Google Scholar]
- 39.Kelly RP, Port JA, Yamahara KM & Crowder LB, 2014. Using environmental DNA to census marine fishes in a large mesocosm. PLoS ONE 9: e86175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kentucky Department for Environmental Protection, Division of Water, 2016. Standard operating procedures. http://water.ky.gov/Pages/SurfaceWaterSOP.aspx.
- 41.Ko H-L, Wang Y-T, Chiu T-S, Lee M-A, Leu M-Y, Chang K-Z, Chen W-Y & Shao K-T, 2013. Evaluating the accuracy of morphological identification of larval fishes by applying DNA barcoding. PLoS ONE 8: e53451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar S,Stecher G & Tamura K,2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular Biology and Evolution 33: 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leray M & Knowlton N,2016. Censusing marine eukaryotic diversity in the twenty-first century. Philosophical Transactions of the Royal Society B: Biological Sciences 371: 20150331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lepère C,Doimazon I,Hugoni M,Vellet A & Debroas D,2016. Diversity and dynamics of active small microbial eukaryotes in the anoxic zone of a freshwater meromictic lake (Pavin, France). Frontiers in Microbiology 7: 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leung B,Lodge DM, Finnoff D,Shogren JF, Lewis MA & Lamberti G,2002. An ounce of prevention or a pound of cure: bioeconomic risk analysis of invasive species. Proceedings of the Royal Society B—Biological Sciences 269: 2407–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lovell D,Pawlowsky-Glahn V,Egozcue JJ, Marguerat S & Bähler J,2015. Proportionality: a valid alternative to correlation for relative data. PLOS Computational Biology. 10.1371/journal.pcbi.1004075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lynch AJ, Cooke SJ, Deines AM, Bower SD, Bunnell DB, Cowx IG, Nguyen VM, Nohner J,Phouthavong K,Riley B,Rogers MW, Taylor WW, Woelmer W,Youn S-J & Beard TD Jr., 2016. The social, economic, and environmental importance of inland fish and fisheries. Environmental Reviews 24: 115–121. [Google Scholar]
- 48.Martin M,2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet Journal 17: 10–12. [Google Scholar]
- 49.McCune B,& Mefford MJ, 2016. PC-ORD: multivariate analysis of ecological data; Version 7.02. [Google Scholar]
- 50.McMahon GF, & Farmer MC, 2004. Reallocation of federal multipurpose reservoirs: principles, policy and practice. Proceedings of the 2003 Georgia Water Resources Conference, held April 23–24, 2003, at the University of Georgia Hatcher Kathryn J., editor, Institute of Ecology, The University of Georgia, Athens, Georgia. [Google Scholar]
- 51.Merchant SS, Prochnik SE, Vallon O,Harris EH, Karpowicz SJ, Witman GB, Terry A,Salamov A,Fritz-Laylin LK, Maréchal-Drouard L,Marshall WF, Qu L-H, Nelson DR, Sanderfoot AA, Spalding MH, Kapitonov VV, Ren Q,Ferris P,Lindquist E,Shapiro H,Lucas SM, Grimwood J,Schmutz J,Cardol P,Cerutti H,Chanfreau G,Chen C-L, Cognat V,Croft MT, l Dent R,Dutcher S,Fernández E,Fukuzawa H,González-Ballester D,González-Halphen D,Hallmann A,Hanikenne M,Hippler M,Inwood W,Jabbari K,Kalanon M,Kuras R,Lefebvre PA, Lemaire SD, Lobanov AV, Lohr M,Manuell A,Meier I,Mets L,Mittag M,Mittelmeier T,Moroney JV, Moseley J,Napoli C,Nedelcu AM, Niyogi K,Novoselov SV, Paulsen IT, Pazour G,Purton S,Ral J-P, Riaño-Pachón DM, Riekhof W,Rymarquis L,Schroda M,Stern D,Umen J,Willows R,Wilson N,Lana Zimmer S,Allmer J,Balk J,Bisova K,Chen C-J, Elias M,Gendler K,Hauser C,Rose Lamb M,Ledford H,Long JC, Minagawa J,Page MD, Pan J,Pootakham W,Roje S,Rose A,Stahlberg E,Terauchi AM, Yang P,Ball S,Bowler C,Dieckmann CL, Gladyshev VN, Green P,Jorgensen R,Mayfield S,Mueller-Roeber B,Rajamani S,Sayre RT, Brokstein P,Dubchak I,Goodstein D,Hornick L,Huang YW, Jhaveri J,Luo Y,Martínez D,Chi W,Ngau A,Otillar B,Poliakov A,Porter A,Szajkowski L,Werner G,Zhou K,Grigoriev IV, Rokhsar DS, & Grossman AR, 2007. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318: 245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Naigaga I,Kaiser H,Muller WJ, Ojok L,Mbabazi D,Magezi G,& Muhumuza E,2011. Fish as bioindicators in aquatic environmental pollution assessment: a case study in Lake Victoria wetlands, Uganda. Physics and Chemistry of the Earth 36: 918–928. [Google Scholar]
- 53.Oksanen J,Blanchet FG, Friendly M,Kindt R,Legendre P,McGlinn D,Minchin PR, O’Hara RB, Simpson GL, Solymos P,Stevens MHH, Szoecs E,& Wagner H,2017. vegan: Community Ecology Package. R package version 2.4–3. https://CRAN.R-project.org/package=vegan. [Google Scholar]
- 54.Pace NR, Stahl DA, Lane DJ, & Olsen GJ, 1986. The analysis of natural microbial populations by ribosomal RNA sequences In Advances in Microbial Ecology (pp. 1–55). Springer, USA. [Google Scholar]
- 55.Patz JA, Daszak P,Tabor GM, Aguirre AA, Pearl M,Epstein J,Wolfe ND, Kilpatrick AM, Foufopoulos J,Molyneux D,Bradley DJ & Members of the Working Group on Land Use Change Disease Emergence, 2004. Unhealthy landscapes: policy recommendations on land use change and infectious disease emergence. Environmental Health Perspectives 112: 1092–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pawlowski J,Lejzerowicz F,Apotheloz-Perret-Gentil L,Visco J & Esling P,2016. Protist metabarcoding and environmental biomonitoring: time for change. European Journal of Protistology 55: 12–25. [DOI] [PubMed] [Google Scholar]
- 57.Piredda R,Tomasino MP, D’Erchia AM, Manzari C,Pesole G,Montresor M,Kooistra WHCF, Sarno D & Zingone A,2017. Diversity and temporal patterns of planktonic protist assemblages at a Mediterranean Long Term Ecological Research site. FEMS Microbiology Ecology. 10.1093/femsec/fiw200. [DOI] [PubMed] [Google Scholar]
- 58.Pochon X,Wood SA, Keeley NB, Lejzerowicz F,Esling P,Drew J & Pawlowski J,2015. Accurate assessment of the impact of salmon farming on benthic sediment enrichment using foraminiferal metabarcoding. Marine Pollution Bulletin 100: 370–382. [DOI] [PubMed] [Google Scholar]
- 59.Pruesse E,Peplies J,& Glöckner FO, 2012. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28: 1823–1829. (Online alignment tool and search engine accessed via https://www.arb-silva.de/aligner/). Accessed 3 June 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rimet F,Vasselon V,Keszte BA & Bouchez A,2018. Do we similarly assess diversity with microscopy and high-throughput sequencing? Case of microalgae in lakes. Organisms Diversity & Evolution 8: 1–12. [Google Scholar]
- 61.Quast C,Pruesse E,Yilmaz P,Gerken J,Schweer T,Yarza P,Peplies J & Glöckner FO, 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research 41: D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.R Core Team, 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- 63.Rees HC, Maddison BC, Middleditch DJ, Patmore JRM & Gough KC, 2014. The detection of aquatic animal species using environmental DNA—a review of eDNA as a survey tool in ecology. Journal of Applied Ecology 51: 1450–1459. [Google Scholar]
- 64.Roller BRK, Stoddard SF & Schmidt TM, 2016. Exploiting rRNA operon copy number to investigate bacterial reproductive strategies. Nature microbiology 1(11): 16160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmidt TM, DeLong EF & Pace NR, 1991. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. Journal of Bacteriology 173: 4371–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schulz R,2004. Field studies on exposure, effects, and risk mitigation of aquatic nonpoint-source insecticide pollution. Journal of Environmental Quality 33: 419–448. [DOI] [PubMed] [Google Scholar]
- 67.Sherr EB & Sherr BF, 2002. Significance of predation by protists in aquatic microbial food webs. Antonie van Leeuwenhoek 81: 293–308. [DOI] [PubMed] [Google Scholar]
- 68.Sherr EB & Sherr BF, 2007. Heterotrophic dinoflagellates: a significant component of microzooplankton biomass and major grazers of diatoms in the sea. Marine Ecology Progress Series 352: 187–197. [Google Scholar]
- 69.Siciliano A,Gesuele R & Guida M,2015. How Daphnia (Cladocera) assays may be used as bioindicators of health effects. Journal of Biodiversity & Endangered Species S1: 005. [Google Scholar]
- 70.Simon M,López-García P,Deschamps P,Moreira D,Restoux G,Bertolino P & Jardillie L,2015. Marked seasonality and high spatial variability of protist communities in shallow freshwater systems. The ISME Journal 9: 1941–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smayda TJ, 2008. Complexity in the eutrophication-harmful algal bloom relationship, with comment on the importance of grazing. Harmful Algae 8: 140–151. [Google Scholar]
- 72.Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM & Herndl GJ, 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proceeding of the National Academy of Sciences USA 103: 12115–12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stoddard SF, Smith BJ, Hein R,Roller BRK, & Schmidt TM, 2014. rrnDB: improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Research 43: D593–D598. 10.1093/nar/gku1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tautz D,Arctander P,Minelli A,Thomas RH & Vogler AP, 2003. A plea for DNA taxonomy. Trends in Ecology & Evolution 18: 70–74. [Google Scholar]
- 75.Techtmann SM, Zhuang M,Campo P,Holder E,Elk M,Hazen TC, Conmy R & Santo Domingo JW, 2017. Corexit 9500 enhances oil biodegradation and changes active bacterial community structure of oil-enriched microcosms. Applied and Environmental Microbiology 83: e03462–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trebitz AS, Hoffman JC, Darling JA, Pilgrim EM, Kelly JR, Brown EA, Chadderton WL, Egan SP, Grey EK, Hashsham SA, Klymus KE, Mahon AR, Ram JL, Schultz MT, Stepien CA & Schardt JC, 2017. Early detection monitoring for aquatic non-indigenous species: optimizing surveillance, incorporating advanced technologies, and identifying research needs. Journal of Environmental Management 202: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.USEPA, 2016. National Lakes Assessment 2012: A Collaborative Survey of Lakes in the United States EPA 841-R-16–113. U.S. Environmental Protection Agency, Washington, DC: https://nationallakesassessment.epa.gov/. [Google Scholar]
- 78.USEPA, 2017. Indicators used in the national aquatic resource surveys. https://www.epa.gov/national-aquatic-resource-surveys/indicators-used-national-aquatic-resource-surveys. Accessed 25 June 2017.
- 79.Valentini A,Taberlet P,Miaud C,Civade R,Herder J,Thomsen PF, Bellemain E,Besnard A,Coissac E,Boyer F,Gaboriaud C,Jean P,Poulet N,Roset N,Copp GH, Geniez P,Pont D,Argillier C,Baudoin JM, Peroux T,Crivelli AJ, Olivier A,Acqueberge M,Le Brun M,Møller PR, Willerslev E & Dejean T,2016. Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Molecular Ecology 25: 929–942. [DOI] [PubMed] [Google Scholar]
- 80.Vasselon V,Rimet F,Tapolczai K & Bouchez A,2017. Assessing ecological status with diatoms DNA metabarcoding: scaling-up on a WFD monitoring network (Mayotte island, France). Ecological Indicators 82: 1–12. [Google Scholar]
- 81.Wagner C & Adrian R,2011. Consequences of changes in thermal regime for plankton diversity and trait composition in a polymictic lake: a matter of temporal scale. Freshwater Biology 56: 1949–1961. Walker PJ & Winton JR, 2010. Emerging viral diseases of fish and shrimp. Veterinary Research 41: 51–75. [Google Scholar]
- 82.White MM & McLaren IA, 2000. Copepod development rates in relation to genome size and 18S rDNA copy number. Genome 43: 750–755. [DOI] [PubMed] [Google Scholar]
- 83.Wilde SB, Johansen JR, Wilde HD, Jiang P,Bartelme BA & Haynie RS, 2014. Aetokthonos hydrillicola gen. et sp. nov.: Epiphytic cyanobacteria on invasive aquatic plants implicated in Avian Vacuolar Myelinopathy. Phytotaxa 181: 243–260. [Google Scholar]
- 84.Woese CR & Fox GE, 1977. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proceedings of the National Academy of Sciences of the United States of America 74: 5088–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wurzbacher C,Fuchs A & Attermeyer K,2017. Shifts among Eukaryota, Bacteria, and Archaea define the vertical organization of a lake sediment. Microbiome 5: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu F-L, Tao S,Dawson RW, Li P-G & Cao J,2001. Lake ecosystem health assessment: indicators and methods. Water Research 35: 3157–3167. [DOI] [PubMed] [Google Scholar]
- 87.Yang J,Zhang X,Xie Y,Song C,Zhang Y,Yu H,& Allen Burton G,2017. Zooplankton community profiling in a eutrophic freshwater ecosystem-Lake Tai Basin by DNA metabarcoding. Scientific Reports 7: 1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yarza P,Yilmaz P,Pruesse E,Glöckner FO, Ludwig W,Schleifer KH, Whitman WB, Euzéby J,Amann R & Rosselló-Móra R,2014. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nature Reviews Microbiology 12: 635–645. [DOI] [PubMed] [Google Scholar]
- 89.Yi Z,Berney C,Hartikainen H,Mahamdallie S,Gardner M,Boenigk J,Cavalier-Smith T & Bass D,2017. High-throughput sequencing of microbial eukaryotes in Lake Baikal reveals ecologically differentiated communities and novel evolutionary radiations. FEMS Microbiology Ecology. 10.1093/femsec/fix073. [DOI] [PubMed] [Google Scholar]
- 90.Yilmaz P,Parfrey LW, Yarza P,Gerken J,Pruesse E,Quast C,Schweer T,Peplies J,Ludwig W & Glöckner FO, 2014. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Research 42: D643–D648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.