

Actin assembly by APC maintains proper organization and dynamics of F-actin at focal adhesions. This, in turn, impacts the organization of other molecular components and the responsiveness of focal adhesions to microtubule capture and autophagosome-induced disassembly.
Abstract
Focal adhesion (FA) turnover depends on microtubules and actin. Microtubule ends are captured at FAs, where they induce rapid FA disassembly. However, actin’s roles are less clear. Here, we use polarization-resolved microscopy, FRAP, live cell imaging, and a mutant of Adenomatous polyposis coli (APC-m4) defective in actin nucleation to investigate the role of actin assembly in FA turnover. We show that APC-mediated actin assembly is critical for maintaining normal F-actin levels, organization, and dynamics at FAs, along with organization of FA components. In WT cells, microtubules are captured repeatedly at FAs as they mature, but once a FA reaches peak maturity, the next microtubule capture event leads to delivery of an autophagosome, triggering FA disassembly. In APC-m4 cells, microtubule capture frequency and duration are altered, and there are long delays between autophagosome delivery and FA disassembly. Thus, APC-mediated actin assembly is required for normal feedback between microtubules and FAs, and maintaining FAs in a state “primed” for microtubule-induced turnover.
Graphical Abstract
Introduction
Directed cell migration is essential for embryonic development, immune surveillance, and tissue repair and regeneration (Weijer, 2009; Bravo-Cordero et al., 2012), and depends on coordinated assembly and turnover of focal adhesions (FAs). FAs are large macromolecular assemblages that link the actin cytoskeleton to the ECM (Ridley et al., 2003; Gardel et al., 2010). FAs initially form at the leading edge of migrating cells as small nascent adhesions. The majority of nascent adhesions are unstable and disappear rapidly; however, a subset grow and mature, polymerize actin stress fibers, move rearward, and then are disassembled (Choi et al., 2008; Gardel et al., 2010; Geiger and Yamada, 2011; Mui et al., 2016).
Microtubules play an important role in FA turnover (Vasiliev et al., 1970; Rinnerthaler et al., 1988). Microtubule plus ends grow along stress fibers to reach FAs, where they are transiently captured and undergo repeated cycles of catastrophe and regrowth/recapture, ultimately leading to FA disassembly (Kaverina et al., 1998, 1999; Krylyshkina et al., 2003; Efimov et al., 2008). However, the timing and duration of microtubule capture events at FAs have not been quantified, nor have these events been correlated with FA maturation. It is also not well understood mechanistically how microtubule capture events induce FA disassembly, although different studies suggest that this involves clathrin-mediated endocytosis, exocytosis of vesicles carrying matrix metalloproteinases, and/or selective autophagy (Ezratty et al., 2005, 2009; Stehbens et al., 2014; Kenific et al., 2016; Sharifi et al., 2016). In the selective autophagy pathway, LC3/ATG8-marked autophagosomes are delivered on microtubules to mature FAs (Mackeh et al., 2013; Kenific et al., 2016), where LC3 interacts with phosphorylated Src and paxillin, leading to autophagic turnover of FAs and paxillin degradation (Sharifi et al., 2016).
Actin is also critical for FA turnover. Formins and Ena/VASP help stimulate FA assembly and maturation (Hotulainen and Lappalainen, 2006; Tojkander et al., 2015, 2018), whereas we recently reported that Adenomatous polyposis coli (APC) promotes FA disassembly (Juanes et al., 2017). APC is a potent actin nucleator in vitro (Okada et al., 2010; Breitsprecher et al., 2012; Jaiswal et al., 2013), and we generated a separation-of-function mutant, APC-m4, that abolishes APC’s actin nucleation activity by altering only two residues in the C-terminal basic domain. Expression of full-length APC-m4 disrupted directional cell migration, and in nonmigrating cells, APC-m4 impaired microtubule-induced FA turnover in nocodazole washout assays (Juanes et al., 2017). However, this study left unanswered (1) whether APC-mediated actin assembly impacts F-actin organization and dynamics at FAs, (2) whether it contributes to FA turnover in migrating cells, and (3) which steps in FA turnover require actin assembly.
Here, we addressed these questions using polarization-resolved fluorescence microscopy, FRAP, super-resolution microscopy, and live cell imaging. Our results show that actin assembly by APC plays a critical role in maintaining proper F-actin organization and dynamics at FAs in migrating cells, and that its loss results in severe delays in FA disassembly stemming from an inability of FAs to respond properly to microtubule capture events.
Results
Actin assembly by APC is required for proper organization of F-actin at FAs
We began by asking how APC-m4 expression affects F-actin organization and dynamics at FAs. For this, we tuned the expression levels of full-length APC-WT and APC-m4 (expressed concurrently, or not, with silencing of endogenous APC; referred to as “ectopic” or “rescue”) to be similar to endogenous APC in U2OS osteosarcoma cells (Fig. S1 A). To assess the “molecular order” of F-actin at FAs (Fig. 1 A, “inside”), and at the stress fibers emanating from FAs (Fig. 1 A, “outside”), we performed polarization-resolved microscopy on fixed cells stained with Alexa Fluor 488–phalloidin. We focused our analysis on mature FAs at the two ends of ventral stress fibers (Hotulainen and Lappalainen, 2006; Tojkander et al., 2015, 2018). In this technique, images are acquired using variable angles of polarized excitation light (Kress et al., 2013; Mavrakis et al., 2014; Loison et al., 2018). Maximum fluorescence is achieved when the polarization angle of the excitation light matches the dipole angles of the fluorophores attached to F-actin. If the fluorophores have the same dipole orientation, and thus a high molecular order, a large change in the signal is detected when the polarization axis of the excitation light is rotated. These measurements yield two parameters of molecular order: mean orientation (ρ) and angular distribution width (Ψ) around the mean orientation (Fig. 1 B). Thus, Ψ is a proxy for the molecular order of actin filaments and contains contributions from both the actin structural order and the angular fluctuations of the fluorophore. Labeling actin with the phalloidin conjugate Alexa Fluor 488 produces a remaining angular fluctuation of ∼90° (Valades Cruz et al., 2016), which imposes a minimum measurable Ψ value. Ψ is therefore represented on a scale from 100° (highest measurable order) to 180° (complete disorder; Fig. 1 B). Under these fixation and staining conditions, the error in Ψ and ρ is only ∼2° (Kress et al., 2013). Previous studies on sarcomeres and contractile actin rings using polarization-resolved microscopy validated that a 5–10° change in Ψ corresponds to a major changes in F-actin organization (Mavrakis et al., 2014; Loison et al., 2018).
Figure 1.

APC-m4 disrupts the molecular order of F-actin at FAs. All data are from U2OS cells. (A) Schematic of FA (gray) and emanating stress fiber (green), highlighting ROIs analyzed by polarization-resolved microscopy. ROIs overlapping with FAs are designated as “inside,” and ROIs just outside of FAs on the stress fiber are designated “outside.” (B) Schematic showing orientational order parameters ρ (mean orientation) and Ψ (mean order or angular aperture). In the cartoon, fluorophores that label actin filaments are depicted as gray ellipsoids. The color bar provides a scale quantifying Ψ for other panels. (C) Representative confocal images of cells grown on collagen micropatterns (H and Y shapes), depleted of endogenous APC and rescued with refractory APC constructs (APC-WT or APC-m4). Cells were fixed and stained with Alexa Fluor 488–phalloidin (F-actin, gray) and phospho-paxillin (P-Paxillin) antibodies (pink). Yellow boxes highlight areas analyzed in D and E. Scale bar, 20 µm. (D) Representative zoom images from boxed region of APC-WT cell in C showing both the inside (In) and outside (Out) ROIs analyzed. Left: Overlay of Alexa Fluor 488–phalloidin (gray) and phospho-paxillin (pink). Remaining panels (left to right) show the color-coded molecular order (Ψ) super-imposed on Alexa Fluor 488–phalloidin (F-actin, gray); intensity-thresholded image of FA (white) superimposed with stick representation of Ψ (encoded in stick color); and mean orientation (ρ) encoded in stick orientation with thresholded image of FAs (gray). Below the image panels are histograms, displaying the Ψ value distribution (in degrees) from the In and Out ROIs combined (<Ψ> = 133.8°), and separately from In (<Ψ> = 132.9°) and Out (<Ψ> = 131.1°). Scale bar, 2 µm. (E) Same as D except for APC-m4 cells. Histograms display the Ψ value distribution from the In and Out ROIs combined (<Ψ> = 143.0°), and separately from In (<Ψ> = 145.2°) and Out (<Ψ> = 140.8°). (F) Total F-actin intensity (from In and Out ROIs combined). (G) Bee swarm plot showing average molecular order (Ψ), separately for inside and outside ROIs. (H) Bee swarm plot showing the SDΨ, separately for inside and outside ROIs. (I) Bee swarm plot showing the SDρ, separately for inside and outside ROIs. Data in panels F–I averaged from three independent experiments. APC-WT (n = 25 ROIs) and APC-m4 (n = 31 ROIs) from n ≥ 15 cells per condition. Error bars, SD. Statistical significance calculated by nonparametric Mann–Whitney two-tailed Student’s t test: ***, P < 0.001; **, P < 0.01; *, P < 0.05; n.s., not significant; AU, arbitrary unit.
We performed polarization-resolved microscopy on micropatterned cells (Fig. 1, C–I) and nonpatterned cells (Fig. S1, B–E; and Fig. S2). The use of patterned cells eliminates variability in cell size and shape, and minimizes heterogeneity in cell architecture between cells in a population (Théry et al., 2006). We silenced endogenous APC and rescued with APC-WT or APC-m4. In APC-m4 rescue cells, F-actin levels in stress fibers and at FAs were significantly diminished (Fig. 1 F), and both regions of interest (ROIs; inside and outside of FAs) were highly disordered compared with APC-WT cells (Fig. 1, D, E, and G). In addition, we determined the SD of Ψ (SDΨ) and the SD of ρ (SDρ; Fig. 1, H and I) in order to assess the uniformity of disorder within the two ROIs. An increase in SD for both Ψ and ρ is the signature of increased disorder at the scale over which the SD is measured, which is a few microns. We observed low SDs for Ψ and ρ in both APC-WT and APC-m4 cells, indicating that the degree of molecular order in F-actin is fairly consistent throughout the entire region analyzed. Similar results were obtained for nonmicropatterned cells (Fig. S1, C–E; and Fig. S2), and cells ectopically expressing APC-WT and APC-m4 (Fig. S2). Thus, APC-m4 expression decreases the molecular order (alignment and/or orientation) of F-actin at FAs and proximal regions of stress fibers.
Actin assembly by APC is required for normal actin dynamics at FAs
We next performed FRAP experiments on U2OS cells cotransfected with GFP-actin and mCherry-zyxin, depleted of endogenous APC by RNAi, and rescued by APC-WT or APC-m4. After photobleaching, we monitored recovery of GFP-actin fluorescence at FA-ventral stress fiber junctions (Fig. 2 A), and at regions of ventral stress fibers at least 5 µm away from FAs (Fig. 2 B). At both locations, there was no statistical difference in the recovery halftime (Fig. 2, C and E); however, the fraction that does not recover (immobile fraction) was much larger at FA-stress fiber junctions in APC-m4 compared with APC-WT cells (Fig. 2 D). In contrast, at locations on stress fibers farther away from FAs, there was no significant difference in the immobile fractions (Fig. 2 F). Similar results were obtained for control (scramble) cells and APC-WT rescue cells, demonstrating that ectopic expression of APC-WT does not alter actin dynamics (Fig. 2 and Fig. S3). Similar results were observed for cells depleted of endogenous APC (no rescue) and cells depleted of APC and rescued with APC-m4 (Fig. 2 and Fig. S3). Therefore, APC-mediated actin assembly contributes to normal actin turnover dynamics at or near FAs, but not farther away on stress fibers.
Figure 2.

APC-m4 alters actin dynamics at FAs. U2OS cells were depleted of endogenous APC and rescued with refractory APC constructs (APC-WT or APC-m4) along with plasmids expressing GFP-actin and mCherry-zyxin. (A) FRAP analysis, in which ROI were selected where GFP-actin and mCherry-zyxin signals overlap (see orange box in cartoon). ROIs were then bleached and monitored for GFP-actin fluorescence recovery. Graphs show mean recovery profiles normalized to zero after bleaching. Data averaged from three independent experiments (n = 30 ROIs from n = 15 cells per condition). (B) FRAP experiments as in A except that ROIs were selected along stress fibers at a distance (>5 µm) from FAs (see orange box in cartoon). Graphs show mean recovery profiles normalized to zero after bleaching. Data averaged from three independent experiments (n = 30 ROIs from n = 15 cells per condition). (C) Average time to 50% maximal recovery for experiments in A. Error bars, SEM. Statistical significance calculated by nonparametric Mann–Whitney two-tailed Student’s t test: n.s., not significant. (D) Average immobile fraction (does not recover in observation window) for experiments in A. Error bars, SEM. Statistical significance calculated by nonparametric Mann–Whitney two-tailed Student’s t test: *, P < 0.05. (E) Average time to 50% maximal recovery for experiments in B. Error bars, SEM. Statistical significance calculated by nonparametric Mann–Whitney two-tailed Student’s t test: n.s., not significant. (F) Average immobile fraction for experiments in B. Error bars, SEM. Statistical significance calculated by nonparametric Mann–Whitney two-tailed Student’s t test: n.s., not significant.
Actin assembly by APC is required to maintain proper density of FA components
Actin maintains the alignment and organization of FA components (Swaminathan and Waterman, 2016; Swaminathan et al., 2017; Kumar et al., 2018) and is required for the recruitment of Src kinase to FAs (Fincham et al., 1996). Src binds to phospho-FAK tyrosine kinase, which leads to Src activation, and further phosphorylation of FAK, initiating a cascade of signaling events that result in FA disassembly (Fincham and Frame, 1998; Li et al., 2002). Therefore, we asked whether APC-m4 alters cellular levels and/or organization of these components at FAs. By immunostaining, there was a decrease in the levels of endogenous active Src, phospho-paxillin, and phospho-FAK at the plasma membrane in APC-m4 cells and a decrease in their total “densities” (fluorescence intensity divided by cell area; Fig. 3, A–D). In addition, super-resolution imaging revealed that the densities of these components were each reduced at FAs in APC-m4 cells (Fig. 3, E–H). Further, the densities of GFP-paxillin, mCherry-zyxin, and vinculin were each reduced at FAs in APC-m4 cells (Fig. 3 I). Thus, APC-mediated actin assembly is required to maintain proper spatial organization of FA components.
Figure 3.

APC-m4 decreases the levels and/or densities of key molecular components at FAs. All data are from MDA-MB-231 cells expressing APC constructs (APC-WT or APC-m4), using fixed or live cell imaging as indicated. (A) Representative immunostaining of endogenous active phospho-Src detected by confocal imaging. Scale bar, 40 µm. Green boxed regions correspond to zoom panels (right; scale bar, 10 µm), which highlight the localization of phospho-Src at the cell periphery. (B) Representative immunostaining of endogenous active phospho-paxillin detected by confocal imaging. Scale bar, 25 µm. Green boxed regions correspond to zoom panels (right; scale bar, 5 µm), which highlight the localization of phospho-paxillin at the cell periphery. (C) Representative immunostaining of endogenous active phospho-FAK tyrosine kinase detected by confocal imaging. Scale bar, 25 µm. Green boxed regions correspond to zoom panels (right; scale bar, 5 µm), which highlight the localization of phospho-FAK at the cell periphery. (D) Densities of endogenous phospho-Src, phospho-paxillin, and phospho-FAK determined from cell images as in A–C. Data averaged from three independent experiments. n ≥ 56 cells for phospho-Src, n = 35 cells for phospho-paxillin, and n ≥ 103 cells for phospho-FAK per condition. Error bars, SEM. Statistical significance calculated by nonparametric Mann–Whitney two-tailed Student’s t test: ****, P < 0.0001. (E) Representative SIM images of cells immunostained for phospho-Src (green) and paxillin (pink). Scale bar, 5 µm. White boxed regions correspond to zoom panels (right; scale bar, 2 µm), highlighting the localization of phospho-Src and paxillin at FAs. (F) Representative SIM images of cells immunostained for phospho-paxillin (green) and paxillin (pink). Scale bar, 5 µm. White boxed regions correspond to zoom panels (right; scale bar, 2 µm), highlighting the localization of phospho-paxillin and paxillin at FAs. (G) Representative SIM images of cells immunostained for phospho-FAK (green) and paxillin (pink). Scale bar, 5 µm. White boxed regions correspond to zoom panels (right; scale bar, 2 µm), highlighting the localization of phospho-paxillin and paxillin at FAs. (H) Density of phospho-Src, phospho-paxillin, and phospho-FAK staining at individual FAs from cell images as in E–G. Data averaged from two independent experiments. n = 50 FAs total from 15 cells per condition. Error bars, SEM. Statistical significance calculated by nonparametric Mann–Whitney two-tailed Student’s t test: ****, P < 0.0001; **, P < 0.01. (I) Densities of signals at FAs for different components: GFP-paxillin and mCherry-zyxin densities were measured from cell images captured by TIRF microscopy; vinculin densities were measured from immunofluorescence images captured by confocal microscopy. Data averaged from three independent experiments. n = 34–159 FAs from n > 10 cells per condition. Error bars, SEM. Statistical significance calculated by nonparametric Mann–Whitney two-tailed Student’s t test: ****, P < 0.0001; ***, P < 0.001.
Loss of APC-mediated actin nucleation in migrating cells slows the rate of FA disassembly
To address how APC-m4 affects FA assembly and/or disassembly kinetics in migrating cells, we performed live imaging on motile human breast cancer MDA-MB-231 cells expressing APC-WT or APC-m4 at levels similar to endogenous APC (Fig. S4, A and B). Cells above were transfected with mCherry-zyxin to mark FAs and imaged by total internal reflection fluorescence (TIRF) microscopy (Fig. 4, A and B; and Videos 1 and 2). All FAs that grew to >2 µm in length were monitored for FA assembly kinetics, from the time of first appearance of mCherry-zyxin signal to time of maximal intensity (Fig. 4 C). Average FA assembly time was not significantly different in APC-WT (4.8 ± 1.5 min) versus APC-m4 cells (4.8 ± 1.7 min; P = 0.68). However, APC-m4 cells showed dramatically slower FA disassembly kinetics, defined as the time from maximum mCherry-zyxin fluorescence (peak FA maturity) to complete disappearance (Fig. 4 D). Average FA disassembly time was seven times longer in APC-m4 (17.5 ± 2.8 min) compared with APC-WT cells (2.5 ± 1.2 min; P < 0.001). Since APC-m4 expression results in enlarged FAs, we also measured FA assembly and disassembly rates, which are independent of FA size. To accomplish this, we used the automatized FA Analysis Server (FAAS) method (Berginski et al., 2011; Berginski and Gomez, 2013). This analysis revealed no significant difference in the rate of FA assembly between APC-WT and APC-m4 cells. However, FA disassembly rates were greatly reduced in APC-m4 cells (Fig. 4 E). No statistical differences were observed in FA assembly or disassembly rates between FAs located at the leading versus trailing edges of the same cell type (APC-WT or APC-m4; Fig. 4, F and G).
Figure 4.

APC-m4 slows FA disassembly. All data are from live TIRF microscopy imaging of migrating MDA-MB-231 cells expressing APC-WT or APC-m4, and either control-RNAi treated (scramble) or Dia1-silenced. (A) Representative image of an APC-WT cell (left; scale bar, 25 µm) showing FAs marked with mCherry-zyxin. The four FAs marked by yellow boxes and numbered correspond to the montages (right). Montages show time points from time-lapse imaging of FA assembly and disassembly. Time = 0 represents FA maximum size (peak fluorescence intensity). Scale bar (in time-lapse montage), 3 µm. (B) Same as in A except APC-m4 cell and corresponding FAs. (C) Histograms showing the distributions of times for individual FAs to complete assembly (from the time of initial appearance of mCherry-zyxin signal to time of peak intensity). Bin width, 2 min. Data from live imaging experiments as in A and B. n = 100 FAs per condition from n = 10 cells per condition. (D) Overlaid histograms showing the distributions of times for individual FAs to complete disassembly (starting from the time of peak fluorescence intensity to complete disappearance of signal). Bin width, 2 min. Data from live imaging experiments as in A and B. n = 840 FAs (APC-WT) and n = 961 FAs (APC-m4), each from n > 20 cells per condition. (E) Box and whisker plots showing data points from all regions of the cell used to determine global FA assembly and disassembly rates. Data are from live imaging experiments as in A and B, and were analyzed with the webtool FAAS. APC-WT (n = 286 FAs for assembly rate and n = 312 FAs for disassembly rate) and APC-m4 (n = 208 FAs for assembly rate and n = 354 FAs for disassembly rate), from n ≥ 8 cells per condition. Statistical significance calculated by ordinary one-way ANOVA Holm–Sidak multiple comparisons test: ****, P < 0.0001; n.s., not significant. (F and G) Box and whisker plots showing data points used to determine FA assembly (F) and disassembly (G) rates at the leading edge (front) or trailing edge (rear) of cells. Data are from live imaging experiments as in A and B, and were analyzed with the webtool FAAS. n = 200 FAs from n ≥ 8 cells per condition. Statistical significance calculated by ordinary one-way ANOVA Holm–Sidak multiple comparisons test: ****, P < 0.0001; n.s., not significant. (H) Box and whisker plots showing data points from all regions of the cell used to determine global FA assembly and disassembly rates for mock (scramble)–treated or Dia1-silenced cells. Data from live imaging experiments were analyzed with the webtool FAAS. Scramble (n = 499 FAs for assembly rate, and n = 344 FAs for disassembly rate) and si-Dia (n = 667 FAs for assembly rate, and n = 628 FAs for disassembly rate), from n ≥ 12 cells per condition. Statistical significance calculated by ordinary one-way ANOVA Holm–Sidak multiple comparisons test: *, P < 0.05; n.s., not significant. (I) Total F-actin levels in cells determined by phalloidin staining. Data averaged from three independent experiments. n = 40–100 cells per condition. Error bars, SEM. Statistical significance calculated by ordinary one-way ANOVA Holm–Sidak multiple comparisons test: ****, P < 0.0001; n.s., not significant.
To test the specificity of APC’s effects on FA disassembly, we also genetically disrupted a different actin nucleator, the formin Dia1, which assembles actin at FAs and promotes FA maturation (Fessenden et al., 2018). FAAS analysis showed that RNAi silencing of Dia1 (si-Dia1) only modestly slowed rates of FA assembly and disassembly (Fig. 4). Importantly, its effects on FA disassembly were not nearly as severe as the effects of APC-m4. Moreover, APC-m4 and si-Dia1 each led to an ∼30% reduction in total F-actin levels in cells (Fig. 4 I), as previously reported (Carramusa et al., 2007; Oakes et al., 2012; Rao and Zaidel-Bar, 2016; Juanes et al., 2017). Thus, the effects of APC-m4 on FA turnover do not appear to arise from a general loss of actin assembly in cells, but rather from a specific disruption of APC-mediated actin nucleation at FAs. Importantly, these results do not exclude the possibility that additional actin assembly-promoting factors (e.g., Arp2/3 complex, ENA/VASP, or other formins) also contribute to FA turnover.
How loss of actin assembly impacts microtubule capture at FAs
We considered how changes in F-actin organization and dynamics caused by APC-m4 might impact microtubule capture at FAs, which induces FA disassembly (Kaverina et al., 1998, 1999; Krylyshkina et al., 2003; Efimov et al., 2008). Microtubule capture was monitored by live TIRF imaging using mCherry-zyxin to mark FAs and 3xGFP-EMBD to mark microtubules (Fig. 5 A and Video 3). It has been shown that microtubule plus ends grow to reach FAs, where they are transiently captured, then retract, regrow, and are captured several more times preceding FA disassembly (Kaverina et al., 1998, 1999; Krylyshkina et al., 2003; Efimov et al., 2008). We observed similar microtubule behavior in MDA-MB-231 cells, and a comparison of APC-WT and APC-m4 cells revealed no significant difference in the percentage of FAs that experienced microtubule capture (Fig. 5 B), or in the frequency of microtubule capture events during FA assembly, and until peak maturation of the FA (Fig. 5 C). However, once FAs reached full maturity, there were almost twice as many microtubule capture events in APC-m4 cells (5.6 ± 3.9 times) compared with APC-WT cells (3.0 ± 1.6 times; Fig. 5 C). Further, microtubules paused almost three times longer at mature FAs in APC-m4 (76.0 ± 7.9 s) compared with APC-WT cells (26.0 ± 3.2 s; Fig. 5 D). This suggests a disruption in the normal feedback communication between microtubules and FAs. We also asked whether microtubule ends are captured in specific regions of FAs: (1) proximal third (relative to microtubule arrival); (2) central third; and (3) distal third. In APC-WT cells, there was no statistical difference in docking sites; however, APC-m4 cells showed a bias for microtubule docking in zone 3 (Fig. 5 E).
Figure 5.

APC-m4 increases the number and duration of microtubule capture events at FAs. All data are from live cell TIRF imaging of migrating MDA-MB-231 cells expressing APC constructs (APC-WT or APC-m4), along with a microtubule marker (3xGFP-EMDB) and FA marker (mCherry-zyxin). (A) Representative time-lapse imaging of microtubules (3xGFP-EMDB; cyan) and FAs (mCherry-zyxin; pink). Scale bar, 4 µm. (B) Percentage of mature FAs (per cell) that capture microtubule plus ends during the 30-min observation window. Data averaged from two experiments as in A; n = 20 cells per condition. Error bars, SD. Statistical significance calculated by nonparametric Mann–Whitney two-tailed Student’s t test: P = 0.9365; n.s., not significant. (C) Scatter plots showing the number of microtubule capture events at individual FAs during FA assembly and disassembly phases (see legends for Fig. 4, C and D, for definitions of phases) from experiments as in A. Data averaged from two experiments; n = 11 FAs per condition from n > 5 cells per condition. Error bars, SEM. Statistical significance calculated by ordinary one-way ANOVA Sidak’s multiple comparisons test: *, P < 0.05; n.s., not significant. (D) Scatter plot showing the durations of microtubule capture events at FAs from experiments as in A. Data averaged from three experiments; n = 50 microtubule capture events per condition from n > 10 cells per condition. Error bars, SEM. Statistical significance calculated by nonparametric Mann–Whitney two-tailed Student’s t test: ****, P < 0.0001. (E) Percentage of microtubule ends that are captured at different regions or “zones” of a FA, from experiments as in A: zone 1, proximal third of FA (relative to microtubule arrival); zone 2, central third; zone 3, distal third. Data averaged from three experiments; n = 90 microtubule capture events per condition from n > 15 cells per condition. Error bars, SEM. Statistical significance calculated by one-way ANOVA Holm–Sidak multiple comparisons test: **, P < 0.01; n.s., not significant.
In addition, we calculated the frequency of microtubule visits at FAs, which revealed a striking difference between APC-WT and APC-m4 cells (0.85 min−1 and 0.35 min−1, respectively). Thus, microtubule visits are more frequent in APC-WT cells. On the other hand, the average time between microtubule visits was about three times longer in APC-m4 cells compared with APC-WT cells (76 and 24 s, respectively). Altogether, microtubules were docked at mature FAs approximately half of the time in both APC-WT and APC-m4 cells, but in APC-m4 cells, the mature FAs had longer lifetimes. Because FAs take longer to disassemble in APC-m4 cells, there were more microtubule visits. In summary, microtubule capture is less efficient in APC-m4 cells, which may also explain the altered zone-preference of microtubule docking in the mutant.
Autophagosome delivery and dynamics at FAs
Selective autophagy has recently been shown to play a critical role in FA turnover (Kenific et al., 2016; Sharifi et al., 2016). In selective autophagy, double-membrane compartments called autophagosomes engulf cytoplasmic material and fuse with lysosomes to degrade and recycle their contents (Kaur and Debnath, 2015; Dikic and Elazar, 2018). Autophagosomes (marked by GFP-LC3/ATG8) are delivered to FAs on microtubules just before FA disassembly, where autophagosome components LC3 and NBR1 interact with FA components, including phospho-paxillin (which is phosphorylated by Src kinase; Fass et al., 2006; Köchl et al., 2006; Kenific and Debnath, 2016; Sharifi et al., 2016). How the arrival of an LC3-positive autophagosome induces FA disassembly remains unclear, but may involve autophagosome-mediated removal of phosphorylated FA components, including paxillin, triggering a cascade that leads to FA disassembly (Kenific et al., 2016).
To investigate whether APC-m4 affects selective autophagy at FAs, we monitored autophagosome (marked with GFP-LC3) dynamics at mature FAs (marked with mCherry-zyxin) in APC-WT versus APC-m4 cells (Fig. 6 A and Video 4). In APC-WT cells, autophagosomes were almost never delivered to a FA until it had reached peak maturity, as previously reported (Kenific et al., 2016). Further, most mature FAs experienced a single autophagosome visit (Fig. 6, B and C), which lasted <40 s (Fig. 6 D), and then the FA rapidly disassembled (Fig. 6, E and F). In contrast, in APC-m4 cells, autophagosomes were delivered multiple times to mature FAs (Fig. 6 C), and the duration of visits increased (Fig. 6 D). Further, there was a large delay between arrival of the first autophagosome and completion of FA disassembly (Fig. 6, E and F). We also monitored dynamics of the NBR1 receptor, which helps target LC3 to FAs (Kenific et al., 2016). APC-WT and APC-m4 cells showed similar targeting and dwell times of GFP-NBR1 at FAs (Fig. 6, G and H). Further, there was no difference in LC3 coimmunoprecipitation with NBR1 (Fig. 6 I). Thus, APC-m4 does not slow FA turnover by altering NBR1 dynamics or interfering with NBR1–LC3 interactions.
Figure 6.

APC-m4 alters autophagosome dynamics at FAs. All data are from live cell TIRF imaging of migrating MDA-MB-231 cells expressing APC constructs (APC-WT or APC-m4), along with markers for autophagosomes (GFP-LC3) and FAs (mCherry-zyxin). For all panels, data are averaged from at least three experiments. (A) Representative time-lapse imaging showing autophagosomes (GFP-LC3, cyan) and FAs (mCherry-zyxin, pink). Time = 0 corresponds to maximum mCherry-zyxin fluorescence intensity (peak FA growth). Scale bar, 3 µm. (B) Percentage of mature FAs targeted by autophagosomes, analyzed from experiments as in A. n > 800 FAs per condition from n ≥ 20 cells per condition. Error bars, SEM. Statistical significance calculated by one-way ANOVA Dunn’s multiple comparisons test: n.s., not significant. (C) Histograms showing distributions of mature FAs targeted by autophagosomes in the 30-min observation window, from experiments as in A. n = 40 FAs from n > 5 cells per condition. (D) Scatter plot showing dwell times of autophagosomes at FAs, analyzed from experiments as in A. n ≥ 42 autophagosomes per condition from n > 10 cells per condition. Error bars, SEM. Statistical significance calculated by nonparametric Mann–Whitney two-tailed Student’s t test: ****, P < 0.0001. (E) Scatter plot showing time after first appearance of an autophagosome at the FA to complete FA disassembly, analyzed from experiments as in A. n = 31 FAs (APC-WT) or n = 50 FAs (APC-m4) from n > 10 cells per condition. Error bars, SEM. Statistical significance calculated by nonparametric Mann–Whitney two-tailed Student’s t test: ****, P < 0.0001. (F) Overlaid histograms showing time elapsed from peak FA maturity (time = 0) to arrival of autophagosome during the 40-min observation window. Negative numbers correspond to rare events in which autophagosomes arrive before FA peak maturation. Data are analyzed from experiments as in A. n = 20 FAs from n > 5 cells per condition. (G) Percentage of mature FAs targeted by GFP-NBR1 receptor, analyzed from live imaging experiments as in A, except using cells expressing mCherry-zyxin and GFP-NBR1. n = 100 FAs (from 15 cells) per condition. Error bars, SEM. Statistical significance calculated by nonparametric Mann–Whitney two-tailed Student’s t test: n.s., not significant. (H) Scatter plot showing dwell times of GFP-NBR1 interactions with FAs, analyzed from live imaging experiments as in G. n = 40 GFP-NBR1 visits to FAs (from n = 10 cells) per condition. Error bars, SEM. Statistical significance calculated by nonparametric Mann–Whitney two-tailed Student’s t test: n.s., not significant. (I) Coimmunoprecipitation of endogenous NBR1 with GFP-LC3, pulled down using GFP–Trap-A agarose beads. Cells transfected with empty vector (expressing GFP alone instead of GFP-LC3) serve as a negative control.
Finally, we tested whether APC-m4 alters general autophagy. However, APC-WT and APC-m4 cells showed no significant difference in endogenous LC3 levels, the ratio of lipid-conjugated LC3 (LC3-II) to nonlipidated LC3 (LC3-I), or autophagosome maturation and eventual fusion with lysosomes (Fig. S5).
Discussion
The question that launched our study was how actin assembly contributes to FA turnover in migrating cells. Recent electron microscopy and polarimetry studies have shown that actin filaments and other components of FAs are densely packed and aligned, with F-actin forming the uppermost layer of a tiered structure ∼120 nm above the membrane (Nordenfelt et al., 2017; Swaminathan et al., 2017; Kumar et al., 2018). These actin structures undergo polarized flux, in which new actin subunits are incorporated at the FA, where the barbed ends are located, and move into the connecting stress fibers in a poleward fashion (Cramer et al., 1997). Our analysis using polarization-resolved microscopy corroborate these findings and show that there is a high degree of molecular order in the F-actin at FAs in APC-WT cells. However, in APC-m4 cells, FAs showed substantially reduced levels of F-actin and increased molecular disorder. Further, our FRAP analysis revealed that F-actin at FAs is less dynamic in APC-m4 cells. Given that the APC-m4 mutant disrupts APC-mediated actin nucleation, without impairing APC’s binding and bundling of F-actin, or APC’s binding and bundling of microtubules (Juanes et al., 2017), these results demonstrate that actin assembly by APC is critical for maintaining proper levels, organization, and dynamics of F-actin at FAs.
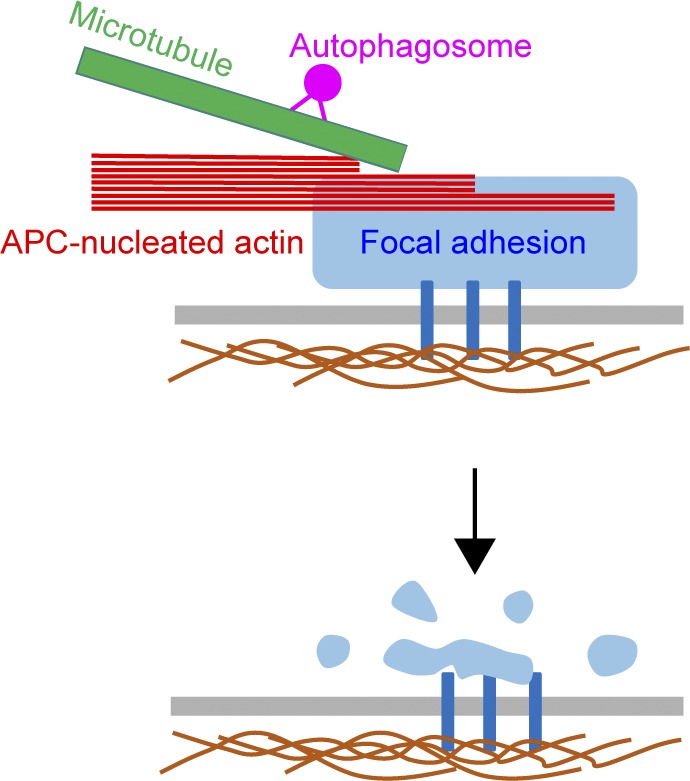
Using live imaging, we also compared microtubule capture, autophagosome delivery, and FA disassembly kinetics at FAs in APC-m4 and APC-WT cells. Our data reveal which steps in FA turnover are influenced by APC-mediated actin assembly (Fig. 7). In APC-WT cells, microtubules are captured at FAs and somehow sense the maturation state of the FA, responding by delivering an autophagosome and then retracting. All of these events occur, on average, within 26 s (the dwell time of microtubules at FAs), consistent with tight feedback between microtubules and FAs (Akhmanova et al., 2009). By comparison, in APC-m4 cells, there is a threefold decrease in the frequency of microtubule visits and a threefold increase in their dwell times at FAs. These observations suggest a deficiency in microtubule capture and a delay in microtubule retraction in APC-m4 cells, accompanied by delays between autophagosome arrival and completion of FA disassembly. Together, this suggests that APC is required to maintain FAs in a state primed for microtubule- and autophagosome-induced disassembly. Previous studies showed that interactions between autophagosomes and phospho-Src, phospho-paxillin, and zyxin are critical for FA turnover (Sandilands et al., 2011; Kenific et al., 2016; Sharifi et al., 2016). APC-m4 cells showed reduced densities of phospho-Src, phospho-paxillin, phospho-FAK, paxillin, zyxin, and vinculin at FAs. Thus, actin assembly by APC is critically required to maintain normal levels and tight packing of these FA components, which may be required for FAs to respond properly to autophagosome arrival and rapidly disassemble.
Figure 7.

Working model for the ordered events in FA turnover and the role of APC-mediated actin assembly. FA assembly (top panels) is similar in APC-WT and APC-m4 cells, with FAs growing for an average of 4.8 min before reaching peak maturity (maximum size). During this phase, microtubule plus ends are repeatedly captured at FAs and then retract, but rarely does this result in delivery of an autophagosome to the FA. However, FA disassembly (the time from peak maturity to complete disappearance) is strikingly different in APC-WT versus APC-m4 cells (bottom panels), taking approximately sevenfold longer in APC-m4 cells (17.5 versus 2.5 min). In APC-m4 cells, microtubule capture events are approximately three times longer than in APC-WT cells (76 versus 26 s), and autophagosome dwell times at FAs are approximately twice as long (72 versus 36 s). However, the frequency of microtubule visits is reduced in APC-m4 cells, as the average time between microtubule visits is increased. In APC-WT cells, once the FA reaches full maturity (stops growing), the next microtubule capture event typically results in autophagosome delivery, leading to rapid disassembly of the FA. However, in APC-m4 cells, microtubule capture and autophagosome delivery occur repeatedly at mature FAs as they slowly disassemble. Zooms in lower panels depict the spatial organization of F-actin and FA components. In APC-m4 cells, F-actin levels and organization are reduced, and the density of FA components is reduced (see zooms in lower panels). Black arrows on the left sides of panels indicate normal timing of events in FA assembly and disassembly, while red arrows indicate prolonged events in FA disassembly in APC-m4 cells. The large red arrow on the right indicates an increase in the number of times specific steps in FA turnover are repeated in APC-m4 cells. MT, microtubule; PM, plasma membrane; SF, stress fiber.
Finally, our data directly address the in vivo role of APC as an actin nucleator. APC is well known as a microtubule regulator, with less consideration given to its high-affinity interactions with G-actin and F-actin and its potent actin nucleation activity (Moseley et al., 2007; Okada et al., 2010; Breitsprecher et al., 2012; Jaiswal et al., 2013; Juanes et al., 2017). However, in most cell types, APC is found primarily at actin-rich regions of the cell cortex, and localizes only to a small subset of the microtubule plus ends (Mimori-Kiyosue et al., 2000; Barth et al., 2002; Mogensen et al., 2002; Kita et al., 2006). Further, Drosophila melanogaster APC is critical for assembling in vivo actin structures that drive a number of important actin-based physiological processes (Webb et al., 2009; Zhou et al., 2011; De Graeve et al., 2012; Molinar-Inglis et al., 2018). Our current results combined with our previous observations (Juanes et al., 2017) demonstrate that human APC’s actin nucleation activity plays a critical role in maintaining proper F-actin levels, organization, and dynamics at FAs, which is required for FA turnover and directed cell migration. Thus, APC is a bona fide actin nucleator in vivo, and this may be one of its chief cytoskeletal roles.
Materials and methods
Key resources
All resources used in this study are listed in Table 1.
Cell culture, RNAi silencing, and transfection of plasmids
Human osteosarcoma (U2OS and HTB96) and human breast cancer (MDA-MB-231 and HTB-26) cell lines were obtained directly from American Type Culture Collection (ATCC), where their identities were authenticated by short tandem repeat DNA profiling and they were tested for mycoplasma contamination. Cell lines were used for a maximum of 25 passages. All cells were grown in DMEM (GIBCO BRL Life Technologies) supplemented with 200 mM L-glutamine (Thermo Fisher Scientific) and 10% FBS (Sigma-Aldrich) at 37°C and 5% CO2.
All cell culture experiments were performed in six-well dishes, seeding each well with ∼60,000 cells. To silence endogenous APC or Dia1, cells were transfected 8–12 h after seeding with 50 nM RNAi oligos (for APC) using Lipofectamine 3000 (Thermo Fisher Scientific), or with 30 nM RNAi oligos (for Dia1) using Lipofectamine RNAiMAX (Thermo Fisher Scientific), according to the manufacturer’s instructions. RNAi oligos were directed against the human APC coding region (si-APC): 5′-GGAUCUGUAUCAAGCCGUUTT-3′ sense and 5′-AACGGCUUGAUACAGAUCCTT-3′ antisense (Invitrogen); or the 3′ UTR of the human Dia1 (si-Dia1): 5′-CUGUUAAUAAAGCAUUGAAUU-3′ sense and 5′-AAUUCAAUGCUUUAUUAACAG-3′ antisense (Integrated DNA Technologies). Cells were transfected in parallel with control “scramble” RNAi oligos: 5′-CAGUCGCGUUUGCGACUGG-3′, with dTdT 3′ overhangs. For APC rescue experiments, cells were transfected with rescue plasmids 12–16 h after silencing using Lipofectamine 3000, and then 24 h later the cells were collected for Western blotting, fixed for immunofluorescence, or used for live imaging. For all experiments where RNAi-resistant full-length APC-WT or APC-m4 rescue plasmids were introduced into cells, with or without depletion of endogenous APC, 600 ng of plasmid was used for transfections. For all live cell imaging, fixed-cell imaging, and immunoprecipitation experiments, plasmids were transfected as above into one of the same two cell lines and used 12–16 h after transfection, as described in detail below for each experiment.
FA turnover assays and analysis
MDA-MB-231 cells were transfected with 600 ng of APC plasmids (WT or m4) and 800 ng of plasmid expressing either mCherry-zyxin (55166; Addgene) or GFP-paxillin (50529; Addgene) in six-well plates. 12–16 h after transfection, cells were replated on collagen-coated glass-bottom dishes and allowed to adhere for 3–4 h. Immediately before imaging, the medium was replaced with DMEM containing high glucose and 25 mM Hepes (pH 7.4; GIBCO BRL Life Technologies), supplemented with 10% FBS, 20 mM L-glutamine, and 1 mM sodium pyruvate. Transfection efficiencies were >90% for mCherry-zyxin. Cells were maintained at 37°C using an Ibidi heated stage and imaged by TIRF microscopy on a Ti200 inverted microscope (Nikon Instruments) equipped with 100-mW solid-state lasers (Agilent Technologies), a CFI Apo 60× 1.49 NA oil-immersion TIRF objective (Nikon Instruments), an electron-multiplying charge-coupled device (EMCCD) camera with a pixel size of 0.267 µm (Andor Ixon), and an additional 1.5× zoom module (Nikon Instruments). Focus was maintained using the Perfect Focus System (Nikon Instruments). Images were captured every 10 s for 40–45 min (10–20-ms exposure at 488-nm excitation and 10% laser power) using Elements software (version 4.30.02; Nikon Instruments). Images were analyzed in Fiji. For comparing the kinetics of mCherry-zyxin to follow FA turnover in cells expressing APC (WT or m4; Fig. 4), only those FAs >2 µm, and that could be tracked from their initial formation through complete disassembly during the observation window, were included in the analysis. To track FAs over time, the Bezier ROI tool was used to manually outline individual FAs. Outlines were redrawn in each frame as necessary, since the FA could change in both size and location, as previously described (Kenific et al., 2016). FA fluorescence intensities were measured in Fiji for each individual FA. To compare the kinetics of FA assembly phase in cells expressing APC-WT or APC-m4, 100 individual FAs per condition were monitored from the appearance of the FA signal to the point of FA maximum fluorescence intensity, and distributions were plotted in GraphPad Prism 6.0c (Fig. 4 C). Maximum intensity, where a FA is fully assembled and “mature” (primed for disassembly), was set to time zero. To compare the kinetics of FA disassembly for cells expressing APC (WT or m4), over 800 individual mature FAs per condition were monitored from the point of FA maximum fluorescence intensity (mature) to complete disappearance, and distributions were plotted side by side in GraphPad Prism 6.0c (Fig. 4 D). To determine the rates of FA assembly and disassembly, which are independent of FA size, we used the FAAS method (Berginski et al., 2011; Berginski and Gomez, 2013). FAAS provides an unbiased and automated image processing pipeline, using submitted images (Berginski and Gomez, 2013). Rates from 208–667 FAs from whole cells, or 200 FAs from the leading versus trailing edge of cells (from different conditions as listed in figure legends), were analyzed using FAAS, and the distributions were plotted in GraphPad Prism 6.0c (Fig. 4, E–H; and Fig. S5 C).
Live cell imaging of microtubule interactions with FAs
For live imaging of microtubules and FAs, MDA-MB-231 cells were cotransfected as described in six-well plates with three different plasmids: 200 ng of a plasmid expressing the microtubule-binding domain of E-MAP-115 (ensconsin) fused to three copies of EGFP (3×GFP-EMTB; 26742; Addgene), 400 ng of a plasmid expressing mCherry-zyxin, and 600 ng of the plasmid expressing full-length APC (WT or m4). 12–16 h after transfection, cells were replated on collagen-coated glass-bottom dishes and allowed to adhere for 3–4 h. Immediately before imaging, the medium was replaced with DMEM containing high glucose and 25 mM Hepes (pH 7.4; GIBCO BRL Life Technologies), supplemented with 10% FBS, 20 mM L-glutamine, and 1 mM sodium pyruvate. Transfection efficiencies were >80% for each plasmid. Live cell imaging by TIRF microscopy was performed as described above for FA turnover assays. Images were captured every 5 s for a total of 30 min (10 ms exposure at 488 nm excitation; 15–20 ms exposure at 543 nm excitation, both at 10% laser power) using NIS Elements software (version 4.30.02). Image analysis was performed in ImageJ. To quantify number of interactions, duration and location (within FAs) of microtubule plus end interactions with FAs (Fig. 5, B–E), individual microtubules were tracked during a 30-min observation window. Results were plotted in GraphPad Prism 6.0c.
Live cell imaging of autophagosome dynamics at FAs
To monitor autophagosome dynamics, MDA-MB-231 cells were transfected as above in six-well plates with three different plasmids: 400 ng of a plasmid expressing mCherry-zyxin, 500 ng of a plasmid expressing GFP-LC3 (to visualize autophagosomes; 11546; Addgene), and 600 ng of a plasmid expressing full-length APC (WT or m4). 12–16 h after transfection, cells were replated on collagen-coated glass-bottom dishes and allowed to adhere for 3–4 h. Immediately before imaging, the medium was replaced with DMEM containing high glucose and 25 mM Hepes (pH 7.4; GIBCO BRL Life Technologies), supplemented with 10% FBS, 20 mM L-glutamine, and 1 mM sodium pyruvate before imaging. Transfection efficiencies were >80% for each plasmid. Live cell imaging by TIRF was performed as above. Images were captured every 10 s for 35–45 min (10 ms exposure at 488 nm excitation; 15–20 ms exposure at 543 nm excitation, both at 10% laser power) using Nikon Elements software (version 4.30.02). Image analysis was performed in ImageJ. To analyze GFP-LC3 targeting at FAs (Fig. 6, B and C), we first identified FAs (marked by mCherry-zyxin), and then quantified the number of times GFP-LC3 vesicles interacted with that FA (signals colocalized) during the observation window, and the durations of the interactions (Fig. 6 D). We also measured the time from first GFP-LC3 interaction with a FA to disassembly of the FA (Fig. 6 E) by cotracking mCherry-zyxin signal (and its disappearance). Results were plotted in GraphPad Prism 6.0c.
To monitor autophagosome maturation, i.e., traffic of autophagosomes and fusion with lysosomes (also called autophagic flux), MDA-MB-231 cells were transfected as described in six-well plates with 600 ng of a plasmid expressing full-length APC (WT or m4). 12–16 h after transfection, the medium was replaced with serum-free media (Thermo Fisher Scientific) supplemented with 20 mM Hepes (pH 7.4) and 20 mM L-glutamine. Cells were then mixed with Premo Autophagy Tandem Sensor RFP–GFP–LC3B (P36239; Thermo Fisher Scientific) and incubated for 16 h as described in the manufacturer’s instructions. The RFP-GFP-LC3B chimera contains an acid-sensitive GFP and an acid-resistant RFP, allowing one to distinguish between LC3B-positive autophagosomes (green/yellow) and LC3B-positive autophagolysosomes (red only; Kimura et al., 2007). To block autophagy, cells were treated with 2 µM Pepstatin A (US Biological) at the time of adding the RFP-GFP-LC3B sensor. Alternatively, where indicated, autophagy was inhibited 3 h before imaging with 100 nM Bafilomycin A1 (Sigma-Aldrich), a V-ATPase inhibitor that blocks lysosomal acidification and autophagosome–lysosome fusion (Mauvezin et al., 2015). Live cell imaging by TIRF was performed as above. Images were captured with 10 ms exposure at 488 nm excitation and 10 ms at 543 nm excitation (both at 10% laser power) using Nikon Elements software (version 4.30.02). Image analysis was performed in ImageJ. To quantify autophagosome maturation (Fig. S5 C), we calculated the ratio of fluorescence intensity (543/488 wavelength) for each vesicle by tracing cells within the images, and then quantifying the integrated fluorescence intensities (for each separate channel) of all vesicles (threshold >10 pixels) in an automated fashion using a custom script in ImageJ, after background subtraction from both channels. To monitor autophagosome fusion with autolysosomes (Fig. S5 D), MDA-MB-231 cells were transfected as described in collagen-coated glass-bottom dishes with three different plasmids: 500 ng of a plasmid expressing GFP-LC3 (autophagosomes marker), 500 ng of a plasmid expressing mCherry-LAMP1 (lysosomal-associated membrane protein 1, a lysosome/late endosome marker; 61524; Addgene), and 600 ng of a plasmid expressing full-length APC (WT or m4). 12–16 h after transfection, and immediately before imaging, the medium was replaced with DMEM containing high glucose and 25 mM Hepes (pH 7.4; GIBCO BRL Life Technologies), supplemented with 10% FBS, 20 mM L-glutamine, and 1 mM sodium pyruvate. Live imaging by TIRF was performed as above. Images were captured at 10-ms exposure at 488-nm excitation and 10-ms exposure at 543-nm excitation (both at 10% laser power) using Nikon Elements software (version 4.30.02). Image analysis (colocalization) was performed in ImageJ.
Live cell imaging of GFP-NBR1 at FAs
To monitor GFP-NBR1 dynamics at FAs, MDA-MB-231 cells were cotransfected as described above in six-well plates with three different plasmids: 1 µg of a plasmid expressing GFP-NBR1 (74202; Addgene), 800 ng of a plasmid expressing mCherry-zyxin, and 600 ng of a plasmid expressing full-length APC (WT or m4). 12–16 h after transfection, cells were replated and imaged as above (for GFP-LC3 dynamics at FAs), except that images were acquired every 10 s for 30 min. Image analysis was performed in Fiji. NBR1 vesicles were tracked manually over time to quantify the percentage that interacts with FAs and the duration of their interactions at FAs (Fig. 6, G and H). Results were plotted in GraphPad Prism 6.0c.
FRAP
For FRAP analysis, U2OS cells were depleted of endogenous APC and rescued with plasmids expressing full-length APC (WT or m4). Cells (4 × 105 per well) were sequentially transfected as described in six-well plates. They were first transfected 12 h after seeding, with 50 nM RNAi oligonucleotides (scramble or siRNA against human APC). Then, 24 h after initial transfection, they were transfected with three different plasmids: 400 ng of a plasmid expressing GFP-actin, 600 ng of a plasmid expressing mCherry-zyxin, and 600 ng of a plasmid expressing RNAi-resistant full-length APC-WT or APC-m4. Then, 48 h after initial transfection, just before live imaging and FRAP, cells were washed and replenished with DMEM containing high glucose and 25 mM Hepes (GIBCO BRL Life Technologies) supplemented with 10% FBS, 20 mM L-glutamine, and 1 mM sodium pyruvate. During live imaging, cells were maintained at 37°C with 5% CO2 using a Zeiss unit temperature and CO2 controller module, and a heating insert adapted to a Zeiss Axio Observer Z1 microscope equipped with a CSU-X1 spinning disk (Yokogawa). Images were captured using an α Plan-Apochromat 63× (NA 1.46) oil objective. The FRAP was controlled using an iLas2 module (Roper Scientific SAS) driven by MetaMorph Software 7.7.8 (Molecular Devices). To select ROIs (1 × 1 µm) for FRAP, we first imaged GFP-actin and mCherry-zyxin using lasers at 488 and 561 nm, respectively (10% laser power). For FRAP measurement of actin dynamics at ROIs (Fig. 2 and Fig. S3), 10 prebleach images were first acquired at 400-ms intervals, and then the ROI was bleached using the 488-nm laser (100 mW) for 700 ms (at 100% laser power). After photobleaching, images were acquired every 400 ms for 24 s, and then every 3 s for 276 s (total observation window, 5 min). For fluorescence recovery analysis, images were first analyzed with MetaMorph software 7.7.8 (Molecular Devices), as described previously (Lorente et al., 2014). The signal measured in the ROI was corrected for acquisition photobleaching and fluctuations of whole fluorescence following a double normalization method, calculated as follows: recovery of fluorescence intensity = It/I0 × T0/Tt, where It is the average intensity in ROI at time t; I0 is the average intensity of the ROI during the prebleaching period; T0 is the intensity during prebleaching of the nonbleached area (mean fluorescence intensity from at least two nonbleached ROI neighboring areas and from three different cells); and Tt is the intensity at time t of this area. The introduction of the correction factor (T0/Tt) accounts for possible small fluctuations in total fluorescence intensity caused by the bleaching itself, and yields a more accurate estimate of the measured fluorescence in the ROI (Phair et al., 2004). The immobile fraction was determined from normalized recovery fluorescence after bleach graphs: (1 − Fend), where Fend is ROI mean intensity at the steady-state (mean from the last four time points on the graph). Recovery halftimes were defined as the time after bleaching required to reach half-maximal fluorescence recovery. Maximum recovery was defined as the mean intensity of the last four data points. Data were plotted in GraphPad Prism 6.0.
Immunostaining of fixed cells
For immunostaining, cells were transfected and replated as described above on 3 × 1 × 1-mm glass coverslips (VWR International), which had been acid-washed and coated with collagen I (Advanced BioMatrix). Cells were next fixed for 15 min with 4% paraformaldehyde in 1× PBS (2.7 mM KCl, 1.8 mM KH2PO4, 10 mM Na2HPO4, and 140 mM NaCl, pH 7.4) at room temperature, and then permeabilized for 15 min in 1× PBS plus 0.5% Triton X-100 and 0.3 M glycine.
To image F-actin or endogenously expressed proteins (phospho-Src, phospho-paxillin, phospho-FAK, paxillin, vinculin, and LC3), cells were fixed and permeabilized as above, and blocked for 1 h at room temperature in 1% BSA dissolved in 1× PBS and 0.1% vol/vol Tween-20 (PBST). For F-actin staining, cells were incubated for 1 h with 1:1,000 Phalloidin-568 (A-12380; Thermo Fisher Scientific). For immunostaining, cells were incubated for 12 h at 4°C with primary antibody: 1:400 rabbit anti-phospho Src (Tyr416, clone 2N8; 04–857; EMD Millipore), 1:500 rabbit anti-phospho-paxillin (Tyr118; PP4501; ECM Biosciences), 1:500 rabbit anti-phospho-FAK (Tyr397, clone 141-9; 44-625G; Invitrogen), 1:500 mouse anti-paxillin (clone 5H11; AHO0492; Invitrogen), 1:1,000 mouse anti-vinculin (V9131; Sigma-Aldrich), or 1:1,000 rabbit anti-LC3 (ab6556; Novus Biologicals). Next, coverslips were washed three times with 1× PBST and incubated for 1 h at room temperature with secondary antibody: 1:1,000 donkey anti-rabbit Alexa Fluor 488 (A-21206; Thermo Fisher Scientific), 1:1,000 goat anti-rabbit Alexa Fluor 555 (A-21428; Thermo Fisher Scientific), or 1:1,000 donkey anti-mouse Alexa Fluor 568 (A-10037; Thermo Fisher Scientific). Then, coverslips were washed three times with PBS plus 0.1% Tween-20 and once with PBS, and then mounted with AquaMount (Thermo Fisher Scientific). Cells were imaged on an i-E upright confocal microscope (Nikon Instruments) equipped with a CSU-W1 spinning disk head (Yokogawa), 100-mW solid-state lasers, an emission tuner for 488- and 561-nm wavelengths, 60× oil objective (NA 1.4; Nikon Instruments), and an Ixon 897 Ultra-CCD camera (Andor Technology). Images were captured as stacks (nine planes, 0.5 µm steps) at 15–20% laser power, with 100–200 ms exposures sequentially at 488 and 561 nm using Nikon Elements software (version 4.30.02). Fiji was used to generate maximum intensity projections and calculate raw integrated fluorescence values of endogenous phospho-Src, phospho-paxillin, phospho-FAK, vinculin, or LC3 levels in cells and/or at FAs (Fig. 3, D, H, and I; and Fig. S5 B).
For structured illumination microscopy (SIM) imaging (Fig. 3, E–G), cells were treated as above for immunofluorescence, except we used H-precision coverslips (Azer Scientific) and Prolong Gold Diamond Antifade (Thermo Fisher Scientific) to mount slides 24–48 h before imaging. Images were collected at room temperature on an N-SIM instrument (Nikon Instruments) equipped with a SR Apo TIRF AC 100xHx1.49 NA oil immersion objective, a LU-N3-SIM laser unit, and an ORCA-flash4.0 CMOS camera (Hamamatsu Photonics). For imaging phospho-Src, phospho-paxillin, or phospho-FAK in combination with paxillin (antibody dilutions and secondary antibodies as above), 3D-SIM image slices were acquired at the central plane of the cell (one single image, without Z-interval). Images were captured with 400-ms exposure times (70% laser power). Images were acquired using a violet-to-red diffraction grating at three angles and five phases of illumination, producing 15 raw images per Z-position, and reconstructed with the default slice reconstruction setting in NIS Elements (Nikon Instruments). All images comparing APC-WT and APC-m4 cells were acquired and processed identically in Fiji. Data were plotted using GraphPad Prism 6.0c.
Polarization-resolved fluorescence microscopy
To determine the molecular orientation and order of F-actin in stress fibers connected to FAs (Figs. 1, S1, and S2), U2OS cells were grown, transfected, fixed, and processed for immunofluorescence as above. For experiments using micropatterned cells (Fig. 1, C–I), transfected cells were replated for 10 h on collagen-precoated micropatterned glass coverslips (10-900-00; Starter’s CYTOO; Théry et al., 2006), and then fixed. Fixed cells were incubated for 12 h with 1:1,000 rabbit anti-phospho-paxillin (Tyr-118; PP4501; ECM Biosciences), washed three times with 1× PBST, and incubated for 1 h at room temperature with 1:1,000 goat anti-rabbit Alexa Fluor 555 (A-21428; Thermo Fisher Scientific) and 1:40 (165 nM) Alexa Fluor 488–phalloidin (A12379; Invitrogen). After three washes with PBS plus 0.1% Triton X-100, and one wash with PBS, coverslips were mounted with Aqua-Mount (Thermo Fisher Scientific) and imaged by polarization-resolved fluorescence microscopy at room temperature. The optical setup for polarization-resolved fluorescence microscopy, described in detail in Wang et al. (2013), is based on a confocal spinning disk unit (CSU-X1-M1; Yokogawa) connected to the side-port of an inverted microscope (Eclipse Ti-U; Nikon Instruments). The excitation is provided by a polarized 488 nm continuous laser (Sapphire 488–200; Coherent) in which power is controlled. The laser beam is sent into an electro-optic modulator (Pockels cell; 28-NP; Quantum Technology) followed by a quarter wave plate (WPQ05M-488; Thorlabs) for production of a linear rotating polarization. A polarization distortion compensator is used to compensate for ellipticity and diattenuation produced by the optics in the excitation path toward the microscope objective. The compensator is set to diattenuation and dichroism values that are initially measured using a calibration procedure described in Wang et al. (2013). The beam is then expanded using a 10× telescope (BE10; Thorlabs) and sent directly sent to the microlens array of the confocal scanner unit by reflection in its dichroic mirror (Di01-T405/488/568/647; Semrock). An objective lens (Nikon Instruments Plan Apo VC 60×, NA = 1.2, water immersion) is used for excitation and light collection. The tube lens of the microscope is magnified by 1.5. An additional magnifier is used (2×) for the final image in the EMCCD camera (iXon 888; 1,024 × 1,024 pixels; Andor), resulting in a pixel size of 72 nm. The image is filtered (bandpass 525/50). The microlens and pinhole array of the confocal scanner unit disks rotate synchronously at a speed of 1,800 rpm, while the EMCCD and electro-optic modulator are synchronized to ensure a fast stack recording for a given number of incident polarization (Wang et al., 2013). A frame rate of 50 ms per image and 10 polarization angles measured leads to a typical rate of one polarization stack recorded per second. Each polarization stack recording is followed by a measurement of the fluorescence image taken with the same optical system, using an excitation wavelength of 640 nm (emission filter 675/50), in order to superimpose the actin order information with the presence of FAs in the cell.
Data analysis for polarization-resolved fluorescence microscopy
Inside the confocal volume, each Alexa Fluor 488–phalloidin molecule exhibits an absorption dipole vector μabs with an orientation (θ,φ) in 3D. Fluorescence is generated from the coupling of these dipoles with the incident linear polarization E(α), which makes an angle α with the horizontal axis x of the sample plane. The recorded fluorescence intensity is proportional to the absorption probability . The total intensity, built up from the incoherent emission from all molecules during the time of the measurement over which they might fluctuate in orientation, results in an angular integration over all angles explored in time and space: . This signal is modulated in α when the absorption dipoles of the fluorescence probes are aligned, e.g., when they do not experience an isotropic distribution. We assume that the orientations explored by molecular dipoles are constrained within an angular cone of total aperture angle ψ, oriented in the sample plane along the direction ρ relative to x (Fig. 1 B). Physically, ψ determines the degree of angular variations present within the focal spot at a given pixel position. This angle, denoted “molecular order,” encompasses the orientation variations among probes (related to the static organization of actin filaments) as well as their time angular fluctuations, integrated during the imaging integration time. ρ determines the preferential orientation of the probes. Thus, ρ and ψ permit quantification of the complete information on molecular organization at each pixel of an image. In practice, (ρ,ψ) are deduced from the measurement of the intensity modulation I(α). This is done by decomposing the dependence of the intensity I(α) as a function of (ρ,ψ) in a modulation form: ; a2(ρ,ψ) and b2(ρ,ψ) are the intensity modulation coefficients. The retrieval of (ρ,ψ) is computed numerically from the measurement of (a2,b2) at each pixel position, accounting for possible polarization distortions (Kress et al., 2013). In practice, these coefficients are measured from the computation of
and
using
as the total intensity and αk as the angles used for the polarization-resolved measurements (typically k = 1 . . . 10 and αk = 0° . . . 180°). (ρ,ψ) are finally represented in a map which combines molecular order and orientation.
For analyzing the molecular organization of actin at FAs, ventral stress fibers were selected, because the majority of mature FAs are found at their two ends. Also, ventral stress fibers do not “cross over” (like dorsal stress fibers and arcs do, which would interfere with the polarization-resolved microscopy analysis). The regions of actin stress fibers on which the polarization analysis is performed are selected based on (1) a segmentation step and (2) a selection or ROI where a FA is present. The segmentation step uses “filament sensor,” a tool recently developed in MATLAB for segmenting filament shapes in a 2D image (Eltzner et al., 2015), based on the filtering of straight contrasted features of an image, e.g., lines of typically several micrometers in length and several pixels in width (300 nm to 1 µm). Only pixels selected by filament sensor are treated as relevant for polarization analysis. The second step in the process, selection of ROI, is based on a pure threshold of the FA intensity image, using a manual choice of thresholding conditions to ensure that the binary mask defining an ROI resembles, at best, the real size of the FA. The result of this segmentation is the selection of typically 5–10 ROIs per cell, each containing one stress fiber that overlaps at its end with a FA. From this segmentation, actin signal pixels that overlap with FA regions are considered as regions named “in,” e.g., actin elements present in the FA, while actin signal pixels that do not overlap with a FA are called “out,” for pixels outside the FA region (but within the stress fiber connected to the FA). For the analysis, we used two different patterns (Y and H) to help ensure that different patterns do not change the outcome, i.e., the differences between APC-WT and APC-m4. We pooled the data from both patterns and analyzed them together. For APC-WT cells, we analyzed n = 25 FA-stress fibers from patterned cells (17 from Y patterns and 8 from H patterns). For APC-m4 cells, we analyzed n = 31 FA-stress fibers from patterned cells (18 from Y patterns and 13 from H patterns). The representation of both Ψ and ρ parameters is combined in a graph where the processed pixels are overlapped with a line, or “stick”; Ψ is indicated by the color of the stick and ρ is indicated by the orientation of the stick. These “stick images” are then overlapped with the intensity images, of either F-actin or FAs.
Different parameters are calculated within each selected ROI (the “in” and “out” regions of the ROI, defined above). The <Ψ> and <ρ> are the averaged values of the Ψ and ρ parameters over the number of pixels present in the ROI. SDΨ and SDρ are the SD values of the Ψ and ρ parameters. While <Ψ> represents the average molecular order in a given population of pixels, SDΨ and SDρ represent the variations around the average for Ψ and ρ, and therefore permit the quantification of heterogeneity within the ROI. Once these values are collected for all ROIs, the averages are calculated and plotted with SD values (error bars) on bee swarm plots.
Western blotting and coimmunoprecipitation
Western blotting was used to compare levels of endogenous APC in cells expressing APC (WT or m4; Fig. S1, A and B; and Fig. S4 A), levels of endogenous Dia1 in silenced or control cells (Fig. S4 D), and ratio of lipid-conjugated LC3 (LC3-II) to nonlipidated LC3 (LC3-I; Streeter et al., 2016) in cells expressing APC (WT or m4; Fig. S5 A). In each case, cells were pelleted and resuspended in lysis buffer (150 mM NaCl, 1.0% NP-40, 1.0% sodium deoxycholate, 1% SDS, 50 mM Tris, pH 7.5, 2 mM EDTA, 0.2 mM sodium orthovanadate, 20 mM β-glycerophosphate, 50 mM sodium fluoride, 1 mM PMSF, 1 mM DTT, and 1× Roche complete protease inhibitor mixture), and incubated for 30 min at 4°C with vortexing every 5 min. Lysate samples were precleared by centrifugation at 15,300 × g for 30 min at 4°C and quantified by Bradford assay, and equal amounts of total protein were immunoblotted. Blots were probed with 1:300 rabbit anti-APC (ab15270; Abcam), 1:1,000 rabbit anti-LC3 (NB100-2220SS; Novus Biologicals), 1:500 mouse anti-Dia1 (E-4; sc-373807; Santa Cruz Biotechnology), 1:2,000 rabbit anti-GFP (ab6556; Abcam), or 1:2,000 rabbit/human anti-GADPH (ab9489; Abcam), washed, and probed with infrared dye–conjugated secondary antibodies (Rockland Immunochemicals). Protein levels were detected and quantified by band densitometry using a LI-COR Odyssey Infrared Imaging System (LI-COR Biotechnology).
For immunoprecipitations (Fig. 6 I and Fig. S5 E), cells were transfected as described above with plasmids expressing full-length APC (WT or m4), GFP-LC3 (11546; Addgene), GFP-LAMP1 (34831; Addgene), and/or GFP empty vector (54522; Addgene). 12 h after transfection, cells were pelleted and resuspended in lysis buffer (150 mM NaCl, 0.1% NP-40, 50 mM Tris-Cl, pH 7.5, 1 mM EDTA, 10% glycerol, 1 mM sodium orthovanadate, 60 mM β-glycerophosphate, 50 mM sodium fluoride, 1 mM PMSF, and 1× Roche complete protease inhibitor mixture), and then incubated for 30 min at 4°C with vortexing every 2 min. Samples were precleared by centrifugation at 15,300 × g for 30 min at 4°C and quantified by Bradford assay. Equal amounts of total protein (cleared lysate) were incubated for 2 h at 4°C with 20 µl GFP–Trap-A agarose beads (gta-20; ChromoTek) in a reaction volume of 500 µl. Beads were washed four times in lysis buffer without detergent, and then incubated with Laemmli buffer for 5 min at 95°C and immunoblotted. Blots were probed sequentially with 1:2,000 rabbit anti-GFP (ab6556; Abcam), 1:1,000 mouse/human anti-NBR1 (H00004077-A01; Abnova), and 1:1,000 rabbit anti-LC3 (NB100-2220SS; Novus Biologicals) antibodies, washed, probed with secondary antibodies as above, and detected by infrared imaging as above.
Quantification and statistical analysis
All experiments were repeated multiple times, as indicated in the legends for each figure. In each case the data were pooled and averaged, and errors (SD or SEM) were calculated using GraphPad Prism (version 6.0c; GraphPad Software). Figure legends list the n values and error bars (SD or SEM) for each experiment. Data were tested for normality using the D’Agostino–Pearson omnibus normality test, and statistical significance was calculated using ordinary one-way ANOVA Sidak’s multiple comparisons test (Fig. 5 C), ordinary one-way ANOVA Holm–Sidak multiple comparisons test (Fig. 4, E–I; Fig. 5 E; Fig. S1 A; Fig. S4, A and D; and Fig. S5 C), one-way ANOVA Dunn’s multiple comparisons test (Fig. 6 B), or nonparametric Mann–Whitney two-tailed Student’s t test (for all other experiments) in Prism software version 6.0c (GraphPad Software). Differences were considered significant if the P value was <0.05 (*), <0.01 (**), <0.001 (***), or <0.0001 (****), as reported in the legend for each figure.
Table 1. Key resources.
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Rabbit anti-phospho Src (Tyr416; clone 2N8) | EMD Millipore | 04-857 |
| Rabbit anti-phospho FAK (Tyr397, clone 141-9) | Invitrogen | 44-625G |
| Rabbit anti-phospho-paxillin (Tyr-118) | ECM Biosciences | PP4501 |
| Mouse anti-paxillin (clone 5H11) | Invitrogen | IAH00492 |
| Mouse anti-vinculin | Sigma-Aldrich | V9131 |
| Rabbit anti-APC | Abcam | ab1527 |
| Mouse anti-Dia1 (E-4) | Santa Cruz Biotechnology | sc-373807 |
| Rabbit anti-LC3 | Novus Biologicals | NB100-2220SS |
| Donkey anti-rabbit Alexa Fluor 488 | Thermo Fisher Scientific | A-21206 |
| Goat anti-rabbit Alexa Fluor 555 | Thermo Fisher Scientific | A-21428 |
| Donkey anti-mouse Alexa Fluor 568 | Thermo Fisher Scientific | A-10037 |
| Rabbit anti-GFP | Abcam | ab6556 |
| Rabbit/human anti-GADPH | Abcam | ab9485 |
| Mouse/human anti-NBR1 | Abnova | H00004077-A01 |
| Donkey anti-rabbit IRDye 680RD | Rockland Immunochemicals | 926-68073 |
| Mouse anti-rabbit IRDye 680RD | Rockland Immunochemicals | 926-32220 |
| Bacterial and virus strains | ||
| XL Blue competent cells | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Alexa Fluor 568–phalloidin | Thermo Fisher Scientific | A12380 |
| 200 mM L-glutamine | Thermo Fisher Scientific | 25030-081 |
| DMEM | Thermo Fisher Scientific | 11995-073 |
| FBS | Sigma-Aldrich | F9423 |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000-015 |
| Lipofectamine RNAiMAX | Thermo Fisher Scientific | 13778075 |
| Hepes | Sigma-Aldrich | H4034 |
| 100 mM sodium pyruvate | Thermo Fisher Scientific | 11360070 |
| Opti-MEM reduced serum media | Thermo Fisher Scientific | 31985-088 |
| Bafilomycin A | Sigma Aldrich | SML1661 |
| Pepstatin A | US Biological | P3280 |
| Collagen I | Advanced BioMatrix | CB-40236 |
| Formaldehyde 37% | Sigma-Aldrich | 252549 |
| Experimental models: Cell lines | ||
| Human osteosarcoma cells: U2OS | ATCC | HTB96 |
| Human breast cancer cells: MDA-MB-231 | ATCC | HTB-26 |
| Oligonucleotides | ||
| siRNA targeting sequence against the human APC: 5′-GGAUCUGUAUCAAGCCGUUTT-3′ sense and 5′-AACGGCUUGAUACAGAUCCTT-3′ antisense | Juanes et al., 2017 | N/A |
| siRNA targeting sequence against the human Dia1: 5′-CUGUUAAUAAAGCAUUGAAUU-3′ sense and 5′-AAUUCAAUGCUUUAUUAACAG-3′ antisense | This paper | N/A |
| Stealth siRNA-control “scramble” oligos: 5′-CAGUCGCGUUUGCGACUGG-3′ | Invitrogen | 12935-200 |
| Recombinant DNA | ||
| Plasmid: APC-WT | Juanes et al., 2017 | Bruce Goode-US |
| Plasmid: APC-m4 | Juanes et al., 2017 | Bruce Goode-US |
| Plasmid: mCherry-zyxin | Kenific et al., 2016 | Addgene 55166 |
| Plasmid: pRK-GFP-paxillin | Chen et al., 2013 | Addgene 50529 |
| Plasmid: 3×GFP-EMTB | Miller and Bement, 2009 | Addgene 26742 |
| Plasmid: GFP-LC3 | Jackson et al., 2005 | Addgene 11546 |
| Plasmid: GFP-NBR1 | Kenific et al., 2016 | Addgene 74202 |
| Plasmid: mCherry-LAMP1 | Miyamoto et al., 2015 | Addgene 61524 |
| Plasmid: GFP-LAMP1 | Falcón-Pérez et al., 2005 | Addgene 34831 |
| Plasmid: GFP-actin | Svitkina et al., 2003 | Tatyana Svitkina-US |
| Plasmid: GFP empty vector | This paper | Addgene 54522 |
| Premo Autophagy Tandem Sensor RFP-GFP-LC3B | Thermo Fisher Scientific | P36239 |
| Software and algorithms | ||
| Fiji/ImageJ | Schindelin et al., 2012 | N/A |
| NIS Elements software, version 4.30.02 | Nikon Instruments | N/A |
| Metamorph Software 7.7.8 | Molecular Devices | N/A |
| MATLAB 2017 | MathWorks | N/A |
| FAAS | Berginski et al., 2011; Berginski and Gomez, 2013 | N/A |
| GraphPad Prism 6.0c | GraphPad Software | N/A |
| Illustrator CS6 | Adobe Systems | N/A |
| Filament sensor | Eltzner et al., 2015 | N/A |
| MATLAB (polarization-resolved fluorescence analysis algorithm) | MathWorks (Kress et al., 2013) and this paper | N/A |
| Labview (acquisition software for polarization-resolved fluorescence imaging) | National Instruments (Wang et al., 2013) | N/A |
| Odyssey Infrared Imaging System | LI-COR Biotechnology | N/A |
| Other | ||
| AquaMount mounting media | Thermo Fisher Scientific | 14-390-5 |
| Prolong Diamond Antifade Mounting | Thermo Fisher Scientific | P-36961 |
| GFP–Trap-A agarose beads | ChromoTek | gta-20 |
| Micro cover glasses 22 × 22 mm | VWR International PA | 48366-067 |
| Circular Round cover glasses 0.15 mm | Thermo Fisher Scientific | 12-545-80 |
| High precision cover glasses 1.5H 22 × 22 mm | Azer Scientific | ES0107052 |
| Dishes with 35-mm glass bottom | Thermo Fisher Scientific | P-35-1.5-14-C |
| Glass pattern coverslips (starter’s CYTOO) | CYTOO | 10-900-00 |
N/A, not applicable.
Online supplemental material
Fig. S1 A shows levels of APC and GAPDH by Western blot and ratio of APC:GADPH. Fig. S1 B shows a schematic of FA and stress fiber indicating the “inside” FAs (In) and “outside” FAs (Out) ROIs quantified for the analysis of the molecular disorder of F-actin. Fig. S1 C shows representative images of cells stained for phospho-paxillin and F-actin from polarization-resolved microscopy experiments. Fig. S1, D and E, shows zooms of FA-stress fiber regions from the representative cells in Fig. S1 C. Fig. S2 shows total levels of F-actin, molecular disorder (Ψ), and SD of Ψ and of ρ. Fig. S3, A and B, shows FRAP recovery curves of GFP-actin after bleaching of “inside” and “outside” regions of stress fibers. Fig. S4 A shows levels of APC and GAPDH by Western blotting. Fig. S4 B shows the ratio of APC:GADPH from Western blots. Fig. S4 C shows levels of Dia1 and GAPDH by Western blotting. Fig. S4 D shows the ratio of endogenous Dia1:GADPH from Western blots. Fig. S5 shows that APC-m4 expression does not alter general autophagy in cells. Fig. S5 A shows endogenous levels of LC3-I, LC3-II, and GADPH in cells, and the ratio of LC3-II:LC3-I from Western blots. Fig. S5 B shows levels of endogenous LC3 determined by immunostaining. Fig. S5 C shows that APC-m4 expression does not alter autophagosome trafficking and fusion with lysosomes, indicated by the ratio of RFP/GFP-LC3 fluorescence intensity per LC3-autophagosome. Fig. S5 D shows colocalization of GFP-LC3 (autophagosomes) and mCherry-LAMP1 (lysosomes) by live cell imaging. Fig. S5 E shows coimmunoprecipitation of endogenous LC3-II with GFP-LAMP1. Video 1 and Video 2 (related to Fig. 4) show FAs in migrating cells. Video 3 (related to Fig. 5) shows microtubules and FAs. Video 4 (related to Fig. 6) shows autophagosomes and FAs in migrating cells.
Supplementary Material
Acknowledgments
We are grateful to Julian Eskin for graphical assistance in preparing the model and videos. We also thank Denise Hilton, Gregory Hoeprich, Luther Pollard, Pascal Verdier-Pinard, Shashank Shekhar, Qing Tang, ShiYu Wang, David Waterman, and Ronen Zaidel-Bar for helpful discussions and/or comments on the manuscript, and to Caio Rimoli for assistance on data analysis.
This work was supported by grants from the Agence Nationale de la Recherche (ANR-16-CE11-008) to A. Badache and from the National Institutes of Health (GM083137 and GM098143) to B.L. Goode.
The authors declare no competing financial interests.
Author contributions: B.L. Goode and M.A. Juanes conceived of this study and designed experiments; M.A. Juanes and D. Isnardon performed FRAP experiments; M.A. Juanes performed the rest of experiments; M.A. Juanes, A. Badache, M. Mavrakis, and S. Brasselet analyzed data; and B.L. Goode and M.A. Juanes wrote the manuscript.
References
- Akhmanova A., Stehbens S.J., and Yap A.S.. 2009. Touch, grasp, deliver and control: functional cross-talk between microtubules and cell adhesions. Traffic. 10:268–274. 10.1111/j.1600-0854.2008.00869.x [DOI] [PubMed] [Google Scholar]
- Barth A.I.M., Siemers K.A., and Nelson W.J.. 2002. Dissecting interactions between EB1, microtubules and APC in cortical clusters at the plasma membrane. J. Cell Sci. 115:1583–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berginski M.E., and Gomez S.M.. 2013. The Focal Adhesion Analysis Server: a web tool for analyzing focal adhesion dynamics. F1000 Res. 2:68 10.12688/f1000research.2-68.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berginski M.E., Vitriol E.A., Hahn K.M., and Gomez S.M.. 2011. High-resolution quantification of focal adhesion spatiotemporal dynamics in living cells. PLoS One. 6:e22025 10.1371/journal.pone.0022025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Cordero J.J., Hodgson L., and Condeelis J.. 2012. Directed cell invasion and migration during metastasis. Curr. Opin. Cell Biol. 24:277–283. 10.1016/j.ceb.2011.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitsprecher D., Jaiswal R., Bombardier J.P., Gould C.J., Gelles J., and Goode B.L.. 2012. Rocket launcher mechanism of collaborative actin assembly defined by single-molecule imaging. Science. 336:1164–1168. 10.1126/science.1218062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carramusa L., Ballestrem C., Zilberman Y., and Bershadsky A.D.. 2007. Mammalian diaphanous-related formin Dia1 controls the organization of E-cadherin-mediated cell-cell junctions. J. Cell Sci. 120:3870–3882. 10.1242/jcs.014365 [DOI] [PubMed] [Google Scholar]
- Chen Y., Pasapera A.M., Koretsky A.P., and Waterman C.M.. 2013. Orientation-specific responses to sustained uniaxial stretching in focal adhesion growth and turnover. Proc. Natl. Acad. Sci. USA. 110:E2352–E2361. 10.1073/pnas.1221637110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C.K., Vicente-Manzanares M., Zareno J., Whitmore L.A., Mogilner A., and Horwitz A.R.. 2008. Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat. Cell Biol. 10:1039–1050. 10.1038/ncb1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer L.P., Siebert M., and Mitchison T.J.. 1997. Identification of novel graded polarity actin filament bundles in locomoting heart fibroblasts: implications for the generation of motile force. J. Cell Biol. 136:1287–1305. 10.1083/jcb.136.6.1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Graeve F.M., Van de Bor V., Ghiglione C., Cerezo D., Jouandin P., Ueda R., Shashidhara L.S., and Noselli S.. 2012. Drosophila apc regulates delamination of invasive epithelial clusters. Dev. Biol. 368:76–85. 10.1016/j.ydbio.2012.05.017 [DOI] [PubMed] [Google Scholar]
- Dikic I., and Elazar Z.. 2018. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19:349–364. 10.1038/s41580-018-0003-4 [DOI] [PubMed] [Google Scholar]
- Efimov A., Schiefermeier N., Grigoriev I., Ohi R., Brown M.C., Turner C.E., Small J.V., and Kaverina I.. 2008. Paxillin-dependent stimulation of microtubule catastrophes at focal adhesion sites. J. Cell Sci. 121:196–204. 10.1242/jcs.012666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltzner B., Wollnik C., Gottschlich C., Huckemann S., and Rehfeldt F.. 2015. The filament sensor for near real-time detection of cytoskeletal fiber structures. PLoS One. 10:e0126346 10.1371/journal.pone.0126346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezratty E.J., Partridge M.A., and Gundersen G.G.. 2005. Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat. Cell Biol. 7:581–590. 10.1038/ncb1262 [DOI] [PubMed] [Google Scholar]
- Ezratty E.J., Bertaux C., Marcantonio E.E., and Gundersen G.G.. 2009. Clathrin mediates integrin endocytosis for focal adhesion disassembly in migrating cells. J. Cell Biol. 187:733–747. 10.1083/jcb.200904054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcón-Pérez J.M., Nazarian R., Sabatti C., and Dell’Angelica E.C.. 2005. Distribution and dynamics of Lamp1-containing endocytic organelles in fibroblasts deficient in BLOC-3. J. Cell Sci. 118:5243–5255. 10.1242/jcs.02633 [DOI] [PubMed] [Google Scholar]
- Fass E., Shvets E., Degani I., Hirschberg K., and Elazar Z.. 2006. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J. Biol. Chem. 281:36303–36316. 10.1074/jbc.M607031200 [DOI] [PubMed] [Google Scholar]
- Fessenden T.B., Beckham Y., Perez-Neut M., Ramirez-San Juan G., Chourasia A.H., Macleod K.F., Oakes P.W., and Gardel M.L.. 2018. Dia1-dependent adhesions are required by epithelial tissues to initiate invasion. J. Cell Biol. 217:1485–1502. 10.1083/jcb.201703145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham V.J., and Frame M.C.. 1998. The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO J. 17:81–92. 10.1093/emboj/17.1.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham V.J., Unlu M., Brunton V.G., Pitts J.D., Wyke J.A., and Frame M.C.. 1996. Translocation of Src kinase to the cell periphery is mediated by the actin cytoskeleton under the control of the Rho family of small G proteins. J. Cell Biol. 135:1551–1564. 10.1083/jcb.135.6.1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardel M.L., Schneider I.C., Aratyn-Schaus Y., and Waterman C.M.. 2010. Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 26:315–333. 10.1146/annurev.cellbio.011209.122036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B., and Yamada K.M.. 2011. Molecular architecture and function of matrix adhesions. Cold Spring Harb. Perspect. Biol. 3:a005033 10.1101/cshperspect.a005033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P., and Lappalainen P.. 2006. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J. Cell Biol. 173:383–394. 10.1083/jcb.200511093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson W.T., Giddings T.H. Jr., Taylor M.P., Mulinyawe S., Rabinovitch M., Kopito R.R., and Kirkegaard K.. 2005. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156 10.1371/journal.pbio.0030156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal R., Stepanik V., Rankova A., Molinar O., Goode B.L., and McCartney B.M.. 2013. Drosophila homologues of adenomatous polyposis coli (APC) and the formin diaphanous collaborate by a conserved mechanism to stimulate actin filament assembly. J. Biol. Chem. 288:13897–13905. 10.1074/jbc.M113.462051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juanes M.A., Bouguenina H., Eskin J.A., Jaiswal R., Badache A., and Goode B.L.. 2017. Adenomatous polyposis coli nucleates actin assembly to drive cell migration and microtubule-induced focal adhesion turnover. J. Cell Biol. 216:2859–2875. 10.1083/jcb.201702007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur J., and Debnath J.. 2015. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 16:461–472. 10.1038/nrm4024 [DOI] [PubMed] [Google Scholar]
- Kaverina I., Rottner K., and Small J.V.. 1998. Targeting, capture, and stabilization of microtubules at early focal adhesions. J. Cell Biol. 142:181–190. 10.1083/jcb.142.1.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaverina I., Krylyshkina O., and Small J.V.. 1999. Microtubule targeting of substrate contacts promotes their relaxation and dissociation. J. Cell Biol. 146:1033–1044. 10.1083/jcb.146.5.1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenific C.M., and Debnath J.. 2016. NBR1-dependent selective autophagy is required for efficient cell-matrix adhesion site disassembly. Autophagy. 12:1958–1959. 10.1080/15548627.2016.1212789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenific C.M., Stehbens S.J., Goldsmith J., Leidal A.M., Faure N., Ye J., Wittmann T., and Debnath J.. 2016. NBR1 enables autophagy-dependent focal adhesion turnover. J. Cell Biol. 212:577–590. 10.1083/jcb.201503075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S., Noda T., and Yoshimori T.. 2007. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 3:452–460. 10.4161/auto.4451 [DOI] [PubMed] [Google Scholar]
- Kita K., Wittmann T., Näthke I.S., and Waterman-Storer C.M.. 2006. Adenomatous polyposis coli on microtubule plus ends in cell extensions can promote microtubule net growth with or without EB1. Mol. Biol. Cell. 17:2331–2345. 10.1091/mbc.e05-06-0498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köchl R., Hu X.W., Chan E.Y.W., and Tooze S.A.. 2006. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 7:129–145. 10.1111/j.1600-0854.2005.00368.x [DOI] [PubMed] [Google Scholar]
- Kress A., Wang X., Ranchon H., Savatier J., Rigneault H., Ferrand P., and Brasselet S.. 2013. Mapping the local organization of cell membranes using excitation-polarization-resolved confocal fluorescence microscopy. Biophys. J. 105:127–136. 10.1016/j.bpj.2013.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylyshkina O., Anderson K.I., Kaverina I., Upmann I., Manstein D.J., Small J.V., and Toomre D.K.. 2003. Nanometer targeting of microtubules to focal adhesions. J. Cell Biol. 161:853–859. 10.1083/jcb.200301102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Anderson K.L., Swift M.F., Hanein D., Volkmann N., and Schwartz M.A.. 2018. Local Tension on Talin in Focal Adhesions Correlates with F-Actin Alignment at the Nanometer Scale. Biophys. J. 115:1569–1579. 10.1016/j.bpj.2018.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Okura M., and Imamoto A.. 2002. Focal adhesions require catalytic activity of Src family kinases to mediate integrin-matrix adhesion. Mol. Cell. Biol. 22:1203–1217. 10.1128/MCB.22.4.1203-1217.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loison O., Weitkunat M., Kaya-Çopur A., Nascimento Alves C., Matzat T., Spletter M.L., Luschnig S., Brasselet S., Lenne P.-F., and Schnorrer F.. 2018. Polarization-resolved microscopy reveals a muscle myosin motor-independent mechanism of molecular actin ordering during sarcomere maturation. PLoS Biol. 16:e2004718 10.1371/journal.pbio.2004718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorente G., Syriani E., and Morales M.. 2014. Actin filaments at the leading edge of cancer cells are characterized by a high mobile fraction and turnover regulation by profilin I. PLoS One. 9:e85817 10.1371/journal.pone.0085817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackeh R., Perdiz D., Lorin S., Codogno P., and Poüs C.. 2013. Autophagy and microtubules - new story, old players. J. Cell Sci. 126:1071–1080. 10.1242/jcs.115626 [DOI] [PubMed] [Google Scholar]
- Mauvezin C., Nagy P., Juhász G., and Neufeld T.P.. 2015. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 6:7007 10.1038/ncomms8007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis M., Azou-Gros Y., Tsai F.-C., Alvarado J., Bertin A., Iv F., Kress A., Brasselet S., Koenderink G.H., and Lecuit T.. 2014. Septins promote F-actin ring formation by crosslinking actin filaments into curved bundles. Nat. Cell Biol. 16:322–334. 10.1038/ncb2921 [DOI] [PubMed] [Google Scholar]
- Miller A.L., and Bement W.M.. 2009. Regulation of cytokinesis by Rho GTPase flux. Nat. Cell Biol. 11:71–77. 10.1038/ncb1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimori-Kiyosue Y., Shiina N., and Tsukita S.. 2000. Adenomatous polyposis coli (APC) protein moves along microtubules and concentrates at their growing ends in epithelial cells. J. Cell Biol. 148:505–518. 10.1083/jcb.148.3.505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T., Rho E., Sample V., Akano H., Magari M., Ueno T., Gorshkov K., Chen M., Tokumitsu H., Zhang J., and Inoue T.. 2015. Compartmentalized AMPK signaling illuminated by genetically encoded molecular sensors and actuators. Cell Reports. 11:657–670. 10.1016/j.celrep.2015.03.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen M.M., Tucker J.B., Mackie J.B., Prescott A.R., and Näthke I.S.. 2002. The adenomatous polyposis coli protein unambiguously localizes to microtubule plus ends and is involved in establishing parallel arrays of microtubule bundles in highly polarized epithelial cells. J. Cell Biol. 157:1041–1048. 10.1083/jcb.200203001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinar-Inglis O., Oliver S.L., Rudich P., Kunttas E., and McCartney B.M.. 2018. APC2 associates with the actin cortex through a multipart mechanism to regulate cortical actin organization and dynamics in the Drosophila ovary. Cytoskeleton (Hoboken). 75:323–335. 10.1002/cm.21471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley J.B., Bartolini F., Okada K., Wen Y., Gundersen G.G., and Goode B.L.. 2007. Regulated binding of adenomatous polyposis coli protein to actin. J. Biol. Chem. 282:12661–12668. 10.1074/jbc.M610615200 [DOI] [PubMed] [Google Scholar]
- Mui K.L., Chen C.S., and Assoian R.K.. 2016. The mechanical regulation of integrin-cadherin crosstalk organizes cells, signaling and forces. J. Cell Sci. 129:1093–1100. 10.1242/jcs.183699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordenfelt P., Moore T.I., Mehta S.B., Kalappurakkal J.M., Swaminathan V., Koga N., Lambert T.J., Baker D., Waters J.C., Oldenbourg R., et al. . 2017. Direction of actin flow dictates integrin LFA-1 orientation during leukocyte migration. Nat. Commun. 8:2047 10.1038/s41467-017-01848-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes P.W., Beckham Y., Stricker J., and Gardel M.L.. 2012. Tension is required but not sufficient for focal adhesion maturation without a stress fiber template. J. Cell Biol. 196:363–374. 10.1083/jcb.201107042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K., Bartolini F., Deaconescu A.M., Moseley J.B., Dogic Z., Grigorieff N., Gundersen G.G., and Goode B.L.. 2010. Adenomatous polyposis coli protein nucleates actin assembly and synergizes with the formin mDia1. J. Cell Biol. 189:1087–1096. 10.1083/jcb.201001016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phair R.D., Gorski S.A., and Misteli T.. 2004. Measurement of dynamic protein binding to chromatin in vivo, using photobleaching microscopy. Methods Enzymol. 375:393–414. 10.1016/S0076-6879(03)75025-3 [DOI] [PubMed] [Google Scholar]
- Rao M.V., and Zaidel-Bar R.. 2016. Formin-mediated actin polymerization at cell-cell junctions stabilizes E-cadherin and maintains monolayer integrity during wound repair. Mol. Biol. Cell. 27:2844–2856. 10.1091/mbc.e16-06-0429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A.J., Schwartz M.A., Burridge K., Firtel R.A., Ginsberg M.H., Borisy G., Parsons J.T., and Horwitz A.R.. 2003. Cell migration: integrating signals from front to back. Science. 302:1704–1709. 10.1126/science.1092053 [DOI] [PubMed] [Google Scholar]
- Rinnerthaler G., Geiger B., and Small J.V.. 1988. Contact formation during fibroblast locomotion: involvement of membrane ruffles and microtubules. J. Cell Biol. 106:747–760. 10.1083/jcb.106.3.747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandilands E., Serrels B., McEwan D.G., Morton J.P., Macagno J.P., McLeod K., Stevens C., Brunton V.G., Langdon W.Y., Vidal M., et al. . 2011. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat. Cell Biol. 14:51–60. 10.1038/ncb2386 [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. . 2012. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi M.N., Mowers E.E., Drake L.E., Collier C., Chen H., Zamora M., Mui S., and Macleod K.F.. 2016. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Reports. 15:1660–1672. 10.1016/j.celrep.2016.04.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehbens S.J., Paszek M., Pemble H., Ettinger A., Gierke S., and Wittmann T.. 2014. CLASPs link focal-adhesion-associated microtubule capture to localized exocytosis and adhesion site turnover. Nat. Cell Biol. 16:561–573. 10.1038/ncb2975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streeter A., Menzies F.M., and Rubinsztein D.C.. 2016. LC3-II Tagging and Western Blotting for Monitoring Autophagic Activity in Mammalian Cells. Methods Mol. Biol. 1303:161–170. 10.1007/978-1-4939-2627-5_8 [DOI] [PubMed] [Google Scholar]
- Svitkina T.M., Bulanova E.A., Chaga O.Y., Vignjevic D.M., Kojima S., Vasiliev J.M., and Borisy G.G.. 2003. Mechanism of filopodia initiation by reorganization of a dendritic network. J. Cell Biol. 160:409–421. 10.1083/jcb.200210174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan V., and Waterman C.M.. 2016. The molecular clutch model for mechanotransduction evolves. Nat. Cell Biol. 18:459–461. 10.1038/ncb3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan V., Kalappurakkal J.M., Mehta S.B., Nordenfelt P., Moore T.I., Koga N., Baker D.A., Oldenbourg R., Tani T., Mayor S., et al. . 2017. Actin retrograde flow actively aligns and orients ligand-engaged integrins in focal adhesions. Proc. Natl. Acad. Sci. USA. 114:10648–10653. 10.1073/pnas.1701136114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry M., Pépin A., Dressaire E., Chen Y., and Bornens M.. 2006. Cell distribution of stress fibres in response to the geometry of the adhesive environment. Cell Motil. Cytoskeleton. 63:341–355. 10.1002/cm.20126 [DOI] [PubMed] [Google Scholar]
- Tojkander S., Gateva G., Husain A., Krishnan R., and Lappalainen P.. 2015. Generation of contractile actomyosin bundles depends on mechanosensitive actin filament assembly and disassembly. eLife. 4:e06126 10.7554/eLife.06126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tojkander S., Ciuba K., and Lappalainen P.. 2018. Camkk2 regulates mechanosensitive assembly of contractile actin stress fibers. Cell Reports. 24:11–19. 10.1016/j.celrep.2018.06.011 [DOI] [PubMed] [Google Scholar]
- Valades Cruz C.A., Shaban H.A., Kress A., Bertaux N., Monneret S., Mavrakis M., Savatier J., and Brasselet S.. 2016. Quantitative nanoscale imaging of orientational order in biological filaments by polarized superresolution microscopy. Proc. Natl. Acad. Sci. USA. 113:E820–E828. 10.1073/pnas.1516811113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliev J.M., Gelfand I.M., Domnina L.V., Ivanova O.Y., Komm S.G., and Olshevskaja L.V.. 1970. Effect of colcemid on the locomotory behaviour of fibroblasts. J. Embryol. Exp. Morphol. 24:625–640. [PubMed] [Google Scholar]
- Wang X., Kress A., Brasselet S., and Ferrand P.. 2013. High frame-rate fluorescence confocal angle-resolved linear dichroism microscopy. Rev. Sci. Instrum. 84:053708 10.1063/1.4807318 [DOI] [PubMed] [Google Scholar]
- Webb R.L., Zhou M.-N., and McCartney B.M.. 2009. A novel role for an APC2-Diaphanous complex in regulating actin organization in Drosophila. Development. 136:1283–1293. 10.1242/dev.026963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weijer C.J. 2009. Collective cell migration in development. J. Cell Sci. 122:3215–3223. 10.1242/jcs.036517 [DOI] [PubMed] [Google Scholar]
- Zhou M.-N., Kunttas-Tatli E., Zimmerman S., Zhouzheng F., and McCartney B.M.. 2011. Cortical localization of APC2 plays a role in actin organization but not in Wnt signaling in Drosophila. J. Cell Sci. 124:1589–1600. 10.1242/jcs.073916 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.