

One third of the eukaryotic proteome matures in the secretory pathway. Sun and Brodsky describe the machinery that maintains protein fidelity and how its actions are coordinated.
Abstract
Protein folding is inherently error prone, especially in the endoplasmic reticulum (ER). Even with an elaborate network of molecular chaperones and protein folding facilitators, misfolding can occur quite frequently. To maintain protein homeostasis, eukaryotes have evolved a series of protein quality-control checkpoints. When secretory pathway quality-control pathways fail, stress response pathways, such as the unfolded protein response (UPR), are induced. In addition, the ER, which is the initial hub of protein biogenesis in the secretory pathway, triages misfolded proteins by delivering substrates to the proteasome or to the lysosome/vacuole through ER-associated degradation (ERAD) or ER-phagy. Some misfolded proteins escape the ER and are instead selected for Golgi quality control. These substrates are targeted for degradation after retrieval to the ER or delivery to the lysosome/vacuole. Here, we discuss how these guardian pathways function, how their activities intersect upon induction of the UPR, and how decisions are made to dispose of misfolded proteins in the secretory pathway.
Introduction
In eukaryotes, approximately one third of the proteome is synthesized at the ER. These proteins enter the ER through a protein translocon in an unfolded state (Rapoport et al., 2017). To gain activity and traffic to their final destinations, proteins residing in the secretory pathway must fold into tertiary and, in some cases, quaternary structures. To this end, myriad chaperones are recruited to stabilize folding intermediates and overcome energy barriers (Balchin et al., 2016). Protein folding in the ER is also guided by posttranslational modifications, including glycosylation, disulfide bond formation, proline isomerization, and lipidation (Braakman and Hebert, 2013; Cherepanova et al., 2016).
Despite the enormous investment of cellular resources dedicated to protein folding, 12–15% of newly synthesized polypeptides in human cells and 1–5% in yeast are cotranslationally eliminated by the ubiquitin-proteasome system (Duttler et al., 2013; Wang et al., 2013). The percentage of newly synthesized proteins in the ER degraded posttranslationally might be even higher, since these substrates may translocate inefficiently into the ER, may fail to be posttranslationally modified, or, for membrane proteins, may be unable to achieve their proper membrane topology (Trombetta and Parodi, 2003; Chen et al., 2011; Shao and Hegde, 2016). These problems are exacerbated if proteins harbor disease-causing mutations that compromise protein biogenesis (Guerriero and Brodsky, 2012; Tao and Conn, 2018).
To counteract protein misfolding and maintain protein homeostasis, or “proteostasis,” in the secretory pathway, multiple quality control (QC) mechanisms exist: ER-associated degradation (ERAD), ER-phagy, Golgi QC (GQC), and plasma membrane QC (PMQC). Each process is regulated to some degree by the unfolded protein response (UPR) and/or the heat shock response, which adjust protein synthesis, chaperone levels, and the activity of protein degradation pathways (Travers et al., 2000; Li et al., 2017; Preissler and Ron, 2018; Karagöz et al., 2019). Some misfolded proteins are subject to more than one QC pathway, and it is becoming clear that QC mechanisms can cooperate or act preferentially on some substrates over others. Regardless, if QC pathways fail, then cellular homeostasis is compromised, and cell and organism death may occur (Chen et al., 2011; Wolff et al., 2014; Schneider and Bertolotti, 2015).
In this review, we first outline the molecular mechanisms that define ERAD, ER-phagy, and GQC and then highlight the decisions that triage misfolded proteins for ERAD versus post-ER quality control (ERQC). We next discuss the interplay between ER and two post-ERQC pathways, i.e., proteasome-dependent GQC and lysosome/vacuole-dependent GQC.
ERAD: The first line of defense
Identification of ERAD substrates
To protect against the transport of misfolded proteins through the secretory pathway, it was hypothesized that an unknown ER-resident protease could destroy misfolded proteins (reviewed in Needham and Brodsky, 2013). Subsequent studies instead revealed the contributions of the cytosolic ubiquitin-proteasome system (Sommer and Jentsch, 1993; Jensen et al., 1995; Ward et al., 1995; Hampton et al., 1996; Hiller et al., 1996; McCracken and Brodsky, 1996; Werner et al., 1996). Thus, the ER membrane establishes a physical barrier that separates the ER folding and degradation machineries, protecting folding intermediates from premature degradation. Despite the immense structural and topological diversity of proteins that enter the secretory pathway, work primarily in the budding yeast, Saccharomyces cerevisae, revealed a conserved principle under which ERAD operates: misfolded proteins are recognized and ubiquitinated through either the ERAD-L (ERAD of substrates with misfolded lesions within the ER lumen), ERAD-C (cytoplasm), or ERAD-M (membrane) pathways, depending on the location of the folding lesion (Huyer et al., 2004; Vashist and Ng, 2004; Carvalho et al., 2006; Denic et al., 2006). Key players required for substrate ubiquitination are the E3 ubiquitin ligases, which in yeast are Hrd1 (ERAD-L/ERAD-M) and Doa10 (ERAD-C). However, the assignment of mammalian ERAD substrates to three ERAD classes is less straightforward due to the existence of the numerous E3 ligases that work individually and in concert (Bernasconi et al., 2010a; Christianson and Ye, 2014). Nevertheless, common in both yeast and mammalian cells are chaperone- and lectin-mediated substrate recognition, energy-dependent substrate retrotranslocation from the ER, polyubiquitination, and proteasome-mediated degradation (Fig. 1).
Figure 1.

The three branches of the ERAD pathway in yeast. Lower panels in A, B, and C define the different steps during ERAD-L, ERAD-M, and ERAD-C, respectively. (A) ERAD-L substrates containing lumenal folding lesions (red star) and N-linked glycans (gray diamond) are recognized and processed by an enzyme cascade to generate an ERAD-targeting glycan (yellow diamond). Chaperones (e.g., Kar2) and lectins (e.g., Yos9) capture the substrate for transfer to the Hrd1 complex for retrotranslocation-coupled ubiquitination (purple triangle). (B) ERAD-M substrates containing a membrane-embedded folding lesion (red star) are recognized and ubiquitinated by the Hrd1 complex. (C) ERAD-C substrates containing cytosolic folding lesions are instead recognized by cytosolic chaperones (e.g., Ydj1/Hsp40 and Ssa1/Hsp70), which bridge the Doa10 ubiquitin ligase to an ERAD substrate. The three ERAD branches converge at a Cdc48-complex–dependent retrotranslocation step (D, top). The Cdc48 complex also contains Ufd1/Npl4, which interacts with ubiquitin (purple triangle). Following retrotranslocation, substrates are escorted to the 26S proteasome for degradation with the help of the Ras23 and Dsk2 shuttling factors.
The best-characterized ERAD substrates are misfolded ERAD-L glycoproteins, for which recognition commonly requires both an N-linked glycan and a protein determinant. During glycoprotein folding, a core glycan, Glc3Man9GlcNAc2, is first appended to an asparagine side chain in the N-X-(S/T) consensus motif (Helenius and Aebi, 2004). The glycan is subsequently trimmed in concert with chaperones and lectins, such as calnexin, that monitor glycoprotein folding (Anelli and Sitia, 2008; Lamriben et al., 2016). Specifically, the three terminal glucose residues are cleaved sequentially by glucosidase I and II, generating Man9GlcNAc2 (Jakob et al., 1998; Hitt and Wolf, 2004). Another lectin, known as malectin, binds the species containing two glucoses and helps prevent the secretion of immature glycoproteins (Schallus et al., 2008). Further trimming by mannosidase I (Mns1) of the glucose-free species removes the α-1,2-mannose from the B-branch in the glycan, yielding Man8GlcNAc2 (Camirand et al., 1991; Jakob et al., 1998). In yeast, the Man8GlcNAc2 glycan is potentially shared by both folded proteins that will exit the ER in COPII (coat promoter-II) vesicles along with misfolded proteins, indicating that Mns1 fails to discriminate between these proteins (Gauss et al., 2011). To specifically generate an ERAD glycan signal, an exposed α-1,6-mannose is generated by Htm1 (EDEM in mammals; Oda et al., 2003). In mammals, there are three Htm1 orthologues, EDEM1, EDEM2, and EDEM3, which function sequentially to convert Man9GlcNAc2 to Man7GlcNAc2 (which exposes α-1,6-mannose) in a two-step process (Ninagawa et al., 2014). In mammals, more extensive mannose trimming by other mannosidases has also been observed and may take place in QC vesicles (Shenkman and Lederkremer, 2019). These misfolded glycoproteins are captured by the Yos9 (OS-9 and XTP-3B in mammals) lectins, which also bind to hydrophobic motifs and deliver substrates to Hrd3 (Sel1L in mammals; van der Goot et al., 2018). Hrd3/Sel1L is a core component of the Hrd1 complex (Fig. 1; Plemper et al., 1999; Denic et al., 2006; Xie et al., 2009).
In addition to a glycan, a feature in most terminally misfolded proteins and folding intermediates is an exposed hydrophobic patch that is otherwise buried in native proteins. Besides assisting substrate delivery to Hrd3 as will be discussed, the solubility of these proteins is maintained by ER-resident molecular chaperones, including an Hsp70 (Kar2 in yeast or BiP in mammals), along with Hsp40 partners (Jakob et al., 1998; Brodsky et al., 1999; Nishikawa et al., 2001; Gauss et al., 2011). The exposure of an unpaired Cys also leads to retention in the ER (Reddy et al., 1996), thereby preventing the formation of potentially toxic disulfide-linked aggregates in later secretory pathway compartments.
More recent work suggests the involvement of another glycan-dependent event during ERAD, O-mannosylation (Xu et al., 2013; Xu and Ng, 2015a,b). In yeast, this modification on Ser/Thr residues helps terminate futile folding cycles. Both N-glycan trimming and O-mannosylation are slow, which allows substrates to linger in the ER. In principle, this provides time for proteins to fold, but this delay may also present folding intermediates to the ERAD machinery, especially since some chaperones are involved in protein folding and turnover (Preston and Brodsky, 2017). This conundrum was solved by the discovery of the Slp1–Emp65 complex, which protects folding intermediates from premature degradation in yeast (Sun and Brodsky, 2017; Zhang et al., 2017).
After selection, ERAD-L substrates are delivered to the Hrd1 complex, which includes Yos9, Hrd3, Der1, Usa1, and Hrd1 (Fig. 1; Carvalho et al., 2006; Denic et al., 2006). The degradation of ERAD-M substrates utilizes a refined group of Hrd1 complex members (Ruggiano et al., 2014). Since the RING domain in Hrd1, which is required for ubiquitin ligase activity, resides on the cytosolic face of the ER, ERAD-L substrates must be retrotranslocated from the ER (Schoebel et al., 2017). The identity of the proteinaceous “retrotranslocon” has been controversial as will be discussed in the Retrotranslocation and degradation section of this paper.
Another ubiquitin ligase, Doa10, is involved in the turnover of ERAD-C substrates (see below) and can even target select ERAD-M substrates, such as an orphaned member of the translocation channel (Sbh2) that exposes a transmembrane degron (Habeck et al., 2015). Another class of ERAD-M substrates are single-pass membrane proteins, such as unassembled α and β T cell receptor subunits, whose transmembrane domains are marginally hydrophobic. These subunits translocate into the ER lumen, where they are then recognized by BiP before being retrotranslocated (Feige and Hendershot, 2013). Other ERAD-M substrates, including a mutated form of the translocation channel, even mislocalize to the inner nuclear membrane and are recognized and ubiquitinated by the Asi complex before proteasomal degradation. The Asi complex also seems to recognize folding lesions within the transmembrane domain (Foresti et al., 2014; Khmelinskii et al., 2014). Together, ERAD-M substrates with diverse folding lesions are recognized by diverse pathways.
Unlike the examples discussed, some membrane proteins expose misfolded lesions in cytosolic domains. The common feature of these ERAD-C substrates is the exposure of normally buried hydrophobic patches. To prevent protein aggregation, cytosolic chaperones, including Hsp70, Hsp40, nucleotide exchange factors, Hsp90, and small heat shock proteins, are engaged (Huyer et al., 2004; Youker et al., 2004; Ahner et al., 2007; Han et al., 2007; Park et al., 2007; Guerriero et al., 2017; Sun and Brodsky, 2018). Hsp70 and Hsp40 not only prevent protein aggregation but also facilitate the association between an E3 ubiquitin ligase and an ERAD-C substrate (Nakatsukasa et al., 2008). Although it is poorly understood how a single chaperone can dictate different fates (protein maturation versus degradation), prolonged chaperone engagement of ERAD-C substrates might be sufficient to recruit an E3 ubiquitin ligase (i.e., Doa10) to modify and retrotranslocate these substrates.
Substrate ubiquitination
The ubiquitination of ERAD substrates requires a cascade of enzymes, including an E1 ubiquitin-activating enzyme, an E2 ubiquitin-conjugating enzyme, and an E3 ubiquitin ligase (Oh et al., 2018). For the Doa10-dependent ubiquitination of ERAD-C substrates, two E2s, Ubc6 and Ubc7, are involved (Fig. 1). Ubc6 can attach a single ubiquitin not only to Lys but also to hydroxylated amino acids, which act as a primer for Ubc7-dependent polyubiquitination (Weber et al., 2016). In conjunction with Doa10, another ligase that is responsible for N-end rule degradation (Ubr1) contributes to the turnover of select ERAD-C substrates (Stolz et al., 2013; Prasad et al., 2018). In contrast to the E2 requirements for ERAD-C, only Ubc7 appears to be required for ERAD-L and ERAD-M, which works in concert with Hrd1. To this end, Ubc7 is tethered to the ER by Cue1, which facilitates ubiquitin loading (Metzger et al., 2013). For the degradation of an ERAD-L and ERAD-M substrate in mammals, mixed K-48/K-11 polyubiquitin chains are appended, which facilitates interaction with downstream components of the ERAD machinery (Leto et al., 2019). The discrimination of a subset of ERAD substrates, such as CD4, may employ a different mechanism, termed deubiquitinase sharpening. In this case, the degree of deubiquitinase-mediated rescue of potential ERAD substrates, rather than chaperone-mediated E3 activity per se, controls the rate of substrate removal (Zhang et al., 2013). More generally, deubiquitinases regulate the timing of ERAD by trimming polyubiquitin chains on misfolded proteins (Blount et al., 2012) and maintain productive protein folding (Abrami et al., 2008; Feldman and van der Goot, 2009). Nevertheless, because some polytopic membrane proteins in the ERAD-M family may bear multiple folding lesions (Buck and Brodsky, 2018), different branches of the ERAD pathway can cooperate with one another during substrate recognition and ubiquitination. The hierarchy of the decision-making process during the degradation of these more complex substrates is unknown.
Retrotranslocation and degradation
For substrates with lumenal lesions (i.e., those handled by the ERAD-L pathway), Sec61 and Hrd1 are prime candidates for the ERAD retrotranslocon. Based on its function as the ER translocation channel, Sec61 was thought to act in reverse and function as a retrotranslocon. Evidence supporting this hypothesis includes experiments demonstrating interaction between Sec61 and ERAD substrates as well as the proteasome (Wiertz et al., 1996b; Kalies et al., 2005; Schäfer and Wolf, 2009). In addition, Apolipoprotein B is retrotranslocated and targeted for degradation when forward translocation through Sec61 is interrupted (Mitchell et al., 1998), indicating that the two functions for this channel, translocation and retrotranslocation, can be uncoupled. More recent evidence favors the Hrd1 ubiquitin ligase as the retrotranslocon for other ERAD substrates. First, the function of this eight transmembrane protein requires oligomerization, and Hrd1 interacts with substrates at an early retrotranslocation stage (Horn et al., 2009; Carvalho et al., 2010). Additional evidence emerged from a reconstitution assay in which substrate retrotranslocation was recapitulated in proteoliposomes containing only Hrd1. In this case, retrotranslocation also required Hrd1-dependent autoubiquitination (Baldridge and Rapoport, 2016). The identity of Hrd1 as a retrotranslocon was corroborated by a cryo-electron microscopy structure of Hrd1 in complex with Hrd3 (Schoebel et al., 2017). Five of the eight transmembrane segments in Hrd1 form a funnel that extends almost completely from the cytosol to the lumen. Moreover, the aqueous cavity formed by Hrd1 is reminiscent of that formed by Sec61.
Another candidate for the retrotranslocon was Der1, a multispanning membrane protein that helps eliminate some ERAD substrates (Knop et al., 1996; Vashist and Ng, 2004; Mehnert et al., 2014). As a component of the Hrd1 complex, Der1 receives ERAD-L substrates from Yos9/Hrd3 and transfers them to Hrd1 (Horn et al., 2009; Mehnert et al., 2014). In one study, Derlin-1 (the Der1 homologue) facilitated the retrotranslocation of a soluble substrate from mammalian microsomes (Wahlman et al., 2007). However, the substrate is degraded in a ubiquitin-independent manner, which may bypass the requirement for an alternate retrotranslocon (Werner et al., 1996). Other evidence indicates that Der1 intimately contacts ERAD substrates as they exit the ER, perhaps by facilitating transmembrane domain solubilization (Mehnert et al., 2014).
Do ERAD-M substrates also use Hrd1 as the retrotranslocon? The identification and extraction of membrane proteins with folding lesions within the lipid bilayer is challenging. To date, HMG-CoA (hydroxymethylglutaryl coenzyme A) reductase (Hmg2 in yeast) is the best-characterized ERAD-M substrate (Stevenson et al., 2016; Johnson and DeBose-Boyd, 2018). This enzyme catalyzes the rate-limiting step during sterol biosynthesis, and thus, the reductase is stringently regulated by sterols and a sterol-dependent chaperone (Wangeline et al., 2017). The interaction between Hrd1 and Hmg2 appears to be direct (Sato et al., 2009), and for many years, it was assumed that Hrd1 facilitates Hmg2 retrotranslocation. More recent data instead implicate a Der1 homologue, Dfm1, as a retrotranslocon (Neal et al., 2018). In contrast, it remains unclear if ERAD-C substrates are extracted directly from the ER membrane in the absence of a retrotranslocon or whether the Doa10 ubiquitin ligase, which contains 14 transmembrane domains, is used (Kreft et al., 2006; Ravid et al., 2006).
During retrotranslocation from the ER or the ER membrane, lumenal disulfide bonds are broken by a reductase, which facilitates egress through the retrotranslocon (Ushioda et al., 2008). The action of a cis-trans Prolyl isomerase and the removal of an N-linked glycan can also contribute to retrotranslocation and/or degradation efficiency (Bernasconi et al., 2010b; Hirayama et al., 2010). Ubiquitinated substrates are then transferred to the 26S proteasome via the Cdc48 complex (or the p97 complex in mammals; Braun et al., 2002; Jarosch et al., 2002; Ye et al., 2003). Cdc48 recruitment to the ER is facilitated by Ubx2 (Neuber et al., 2005; Schuberth and Buchberger, 2005). Ultimately, the Cdc48 complex provides the driving force for retrotranslocation via ATP hydrolysis (Bays et al., 2001; Ye et al., 2001; Rabinovich et al., 2002) and pulls or segregates ubiquitinated substrates out of the ER membrane and into the cytosol before proteasomal degradation (Bodnar and Rapoport, 2017).
Cdc48 is a member of the ATPases associated with various cellular activities (AAA+ ATPase) family and contains a central pore, which is reminiscent of related protein-unfolding AAA+s, such as Hsp104 and ClpB (Mogk et al., 2015; Gates et al., 2017). During retrotranslocation, polyubiquitinated substrates bind the Cdc48–p97 complex via Ufd1/Npl4, and an unfolded loop in the substrate reaches the central pore in Cdc48–p97 (Blythe et al., 2017; Bodnar and Rapoport, 2017). Upon ATPase-dependent entry into the pore, Ufd1/Npl4 binding is weakened by ubiquitin chain trimming, which facilitates further threading through the pore. Polyubiquitin chain trimming is catalyzed by an associated deubiquitinating enzyme. Released substrates with ubiquitin chains that are too short to be degraded by the proteasome are modified by another Cdc48 cofactor, Ufd2, which is an E4 ubiquitin elongation enzyme (Koegl et al., 1999). A recent study indicates that Cdc48 initiates retrotranslocation via the unfolding of a substrate-appended ubiquitin molecule, which enters the Cdc48 pore (Twomey et al., 2019).
ERAD substrate delivery to the proteasome is facilitated by shuttling factors that contain both ubiquitin- and proteasome-binding domains (Kim et al., 2004; Medicherla et al., 2004). In higher eukaryotes, a chaperone holdase complex, which contains Bag6, associates with retrotranslocated ERAD substrates to prevent aggregation (Wang et al., 2011b). Chaperone holdases are especially critical for retrotranslocated membrane proteins since they contain hydrophobic transmembrane domains and are consequently aggregation prone. Since yeast lack Bag6, Hsp104 facilitates the solubilization and retrotranslocation of aggregation-prone ERAD substrates (Preston et al., 2018; Doonan et al., 2019).
Protein QC versus quantity control
In addition to misfolded proteins, some native proteins deemed unnecessary are turned-over by ERAD. For the quantity control of these substrates, adaptor protein binding, ligand binding, and/or the display of a degron is essential.
As outlined previously, Hmg2 is degraded in response to the accumulation of specific sterols. When sterol levels are high, Hmg2 adopts a conformation that is recognized by Hrd1–Hrd3 (Theesfeld and Hampton, 2013). On the contrary, depleted lipids trigger Apolipoprotein B degradation, since the protein fails to translocate fully into the ER and assemble into a lipoprotein particle (Doonan et al., 2018). An example of ligand-dependent quantity control is the yeast cadmium exporter Pca1, which contains a cytosolic ERAD-targeting signal that becomes exposed in the absence of cadmium. When cadmium levels rise, Pca1 is stable (Adle et al., 2009). An example of adaptor-mediated quantity control is Ypf1, which signals the ERAD of a yeast zinc transporter under zinc-replete conditions (Avci et al., 2014). Another example of adaptor-mediated quantity control occurs when the ERAD pathway is hijacked by a virus. The human cytomegalovirus encodes two membrane adaptors, US2 and US11, which bind newly synthesized major histocompatibility complex (MHC) class I molecules and recruit the ERAD machinery (Wiertz et al., 1996a; Stagg et al., 2009). Cell-surface display of MHC class I is reduced, thereby disabling the host immune response. A similar strategy is used by the HIV-encoded adaptor Vpu, which directs CD4 to the proteasome (Fujita et al., 1997; Schubert et al., 1998). Very recently, a link between cancer and ERAD quantity control was uncovered: the antidiabetic drug metformin signals the destruction of an oncogenic ligand, which consequently attenuates tumor growth (Cha et al., 2018). Together, it is probable that new examples of ERAD-dependent quantity control will emerge, along with additional examples of QC, especially as expanding clinical and genomic databases are leading to the identification of previously undiscovered disease-associated proteins that access the secretory pathway.
ER-phagy: A brother-in-arms for the ERAD pathway
While most misfolded proteins in the ER are efficiently handled by the ERAD pathway, the diameter of the pore in the retrotranslocon may preclude misfolded oligomeric or aggregated proteins from being exported from the ER. Because these substrates are toxic, the ER is equipped with another mechanism to protect homeostasis, ER-phagy, and links between ER-phagy and diseases are continually emerging. One study classified yeast ER-phagy as micro-ER-phagy and macro-ER-phagy (Lipatova and Segev, 2015), and a receptor-mediated degradation pathway for misfolded proteins generated in the ER was recently termed ER-to-lysosome–associated degradation (ERLAD; reviewed in Fregno and Molinari, 2019; Wilkinson, 2019).
The term ER-phagy was first used upon the discovery of ER whorls, which arise after UPR-associated lipid synthesis in yeast (Bernales et al., 2006, 2007). Later studies indicated that ER-phagy is induced by nitrogen starvation, rapamycin treatment, and protein aggregation and uses the core autophagy machinery (Grumati et al., 2018). ER-phagy also disposes of ERAD-resistant misfolded proteins (Fregno and Molinari, 2019). The discovery of various ER-phagy receptors (Fig. 2) highlights the potential for substrate selection. Each receptor contains a domain that facilitates interaction with an autophagy-requiring factor, Atg8/LC3. However, many receptors lack lumenal domains, so their ability to identify lumenal cargo requires an adaptor.
Figure 2.

ER-phagy in yeast and mammals. ER-phagy receptors reside in distinct ER subdomains (yeast, left; mammals, right). ER-phagy receptors concentrate cargo in an ER subdomain via interaction with Atg8 (yeast) or LC3/GABARAP (mammal) in the growing autophagic membrane, the phagophore. ER-phagy substrates are next enclosed in a double-membrane-bound autophagosome, which fuses with the lysosome/vacuole. In yeast, the UPR (black lightning bolt) can give rise to ER whorls, which are nonselectively delivered to the vacuole. In mammals, Sec62 helps reestablish ER homeostasis after UPR induction.
In yeast, Atg40 localizes primarily to the cortical and cytoplasmic ER, resulting in autophagic sequestration of ER subdomains. Atg40 contains an Atg8 interacting motif, allowing for Atg8 engagement of the phagophore membrane (Mochida et al., 2015). The phagophore serves as a precursor to autophagosomes, which ultimately fuse with lysosomes via endosomes and multivesicular bodies (MVBs; see below). Nitrogen starvation or rapamycin enhances Atg40 expression, providing a means to induce ER-phagy. These conditions, along with the expression of an aggregation-prone ERAD substrate, also induce the expression of an alternate COPII subunit, Lst1, which functions with Atg40 to select ER-phagy sites. An Lst1 homologue, SEC24C, similarly marks ER-phagy sites in mammals (Cui et al., 2019).
The identification of ER-phagy receptors in mammals, including FAM134B, RTN3, Sec62, cell-cycle progression gene 1 (CCPG1), ATL3, and TEX264, has advanced our understanding of ER-phagy (Khaminets et al., 2015; Fumagalli et al., 2016; Grumati et al., 2017; Smith et al., 2018; An et al., 2019; Chen et al., 2019; Chino et al., 2019). For example, FAM134B mediates starvation-induced ER-phagy in mammalian cells and localizes at the edge of ER sheets via a reticulon homology domain, promoting membrane curvature during autophagosome formation (Khaminets et al., 2015). The presence of an LC3-interacting region (LIR) motif in FAM134B recruits LC3 (the orthologue of yeast Atg8) and/or GABARAP (Gamma-aminobutyric acid receptor-associated protein) at the limiting membrane of growing phagophores.
An expanding group of misfolded proteins has been identified as FAM134B cargo. Notably, mutations in type I procollagen (PC1) or the Hsp47 collagen chaperone form ERAD-resistant protein aggregates, which are eliminated by ER-phagy or ERLAD in a FAM134B-dependent manner (Ishida et al., 2006; Fregno et al., 2018; Forrester et al., 2019). In at least one case, FAM134B-dependent recognition requires a chaperone-like lectin in the ER, calnexin. This is essential, since FAM134B lacks an ER lumenal domain. Another FAM134B substrate is a misfolded form of the Niemman–Pick type C disease protein, but unlike PC1, this substrate is degraded by both ERAD and ER-phagy (Gelsthorpe et al., 2008; Schultz et al., 2018). Interestingly, FAM134B is cleaved by a protease encoded in the Zika and Dengue viral genomes (Lennemann and Coyne, 2017). It will be important to identify other lumenal factors that contribute to substrate recognition and determine whether additional pathogens modulate ER-phagy efficiency.
The ER-phagy receptor RTN3 also contains a reticulon homology domain (Yang and Strittmatter, 2007). During amino acid starvation, RTN3 oligomerizes to engage and cluster LC3, which forms autophagosomes at ER tubules (Grumati et al., 2017). ATL3 also functions at ER tubules (Liang et al., 2018; Chen et al., 2019). Although ATL3 and RTN3 drive autophagosome formation and interact with different ATG8 family members, RTN3 overexpression restores ER-phagy upon loss of ATL3 (Chen et al., 2019). In contrast, a receptor for “recovER-phagy” is an LIR motif-containing protein, Sec62, which in this case acts independently of its role during protein translocation (Fumagalli et al., 2016). RecovER-phagy resets ER volume and content after ER stress and employs the autophagy core machinery.
Unlike the receptors mentioned above, CCPG1 contains both LIR and FIP200-interacting regions, which bind independently to LC3 as well as FIP200, another component that associates with the autophagy machinery (Smith et al., 2018). During ER stress, CCPG1 expression is up-regulated to remove portions of the peripheral ER to reduce protein aggregation. It is unknown if CCPG1 specifically recognizes aggregated or misfolded proteins.
Recently, TEX264 was identified as a starvation-induced ER-phagy receptor that functions at three-way junctions in the ER (An et al., 2019; Chino et al., 2019). TEX264 contains a long disordered region between its membrane domain and the LIR motif that is required for ER-phagy (Chino et al., 2019). Compared with other identified receptors, TEX264 binds with greater efficiency to LC3 and GABARAP. In addition, TEX264 is expressed more ubiquitously and probably accounts for half of all ER-phagy flux during starvation (Chino et al., 2019). Overall, even though the identification of these ER-phagy receptors has helped define the nature of cargo selection, how the ER-phagy machinery degrades most receptor-bound cargo proteins remains an important open question (Wilkinson, 2019).
Finally, other pathways that handle misfolded ER proteins and use autophagy-associated factors have been identified. For example, α-1 antitrypsin-Z (AT-Z), which is an aggregation-prone and disease-associated substrate, is destroyed by ERAD as well as autophagy in yeast (Kruse et al., 2006). In mammalian cells, AT-Z can be sorted for lysosome-dependent degradation by the ER-phagy receptor FAM134B, but this pathway is distinct from the canonical ER-phagy pathway and thus represents an example of an ERLAD substrate (Fregno et al., 2018). Also, in contrast to the FAM134B-dependent targeting of PC1, another report suggested that collagen was sequestered at ER exit sites marked by COPII components along with components of the autophagy machinery (Omari et al., 2018). Based on image analysis, it was proposed that the lysosome contacts these sites and directly engulfs collagen. The authors suggested that this autophagosome-independent process may involve the recognition of ubiquitinated COPII components that are otherwise used to enlarge COPII vesicles (Jin et al., 2012).
GQC: A means to catch ER escapees
Secretory proteins exit the ER in COPII vesicles through bulk flow or a receptor-mediated process (Barlowe and Helenius, 2016; Gomez-Navarro and Miller, 2016). Although high levels of secretory proteins could overwhelm ER retention mechanisms, autoregulation of ER export monitors and regulates the flux of folded cargos from the ER in mammalian cells (Subramanian et al., 2019). In response to increased secretory protein production, autoregulation of ER export components are activated to facilitate cargo export and attenuate protein synthesis.
The Golgi apparatus, the next organelle encountered by secretory proteins after ER exit, functions as a protein modification factory, chemically altering substrates via glycosylation, acetylation, phosphorylation, sulfation, palmitoylation, and methylation. Some substrates are proteolytically activated. In addition, the Golgi serves as a QC checkpoint (Arvan et al., 2002; Potelle et al., 2015). Collectively, these events have been referred to as post-ERQC, ERLAD, or GQC, although there are some distinctions among these pathways. Indeed, a growing number of misfolded proteins escape ERAD and are delivered to the lysosome/vacuole for degradation after being selected in the Golgi. Based on where they are degraded, we propose that GQC can be differentiated into proteasome-targeted GQC and lysosome/vacuole–targeted GQC (Fig. 3). Regardless of how these events are defined, there are critical open questions in this field of research. Why do some substrates avoid ERAD but are instead targeted for GQC? What are the structural or other features that distinguish between these substrates? What role does ER–Golgi recycling play in substrate degradation? Does the entire population or only a subset of misfolded proteins traffic to the Golgi before being retrieved and degraded? Can drugs be developed to rescue disease-causing proteins that access later steps in the secretory pathway before being degraded?
Figure 3.

The GQC pathway in yeast. Misfolded proteins targeted for GQC are transported to the cis-Golgi via COPII vesicles. Proteasome-targeted GQC: ER chaperones (e.g., Kar2 [in pink]) and retrieval receptors (in blue) in the cis-Golgi bind and retrieve misfolded proteins to the ER in COPI vesicles. These substrates are eliminated by ERAD-L. Lysosome/vacuole–targeted GQC: misfolded proteins that migrate to the trans-Golgi are sorted by the ESCRT/MVB pathway via Vps10 (green) or a ubiquitin ligase (e.g., Tul1). Following sorting, misfolded proteins are delivered to the vacuole.
Beyond the Golgi, select misfolded or dispensable proteins that are instead identified exclusively at the plasma membrane and delivered to the lysosome/vacuole are handled by the PMQC machinery. Interestingly, several elements of the PMQC pathway resemble those used during QC in the ER and Golgi. Due to space considerations, the reader is referred to reviews on this topic (Okiyoneda et al., 2011; MacGurn, 2014).
Proteasome-targeted GQC
Evidence from both yeast and mammalian studies support the involvement of the Golgi during ERAD. In budding yeast, efficient elimination of select ERAD-L substrates requires ER, exit because blocking ER-to-Golgi trafficking inhibited ERAD (Caldwell et al., 2001; Vashist et al., 2001; Taxis et al., 2002; Vashist and Ng, 2004). In fact, Erv29, which cycles between the ER and Golgi, was identified as the COPII vesicle cargo receptor for two such substrates (Caldwell et al., 2001; Vashist et al., 2001). Other misfolded proteins with mutated transmembrane domains are specifically returned to the ER from the Golgi via a retrieval receptor, Rer1 (Letourneur and Cosson, 1998; Sato et al., 2003). Because Erv29 and Rer1 fail to distinguish between folded and misfolded cargo, the escaped ERAD substrates are probably packaged into COPII and retrieved via COPI vesicles (Vashist et al., 2001) along with proteins that normally reside in the ER and must be retrieved.
In mammalian cells, unassembled MHC class I heavy-chain molecules expose polar residues in the transmembrane domain that are otherwise shielded by a partner, β2 microglobulin (Hughes et al., 1997). Instead of being retained in the ER, unassembled MHC class I molecules advance to the cis-Golgi before retrieval and ERAD (Hsu et al., 1991). A similar result was evident for the MHC class II β subunit, which cycles between the Golgi and ER. Accelerated proteasomal degradation of the β subunit upon brefeldin A treatment, which redistributes Golgi components to the ER, is consistent with Golgi transport (Dusseljee et al., 1998). COPI vesicle transport also plays an important role in retrieving unassembled TCRα (T cell receptor alpha subunit) to the ER (Yamamoto et al., 2001). In other cases, the ER chaperone BiP in conjunction with the K/HDEL receptor retrieves aberrant proteins, such as an unassembled TCRα subunit (Hammond and Helenius, 1994; Yamamoto et al., 2001). Consistent with data in yeast, the human orthologue of Rer1 also returns misfolded proteins with lesions in a transmembrane domain to the ER (Yamasaki et al., 2014; Briant et al., 2017). In contrast, when a misfolded GPI-anchored protein leaves the ER and then escapes GQC, it is handled by PMQC but remains bound to ER chaperones (Satpute-Krishnan et al., 2014; Zavodszky and Hegde, 2019).
Regardless of the mechanism by which substrates are retrieved, proteasome-targeted GQC most likely takes place in the cis-Golgi or perhaps in the ER-Golgi intermediate compartment. First, the K/HDEL retrieval receptor in yeast, Rer1, and Erv29 primarily reside in the cis-Golgi under steady-state conditions. Second, several proteasome-targeted GQC substrates fail to acquire Golgi modifications, which occurs in the medial Golgi in mammalian cells (Hsu et al., 1991; Dusseljee et al., 1998; Ito et al., 2002). Because ER retrieval is independent of substrate ubiquitination or the MVB pathway (see below), proteasome-dependent GQC is a gatekeeper that prevents ERAD substrates from entering later steps in the secretory pathway.
Lyosome/vacuole–targeted GQC
The previous use of the term GQC referred to an event that delivers misfolded proteins for lysosomal degradation. In this article, we define lysosome/vacuole–targeted GQC to describe this pathway, one that sorts misfolded proteins for lysosomal or vacuolar degradation in higher cells and in yeast, respectively. Data have primarily arisen from studies in yeast and revealed two routes to steer misfolded proteins from the Golgi to the lysosome/vacuole: receptor-mediated sorting and ubiquitin ligase-mediated sorting.
Receptor-mediated GQC
The proteinase A and carboxypeptidase Y hydrolases are sorted to the vacuole from the Golgi via clathrin-coated vesicles (Bowers and Stevens, 2005), but loss of Vps10 results in secretion (Marcusson et al., 1994; Cooper and Stevens, 1996). In addition to these wild-type cargo, Vps10 also recognizes and sorts misfolded proteins for vacuolar degradation (Hong et al., 1996; Holkeri and Makarow, 1998; Wang and Ng, 2010). Both deletion of VPS10 and overexpression of a misfolded substrate lead to secretion, indicating a saturable process (Hong et al., 1996). How Vps10 recognizes folded versus misfolded proteins remains unknown, but these substrates may engage different sites on Vps10 (Jørgensen et al., 1999).
A similar sorting mechanism was subsequently discovered in mammalian cells, and a key factor in the sorting of some aggregation-prone substrates is sortillin. Sortilin is a Vps10 orthologue that resides primarily in the Golgi and plasma membrane and mediates lysosomal cargo sorting (Nielsen et al., 1999, 2001; Lefrancois et al., 2003; Amengual et al., 2018). Similar to its yeast counterpart, sortilin also regulates the lysosomal sorting of misfolded proteins, such as aggregated GPP130, which is induced by manganese (Tewari et al., 2015; Venkat and Linstedt, 2017). Interestingly, while oligomerization/aggregation is sufficient to trigger lysosomal degradation, trafficking is sortilin independent, suggesting that protein aggregation is necessary, but not sufficient, for sortilin-mediated sorting. The finding that sortilin recognizes protein aggregates in the Golgi for lysosomal delivery is reminiscent of a study in which it was shown that furin aggregation led to lysosomal delivery (Wolins et al., 1997). Similarly, protein aggregation in the trans-Golgi was recently observed, and the aggregates were sequestered in a specific compartment in the endo/lysosomal pathway (Hellerschmied et al., 2019). The structural features and machinery that specify Vps10/sortilin-dependent GQC are unclear.
Ubiquitin ligase–mediated GQC
In yeast, two ubiquitin ligases in the Golgi recognize misfolded proteins for degradation in the lysosome: Tul1 and Rsp5 (Reggiori and Pelham, 2002; Wang et al., 2011c). After ubiquitination, misfolded membrane proteins are ferried concomitant with an inward budding process that forms intraluminal vesicles (Piper and Katzmann, 2007). During this event, ESCRT (endosomal sorting complexes required for transport) is recruited to invaginate the endosomal limiting membrane. ESCRT-0 is first recruited by binding ubiquitin on substrates and directs the assembly of downstream complexes. Next, ESCRT-I clusters ubiquitinated proteins and bridges ESCRT-0 and ESCRT-II. ESCRT-II has high affinity for endosome-enriched phospholipid phosphatidylinositol-3-phsophate and is assembled on the late endosomal membrane. ESCRT-III is then recruited and oligomerizes to induce inward budding and intraluminal vesicle invagination. Finally, an ATPase, Vps4, strips ESCRT components and directs membrane scission (Henne et al., 2011; Christ et al., 2017). Late endosomes with intraluminal vesicles are termed MVBs, which fuse with the lysosome/vacuole for cargo delivery (Piper and Katzmann, 2007).
The substrates for Tul1 contain polar residues in membrane-spanning domains. Tul1 functions with the ubiquitin-conjugating enzyme, Ubc4, to modify cytosolic domains on misfolded substrates, thereby acting as a sorting signal for the MVB pathway. Two wild-type proteins, CPS1 and Phm5, also possess polar residues in the membrane-spanning domain and are similarly ubiquitinated and delivered to the vacuole where they function. The scope of Tul1 substrates is expanded after stress (Dobzinski et al., 2015).
Tul1 was also identified as a component of the Dsc complex in fission yeast, and the organization of this complex mirrors that of the Hrd1 complex (Stewart et al., 2011, 2012). With one exception, each component exists in budding yeast, and the employment of Cdc48 suggests that a unique form of GQC is at play, one that complements the ESCRT and ERAD pathways (Stewart et al., 2011; Schmidt et al., 2019). For example, when ergosterol or oxygen is depleted, the fission yeast Dsc complex binds ubiquitinated SREBP, which drives proteasome recruitment. After selective cleavage, a cytosolic transcriptional factor is produced that activates genes required for lipid metabolism (Stewart et al., 2011). How the Dsc complex recognizes its substrates and how the transcription factor domain in SREBP is protected from being completely degraded are unknown.
Interestingly, specific partners recruit the Tul1-containing complex to different organelles (Li et al., 2015). For example, Gld1 directs the Dsc complex to the Golgi and endosome, whereas Vld1 targets the complex to the vacuolar membrane (Yang et al., 2018). Therefore, Tul1 is a versatile E3 ligase that complexes with different subunits to protect protein fidelity and mediate quantity control at various locations in the late secretory pathway.
Rsp5 is the other versatile ligase involved in lysosome/vacuole–targeted GQC but is primarily known for its role as a trigger for the ubiquitin-dependent down-regulation of membrane proteins at the plasma membrane (Belgareh-Touzé et al., 2008; MacGurn et al., 2012). Rsp5 even clears mislocalized vacuolar membrane proteins (Sardana et al., 2019). Regardless of whether it acts during QC or to regulate membrane proteins, Rsp5 recognizes a PPXY motif in substrates or substrate-binding adaptors. Like Tul1, adaptors residing in different organelles deliver Rsp5 to the plasma membrane, endosomes, the vacuole, or the Golgi in yeast (Belgareh-Touzé et al., 2008; Sardana et al., 2019). For example, Rsp5 is recruited by the Bul1 and Bul2 adaptors to ubiquitinate misfolded membrane proteins for subsequent vacuole delivery (Chang and Fink, 1995; Luo and Chang, 1997; Pizzirusso and Chang, 2004). An obligate GQC substrate, Wsc1*, is another Rsp5 substrate routed from the Golgi to the vacuole (Wang and Ng, 2010; Wang et al., 2011c). The absence of a PPXY motif on Wsc1* suggests the existence of an as-yet-unidentified adaptor. Rsp5 also determines the fate of overexpressed ERAD substrates that escape the ER and advance to the Golgi but are then delivered to the vacuole (Haynes et al., 2002; Spear and Ng, 2003), as well as plasma membrane transporters in response to environmental changes (De Craene et al., 2001; Froissard et al., 2007). Overall, Rsp5 disposes of misfolded membrane proteins in the Golgi and dispensable transporters, the latter of which is reminiscent of ERAD quantity control.
Unlike Rsp5, members of the homologous NEDD4 family of ubiquitin ligases in mammalian cells are involved in neither GQC nor PMQC. Instead, these enzymes induce the endocytosis of plasma membrane proteins and subsequent hand-off to the MVB pathway (Rotin and Kumar, 2009). Channels regulated by NEDD4 and its relatives include the epithelial sodium channel and voltage-gated sodium channels, along with the insulin-like growth factor and TGFβ signaling pathway receptors (Staub et al., 1996; Fotia et al., 2004; Cao et al., 2008; Rougier et al., 2011).
In sum, the Golgi is armed with two QC checkpoints to destroy misfolded proteins that advance beyond the ER: proteasome-targeted GQC, which is initiated in the cis-Golgi, and lysosome/vacuole–targeted GQC, which most likely takes place in the trans-Golgi. Based on ill-defined criteria, GQC substrates are returned for ERAD via cis-Golgi resident receptors or directed for lysosomal degradation, which requires the Vps10/sortillin receptor (e.g., as seen for misfolded lumenal/soluble proteins) or a ubiquitin ligase complex (e.g., as seen for misfolded membrane proteins).
Cooperation among ERAD, ER-phagy, and GQC
Competition and backup pathways
Although the ERAD machinery monitors the fidelity of protein folding and clears most misfolded proteins, substrate selection is restricted by several biochemical properties. Aggregation propensity, the severity of a mutation, the site and nature of a folding lesion, the kinetics and thermodynamics of folding, and the rate at which a protein exits the ER constrain ERAD (Wiseman et al., 2007). In some cases, a substrate may avoid ERAD simply because it fails to bind a specific chaperone (Coughlan et al., 2004; Wang and Ng, 2010). Consequently, ER-phagy and GQC expand both the ability and capacity of the ER to recognize and destroy misfolded proteins.
Substrates for proteasome-targeted GQC or lysosome/vacuole–targeted GQC undergo anterograde trafficking from the ER to Golgi, which often requires an ER exit sequence (Kincaid and Cooper, 2007; Kawaguchi et al., 2010). However, prolonged chaperones engagement (e.g., with BiP and/or calnexin) may distinguish proteasome-targeted GQC substrates from lysosome/vacuole–targeted GQC substrates. Proteasome-targeted GQC substrates possess both a functional ER exit signal and a misfolded lesion that signals ERAD, but lysosome/vacuole–targeted GQC substrates may display more subtle lesions and contain an ER exit signal (Reggiori and Pelham, 2002; Wang and Ng, 2010; Wang et al., 2011c). For misfolded protein substrates that display both an ERAD signal and an ER exit signal, the ERAD and ER exit machineries compete (Kincaid and Cooper, 2007). Conversely, when ERAD is compromised, some misfolded substrates with functional exit signals traffic beyond the ER more efficiently (Spear and Ng, 2003; Kincaid and Cooper, 2007; Wang et al., 2011a). Compromising ERAD activity can also increase ER exit (Kincaid and Cooper, 2007; Wang et al., 2014). Furthermore, under specific conditions when a soluble ERAD substrate is overexpressed, ERAD becomes saturated, resulting in Golgi and then lysosome/vacuole delivery (Spear and Ng, 2003).
Most lysosome/vacuole–targeted GQC substrates contain folding lesions in the lumenal domain or a transmembrane domain. This observation is consistent with the finding that ERAD substrates with lumenal lesions can advance to the Golgi before being returned to the ER, while those with cytosolic folding lesions are retained in the ER (Vashist and Ng, 2004). Cytosolic folding lesions may disturb the presentation of an ER exit signal, and cytosolic chaperones may outcompete components required for COPII-mediated ER exit. Recently, we reported on a novel substrate with a cytosolic folding lesion that is degraded by both ERAD and vacuole-targeted GQC. We also showed that substrate aggregation alters the balance between ERAD and vacuole-dependent degradation in yeast (Sun and Brodsky, 2018).
In comparison to GQC, ER-phagy might handle more severely misfolded substrates that cannot be disposed of by ERAD due to their aggregation state or the absence of an ERAD signal, which in most cases remains mysterious. Protein misfolding and aggregation could also bury ER exit signals, preventing anterograde transport. Because calnexin plays a role in ERAD and recruits FAM134B (Forrester et al., 2019), two distinct QC machineries, ERAD and ERLAD, which can rely on FAM134B and ATG-associated vesicle transport, are linked by calnexin. Other substrates are simultaneously routed for ERAD and ER-phagy (e.g., Schultz et al., 2018; Cunningham et al., 2019). The identification of a detergent-soluble protein degraded in the lysosome, which forms ER puncta that colocalize with an autophagy receptor and requires autophagy–associated proteins, suggests that aggregate-free misfolded substrates are also targeted for ER-phagy (Houck et al., 2014).
The UPR: A unifying principle
When ERAD is unable to maintain ER homeostasis, the UPR is induced. As a result, protein translation is attenuated, the ER expands, and protein folding, ERAD, ER-phagy, and GQC are enhanced (Travers et al., 2000; Yoshida et al., 2003, 2006; Smith and Wilkinson, 2017; Smith et al., 2018; Karagöz et al., 2019). In turn, ERAD controls the levels of some UPR components (Hwang and Qi, 2018), and factor required for the ERAD of glycosylated substrates are induced by the UPR. One such component is stabilized by a partner (Termine et al., 2009). Other ERAD components induced by the UPR include Hrd1, Der1, Hrd3, Ubc7, and Erv29, the last of which transports both folded and misfolded cargo. Although the expression of Rer1, the retrieval receptor for proteasome-targeted GQC, is UPR-independent, the loss of Rer1 induces ER stress in yeast and worms (Ghavidel et al., 2015). Tul1, which is required for receptor-mediated GQC, is also induced by the UPR (Roth et al., 1998; Travers et al., 2000; Reggiori and Pelham, 2002), as is autophagic flux (Senft and Ronai, 2015). In metazoans, the PERK-eIF2α branch of the UPR enhances autophagy (B’chir et al., 2013), and the ER-phagy receptor CCPG1 is induced by ER stress (Smith et al., 2018). A conserved Atg8-binding adaptor, Ubx5, was recently identified that delivers mutated, inactive Cdc48/p97 for autophagic degradation, which would otherwise compromise ERAD (Marshall et al., 2019). Another ERAD component, the proteasome itself, also becomes an autophagy substrate when inactivated (Marshall et al., 2015). Overall, the machineries employed by ERAD, ER-phagy, and GQC are stress inducible, suggesting cooperative orchestration of ER proteostasis (Fig. 4).
Figure 4.
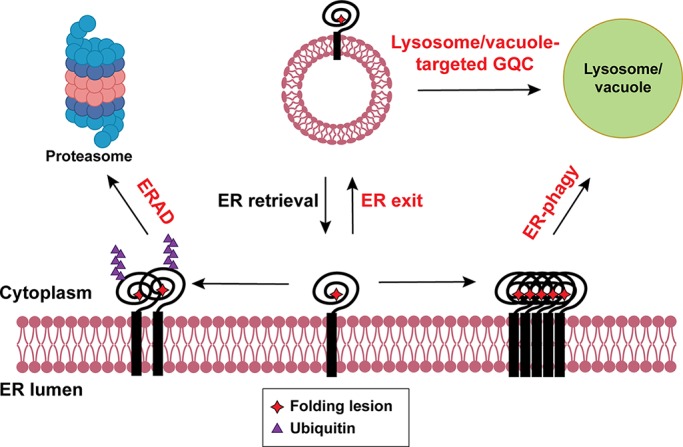
The fates of misfolded proteins in the ER. Most misfolded proteins in the ER are eliminated by ERAD via the ubiquitin-proteasome system. Some misfolded proteins exit the ER and are sorted to the lysosome/vacuole for degradation. Other misfolded proteins are transported to the lysosome/vacuole for degradation via ER-phagy. The three QC machineries act coordinately to safeguard ER proteostasis and are induced by the UPR (defined by red text).
Concluding remarks
We have highlighted key pathways that maintain proteostasis in the early secretory pathway, but our current knowledge base is restricted by the field’s study of select experimental models and a fraction of the universe of substrates that might be targeted by these pathways. With growing interest in the QC field, and the isolation of an expanding cadre of substrates and methods, these limitations will ultimately be overcome. A number of related pathways have also emerged; it was recently demonstrated that a UPR- and chaperone-dependent “reflux” of folded proteins from the ER to the cytosol occurs in yeast (Igbaria et al., 2019).
One open question is whether the UPR is the only mechanism that coordinately regulates the ERAD, ER-phagy, and/or GQC pathways. Downstream responses triggered by the UPR were identified when cells are treated with poisons that severely compromise protein folding, but more subtle and diverse responses are apparent upon the expression of distinct misfolded proteins (Buck et al., 2015; Bergmann et al., 2018). Furthermore, overexpression of TFEB, which increases autophagy and lysosomal function, remedies liver toxicity associated with AT-Z (see above), which is degraded by both ERAD and the lysosome (Pastore et al., 2013). These data suggest that alternate signal transduction pathways might be harnessed to treat diseases associated with early secretory pathway QC. Moreover, silencing distinct chaperones in the ER, which should globally compromise folding, leads to a UPR-independent phenomenon that alters the levels of other chaperones (Eletto et al., 2012). More trivially, inhibition of one pathway has indirect consequences on another pathway. For example, autophagy inhibition increases the levels of p62, a receptor for ubiquitinated autophagy cargo. The resulting rise in p62 delays delivery of proteasome-targeted substrates (Korolchuk et al., 2009).
Another open question is whether the decision between ERAD versus other pathways is made only within the ER. Mutant forms of CFTR, which are associated with cystic fibrosis, use some of the same QC factors for ERAD and PMQC (Okiyoneda et al., 2011). Thus, it is possible that misfolded proteins are identified and tagged with chaperones in the ER, which signals QC events later in the secretory pathway. In addition, components associated with glycan QC can migrate to an ERQC compartment, which is distinct from the ER and potentially isolates the ER folding and degradation machineries (Shenkman and Lederkremer, 2019). In the future, it will be important to determine how this compartment interfaces with the ER-phagy and GQC pathways.
Finally, a critical undertaking will be to translate discoveries to animal models. Some studies have examined how ERAD defects lead to specific pathologies and modulate physiological responses and signaling pathways in mice (Qi et al., 2017). The development of new animal models in which ERAD, ER-phagy, and GQC can be studied is increasingly vital.
Acknowledgments
We apologize to colleagues whose work we were unable to discuss due to character limits. We thank Meir Aridor, Adam Linstedt, Allyson O’Donnell, and Susan Ferro-Novick for critically reading the manuscript.
Work on ERAD in the laboratory is supported by National Institutes of Health grants R01 GM075061, P30 DK079307, and R35 GM131732.
The authors declare no competing financial interests.
References
- Abrami L., Kunz B., Iacovache I., and van der Goot F.G.. 2008. Palmitoylation and ubiquitination regulate exit of the Wnt signaling protein LRP6 from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 105:5384–5389. 10.1073/pnas.0710389105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adle D.J., Wei W., Smith N., Bies J.J., and Lee J.. 2009. Cadmium-mediated rescue from ER-associated degradation induces expression of its exporter. Proc. Natl. Acad. Sci. USA. 106:10189–10194. 10.1073/pnas.0812114106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahner A., Nakatsukasa K., Zhang H., Frizzell R.A., and Brodsky J.L.. 2007. Small heat-shock proteins select deltaF508-CFTR for endoplasmic reticulum-associated degradation. Mol. Biol. Cell. 18:806–814. 10.1091/mbc.e06-05-0458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amengual J., Guo L., Strong A., Madrigal-Matute J., Wang H., Kaushik S., Brodsky J.L., Rader D.J., Cuervo A.M., and Fisher E.A.. 2018. Autophagy Is Required for Sortilin-Mediated Degradation of Apolipoprotein B100. Circ. Res. 122:568–582. 10.1161/CIRCRESAHA.117.311240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H., Ordureau A., Paulo J.A., Shoemaker C.J., Denic V., and Harper J.W.. 2019. TEX264 Is an Endoplasmic Reticulum-Resident ATG8-Interacting Protein Critical for ER Remodeling during Nutrient Stress. Mol. Cell. 74:891–908.e10. 10.1016/j.molcel.2019.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anelli T., and Sitia R.. 2008. Protein quality control in the early secretory pathway. EMBO J. 27:315–327. 10.1038/sj.emboj.7601974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvan P., Zhao X., Ramos-Castaneda J., and Chang A.. 2002. Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic. 3:771–780. 10.1034/j.1600-0854.2002.31102.x [DOI] [PubMed] [Google Scholar]
- Avci D., Fuchs S., Schrul B., Fukumori A., Breker M., Frumkin I., Chen C.Y., Biniossek M.L., Kremmer E., Schilling O., et al. . 2014. The yeast ER-intramembrane protease Ypf1 refines nutrient sensing by regulating transporter abundance. Mol. Cell. 56:630–640. 10.1016/j.molcel.2014.10.012 [DOI] [PubMed] [Google Scholar]
- B’chir W., Maurin A.C., Carraro V., Averous J., Jousse C., Muranishi Y., Parry L., Stepien G., Fafournoux P., and Bruhat A.. 2013. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41:7683–7699. 10.1093/nar/gkt563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balchin D., Hayer-Hartl M., and Hartl F.U.. 2016. In vivo aspects of protein folding and quality control. Science. 353:aac4354 10.1126/science.aac4354 [DOI] [PubMed] [Google Scholar]
- Baldridge R.D., and Rapoport T.A.. 2016. Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell. 166:394–407. 10.1016/j.cell.2016.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe C., and Helenius A.. 2016. Cargo Capture and Bulk Flow in the Early Secretory Pathway. Annu. Rev. Cell Dev. Biol. 32:197–222. 10.1146/annurev-cellbio-111315-125016 [DOI] [PubMed] [Google Scholar]
- Bays N.W., Wilhovsky S.K., Goradia A., Hodgkiss-Harlow K., and Hampton R.Y.. 2001. HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol. Biol. Cell. 12:4114–4128. 10.1091/mbc.12.12.4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgareh-Touzé N., Léon S., Erpapazoglou Z., Stawiecka-Mirota M., Urban-Grimal D., and Haguenauer-Tsapis R.. 2008. Versatile role of the yeast ubiquitin ligase Rsp5p in intracellular trafficking. Biochem. Soc. Trans. 36:791–796. 10.1042/BST0360791 [DOI] [PubMed] [Google Scholar]
- Bergmann T.J., Fregno I., Fumagalli F., Rinaldi A., Bertoni F., Boersema P.J., Picotti P., and Molinari M.. 2018. Chemical stresses fail to mimic the unfolded protein response resulting from luminal load with unfolded polypeptides. J. Biol. Chem. 293:5600–5612. 10.1074/jbc.RA117.001484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S., McDonald K.L., and Walter P.. 2006. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 4:e423 10.1371/journal.pbio.0040423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S., Schuck S., and Walter P.. 2007. ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy. 3:285–287. 10.4161/auto.3930 [DOI] [PubMed] [Google Scholar]
- Bernasconi R., Galli C., Calanca V., Nakajima T., and Molinari M.. 2010a Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J. Cell Biol. 188:223–235. 10.1083/jcb.200910042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernasconi R., Soldà T., Galli C., Pertel T., Luban J., and Molinari M.. 2010b Cyclosporine A-sensitive, cyclophilin B-dependent endoplasmic reticulum-associated degradation. PLoS One. 5:e13008 10.1371/journal.pone.0013008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount J.R., Burr A.A., Denuc A., Marfany G., and Todi S.V.. 2012. Ubiquitin-specific protease 25 functions in Endoplasmic Reticulum-associated degradation. PLoS One. 7:e36542 10.1371/journal.pone.0036542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blythe E.E., Olson K.C., Chau V., and Deshaies R.J.. 2017. Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP•NPLOC4•UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proc. Natl. Acad. Sci. USA. 114:E4380–E4388. 10.1073/pnas.1706205114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar N.O., and Rapoport T.A.. 2017. Molecular Mechanism of Substrate Processing by the Cdc48 ATPase Complex. Cell. 169:722–735.e729. 10.1016/j.cell.2017.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers K., and Stevens T.H.. 2005. Protein transport from the late Golgi to the vacuole in the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta. 1744:438–454. 10.1016/j.bbamcr.2005.04.004 [DOI] [PubMed] [Google Scholar]
- Braakman I., and Hebert D.N.. 2013. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 5:a013201 10.1101/cshperspect.a013201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S., Matuschewski K., Rape M., Thoms S., and Jentsch S.. 2002. Role of the ubiquitin-selective CDC48(UFD1/NPL4 )chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J. 21:615–621. 10.1093/emboj/21.4.615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briant K., Johnson N., and Swanton E.. 2017. Transmembrane domain quality control systems operate at the endoplasmic reticulum and Golgi apparatus. PLoS One. 12:e0173924 10.1371/journal.pone.0173924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky J.L., Werner E.D., Dubas M.E., Goeckeler J.L., Kruse K.B., and McCracken A.A.. 1999. The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J. Biol. Chem. 274:3453–3460. 10.1074/jbc.274.6.3453 [DOI] [PubMed] [Google Scholar]
- Buck T.M., and Brodsky J.L.. 2018. Epithelial sodium channel biogenesis and quality control in the early secretory pathway. Curr. Opin. Nephrol. Hypertens. 27:364–372. 10.1097/MNH.0000000000000438 [DOI] [PubMed] [Google Scholar]
- Buck T.M., Jordan R., Lyons-Weiler J., Adelman J.L., Needham P.G., Kleyman T.R., and Brodsky J.L.. 2015. Expression of three topologically distinct membrane proteins elicits unique stress response pathways in the yeast Saccharomyces cerevisiae. Physiol. Genomics. 47:198–214. 10.1152/physiolgenomics.00101.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell S.R., Hill K.J., and Cooper A.A.. 2001. Degradation of endoplasmic reticulum (ER) quality control substrates requires transport between the ER and Golgi. J. Biol. Chem. 276:23296–23303. 10.1074/jbc.M102962200 [DOI] [PubMed] [Google Scholar]
- Camirand A., Heysen A., Grondin B., and Herscovics A.. 1991. Glycoprotein biosynthesis in Saccharomyces cerevisiae. Isolation and characterization of the gene encoding a specific processing alpha-mannosidase. J. Biol. Chem. 266:15120–15127. [PubMed] [Google Scholar]
- Cao X.R., Lill N.L., Boase N., Shi P.P., Croucher D.R., Shan H., Qu J., Sweezer E.M., Place T., Kirby P.A., et al. . 2008. Nedd4 controls animal growth by regulating IGF-1 signaling. Sci. Signal. 1:ra5 10.1126/scisignal.1160940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P., Goder V., and Rapoport T.A.. 2006. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 126:361–373. 10.1016/j.cell.2006.05.043 [DOI] [PubMed] [Google Scholar]
- Carvalho P., Stanley A.M., and Rapoport T.A.. 2010. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 143:579–591. 10.1016/j.cell.2010.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha J.H., Yang W.H., Xia W., Wei Y., Chan L.C., Lim S.O., Li C.W., Kim T., Chang S.S., Lee H.H., et al. . 2018. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell. 71:606–620.e607. 10.1016/j.molcel.2018.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A., and Fink G.R.. 1995. Targeting of the yeast plasma membrane [H+]ATPase: a novel gene AST1 prevents mislocalization of mutant ATPase to the vacuole. J. Cell Biol. 128:39–49. 10.1083/jcb.128.1.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Retzlaff M., Roos T., and Frydman J.. 2011. Cellular strategies of protein quality control. Cold Spring Harb. Perspect. Biol. 3:a004374 10.1101/cshperspect.a004374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Xiao Y., Chai P., Zheng P., Teng J., and Chen J.. 2019. ATL3 Is a Tubular ER-Phagy Receptor for GABARAP-Mediated Selective Autophagy. Curr. Biol. 29:846–855.e846. 10.1016/j.cub.2019.01.041 [DOI] [PubMed] [Google Scholar]
- Cherepanova N., Shrimal S., and Gilmore R.. 2016. N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 41:57–65. 10.1016/j.ceb.2016.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chino H., Hatta T., Natsume T., and Mizushima N.. 2019. Intrinsically Disordered Protein TEX264 Mediates ER-phagy. Mol. Cell. 74:909–921.e6. 10.1016/j.molcel.2019.03.033 [DOI] [PubMed] [Google Scholar]
- Christ L., Raiborg C., Wenzel E.M., Campsteijn C., and Stenmark H.. 2017. Cellular Functions and Molecular Mechanisms of the ESCRT Membrane-Scission Machinery. Trends Biochem. Sci. 42:42–56. 10.1016/j.tibs.2016.08.016 [DOI] [PubMed] [Google Scholar]
- Christianson J.C., and Ye Y.. 2014. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat. Struct. Mol. Biol. 21:325–335. 10.1038/nsmb.2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A.A., and Stevens T.H.. 1996. Vps10p cycles between the late-Golgi and prevacuolar compartments in its function as the sorting receptor for multiple yeast vacuolar hydrolases. J. Cell Biol. 133:529–541. 10.1083/jcb.133.3.529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlan C.M., Walker J.L., Cochran J.C., Wittrup K.D., and Brodsky J.L.. 2004. Degradation of mutated bovine pancreatic trypsin inhibitor in the yeast vacuole suggests post-endoplasmic reticulum protein quality control. J. Biol. Chem. 279:15289–15297. 10.1074/jbc.M309673200 [DOI] [PubMed] [Google Scholar]
- Cui Y., Parashar S., Zahoor M., Needham P.G., Mari M., Zhu M., Chen S., Ho H.C., Reggiori F., Farhan H., et al. . 2019. A COPII subunit acts with an autophagy receptor to target endoplasmic reticulum for degradation. Science. 365:53–60. 10.1126/science.aau9263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C.N., Williams J.M., Knupp J., Arunagiri A., Arvan P., and Tsai B.. 2019. Cells Deploy a Two-Pronged Strategy to Rectify Misfolded Proinsulin Aggregates. Mol. Cell. 75:442–456.e444. 10.1016/j.molcel.2019.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Craene J.O., Soetens O., and Andre B.. 2001. The Npr1 kinase controls biosynthetic and endocytic sorting of the yeast Gap1 permease. J. Biol. Chem. 276:43939–43948. 10.1074/jbc.M102944200 [DOI] [PubMed] [Google Scholar]
- Denic V., Quan E.M., and Weissman J.S.. 2006. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 126:349–359. 10.1016/j.cell.2006.05.045 [DOI] [PubMed] [Google Scholar]
- Dobzinski N., Chuartzman S.G., Kama R., Schuldiner M., and Gerst J.E.. 2015. Starvation-Dependent Regulation of Golgi Quality Control Links the TOR Signaling and Vacuolar Protein Sorting Pathways. Cell Reports. 12:1876–1886. 10.1016/j.celrep.2015.08.026 [DOI] [PubMed] [Google Scholar]
- Doonan L.M., Fisher E.A., and Brodsky J.L.. 2018. Can modulators of apolipoproteinB biogenesis serve as an alternate target for cholesterol-lowering drugs? Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 1863:762–771. 10.1016/j.bbalip.2018.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doonan L.M., Guerriero C.J., Preston G.M., Buck T.M., Khazanov N., Fisher E.A., Senderowitz H., and Brodsky J.L.. 2019. Hsp104 facilitates the endoplasmic-reticulum-associated degradation of disease-associated and aggregation-prone substrates. Protein Sci. 28:1290–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusseljee S., Wubbolts R., Verwoerd D., Tulp A., Janssen H., Calafat J., and Neefjes J.. 1998. Removal and degradation of the free MHC class II beta chain in the endoplasmic reticulum requires proteasomes and is accelerated by BFA. J. Cell Sci. 111:2217–2226. [DOI] [PubMed] [Google Scholar]
- Duttler S., Pechmann S., and Frydman J.. 2013. Principles of cotranslational ubiquitination and quality control at the ribosome. Mol. Cell. 50:379–393. 10.1016/j.molcel.2013.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eletto D., Maganty A., Eletto D., Dersh D., Makarewich C., Biswas C., Paton J.C., Paton A.W., Doroudgar S., Glembotski C.C., and Argon Y.. 2012. Limitation of individual folding resources in the ER leads to outcomes distinct from the unfolded protein response. J. Cell Sci. 125:4865–4875. 10.1242/jcs.108928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige M.J., and Hendershot L.M.. 2013. Quality control of integral membrane proteins by assembly-dependent membrane integration. Mol. Cell. 51:297–309. 10.1016/j.molcel.2013.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman M., and van der Goot F.G.. 2009. Novel ubiquitin-dependent quality control in the endoplasmic reticulum. Trends Cell Biol. 19:357–363. 10.1016/j.tcb.2009.05.005 [DOI] [PubMed] [Google Scholar]
- Foresti O., Rodriguez-Vaello V., Funaya C., and Carvalho P.. 2014. Quality control of inner nuclear membrane proteins by the Asi complex. Science. 346:751–755. 10.1126/science.1255638 [DOI] [PubMed] [Google Scholar]
- Forrester A., De Leonibus C., Grumati P., Fasana E., Piemontese M., Staiano L., Fregno I., Raimondi A., Marazza A., Bruno G., et al. . 2019. A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J. 38:e99847 10.15252/embj.201899847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotia A.B., Ekberg J., Adams D.J., Cook D.I., Poronnik P., and Kumar S.. 2004. Regulation of neuronal voltage-gated sodium channels by the ubiquitin-protein ligases Nedd4 and Nedd4-2. J. Biol. Chem. 279:28930–28935. 10.1074/jbc.M402820200 [DOI] [PubMed] [Google Scholar]
- Fregno I., and Molinari M.. 2019. Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways. Crit. Rev. Biochem. Mol. Biol. 54:153–163. 10.1080/10409238.2019.1610351 [DOI] [PubMed] [Google Scholar]
- Fregno I., Fasana E., Bergmann T.J., Raimondi A., Loi M., Soldà T., Galli C., D’Antuono R., Morone D., Danieli A., et al. . 2018. ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J. 37:e99259 10.15252/embj.201899259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froissard M., Belgareh-Touzé N., Dias M., Buisson N., Camadro J.M., Haguenauer-Tsapis R., and Lesuisse E.. 2007. Trafficking of siderophore transporters in Saccharomyces cerevisiae and intracellular fate of ferrioxamine B conjugates. Traffic. 8:1601–1616. 10.1111/j.1600-0854.2007.00627.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K., Omura S., and Silver J.. 1997. Rapid degradation of CD4 in cells expressing human immunodeficiency virus type 1 Env and Vpu is blocked by proteasome inhibitors. J. Gen. Virol. 78:619–625. 10.1099/0022-1317-78-3-619 [DOI] [PubMed] [Google Scholar]
- Fumagalli F., Noack J., Bergmann T.J., Cebollero E., Pisoni G.B., Fasana E., Fregno I., Galli C., Loi M., Soldà T., et al. . 2016. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 18:1173–1184. 10.1038/ncb3423 [DOI] [PubMed] [Google Scholar]
- Gates S.N., Yokom A.L., Lin J., Jackrel M.E., Rizo A.N., Kendsersky N.M., Buell C.E., Sweeny E.A., Mack K.L., Chuang E., et al. . 2017. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science. 357:273–279. 10.1126/science.aan1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauss R., Kanehara K., Carvalho P., Ng D.T., and Aebi M.. 2011. A complex of Pdi1p and the mannosidase Htm1p initiates clearance of unfolded glycoproteins from the endoplasmic reticulum. Mol. Cell. 42:782–793. 10.1016/j.molcel.2011.04.027 [DOI] [PubMed] [Google Scholar]
- Gelsthorpe M.E., Baumann N., Millard E., Gale S.E., Langmade S.J., Schaffer J.E., and Ory D.S.. 2008. Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Biol. Chem. 283:8229–8236. 10.1074/jbc.M708735200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavidel A., Baxi K., Ignatchenko V., Prusinkiewicz M., Arnason T.G., Kislinger T., Carvalho C.E., and Harkness T.A.. 2015. A Genome Scale Screen for Mutants with Delayed Exit from Mitosis: Ire1-Independent Induction of Autophagy Integrates ER Homeostasis into Mitotic Lifespan. PLoS Genet. 11:e1005429 10.1371/journal.pgen.1005429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Navarro N., and Miller E.A.. 2016. COP-coated vesicles. Curr. Biol. 26:R54–R57. 10.1016/j.cub.2015.12.017 [DOI] [PubMed] [Google Scholar]
- Grumati P., Morozzi G., Hölper S., Mari M., Harwardt M.I., Yan R., Müller S., Reggiori F., Heilemann M., and Dikic I.. 2017. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife. 6:e25555 10.7554/eLife.25555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumati P., Dikic I., and Stolz A.. 2018. ER-phagy at a glance. J. Cell Sci. 131:jcs217364 10.1242/jcs.217364 [DOI] [PubMed] [Google Scholar]
- Guerriero C.J., and Brodsky J.L.. 2012. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 92:537–576. 10.1152/physrev.00027.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero C.J., Reutter K.R., Augustine A.A., Preston G.M., Weiberth K.F., Mackie T.D., Cleveland-Rubeor H.C., Bethel N.P., Callenberg K.M., Nakatsukasa K., et al. . 2017. Transmembrane helix hydrophobicity is an energetic barrier during the retrotranslocation of integral membrane ERAD substrates. Mol. Biol. Cell. 28:2076–2090. 10.1091/mbc.e17-03-0184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habeck G., Ebner F.A., Shimada-Kreft H., and Kreft S.G.. 2015. The yeast ERAD-C ubiquitin ligase Doa10 recognizes an intramembrane degron. J. Cell Biol. 209:261–273. 10.1083/jcb.201408088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C., and Helenius A.. 1994. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J. Cell Biol. 126:41–52. 10.1083/jcb.126.1.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton R.Y., Gardner R.G., and Rine J.. 1996. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell. 7:2029–2044. 10.1091/mbc.7.12.2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S., Liu Y., and Chang A.. 2007. Cytoplasmic Hsp70 promotes ubiquitination for endoplasmic reticulum-associated degradation of a misfolded mutant of the yeast plasma membrane ATPase, PMA1. J. Biol. Chem. 282:26140–26149. 10.1074/jbc.M701969200 [DOI] [PubMed] [Google Scholar]
- Haynes C.M., Caldwell S., and Cooper A.A.. 2002. An HRD/DER-independent ER quality control mechanism involves Rsp5p-dependent ubiquitination and ER-Golgi transport. J. Cell Biol. 158:91–101. 10.1083/jcb.200201053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A., and Aebi M.. 2004. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73:1019–1049. 10.1146/annurev.biochem.73.011303.073752 [DOI] [PubMed] [Google Scholar]
- Hellerschmied D., Serebrenik Y.V., Shao L., Burslem G.M., and Crews C.M.. 2019. Protein Folding State-dependent Sorting at the Golgi Apparatus. Mol. Biol. Cell. 30:2296–2308. 10.1091/mbc.E19-01-0069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henne W.M., Buchkovich N.J., and Emr S.D.. 2011. The ESCRT pathway. Dev. Cell. 21:77–91. 10.1016/j.devcel.2011.05.015 [DOI] [PubMed] [Google Scholar]
- Hiller M.M., Finger A., Schweiger M., and Wolf D.H.. 1996. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 273:1725–1728. 10.1126/science.273.5282.1725 [DOI] [PubMed] [Google Scholar]
- Hirayama H., Seino J., Kitajima T., Jigami Y., and Suzuki T.. 2010. Free oligosaccharides to monitor glycoprotein endoplasmic reticulum-associated degradation in Saccharomyces cerevisiae. J. Biol. Chem. 285:12390–12404. 10.1074/jbc.M109.082081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitt R., and Wolf D.H.. 2004. DER7, encoding alpha-glucosidase I is essential for degradation of malfolded glycoproteins of the endoplasmic reticulum. FEMS Yeast Res. 4:815–820. 10.1016/j.femsyr.2004.04.002 [DOI] [PubMed] [Google Scholar]
- Holkeri H., and Makarow M.. 1998. Different degradation pathways for heterologous glycoproteins in yeast. FEBS Lett. 429:162–166. 10.1016/S0014-5793(98)00586-9 [DOI] [PubMed] [Google Scholar]
- Hong E., Davidson A.R., and Kaiser C.A.. 1996. A pathway for targeting soluble misfolded proteins to the yeast vacuole. J. Cell Biol. 135:623–633. 10.1083/jcb.135.3.623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn S.C., Hanna J., Hirsch C., Volkwein C., Schütz A., Heinemann U., Sommer T., and Jarosch E.. 2009. Usa1 functions as a scaffold of the HRD-ubiquitin ligase. Mol. Cell. 36:782–793. 10.1016/j.molcel.2009.10.015 [DOI] [PubMed] [Google Scholar]
- Houck S.A., Ren H.Y., Madden V.J., Bonner J.N., Conlin M.P., Janovick J.A., Conn P.M., and Cyr D.M.. 2014. Quality control autophagy degrades soluble ERAD-resistant conformers of the misfolded membrane protein GnRHR. Mol. Cell. 54:166–179. 10.1016/j.molcel.2014.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu V.W., Yuan L.C., Nuchtern J.G., Lippincott-Schwartz J., Hammerling G.J., and Klausner R.D.. 1991. A recycling pathway between the endoplasmic reticulum and the Golgi apparatus for retention of unassembled MHC class I molecules. Nature. 352:441–444. 10.1038/352441a0 [DOI] [PubMed] [Google Scholar]
- Hughes E.A., Hammond C., and Cresswell P.. 1997. Misfolded major histocompatibility complex class I heavy chains are translocated into the cytoplasm and degraded by the proteasome. Proc. Natl. Acad. Sci. USA. 94:1896–1901. 10.1073/pnas.94.5.1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyer G., Piluek W.F., Fansler Z., Kreft S.G., Hochstrasser M., Brodsky J.L., and Michaelis S.. 2004. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J. Biol. Chem. 279:38369–38378. 10.1074/jbc.M402468200 [DOI] [PubMed] [Google Scholar]
- Hwang J., and Qi L.. 2018. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 43:593–605. 10.1016/j.tibs.2018.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igbaria A., Merksamer P.I., Trusina A., Tilahun F., Johnson J.R., Brandman O., Krogan N.J., Weissman J.S., and Papa F.R.. 2019. Chaperone-mediated reflux of secretory proteins to the cytosol during endoplasmic reticulum stress. Proc. Natl. Acad. Sci. USA. 116:11291–11298. 10.1073/pnas.1904516116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y., Kubota H., Yamamoto A., Kitamura A., Bächinger H.P., and Nagata K.. 2006. Type I collagen in Hsp47-null cells is aggregated in endoplasmic reticulum and deficient in N-propeptide processing and fibrillogenesis. Mol. Biol. Cell. 17:2346–2355. 10.1091/mbc.e05-11-1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M., Amizuka N., Ozawa H., and Oda K.. 2002. Retention at the cis-Golgi and delayed degradation of tissue-non-specific alkaline phosphatase with an Asn153-->Asp substitution, a cause of perinatal hypophosphatasia. Biochem. J. 361:473–480. 10.1042/bj3610473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob C.A., Burda P., Roth J., and Aebi M.. 1998. Degradation of misfolded endoplasmic reticulum glycoproteins in Saccharomyces cerevisiae is determined by a specific oligosaccharide structure. J. Cell Biol. 142:1223–1233. 10.1083/jcb.142.5.1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosch E., Taxis C., Volkwein C., Bordallo J., Finley D., Wolf D.H., and Sommer T.. 2002. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol. 4:134–139. 10.1038/ncb746 [DOI] [PubMed] [Google Scholar]
- Jensen T.J., Loo M.A., Pind S., Williams D.B., Goldberg A.L., and Riordan J.R.. 1995. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 83:129–135. 10.1016/0092-8674(95)90241-4 [DOI] [PubMed] [Google Scholar]
- Jin L., Pahuja K.B., Wickliffe K.E., Gorur A., Baumgärtel C., Schekman R., and Rape M.. 2012. Ubiquitin-dependent regulation of COPII coat size and function. Nature. 482:495–500. 10.1038/nature10822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B.M., and DeBose-Boyd R.A.. 2018. Underlying mechanisms for sterol-induced ubiquitination and ER-associated degradation of HMG CoA reductase. Semin. Cell Dev. Biol. 81:121–128. 10.1016/j.semcdb.2017.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen M.U., Emr S.D., and Winther J.R.. 1999. Ligand recognition and domain structure of Vps10p, a vacuolar protein sorting receptor in Saccharomyces cerevisiae. Eur. J. Biochem. 260:461–469. 10.1046/j.1432-1327.1999.00176.x [DOI] [PubMed] [Google Scholar]
- Kalies K.U., Allan S., Sergeyenko T., Kröger H., and Römisch K.. 2005. The protein translocation channel binds proteasomes to the endoplasmic reticulum membrane. EMBO J. 24:2284–2293. 10.1038/sj.emboj.7600731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagöz G.E., Acosta-Alvear D., and Walter P.. 2019. The Unfolded Protein Response: Detecting and Responding to Fluctuations in the Protein-Folding Capacity of the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 11:a033886 10.1101/cshperspect.a033886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S., Hsu C.L., and Ng D.T.. 2010. Interplay of substrate retention and export signals in endoplasmic reticulum quality control. PLoS One. 5:e15532 10.1371/journal.pone.0015532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A., Heinrich T., Mari M., Grumati P., Huebner A.K., Akutsu M., Liebmann L., Stolz A., Nietzsche S., Koch N., et al. . 2015. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 522:354–358. 10.1038/nature14498 [DOI] [PubMed] [Google Scholar]
- Khmelinskii A., Blaszczak E., Pantazopoulou M., Fischer B., Omnus D.J., Le Dez G., Brossard A., Gunnarsson A., Barry J.D., Meurer M., et al. . 2014. Protein quality control at the inner nuclear membrane. Nature. 516:410–413. 10.1038/nature14096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I., Mi K., and Rao H.. 2004. Multiple interactions of rad23 suggest a mechanism for ubiquitylated substrate delivery important in proteolysis. Mol. Biol. Cell. 15:3357–3365. 10.1091/mbc.e03-11-0835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kincaid M.M., and Cooper A.A.. 2007. Misfolded proteins traffic from the endoplasmic reticulum (ER) due to ER export signals. Mol. Biol. Cell. 18:455–463. 10.1091/mbc.e06-08-0696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M., Finger A., Braun T., Hellmuth K., and Wolf D.H.. 1996. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 15:753–763. 10.1002/j.1460-2075.1996.tb00411.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koegl M., Hoppe T., Schlenker S., Ulrich H.D., Mayer T.U., and Jentsch S.. 1999. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell. 96:635–644. 10.1016/S0092-8674(00)80574-7 [DOI] [PubMed] [Google Scholar]
- Korolchuk V.I., Mansilla A., Menzies F.M., and Rubinsztein D.C.. 2009. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell. 33:517–527. 10.1016/j.molcel.2009.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreft S.G., Wang L., and Hochstrasser M.. 2006. Membrane topology of the yeast endoplasmic reticulum-localized ubiquitin ligase Doa10 and comparison with its human ortholog TEB4 (MARCH-VI). J. Biol. Chem. 281:4646–4653. 10.1074/jbc.M512215200 [DOI] [PubMed] [Google Scholar]
- Kruse K.B., Brodsky J.L., and McCracken A.A.. 2006. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol. Biol. Cell. 17:203–212. 10.1091/mbc.e04-09-0779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamriben L., Graham J.B., Adams B.M., and Hebert D.N.. 2016. N-Glycan-based ER Molecular Chaperone and Protein Quality Control System: The Calnexin Binding Cycle. Traffic. 17:308–326. 10.1111/tra.12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrancois S., Zeng J., Hassan A.J., Canuel M., and Morales C.R.. 2003. The lysosomal trafficking of sphingolipid activator proteins (SAPs) is mediated by sortilin. EMBO J. 22:6430–6437. 10.1093/emboj/cdg629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennemann N.J., and Coyne C.B.. 2017. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy. 13:322–332. 10.1080/15548627.2016.1265192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto D.E., Morgens D.W., Zhang L., Walczak C.P., Elias J.E., Bassik M.C., and Kopito R.R.. 2019. Genome-wide CRISPR Analysis Identifies Substrate-Specific Conjugation Modules in ER-Associated Degradation. Mol. Cell. 73:377–389.e311. 10.1016/j.molcel.2018.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneur F., and Cosson P.. 1998. Targeting to the endoplasmic reticulum in yeast cells by determinants present in transmembrane domains. J. Biol. Chem. 273:33273–33278. 10.1074/jbc.273.50.33273 [DOI] [PubMed] [Google Scholar]
- Li J., Labbadia J., and Morimoto R.I.. 2017. Rethinking HSF1 in Stress, Development, and Organismal Health. Trends Cell Biol. 27:895–905. 10.1016/j.tcb.2017.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Koshi T., and Emr S.D.. 2015. Membrane-anchored ubiquitin ligase complex is required for the turnover of lysosomal membrane proteins. J. Cell Biol. 211:639–652. 10.1083/jcb.201505062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J.R., Lingeman E., Ahmed S., and Corn J.E.. 2018. Atlastins remodel the endoplasmic reticulum for selective autophagy. J. Cell Biol. 217:3354–3367. 10.1083/jcb.201804185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipatova Z., and Segev N.. 2015. A Role for Macro-ER-Phagy in ER Quality Control. PLoS Genet. 11:e1005390 10.1371/journal.pgen.1005390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W., and Chang A.. 1997. Novel genes involved in endosomal traffic in yeast revealed by suppression of a targeting-defective plasma membrane ATPase mutant. J. Cell Biol. 138:731–746. 10.1083/jcb.138.4.731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGurn J.A. 2014. Garbage on, garbage off: new insights into plasma membrane protein quality control. Curr. Opin. Cell Biol. 29:92–98. 10.1016/j.ceb.2014.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGurn J.A., Hsu P.C., and Emr S.D.. 2012. Ubiquitin and membrane protein turnover: from cradle to grave. Annu. Rev. Biochem. 81:231–259. 10.1146/annurev-biochem-060210-093619 [DOI] [PubMed] [Google Scholar]
- Marcusson E.G., Horazdovsky B.F., Cereghino J.L., Gharakhanian E., and Emr S.D.. 1994. The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10 gene. Cell. 77:579–586. 10.1016/0092-8674(94)90219-4 [DOI] [PubMed] [Google Scholar]
- Marshall R.S., Li F., Gemperline D.C., Book A.J., and Vierstra R.D.. 2015. Autophagic Degradation of the 26S Proteasome Is Mediated by the Dual ATG8/Ubiquitin Receptor RPN10 in Arabidopsis. Mol. Cell. 58:1053–1066. 10.1016/j.molcel.2015.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall R.S., Hua Z., Mali S., McLoughlin F., and Vierstra R.D.. 2019. ATG8-Binding UIM Proteins Define a New Class of Autophagy Adaptors and Receptors. Cell. 177:766–781.e724. 10.1016/j.cell.2019.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken A.A., and Brodsky J.L.. 1996. Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J. Cell Biol. 132:291–298. 10.1083/jcb.132.3.291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicherla B., Kostova Z., Schaefer A., and Wolf D.H.. 2004. A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Rep. 5:692–697. 10.1038/sj.embor.7400164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehnert M., Sommer T., and Jarosch E.. 2014. Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat. Cell Biol. 16:77–86. 10.1038/ncb2882 [DOI] [PubMed] [Google Scholar]
- Metzger M.B., Liang Y.H., Das R., Mariano J., Li S., Li J., Kostova Z., Byrd R.A., Ji X., and Weissman A.M.. 2013. A structurally unique E2-binding domain activates ubiquitination by the ERAD E2, Ubc7p, through multiple mechanisms. Mol. Cell. 50:516–527. 10.1016/j.molcel.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D.M., Zhou M., Pariyarath R., Wang H., Aitchison J.D., Ginsberg H.N., and Fisher E.A.. 1998. Apoprotein B100 has a prolonged interaction with the translocon during which its lipidation and translocation change from dependence on the microsomal triglyceride transfer protein to independence. Proc. Natl. Acad. Sci. USA. 95:14733–14738. 10.1073/pnas.95.25.14733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida K., Oikawa Y., Kimura Y., Kirisako H., Hirano H., Ohsumi Y., and Nakatogawa H.. 2015. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 522:359–362. 10.1038/nature14506 [DOI] [PubMed] [Google Scholar]
- Mogk A., Kummer E., and Bukau B.. 2015. Cooperation of Hsp70 and Hsp100 chaperone machines in protein disaggregation. Front. Mol. Biosci. 2:22 10.3389/fmolb.2015.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K., Huyer G., Michaelis S., and Brodsky J.L.. 2008. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 132:101–112. 10.1016/j.cell.2007.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S., Jaeger P.A., Duttke S.H., Benner C., Glass C.K., Ideker T., and Hampton R.Y.. 2018. The Dfm1 Derlin Is Required for ERAD Retrotranslocation of Integral Membrane Proteins. Mol. Cell. 69:915 10.1016/j.molcel.2018.02.014 [DOI] [PubMed] [Google Scholar]
- Needham P.G., and Brodsky J.L.. 2013. How early studies on secreted and membrane protein quality control gave rise to the ER associated degradation (ERAD) pathway: the early history of ERAD. Biochim. Biophys. Acta. 1833:2447–2457. 10.1016/j.bbamcr.2013.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuber O., Jarosch E., Volkwein C., Walter J., and Sommer T.. 2005. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat. Cell Biol. 7:993–998. 10.1038/ncb1298 [DOI] [PubMed] [Google Scholar]
- Nielsen M.S., Jacobsen C., Olivecrona G., Gliemann J., and Petersen C.M.. 1999. Sortilin/neurotensin receptor-3 binds and mediates degradation of lipoprotein lipase. J. Biol. Chem. 274:8832–8836. 10.1074/jbc.274.13.8832 [DOI] [PubMed] [Google Scholar]
- Nielsen M.S., Madsen P., Christensen E.I., Nykjaer A., Gliemann J., Kasper D., Pohlmann R., and Petersen C.M.. 2001. The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. EMBO J. 20:2180–2190. 10.1093/emboj/20.9.2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninagawa S., Okada T., Sumitomo Y., Kamiya Y., Kato K., Horimoto S., Ishikawa T., Takeda S., Sakuma T., Yamamoto T., and Mori K.. 2014. EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol. 206:347–356. 10.1083/jcb.201404075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa S.I., Fewell S.W., Kato Y., Brodsky J.L., and Endo T.. 2001. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J. Cell Biol. 153:1061–1070. 10.1083/jcb.153.5.1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y., Hosokawa N., Wada I., and Nagata K.. 2003. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science. 299:1394–1397. 10.1126/science.1079181 [DOI] [PubMed] [Google Scholar]
- Oh E., Akopian D., and Rape M.. 2018. Principles of Ubiquitin-Dependent Signaling. Annu. Rev. Cell Dev. Biol. 34:137–162. 10.1146/annurev-cellbio-100617-062802 [DOI] [PubMed] [Google Scholar]
- Okiyoneda T., Apaja P.M., and Lukacs G.L.. 2011. Protein quality control at the plasma membrane. Curr. Opin. Cell Biol. 23:483–491. 10.1016/j.ceb.2011.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omari S., Makareeva E., Roberts-Pilgrim A., Mirigian L., Jarnik M., Ott C., Lippincott-Schwartz J., and Leikin S.. 2018. Noncanonical autophagy at ER exit sites regulates procollagen turnover. Proc. Natl. Acad. Sci. USA. 115:E10099–E10108. 10.1073/pnas.1814552115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.H., Bolender N., Eisele F., Kostova Z., Takeuchi J., Coffino P., and Wolf D.H.. 2007. The cytoplasmic Hsp70 chaperone machinery subjects misfolded and endoplasmic reticulum import-incompetent proteins to degradation via the ubiquitin-proteasome system. Mol. Biol. Cell. 18:153–165. 10.1091/mbc.e06-04-0338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore N., Blomenkamp K., Annunziata F., Piccolo P., Mithbaokar P., Maria Sepe R., Vetrini F., Palmer D., Ng P., Polishchuk E., et al. . 2013. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol. Med. 5:397–412. 10.1002/emmm.201202046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper R.C., and Katzmann D.J.. 2007. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 23:519–547. 10.1146/annurev.cellbio.23.090506.123319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzirusso M., and Chang A.. 2004. Ubiquitin-mediated targeting of a mutant plasma membrane ATPase, Pma1-7, to the endosomal/vacuolar system in yeast. Mol. Biol. Cell. 15:2401–2409. 10.1091/mbc.e03-10-0727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plemper R.K., Bordallo J., Deak P.M., Taxis C., Hitt R., and Wolf D.H.. 1999. Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J. Cell Sci. 112:4123–4134. [DOI] [PubMed] [Google Scholar]
- Potelle S., Klein A., and Foulquier F.. 2015. Golgi post-translational modifications and associated diseases. J. Inherit. Metab. Dis. 38:741–751. 10.1007/s10545-015-9851-7 [DOI] [PubMed] [Google Scholar]
- Prasad R., Xu C., and Ng D.T.W.. 2018. Hsp40/70/110 chaperones adapt nuclear protein quality control to serve cytosolic clients. J. Cell Biol. 217:2019–2032. 10.1083/jcb.201706091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissler S., and Ron D.. 2018. Erratum: Early Events in the Endoplasmic Reticulum Unfolded Protein Response. Cold Spring Harb. Perspect. Biol. 10:a037309 10.1101/cshperspect.a037309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston G.M., and Brodsky J.L.. 2017. The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem. J. 474:445–469. 10.1042/BCJ20160582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston G.M., Guerriero C.J., Metzger M.B., Michaelis S., and Brodsky J.L.. 2018. Substrate Insolubility Dictates Hsp104-Dependent Endoplasmic-Reticulum-Associated Degradation. Mol. Cell. 70:242–253.e246. 10.1016/j.molcel.2018.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L., Tsai B., and Arvan P.. 2017. New Insights into the Physiological Role of Endoplasmic Reticulum-Associated Degradation. Trends Cell Biol. 27:430–440. 10.1016/j.tcb.2016.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich E., Kerem A., Fröhlich K.U., Diamant N., and Bar-Nun S.. 2002. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol. Cell. Biol. 22:626–634. 10.1128/MCB.22.2.626-634.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport T.A., Li L., and Park E.. 2017. Structural and Mechanistic Insights into Protein Translocation. Annu. Rev. Cell Dev. Biol. 33:369–390. 10.1146/annurev-cellbio-100616-060439 [DOI] [PubMed] [Google Scholar]
- Ravid T., Kreft S.G., and Hochstrasser M.. 2006. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 25:533–543. 10.1038/sj.emboj.7600946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P., Sparvoli A., Fagioli C., Fassina G., and Sitia R.. 1996. Formation of reversible disulfide bonds with the protein matrix of the endoplasmic reticulum correlates with the retention of unassembled Ig light chains. EMBO J. 15:2077–2085. 10.1002/j.1460-2075.1996.tb00561.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F., and Pelham H.R.. 2002. A transmembrane ubiquitin ligase required to sort membrane proteins into multivesicular bodies. Nat. Cell Biol. 4:117–123. 10.1038/ncb743 [DOI] [PubMed] [Google Scholar]
- Roth F.P., Hughes J.D., Estep P.W., and Church G.M.. 1998. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat. Biotechnol. 16:939–945. 10.1038/nbt1098-939 [DOI] [PubMed] [Google Scholar]
- Rotin D., and Kumar S.. 2009. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 10:398–409. 10.1038/nrm2690 [DOI] [PubMed] [Google Scholar]
- Rougier J.S., Albesa M., Abriel H., and Viard P.. 2011. Neuronal precursor cell-expressed developmentally down-regulated 4-1 (NEDD4-1) controls the sorting of newly synthesized Ca(V)1.2 calcium channels. J. Biol. Chem. 286:8829–8838. 10.1074/jbc.M110.166520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiano A., Foresti O., and Carvalho P.. 2014. Quality control: ER-associated degradation: protein quality control and beyond. J. Cell Biol. 204:869–879. 10.1083/jcb.201312042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardana R., Zhu L., and Emr S.D.. 2019. Rsp5 Ubiquitin ligase-mediated quality control system clears membrane proteins mistargeted to the vacuole membrane. J. Cell Biol. 218:234–250. 10.1083/jcb.201806094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato B.K., Schulz D., Do P.H., and Hampton R.Y.. 2009. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol. Cell. 34:212–222. 10.1016/j.molcel.2009.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K., Sato M., and Nakano A.. 2003. Rer1p, a retrieval receptor for ER membrane proteins, recognizes transmembrane domains in multiple modes. Mol. Biol. Cell. 14:3605–3616. 10.1091/mbc.e02-12-0777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpute-Krishnan P., Ajinkya M., Bhat S., Itakura E., Hegde R.S., and Lippincott-Schwartz J.. 2014. ER stress-induced clearance of misfolded GPI-anchored proteins via the secretory pathway. Cell. 158:522–533. 10.1016/j.cell.2014.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer A., and Wolf D.H.. 2009. Sec61p is part of the endoplasmic reticulum-associated degradation machinery. EMBO J. 28:2874–2884. 10.1038/emboj.2009.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallus T., Jaeckh C., Fehér K., Palma A.S., Liu Y., Simpson J.C., Mackeen M., Stier G., Gibson T.J., Feizi T., et al. . 2008. Malectin: a novel carbohydrate-binding protein of the endoplasmic reticulum and a candidate player in the early steps of protein N-glycosylation. Mol. Biol. Cell. 19:3404–3414. 10.1091/mbc.e08-04-0354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt O., Weyer Y., Baumann V., Widerin M.A., Eising S., Angelova M., Schleiffer A., Kremser L., Lindner H., Peter M., et al. . 2019. Endosome and Golgi-associated degradation (EGAD) of membrane proteins regulates sphingolipid metabolism. EMBO J. 38 10.15252/embj.2018101433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider K., and Bertolotti A.. 2015. Surviving protein quality control catastrophes--from cells to organisms. J. Cell Sci. 128:3861–3869. 10.1242/jcs.173047 [DOI] [PubMed] [Google Scholar]
- Schoebel S., Mi W., Stein A., Ovchinnikov S., Pavlovicz R., DiMaio F., Baker D., Chambers M.G., Su H., Li D., et al. . 2017. Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature. 548:352–355. 10.1038/nature23314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U., Antón L.C., Bacík I., Cox J.H., Bour S., Bennink J.R., Orlowski M., Strebel K., and Yewdell J.W.. 1998. CD4 glycoprotein degradation induced by human immunodeficiency virus type 1 Vpu protein requires the function of proteasomes and the ubiquitin-conjugating pathway. J. Virol. 72:2280–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth C., and Buchberger A.. 2005. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat. Cell Biol. 7:999–1006. 10.1038/ncb1299 [DOI] [PubMed] [Google Scholar]
- Schultz M.L., Krus K.L., Kaushik S., Dang D., Chopra R., Qi L., Shakkottai V.G., Cuervo A.M., and Lieberman A.P.. 2018. Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat. Commun. 9:3671 10.1038/s41467-018-06115-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senft D., and Ronai Z.A.. 2015. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 40:141–148. 10.1016/j.tibs.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S., and Hegde R.S.. 2016. Target Selection during Protein Quality Control. Trends Biochem. Sci. 41:124–137. 10.1016/j.tibs.2015.10.007 [DOI] [PubMed] [Google Scholar]
- Shenkman M., and Lederkremer G.Z.. 2019. Compartmentalization and Selective Tagging for Disposal of Misfolded Glycoproteins. Trends Biochem. Sci. 44:827–836. 10.1016/j.tibs.2019.04.012 [DOI] [PubMed] [Google Scholar]
- Smith M., and Wilkinson S.. 2017. ER homeostasis and autophagy. Essays Biochem. 61:625–635. 10.1042/EBC20170092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M.D., Harley M.E., Kemp A.J., Wills J., Lee M., Arends M., von Kriegsheim A., Behrends C., and Wilkinson S.. 2018. CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev. Cell. 44:217–232.e211. 10.1016/j.devcel.2017.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer T., and Jentsch S.. 1993. A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature. 365:176–179. 10.1038/365176a0 [DOI] [PubMed] [Google Scholar]
- Spear E.D., and Ng D.T.. 2003. Stress tolerance of misfolded carboxypeptidase Y requires maintenance of protein trafficking and degradative pathways. Mol. Biol. Cell. 14:2756–2767. 10.1091/mbc.e02-11-0717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg H.R., Thomas M., van den Boomen D., Wiertz E.J., Drabkin H.A., Gemmill R.M., and Lehner P.J.. 2009. The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J. Cell Biol. 186:685–692. 10.1083/jcb.200906110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub O., Dho S., Henry P., Correa J., Ishikawa T., McGlade J., and Rotin D.. 1996. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 15:2371–2380. 10.1002/j.1460-2075.1996.tb00593.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson J., Huang E.Y., and Olzmann J.A.. 2016. Endoplasmic Reticulum-Associated Degradation and Lipid Homeostasis. Annu. Rev. Nutr. 36:511–542. 10.1146/annurev-nutr-071715-051030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart E.V., Nwosu C.C., Tong Z., Roguev A., Cummins T.D., Kim D.U., Hayles J., Park H.O., Hoe K.L., Powell D.W., et al. . 2011. Yeast SREBP cleavage activation requires the Golgi Dsc E3 ligase complex. Mol. Cell. 42:160–171. 10.1016/j.molcel.2011.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart E.V., Lloyd S.J., Burg J.S., Nwosu C.C., Lintner R.E., Daza R., Russ C., Ponchner K., Nusbaum C., and Espenshade P.J.. 2012. Yeast sterol regulatory element-binding protein (SREBP) cleavage requires Cdc48 and Dsc5, a ubiquitin regulatory X domain-containing subunit of the Golgi Dsc E3 ligase. J. Biol. Chem. 287:672–681. 10.1074/jbc.M111.317370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A., Besser S., Hottmann H., and Wolf D.H.. 2013. Previously unknown role for the ubiquitin ligase Ubr1 in endoplasmic reticulum-associated protein degradation. Proc. Natl. Acad. Sci. USA. 110:15271–15276. 10.1073/pnas.1304928110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Capalbo A., Iyengar N.R., Rizzo R., di Campli A., Di Martino R., Lo Monte M., Beccari A.R., Yerudkar A., Del Vecchio C., et al. . 2019. Auto-regulation of Secretory Flux by Sensing and Responding to the Folded Cargo Protein Load in the Endoplasmic Reticulum. Cell. 176:1461–1476.e1423. 10.1016/j.cell.2019.01.035 [DOI] [PubMed] [Google Scholar]
- Sun Z., and Brodsky J.L.. 2017. Guardians of the ERAD Galaxy. Cell. 171:267–268. 10.1016/j.cell.2017.09.023 [DOI] [PubMed] [Google Scholar]
- Sun Z., and Brodsky J.L.. 2018. The degradation pathway of a model misfolded protein is determined by aggregation propensity. Mol. Biol. Cell. 29:1422–1434. 10.1091/mbc.E18-02-0117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y.X., and Conn P.M.. 2018. Pharmacoperones as Novel Therapeutics for Diverse Protein Conformational Diseases. Physiol. Rev. 98:697–725. 10.1152/physrev.00029.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxis C., Vogel F., and Wolf D.H.. 2002. ER-golgi traffic is a prerequisite for efficient ER degradation. Mol. Biol. Cell. 13:1806–1818. 10.1091/mbc.01-08-0399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Termine D.J., Moremen K.W., and Sifers R.N.. 2009. The mammalian UPR boosts glycoprotein ERAD by suppressing the proteolytic downregulation of ER mannosidase I. J. Cell Sci. 122:976–984. 10.1242/jcs.037291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari R., Bachert C., and Linstedt A.D.. 2015. Induced oligomerization targets Golgi proteins for degradation in lysosomes. Mol. Biol. Cell. 26:4427–4437. 10.1091/mbc.E15-04-0207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theesfeld C.L., and Hampton R.Y.. 2013. Insulin-induced gene protein (INSIG)-dependent sterol regulation of Hmg2 endoplasmic reticulum-associated degradation (ERAD) in yeast. J. Biol. Chem. 288:8519–8530. 10.1074/jbc.M112.404517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers K.J., Patil C.K., Wodicka L., Lockhart D.J., Weissman J.S., and Walter P.. 2000. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 101:249–258. 10.1016/S0092-8674(00)80835-1 [DOI] [PubMed] [Google Scholar]
- Trombetta E.S., and Parodi A.J.. 2003. Quality control and protein folding in the secretory pathway. Annu. Rev. Cell Dev. Biol. 19:649–676. 10.1146/annurev.cellbio.19.110701.153949 [DOI] [PubMed] [Google Scholar]
- Twomey E.C., Ji Z., Wales T.E., Bodnar N.O., Ficarro S.B., Marto J.A., Engen J.R., and Rapoport T.A.. 2019. Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science. 365:eaax1033. 10.1126/science.aax1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushioda R., Hoseki J., Araki K., Jansen G., Thomas D.Y., and Nagata K.. 2008. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science. 321:569–572. 10.1126/science.1159293 [DOI] [PubMed] [Google Scholar]
- van der Goot A.T., Pearce M.M.P., Leto D.E., Shaler T.A., and Kopito R.R.. 2018. Redundant and Antagonistic Roles of XTP3B and OS9 in Decoding Glycan and Non-glycan Degrons in ER-Associated Degradation. Mol. Cell. 70:516–530.e516. 10.1016/j.molcel.2018.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S., and Ng D.T.. 2004. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol. 165:41–52. 10.1083/jcb.200309132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S., Kim W., Belden W.J., Spear E.D., Barlowe C., and Ng D.T.. 2001. Distinct retrieval and retention mechanisms are required for the quality control of endoplasmic reticulum protein folding. J. Cell Biol. 155:355–368. 10.1083/jcb.200106123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkat S., and Linstedt A.D.. 2017. Manganese-induced trafficking and turnover of GPP130 is mediated by sortilin. Mol. Biol. Cell. 28:2569–2578. 10.1091/mbc.e17-05-0326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlman J., DeMartino G.N., Skach W.R., Bulleid N.J., Brodsky J.L., and Johnson A.E.. 2007. Real-time fluorescence detection of ERAD substrate retrotranslocation in a mammalian in vitro system. Cell. 129:943–955. 10.1016/j.cell.2007.03.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., and Ng D.T.. 2010. Evasion of endoplasmic reticulum surveillance makes Wsc1p an obligate substrate of Golgi quality control. Mol. Biol. Cell. 21:1153–1165. 10.1091/mbc.e09-10-0910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Song W., Brancati G., and Segatori L.. 2011a Inhibition of endoplasmic reticulum-associated degradation rescues native folding in loss of function protein misfolding diseases. J. Biol. Chem. 286:43454–43464. 10.1074/jbc.M111.274332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Durfee L.A., and Huibregtse J.M.. 2013. A cotranslational ubiquitination pathway for quality control of misfolded proteins. Mol. Cell. 50:368–378. 10.1016/j.molcel.2013.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Liu Y., Soetandyo N., Baek K., Hegde R., and Ye Y.. 2011b A ubiquitin ligase-associated chaperone holdase maintains polypeptides in soluble states for proteasome degradation. Mol. Cell. 42:758–770. 10.1016/j.molcel.2011.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Thibault G., and Ng D.T.. 2011c Routing misfolded proteins through the multivesicular body (MVB) pathway protects against proteotoxicity. J. Biol. Chem. 286:29376–29387. 10.1074/jbc.M111.233346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.J., Di X.J., and Mu T.W.. 2014. Using pharmacological chaperones to restore proteostasis. Pharmacol. Res. 83:3–9. 10.1016/j.phrs.2014.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangeline M.A., Vashistha N., and Hampton R.Y.. 2017. Proteostatic Tactics in the Strategy of Sterol Regulation. Annu. Rev. Cell Dev. Biol. 33:467–489. 10.1146/annurev-cellbio-111315-125036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward C.L., Omura S., and Kopito R.R.. 1995. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 83:121–127. 10.1016/0092-8674(95)90240-6 [DOI] [PubMed] [Google Scholar]
- Weber A., Cohen I., Popp O., Dittmar G., Reiss Y., Sommer T., Ravid T., and Jarosch E.. 2016. Sequential Poly-ubiquitylation by Specialized Conjugating Enzymes Expands the Versatility of a Quality Control Ubiquitin Ligase. Mol. Cell. 63:827–839. 10.1016/j.molcel.2016.07.020 [DOI] [PubMed] [Google Scholar]
- Werner E.D., Brodsky J.L., and McCracken A.A.. 1996. Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc. Natl. Acad. Sci. USA. 93:13797–13801. 10.1073/pnas.93.24.13797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiertz E.J., Jones T.R., Sun L., Bogyo M., Geuze H.J., and Ploegh H.L.. 1996a The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 84:769–779. 10.1016/S0092-8674(00)81054-5 [DOI] [PubMed] [Google Scholar]
- Wiertz E.J., Tortorella D., Bogyo M., Yu J., Mothes W., Jones T.R., Rapoport T.A., and Ploegh H.L.. 1996b Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 384:432–438. 10.1038/384432a0 [DOI] [PubMed] [Google Scholar]
- Wilkinson S. 2019. Emerging Principles of Selective ER Autophagy. J. Mol. Biol. 10.1016/j.jmb.2019.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman R.L., Koulov A., Powers E., Kelly J.W., and Balch W.E.. 2007. Protein energetics in maturation of the early secretory pathway. Curr. Opin. Cell Biol. 19:359–367. 10.1016/j.ceb.2007.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff S., Weissman J.S., and Dillin A.. 2014. Differential scales of protein quality control. Cell. 157:52–64. 10.1016/j.cell.2014.03.007 [DOI] [PubMed] [Google Scholar]
- Wolins N., Bosshart H., Küster H., and Bonifacino J.S.. 1997. Aggregation as a determinant of protein fate in post-Golgi compartments: role of the luminal domain of furin in lysosomal targeting. J. Cell Biol. 139:1735–1745. 10.1083/jcb.139.7.1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W., Kanehara K., Sayeed A., and Ng D.T.. 2009. Intrinsic conformational determinants signal protein misfolding to the Hrd1/Htm1 endoplasmic reticulum-associated degradation system. Mol. Biol. Cell. 20:3317–3329. 10.1091/mbc.e09-03-0231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C., and Ng D.T.. 2015a Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Biol. 16:742–752. 10.1038/nrm4073 [DOI] [PubMed] [Google Scholar]
- Xu C., and Ng D.T.. 2015b O-mannosylation: The other glycan player of ER quality control. Semin. Cell Dev. Biol. 41:129–134. 10.1016/j.semcdb.2015.01.014 [DOI] [PubMed] [Google Scholar]
- Xu C., Wang S., Thibault G., and Ng D.T.. 2013. Futile protein folding cycles in the ER are terminated by the unfolded protein O-mannosylation pathway. Science. 340:978–981. 10.1126/science.1234055 [DOI] [PubMed] [Google Scholar]
- Yamamoto K., Fujii R., Toyofuku Y., Saito T., Koseki H., Hsu V.W., and Aoe T.. 2001. The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J. 20:3082–3091. 10.1093/emboj/20.12.3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki A., Hara T., Maejima I., Sato M., Sato K., and Sato K.. 2014. Rer1p regulates the ER retention of immature rhodopsin and modulates its intracellular trafficking. Sci. Rep. 4:5973 10.1038/srep05973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.S., and Strittmatter S.M.. 2007. The reticulons: a family of proteins with diverse functions. Genome Biol. 8:234 10.1186/gb-2007-8-12-234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Arines F.M., Zhang W., and Li M.. 2018. Sorting of a multi-subunit ubiquitin ligase complex in the endolysosome system. eLife. 7:e33116 10.7554/eLife.33116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y., Meyer H.H., and Rapoport T.A.. 2001. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 414:652–656. 10.1038/414652a [DOI] [PubMed] [Google Scholar]
- Ye Y., Meyer H.H., and Rapoport T.A.. 2003. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 162:71–84. 10.1083/jcb.200302169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H., Matsui T., Hosokawa N., Kaufman R.J., Nagata K., and Mori K.. 2003. A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell. 4:265–271. 10.1016/S1534-5807(03)00022-4 [DOI] [PubMed] [Google Scholar]
- Yoshida H., Oku M., Suzuki M., and Mori K.. 2006. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J. Cell Biol. 172:565–575. 10.1083/jcb.200508145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youker R.T., Walsh P., Beilharz T., Lithgow T., and Brodsky J.L.. 2004. Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol. Biol. Cell. 15:4787–4797. 10.1091/mbc.e04-07-0584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavodszky E., and Hegde R.S.. 2019. Misfolded GPI-anchored proteins are escorted through the secretory pathway by ER-derived factors. eLife. 8:e46740 10.7554/eLife.46740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Xu C., Larrimore K.E., and Ng D.T.W.. 2017. Slp1-Emp65: A Guardian Factor that Protects Folding Polypeptides from Promiscuous Degradation. Cell. 171:346–357.e312. 10.1016/j.cell.2017.08.036 [DOI] [PubMed] [Google Scholar]
- Zhang Z.R., Bonifacino J.S., and Hegde R.S.. 2013. Deubiquitinases sharpen substrate discrimination during membrane protein degradation from the ER. Cell. 154:609–622. 10.1016/j.cell.2013.06.038 [DOI] [PMC free article] [PubMed] [Google Scholar]