

Abstract
The high mortality rate of lung squamous cell carcinoma (LUSC) is in part due to the lack of early detection of its biomarkers. The identification of key molecules involved in LUSC is therefore required to improve clinical diagnosis and treatment outcomes. The present study used the microarray datasets GSE31552, GSE6044 and GSE12428 from the Gene Expression Omnibus database to identify differentially expressed genes (DEGs). Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) enrichment analyses were conducted to construct the protein-protein interaction network of DEGs and hub genes module using STRING and Cytoscape. The 67 DEGs identified consisted of 42 upregulated genes and 25 downregulated genes. The pathways predicted by KEGG and GO enrichment analyses of DEGs mainly included cell cycle, cell proliferation, glycolysis or gluconeogenesis, and tetrahydrofolate metabolic process. Further analysis of the University of California Santa Cruz and ONCOMINE databases identified 17 hub genes. Overall, the present study demonstrated hub genes that were closely associated with clinical tissue samples of LUSC, and identified TYMS, CCNB2 and RFC4 as potential novel biomarkers of LUSC. The findings of the present study contribute to an improved understanding of the molecular mechanisms of carcinogenesis and progression of LUSC, and assist with the identification of potential diagnostic and therapeutic targets of LUSC.
Keywords: lung squamous cell carcinoma, differentially expressed genes, microarray, protein-protein interaction, Kyoto Encyclopedia of Genes and Genomes analysis, Gene Ontology enrichment analysis
Introduction
Lung cancer is a disease with the highest morbidity and mortality rates worldwide. It is reported that non-small cell lung cancer (NSCLC) accounts for 85% of the total lung cancer cases worldwide, of which squamous cell lung cancer (LUSC), often with poor prognosis, accounted for 30% of NSCLC in 2017 (1,2). Data has demonstrated that more than 1 in 3 patients with lung adenocarcinoma (LUAD) benefit from molecular-targeted therapies (3). Inhibitors of epidermal growth factor receptor, v-ki-ras2 kirsten rat sarcoma viral oncogene homologue and anaplastic lymphoma receptor tyrosine kinase are some of the few molecules that are targeted in lung cancer therapy (4). However, the application of molecular-targeted therapies in the diagnosis and treatment of LUSC in the clinical setting is very limited. Thus, the identification of biomarkers that are associated with the diagnosis and treatment of LUSC has become one of the main focus areas in research. An increasing number of studies have revealed new genetic changes associated with LUSC, including the oncogenes baculoviral IAP repeat contain 5 (BIRC5) and GAPDH. BIRC5 is an important inhibitor of apoptosis, which serves an important role in carcinogenesis and progression of LUSC (5). Li et al (6) reported a significantly higher expression level of BIRC5 in LUSC tissues compared with normal tissues, indicating the potential of BIRC5 as a target for anti-tumor therapy. On the other hand, GAPDH has been reported to serve a crucial role in regulating glycolysis in cancer cells. GAPDH depletes ATP in cancer cells via the inhibition of glycolysis, which eventually kills cancer cells (7,8). Hence, GAPDH has become a therapeutic target of interest against cancer cells. LUSC accounts for more than 400,000 deaths worldwide each year (2); it is important to highlight that the mortality rate of LUSC is inevitably high, even at early stage, despite several discoveries of potential targets such as BIRC5 and GAPDH. Thus, the investigation of other potential molecular mechanisms associated with LUSC is important.
In recent years, gene microarray and gene chip technologies have developed rapidly, which has provided a theoretical basis for the detection of genetic alterations in cancer cells (9,10). These technologies can be applied to identify differentially expressed genes (DEGs), which can potentially be associated with the carcinogenicity and progression of LUSC. In the present study, in order to avoid false positive results from a single microarray gene expression dataset, three mRNA microarray datasets from the Gene Expression Omnibus (GEO) database platform were downloaded. LUSC tissues and non-cancerous tissues were analyzed in order to identify DEGs. Furthermore, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were conducted and a protein-protein interaction (PPI) network was constructed in order to understand the molecular mechanisms underlying the generation and progression of LUSC. The associations between the hub genes and clinical tissue samples were identified using the University of California Santa Cruz (UCSC) Cancer Genomics Browser and ONCOMINE database. A total of 67 DEGs and 17 hub genes were identified as potential diagnostic and therapeutic biomarkers of LUSC. Five hub genes with the highest node value were selected via CentiScaPe, in which the results from the UCSC and ONCOMINE online clinical databases indicated all five hub genes to be associated with unfavorable prognosis of LUSC. Thymidylate synthetase (TYMS), cyclin B2 (CCNB2) and replication factor C subunit 4 (RFC4) were suggested as potential and novel target genes for the treatment of LUSC.
Materials and methods
Microarray data
GEO (http://www.ncbi.nlm.nih.gov/geo) is an open database of gene expression abundance, consisting of high throughout gene expression data, gene microarrays and gene chips. Three gene expression datasets [GSE31552 (11), GSE6044 (12) and GSE12428 (13)] were downloaded from the GEO (Affymetrix GPL6244 platform, Affymetrix Human Gene 1.0 ST Array; Affymetrix GPL201 platform, Affymetrix Human HG-Focus Target Array; Affymetrix GPL1708 platform, Agilent-012391 Whole Human Genome Oligo Microarray G4112A). The GSE31552 dataset included 25 LUSC tissue samples and 25 non-cancerous samples. GSE6044 included 15 LUSC tissue samples and 5 non-cancerous samples. GSE12428 included 34 LUSC tissue samples and 28 non-cancerous samples.
Identification of DEGs
The DEGs between LUSC and non-cancerous tissue samples were selected by GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r). GEO2R is a GEO online analysis tool, which analyzes differential gene expression among two or more datasets in GEO. The P-values were adopted to screen the DEGs accurately, and the probe sets without corresponding gene symbols during the screening process were removed. LogFC (fold change) >1 or logFC<-1 and P<0.05 were considered as statistically significant.
‘KEGG pathway’ and ‘Gene Oncology (GO)’ enrichment analyses of DEGs
The Database for Annotation, Visualization and Integrated Discovery database (DAVID; http://david.ncifcrf.gov; version 6.8) is an online gene and pathway functional annotation database that contains biological information and also provides analysis tools (14). Biological information can be extracted from the comprehensive set of genes and proteins, which provides functional annotations. The KEGG database can be used to analyze genome information and gene function and study the gene expression information as a whole network (15). GO is a type of bioinformatics tool for annotating genes and analyzing their biological processes. GO enrichment analyses contain three modules of molecular function, cell composition and biological process (16). In order to analyze the function and cell signaling pathways of DEGs, KEGG and GO enrichment analyses were conducted using the DAVID database. KEGG and GO enrichment bubble plots were drawn using online graphics tools Image GP (http://www.ehbio.com/ImageGP/). P<0.05 was considered as statistically significant.
PPI network and hub gene module construction
The PPI network of DEGs was constructed by the online analysis website Search Tool for the Retrieval of Interacting Genes (STRING; http://string-db.org; version 11.0) (17) and the interaction of a combined score >0.4 was considered as statistically significant. Analyzing the function of PPI can provide insights into the mechanisms of disease occurrence and development. Cytoscape (version 3.6.1) is an open bioinformatics software platform that can be used to construct a visual network of molecular interactions (18). The plug-in Molecular Complex Detection (MCODE) (version 1.4.2) of Cytoscape is an APP for detecting densely correlated regions in the PPI networks (19). The gene modules were visualized and graphically displayed with the plug-in MCODE. The selection criteria were as follows: MCODE score >5; node score cut-off, 0.2; degree cut-off, 2; k-score, 2; and Max depth, 100. CentiScaPe (version 2.2), a Cytoscape APP specifically designed to calculate centrality indexes for the selection of the most critical nodes in a network (20). The plug-in CentiScaPe 2.2 was used to identify hub genes for functional analysis with interaction node degrees ≥10.
Hub genes selection and analysis
The hub genes with interaction node degrees ≥10 were screened. The network of the genes and their co-expression genes was constructed by the online platform cBioPortal (http://www.cbioportal.org) (21,22). Hierarchical clustering of hub genes was constructed by online analysis website UCSC Cancer Genomics Browser (http://genome-cancer.ucsc.edu) (23). Heat maps of hub genes expression in three different studies of clinical LUSC samples vs. non-cancerous tissue samples (24–26), and the associations between the expression patterns and tumor stage, overall survival status (the survival rate of patients from diagnosis to the end of the study) and survival status at 5 years (the survival rate five years after diagnosis) were analyzed using the Hou Lung dataset (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE19188) (27), which was obtained from the Oncomine database (http://www.oncomine.com) (28,29).
Results
Identification of DEGs in LUSC
The standard microarrays were obtained from the GEO database platform. Following further analysis using GEO2R, DEGs were identified from the GSE31552 (957), GSE6044 (724) and GSE12428 (745) datasets. The 67 DEGs between the three datasets are presented in a Venn diagram (Fig. 1), consisting of 42 upregulated genes and 25 downregulated genes between LUSC and non-cancerous tissues.
Figure 1.
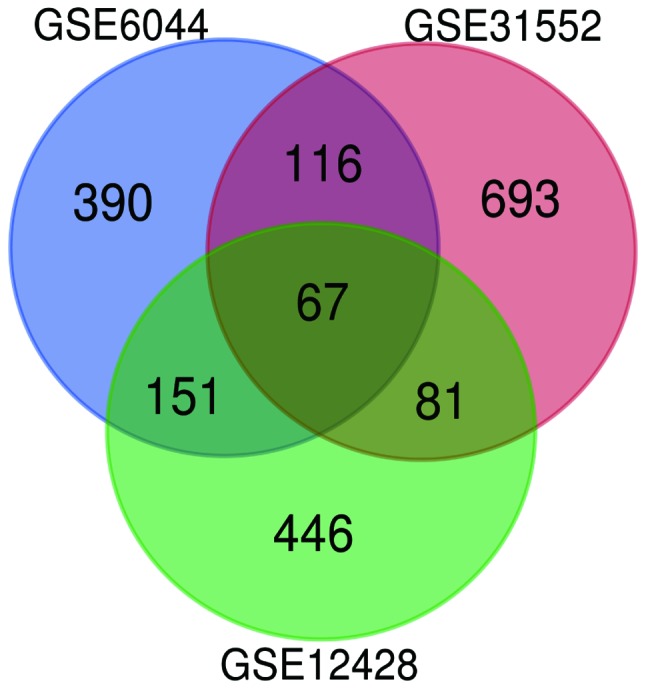
Venn diagram. Genes with |logFC|>1 and P<0.05, among the mRNA expression profiling datasets GSE31552, GSE6044 and GSE12428, were selected as DEGs. The overlap among the three datasets resulted in 67 DEGs.
KEGG and GO enrichment analyses of DEGs
Functional and pathway enrichment analyses of DEGs were conducted by DAVID to obtain the biological classification. The GO enrichment analysis included biological processes (BP), cell component (CC) and molecular function (MF) terms of the DEGs. The results of the KEGG and GO enrichment analyses are presented as bubble plots in Fig. 2. Changes in BP were significantly enriched in ‘cell division’, ‘positive regulation of cell proliferation’, ‘negative regulation of endopeptidase activity’ and ‘tetrahydrofolate metabolic process’ (Fig. 2A). Changes in CC were significantly enriched in ‘extracellular exosome’, ‘extracellular space’, ‘cytosol’ and ‘vesicle’ (Fig. 2B). Changes in MF were mainly enriched in ‘protein binding’, ‘cysteine-type endopeptidase inhibitor activity’, ‘protein binding’ and ‘identical protein binding’ (Fig. 2C). The KEGG pathway analysis was mainly enriched in ‘cell cycle’, ‘glycolysis or gluconeogenesis’, ‘metabolic pathways’ and ‘one carbon pool by folate’ (Fig. 2D).
Figure 2.

KEGG and GO enrichment plots of DEGs. The plots from the GO enrichment analysis of DEGs for (A) biological processes (B) cell and (C) molecular function were obtained using DAVID and drawn using Image GP. (D) KEGG enrichment analysis was performed using DAVID, the bubble plot of KEGG analysis was drawn using Image GP. KEGG, Kyoto Encyclopedia of Genes and Genomes; GO, Gene Ontology; DAVID, Database for Annotation, Visualization and Integrated Discovery.
PPI network and hub gene module construction
In order to identify the hub genes of LUSC, the PPI network of DEGs was analyzed by STRING. The results revealed that most genes interacted with each other and were located in the center of the network, and were closely associated with the surrounding proteins in the network (Fig. 3A). To enhance the accuracy of the results, the PPI network was also analyzed by Cytoscape. The obtained results were in correspondence with the results of STRING (Fig. 3B), and the hub gene module was obtained using MCODE (Fig. 3C).
Figure 3.



PPI network and hub gene module of DEGs. (A) The PPI network was established with the Search Tool for the Retrieval of Interacting Gene tool. (B) The PPI network of DEGs was established using Cytoscape. (C) The hub gene module was generated from the PPI network, consisting of 15 nodes and 84 edges. Upregulated genes are marked in red. Downregulated genes are marked in blue. DEGs, differentially expressed genes; PPI, protein-protein interaction.
Hub gene selection and analysis
In total, 17 genes were regarded as hub genes with degrees ≥10 using CentiScaPe. The names, abbreviations and functions for each of these hub genes are summarized in Table I. The genes associated with the hub genes and their co-expression network were obtained using the cBioPortal online platform by performing interaction analysis (Fig. 4A). The expression of 17 hub genes in LUSC tissues and its association with the severity and prognosis among LUSC patients were further explored using the UCSC and ONCOMINE online databases. Furthermore, a heat map of hierarchical clustering obtained using UCSC demonstrated that the expression of hub genes in LUSC tissues was higher compared with that of non-cancerous samples. However, the expression of hub genes showed no differences with gender (Fig. 4B). The heat map of hub genes expression in clinical LUSC tissue samples and normal tissue samples were analyzed using three different datasets, using the ONCOMINE online platform. The results revealed that most of the hub genes were significantly upregulated in clinical LUSC samples in all the datasets (Fig. 5). The hub genes whose interaction node degree was among the top five were TYMS, CCNB2, RFC4, BIRC5 and GAPDH, indicating their potential role in the processes of carcinogenesis, development and unfavorable prognosis of LUSC. The associations between the upregulated hub genes and tumor stage, overall survival status and survival status at 5 years were also analyzed. The top five genes were associated with high tumor stage, poor overall survival status and poor survival status at 5 years, which suggests their upregulation to be involved in the promotion of tumor progression and poor prognosis (Fig. 6).
Table I.
Full name, abbreviation and function of 17 hub genes with node degree ≥10.
| No. | Gene symbol | Full name | Function |
|---|---|---|---|
| 1 | TYMS | Thymidylate synthetase | DNA synthesis, DNA repair and proliferation of cancer cells. |
| 2 | CCNB2 | Cyclin B2 | Correlated with invasion, metastasis and poor prognosis of various cancer types. |
| 3 | RFC4 | Replication factor C subunit 4 | Associated with poorly differentiated and advanced |
| Tumor-Node-Metastasis stage in multiple cancer types. | |||
| 4 | BIRC5 | Baculoviral IAP repeat containing 5 | Plays a key role in proliferation, apoptosis and angiogenesis of LUSC. |
| 5 | GAPDH | Glyceraldehyde-3-Phosphate dehydrogenase | Elevated GAPDH significantly promotes cell proliferation and migration in LUSC. |
| 6 | CKS1B | CDC28 protein kinase regulatory subunit 1B | Attributes to prognosis, chemoresistance and chemosensitivity in cancer. |
| 7 | MCM6 | Minichromosome maintenance complex component 6 | Initiation of DNA replication and a marker for proliferating cells. |
| 8 | EZH2 | Enhancer of Zeste 2 polycomb repressive complex 2 subunit | Invasion, cell proliferation and adverse prognosis in LUSC. |
| 9 | PTTG1 | Pituitary tumor-transforming 1 | Carcinogenesis, migration, invasion and prognosis. |
| 10 | CDK4 | Cyclin dependent kinase 4 | Regulates cell cycle positively, overexpressed and gene amplified in LUSC. |
| 11 | TPX2 | TPX2, microtubule nucleation factor | Overexpression associated with differentiation grade, stage and metastasis of LUSC. |
| 12 | PRC1 | Protein regulator of cytokinesis 1 | Promotes progression and migration in LUSC. |
| 13 | CKS2 | CDC28 protein kinase regulatory subunit 2 | Inhibit DNA damage response and contribute to tumor cell proliferation in breast cancer |
| 14 | CDC45 | Cell division cycle 45 | Regulator of cell proliferation and associated with S-phase DNA damage. |
| 15 | KPNA2 | Karyopherin subunit alpha 2 | Attributed to cancer cell proliferation and metastasis. |
| 16 | NCAPG | Non-SMC condensin I complex subunit G | Associated with cell cycle, apoptosis and migration in human hepatocellular carcinoma. |
| 17 | UBE2S | Ubiquitin conjugating enzyme E2 S | Involved in the malignant characteristics, mitosis and survival of various types of cancer cells. |
Figure 4.


Co-expression network and heat map of hub genes. (A) The network of hub genes and their co-expression genes were constructed using cBioPortal. Nodes with bold black outline represent hub genes. Nodes with thin black outline represent the co-expression genes. (B) The heat map of hierarchical clustering of hub genes was created with the University of California Santa Cruz Cancer Genomics browser. The samples under the brown bar are normal samples, whereas the samples under the blue bar are lung squamous cell carcinoma samples. The sex under the red bar is male and the sex under the blue bar is female. High expression of genes in samples is marked in red and low expression of genes is marked in blue. PPI, protein-protein interaction.
Figure 5.


ONCOMINE analysis of LUSC vs. normal tissue of hub genes. Heat maps of hub genes expression in clinical LUSC samples vs. normal tissue samples. Hub genes: (A) TYMS, (B) CCNB2, (C) RFC4, (D) BIRC5, (E) GAPDH, (F) CKS1B, (G) MCM6, (H) EZH2, (I) PTTG1, (J) CDK4, (K) TPX2, (L) PRC1, (M) CKS2, (N) CDC45, (O) KPNA2, (P) NCAPG and (Q) UBE2S. The rank for a gene is the median rank for that gene across each of the analyses and the P-value for a gene is its P-value for the median-ranked analysis. LUSC, lung squamous cell carcinoma.
Figure 6.


The associations between upregulation of TYMS, CCNB2, RFC4, BIRC5 and GAPDH and tumor stage, overall survival status and survival status at 5 years in the Hou Lung dataset which was obtained from the ONCOMINE database. (A-C) TYMS, (D-F) CCNB2, (G-I) RFC4, (J-L) BIRC5 and (M-O) GAPDH mRNA expression in LUSC vs. normal lung tissues. LUSC lung squamous cell carcinoma.
Discussion
In recent years, the incidence and mortality of lung cancer has continued to increase rapidly worldwide (30). LUAD and LUSC are the two common types of lung cancer. The incidence of LUSC was reported to be associated with smoking, whereas the treatment of LUSC remains limited compared with that of LUAD. The underlying pathological mechanisms of LUSC at the molecular level are still at the exploration stage (31). Mutations or amplifications of phosphatidylinositol-3 kinases (PI3K), phosphatase and Tensin homolog (PTEN), erythropoietin-producing hepatocellular A2 (EphA2) and liver kinase B1 (LKB1) were reported to be associated with the incidence, progression and prognosis of LUSC (32,33). A study conducted using the Cancer Genome Atlas Research Network demonstrated the dysfunction of NFE2L2, KEAP1, CDKN2A and RB1, and the abnormal structures of their products are associated with the occurrence and development of LUSC (34). The high mortality rate of LUSC is in part due to the lack of early detection of LUSC biomarkers (35). As a result, the identification of key molecules involved in LUSC is required and important for improving clinical efficacy. Microarray is a high-throughput technology in obtaining novel biomarkers, which can provide the basis for further studies on the mechanism of LUSC and clinical targeted therapies at the molecular level.
In the present study, three mRNA microarray datasets were analyzed to identify 67 common DEGs. The DEGs consisted of 42 upregulated and 25 downregulated genes between LUSC tissue samples and normal tissue samples. GO terms and KEGG pathway enrichments were analyzed in order to investigate interactions among the DEGs. The results indicated that the 67 DEGs were significantly enriched in cell cycle, cell proliferation, glycolysis or gluconeogenesis, and tetrahydrofolate metabolic process. Previous studies have illustrated that dysregulations of cell cycle and cell proliferation serve roles in the carcinogenesis and malignant change of LUSC (36–38). In addition, multiple studies have also shown that glycolysis or gluconeogenesis serve important roles in tumor initiation, progression and unfavorable prognosis in cancer (39,40). Furthermore, gene polymorphism of tetrahydrofolate induces a decreased activity of tetrahydrofolate reductase, which affects the normal metabolism of folate in cells, where tetrahydrofolate metabolic disorder is closely associated with tumorigenesis (41,42). The findings of the present study were in accordance with the conclusions of previous studies and showed that GO and KEGG enrichment analyses were significantly enriched in cell cycle, cell proliferation, glycolysis or gluconeogenesis, and tetrahydrofolate metabolic process. A total of 17 DEGs were identified as hub genes with an interaction node degree ≥10. The hub genes whose degrees were among the top five were TYMS, CCNB2, RFC4, BIRC5 and GAPDH, and the PPI network showed that they were directly interacting with each other.
Several studies have suggested that TYMS is a predictive biomarker to test for the effectiveness of pemetrexed used in chemotherapy for treating NSCLC (43,44). Lu et al (45) reported that the expression of TYMS was significantly upregulated among patients with lymph node metastasis. The expression of TYMS was also higher among patients with 5-year recurrence rate. Besides, high expression of TYMS was found in the case of breast cancer, which resulted in increased susceptibility of an individual to the progression of the disease. Gene polymorphisms of TYMS have been reported to have potential in improving the diagnosis, prevention and treatment of breast cancer (46). Hence, several studies are now focusing on the association between gene polymorphic variations of TYMS with various types of cancer (47–49). BIRC5 was upregulated in 76% LUSC samples and the expression in LUSC tissues was significantly higher compared with that in non-cancerous tissues (50). BIRC5 is a potential biomarker or therapeutic target of smoking-associated LUSC (51). The expression of BIRC5 was higher among patients who are smokers compared with non-smokers, and in squamous vs. non-squamous lung tumor (P<0.001). The present study also demonstrated that BIRC5 expression level was negatively associated with the expression of tumor suppressor gene Tp53 (52). GAPDH serves a critical role in inhibiting the process of glycolysis in tumor cells (53). The expression of GAPDH was notably upregulated in LUSC tissues, and an increased level of GAPDH significantly promotes the cell proliferation and migration in LUSC (54). CCNB2 as a member of the cell cyclin protein family, was significantly associated with different staging and metastatic statuses of tumors (P<0.001). Thus, CCNB2 is a potential biomarker for evaluating metastatic status and therapeutic efficacy for cancer patients (55). Additionally, the upregulation of CCNB2 was closely associated with the degree of differentiation, progression, lymph node metastasis, invasion and adverse prognosis in NSCLC (56). CCNB2 has also been found to be upregulated in patients with bladder and colorectal cancer (57). Thus, CCNB2 is a potential diagnostic biomarker and a therapeutic target for LUSC. RFC4 was involved in DNA replication and regulation of cell proliferation and cell cycle. Studies reported an association of RFC4 with cancer progression and worse survival outcome, and the ability to predict response to radiotherapy and neoadjuvant radiotherapy in rectal cancer (58,59). RFC4 has been demonstrated to be associated with several types of cancer, however the underlying carcinogenic mechanism needs to be further explored. The top five hub genes reported in the present study were associated with various types of cancer. Multiple studies have demonstrated that GAPDH and BIRC5 are associated with LUSC (50,54). However, to our knowledge, no previous studies reported TYMS, CCNB2 and RFC4 to be directly associated with LUSC. In the present study, ONCOMINE and UCSC analysis confirmed that the top five hub genes from clinical LUSC samples were significantly upregulated and were all associated with different staging of cancer and survival rate compared to that of other samples. Therefore, TYMS, CCNB2 and RFC4 are potential novel biomarkers of LUSC for further investigation.
Among the other 12 hub genes identified in the present study, MCM6, EZH2, CDK4, TPX2 and PRC1 were previously reported to be associated with LUSC. Minichromosome maintenance (MCM) proteins serve a critical role in cell proliferation and cell cycle. Meanwhile, MCM6 is often associated with poor prognosis, particularly among male patients with LUSC and with a history of smoking (60). The presence of EZH2 was associated with the aggressiveness of cancer development. Behrens et al (61) analyzed 221 LUSC samples and 320 lung adenocarcinomas samples, which revealed significantly higher expression of EZH2 in LUSC compared with that of LUAD (P<0.0001). Cell cycle protein CDK4 has an established association with neoplasia and cancer progression. Recent studies have found that pathways including that of CDK4/6 were frequently altered in LUSC via diverse mechanisms, suggesting CDK4/6 inhibitors as potential target for the treatment of LUSC (62,63). TPX2, which actively participates in the formation of spindle microtubules during mitosis, was significantly associated with cell differentiation and metastatic status of LUSC cells, suggesting its potential as a prognostic predictor of LUSC (64). PRC1, an important protein involved in cytokinesis, plays an important role in microtubule organization in eukaryotes (65). A recent study has implicated the overexpression of PRC1 in LUSC to be associated with increased susceptibility to lymph node metastasis and shorter survival time in patients with LUSC (66).
Literature review revealed that the interaction among LUSC and hub genes CKS1B, PTTG1, CKS2, CDC45, KPNA2, NCAPG and UBE2S has not been widely reported. These genes are potential novel biomarkers and therapeutic targets of LUSC. CKS1B, an adaptor for cyclin-dependent kinases, was shown to be associated with chemoresistance, low chemosensitivity and poor prognosis in cancer. An elevated level of CKS1B has been reported to result in the resistance of cancer cells to bortezomib, and activation of the NEDD8 pathway, which in turn leads to further advancement of cancer. Thus, CKS1B is a potential novel target in multiple myeloma (67). In addition, CKS1B promotes proteasomal degradation and ubiquitination of p27Kip1. Overexpression of CKS1B contributes to an increased turnover rate of p27Kip1 and promotion of cancer cell proliferation, resulting in poor prognosis in many types of cancer (68). PTTG1 functions to regulate transcription, the G-M phase of mitosis and the repair of DNA. Overexpression of PTTG1 has been reported in multiple types of cancer, in which PTTG1 is associated with metabolic processes, such as carcinogenesis, migration, invasion and epithelial-mesenchymal transition in squamous cell carcinomas (69). Recent studies demonstrated that various non-coding RNAs lead to cancer cell growth and metastasis via PTTG1 (70,71). Therefore, PTTG1 is a potential and novel therapeutic target of LUSC tumor growth and metastasis. CKS2, as a cyclin-dependent kinase-interacting protein, serves important roles in regulating cell cycle, inducing apoptosis, as well as regulating cancer cell invasion and metastasis. Upregulation of CKS2 can lead to DNA damage in cells, which can increase the proliferation of cancer cell (72). Therefore, the molecular mechanism by which CKS2 regulates cell cycle and induces cell apoptosis may be critical in investigating diagnostic methods and treatment methods for LUSC. CDC45 is an essential regulator of cell proliferation. An elevated expression of CDC45 is correlated with DNA damage in the S-phase, in which the anti-cancer effect of CDC45 suppressor is mediated by limiting DNA damage during S phase (73). KPNA2, as a member of the Karyopherin α family, actively participates in the process of signal transduction from the extracellular space to the nucleus. Furthermore, the upregulation of KPNA2 is correlated with cancer cell proliferation and metastasis (74). Moreover, several studies reported that KPNA2 is involved in the progression of cancer by regulating nuclear translocation of cancer-associated proteins; which may explain the significantly upregulated expression of KPNA2 in LUSC. NCAPG is a novel mitotic gene and provides novel therapeutic targets for cancer. Goto et al (75) reported that NCAPG, as a target of miR-145-3p, could predict the survival rate of patients with prostate cancer. UBE2S is a central protein in the process of ubiquitination and is associated with the malignancy of various types of cancer (76). Studies have reported the upregulation of UBE2S to enhance the nuclear translocation of β-catenin and induced expression of c-Myc and cyclin D1, suggesting UBE2S as a potential prognostic factor and oncogene in LUSC (77–78).
In conclusion, the present study was conducted in order to identify potential DEGs that may be associated with carcinogenesis or adverse progression of LUSC. A total of 67 DEGs and 17 hub genes were identified, in which hub genes were regarded as promising targets for the diagnosis and treatment of LUSC. Meanwhile, TYMS, CCNB2 and RFC4 were identified as potential novel biomarkers of LUSC. However, the present study was based on bioinformatics methods and no experiments were performed to validate the findings. Therefore, further studies are required to explore the biological association between the genes identified in the present study in LUSC, in order to improve treatments and clinical outcomes of LUSC.
Acknowledgements
Not applicable.
Funding
The present study was financially supported by the National Natural Science Foundation of China (grant no. 81273696).
Availability of data and materials
The datasets used and/or analyzed in the present study are available from the corresponding authors on reasonable request.
Authors' contributions
JM, XZ, HD and SL designed the study. JM and XZ analyzed and interpreted the microarray datasets, and wrote the manuscript. XY and LM made substantial contributions to acquisition and interpretation of data. JM, XG, HY, JC and YL analyzed the data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Youlden DR, Cramb SM, Baade PD. The International Epidemiology of Lung Cancer: Geographical distribution and secular trends. J Thorac Oncol. 2008;3:819–831. doi: 10.1097/JTO.0b013e31818020eb. [DOI] [PubMed] [Google Scholar]
- 2.Lazarus KA, Hadi F, Zambon E, Bach K, Santolla MF, Watson JK, Correia LL, Das M, Ugur R, Pensa S, et al. BCL11A interacts with SOX2 to control the expression of epigenetic regulators in lung squamous carcinoma. Nat Commun. 2018;9:3327. doi: 10.1038/s41467-018-05790-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kris MG, Johnson BE, Kwiatkowski DJ, Iafrate AJ, Wistuba II, Aronson SL, Engelman JA, Shyr Y, Khuri FR, Rudin CM, et al. Identification of driver mutations in tumor specimens from 1,000 patients with lung adenocarcinoma: The NCI's Lung Cancer Mutation Consortium (LCMC) J Clin Oncol. 2011;29:CRA7506. doi: 10.1200/jco.2011.29.18_suppl.cra7506. [DOI] [Google Scholar]
- 4.Chan BA, Hughes BG. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl Lung Cancer Res. 2015;4:36–54. doi: 10.3978/j.issn.2218-6751.2014.05.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vayshlya NA, Zinovyeva MV, Sass AV, Kopantzev EP, Vinogradova TV, Sverdlov ED. Increased expression of BIRC5 in non-small cell lung cancer and esophageal squamous cell carcinoma does not correlate with the expression of its inhibitors SMAC/DIABLO and PML. Mol Biol. 2008;42:579–587. doi: 10.1134/S0026893308040146. [DOI] [PubMed] [Google Scholar]
- 6.Li S, Sun X, Miao S, Liu J, Jiao W. Differential protein-coding gene and long noncoding RNA expression in smoking-related lung squamous cell carcinoma. Thorac Cancer. 2017;8:672–681. doi: 10.1111/1759-7714.12510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ganapathy-Kanniappan S, Geschwind JF, Kunjithapatham R, Buijs M, Vossen JA, Tchernyshyov I, Cole RN, Syed LH, Rao PP, Ota S, Vali M. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is pyruvylated during 3-bromopyruvate mediated cancer cell death. Anticancer Res. 2009;29:4909–4918. [PMC free article] [PubMed] [Google Scholar]
- 8.Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, Keating MJ, Huang P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005;65:613–621. [PubMed] [Google Scholar]
- 9.Dawany NB, Tozeren A. Asymmetric microarray data produces gene lists highly predictive of research literature on multiple cancer types. BMC Bioinformatics. 2010;11:483. doi: 10.1186/1471-2105-11-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawany NB, Dampier WN, Tozeren A. Large-scale integration of microarray data reveals genes and pathways common to multiple cancer types. Int J Cancer. 2011;128:2881–2891. doi: 10.1002/ijc.25854. [DOI] [PubMed] [Google Scholar]
- 11.Lin J, Marquardt G, Mullapudi N, Wang T, Han W, Shi M, Keller S, Zhu C, Locker J, Spivack SD. Lung cancer transcriptomes refined with laser capture microdissection. Am J Pathol. 2014;184:2868–2884. doi: 10.1016/j.ajpath.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rohrbeck A, Neukirchen J, Rosskopf M, Pardillos GG, Geddert H, Schwalen A, Gabbert HE, von Haeseler A, Pitschke G, Schott M, et al. Gene expression profiling for molecular distinction and characterization of laser captured primary lung cancers. J Transl Med. 2008;6:69. doi: 10.1186/1479-5876-6-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boelens MC, van den Berg A, Fehrmann RS, Geerlings M, de Jong WK, te Meerman GJ, Sietsma H, Timens W, Postma DS, Groen HJ. Current smoking-specific gene expression signature in normal bronchial epithelium is enhanced in squamous cell lung cancer. J Pathol. 2009;218:182–191. doi: 10.1002/path.2520. [DOI] [PubMed] [Google Scholar]
- 14.Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, Lempicki RA. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–W175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scardoni G, Tosadori G, Faizan M, Spoto F, Fabbri F, Laudanna C. Biological network analysis with CentiScaPe: Centralities and experimental dataset integration. F1000Res. 2014;3:139. doi: 10.12688/f1000research.4477.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mangan ME, Williams JM, Lathe SM, Karolchik D, Lathe WC., III UCSC genome browser: Deep support for molecular biomedical research. Biotechnol Annu Rev. 2008;14:63–108. doi: 10.1016/S1387-2656(08)00003-3. [DOI] [PubMed] [Google Scholar]
- 24.Hou J, Aerts J, den Hamer B, van Ijcken W, den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens JA, Hoogsteden HC, et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010;5:e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci USA. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wachi S, Yoneda K, Wu R. Interactome-transcriptome analysis reveals the high centrality of genes differentially expressed in lung cancer tissues. Bioinformatics. 2005;21:4205–4208. doi: 10.1093/bioinformatics/bti688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hou J, Aerts J, den Hamer B, van Ijcken W, den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens JA, Hoogsteden HC, et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010;5:e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou GX, Liu P, Yang J, Wen S. Mining expression and progQixing M, Gaochao D, Wenjie X, Anpeng W, Bing C, Weidong M, Lin X and Feng J: Microarray analyses reveal genes related to progression and prognosis of esophageal squamous cell carcinoma. Oncotarget. 2017;8:78838–78850. doi: 10.18632/oncotarget.20232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/S1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 31.Sholl LM, Aisner DL, Varella-Garcia M, Berry LD, Dias-Santagata D, Wistuba II, Chen H, Fujimoto J, Kugler K, Franklin WA, et al. Vol. 10. J Thorac Oncol; 2015. Multi-institutional Oncogenic Driver Mutation Analysis in Lung Adenocarcinoma: The Lung Cancer Mutation Consortium Experience; pp. 768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mantripragada K, Khurshid H. Targeting genomic alterations in squamous cell lung cancer. Front Oncol. 2013;3:195. doi: 10.3389/fonc.2013.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heist RS, Sequist LV, Engelman JA. Genetic changes in squamous cell lung cancer: A review. J Thorac Oncol. 2012;7:924–933. doi: 10.1097/JTO.0b013e31824cc334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cancer Genome Atlas Research Network, corp-author. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brambilla C, Laffaire J, Lantuejoul S, Moro-Sibilot D, Mignotte H, Arbib F, Toffart AC, Petel F, Hainaut P, Rousseaux S, et al. Lung squamous cell carcinomas with basaloid histology represent a specific molecular entity. Clin Cancer Res. 2014;20:5777–5786. doi: 10.1158/1078-0432.CCR-14-0459. [DOI] [PubMed] [Google Scholar]
- 36.Yao W, Bai Y, Li Y, Guo L, Zeng P, Wang Y, Qi B, Liu S, Qin X, Li Y, Zhao B. Upregulation of MALAT-1 and its association with survival rate and the effect on cell cycle and migration in patients with esophageal squamous cell carcinoma. Tumour Biol. 2016;37:4305–4312. doi: 10.1007/s13277-015-4223-3. [DOI] [PubMed] [Google Scholar]
- 37.Lee CH, Lee MK, Kang CD, Kim YD, Park DY, Kim JY, Sol MY, Suh KS. Differential expression of hypoxia inducible factor-1 alpha and tumor cell proliferation between squamous cell carcinomas and adenocarcinomas among operable non-small cell lung carcinomas. J Korean Med Sci. 2003;18:196–203. doi: 10.3346/jkms.2003.18.2.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirano T, Franzén B, Kato H, Ebihara Y, Auer G. Genesis of squamous cell lung carcinoma. Sequential changes of proliferation, DNA ploidy, and p53 expression. Am J Pathol. 1994;144:296–302. [PMC free article] [PubMed] [Google Scholar]
- 39.Liu GM, Zhang YM. Targeting FBPase is an emerging novel approach for cancer therapy. Cancer Cell Int. 2018;18:36. doi: 10.1186/s12935-018-0533-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 41.Keku T, Millikan R, Worley K, Winkel S, Eaton A, Biscocho L, Martin C, Sandler R. 5,10-methylenetetrahydrofolate reductase codon 677 and 1298 polymorphisms and colon cancer in African Americans and whites. Cancer Epidemiol Biomarkers Prev. 2002;11:1611–1621. [PubMed] [Google Scholar]
- 42.Ferlazzo N, Currò M, Zinellu A, Caccamo D, Isola G, Ventura V, Carru C, Matarese G, Ientile R. Influence of MTHFR genetic background on p16 and MGMT methylation in oral squamous cell cancer. Int J Mol Sci. 2017;18:E724. doi: 10.3390/ijms18040724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fujii T, Toyooka S, Ichimura K, Fujiwara Y, Hotta K, Soh J, Suehisa H, Kobayashi N, Aoe M, Yoshino T, et al. ERCC1 protein expression predicts the response of cisplatin-based neoadjuvant chemotherapy in non-small-cell lung cancer. Lung Cancer. 2008;59:377–384. doi: 10.1016/j.lungcan.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 44.Kasai D, Ozasa H, Oguri T, Miyazaki M, Uemura T, Takakuwa O, Kunii E, Ohkubo H, Maeno K, Niimi A. Thymidylate synthase gene copy number as a predictive marker for response to pemetrexed treatment of lung adenocarcinoma. Anticancer Res. 2013;33:1935–1940. [PubMed] [Google Scholar]
- 45.Lu Y, Zhuo C, Cui B, Liu Z, Zhou P, Lu Y, Wang B. TYMS serves as a prognostic indicator to predict the lymph node metastasis in Chinese patients with colorectal cancer. Clin Biochem. 2013;46:1478–1483. doi: 10.1016/j.clinbiochem.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 46.da Silva Nogueira J, Jr, de Lima Marson FA, Sílvia Bertuzzo C. Thymidylate synthase gene (TYMS) polymorphisms in sporadic and hereditary breast cancer. BMC Res Notes. 2012;5:676. doi: 10.1186/1756-0500-5-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shichijo S, Azuma K, Komatsu N, Ito M, Maeda Y, Ishihara Y, Itoh K. Two proliferation-related proteins, TYMS and PGK1, could be new cytotoxic T lymphocyte-directed tumor-associated antigens of HLA-A2+ colon cancer. Clin Cancer Res. 2004;10:5828–5836. doi: 10.1158/1078-0432.CCR-04-0350. [DOI] [PubMed] [Google Scholar]
- 48.Chao YL, Anders CK. TYMS gene polymorphisms in breast cancer patients receiving 5-fluorouracil-based chemotherapy. Clin Breast Cancer. 2018;18:e301–e304. doi: 10.1016/j.clbc.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He Y, Penney ME, Negandhi AA, Parfrey PS, Savas S, Yilmaz YE. XRCC3 Thr241Met and TYMS variable number tandem repeat polymorphisms are associated with time-to-metastasis in colorectal cancer. PLoS One. 2018;13:e0192316. doi: 10.1371/journal.pone.0192316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Knizhnik AV, Kovaleva OB, Laktionov KK, Mochal'nikova VV, Komel'kov AV, Chevkina EM, Zborovskaia IB. Arf6, RalA and BIRC5 protein expression in non small cell lung cancer. Mol Biol (Mosk) 2011;45:307–315. doi: 10.1134/S0026893310061032. (In Russian) [DOI] [PubMed] [Google Scholar]
- 51.Yu X, Zhang Y, Cavazos D, Ma X, Zhao Z, Du L, Pertsemlidis A. miR-195 targets cyclin D3 and survivin to modulate the tumorigenesis of non-small cell lung cancer. Cell Death Dis. 2018;9:193. doi: 10.1038/s41419-017-0219-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Phiboonchaiyanan PP, Petpiroon N, Sritularak B, Chanvorachote P. Phoyunnanin E induces apoptosis of non-small cell lung cancer cells via p53 activation and down-regulation of survivin. Anticancer Res. 2018;38:6281–6290. doi: 10.21873/anticanres.12984. [DOI] [PubMed] [Google Scholar]
- 53.Ganapathy-Kanniappan S. Evolution of GAPDH as a druggable target of tumor glycolysis? Expert Opin Ther Targets. 2018;22:295–298. doi: 10.1080/14728222.2018.1449834. [DOI] [PubMed] [Google Scholar]
- 54.Hao L, Zhou X, Liu S, Sun M, Song Y, Du S, Sun B, Guo C, Gong L, Hu J, et al. Elevated GAPDH expression is associated with the proliferation and invasion of lung and esophageal squamous cell carcinomas. Proteomics. 2015;15:3087–3100. doi: 10.1002/pmic.201400577. [DOI] [PubMed] [Google Scholar]
- 55.Mo ML, Chen Z, Li J, Li HL, Sheng Q, Ma HY, Zhang FX, Hua YW, Zhang X, Sun DQ, et al. Use of serum circulating CCNB2 in cancer surveillance. Int J Biol Markers. 2010;25 doi: 10.5301/JBM.2010.6088. [DOI] [PubMed] [Google Scholar]
- 56.Takashima S, Saito H, Takahashi N, Imai K, Kudo S, Atari M, Saito Y, Motoyama S, Minamiya Y. Strong expression of cyclin B2 mRNA correlates with a poor prognosis in patients with non-small cell lung cancer. Tumour Biol. 2014;35:4257–4265. doi: 10.1007/s13277-013-1556-7. [DOI] [PubMed] [Google Scholar]
- 57.Lei CY, Wang W, Zhu YT, Fang WY, Tan WL. The decrease of cyclin B2 expression inhibits invasion and metastasis of bladder cancer. Urol Oncol. 2016;34:237.e1–e10. doi: 10.1016/j.urolonc.2015.11.011. [DOI] [PubMed] [Google Scholar]
- 58.Wang XC, Yue X, Zhang RX, Liu TY, Pan ZZ, Yang MJ, Lu ZH, Wang ZY, Peng JH, Le LY, et al. Genome-wide RNAi screening identifies RFC4 as a factor that mediates radioresistance in colorectal cancer by facilitating nonhomologous end joining repair. Clin Cancer Res. 2019;25:4567–4579. doi: 10.1158/1078-0432.CCR-18-3735. [DOI] [PubMed] [Google Scholar]
- 59.Xiang J, Fang L, Luo Y, Yang Z, Liao Y, Cui J, Huang M, Yang Z, Huang Y, Fan X, et al. Levels of human replication factor C4, a clamp loader, correlate with tumor progression and predict the prognosis for colorectal cancer. J Transl Med. 2014;12:320. doi: 10.1186/s12967-014-0320-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu YZ, Wang BS, Jiang YY, Cao J, Hao JJ, Zhang Y, Xu X, Cai Y, Wang MR. MCMs expression in lung cancer: Implication of prognostic significance. J Cancer. 2017;8:3641–3647. doi: 10.7150/jca.20777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Behrens C, Solis LM, Lin H, Yuan P, Tang X, Kadara H, Riquelme E, Galindo H, Moran CA, Kalhor N, et al. EZH2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non-small cell lung carcinoma. Clin Cancer Res. 2013;19:6556–6565. doi: 10.1158/1078-0432.CCR-12-3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi R, Li M, Raghavan V, Tam S, Cabanero M, Pham NA, Shepherd FA, Moghal N, Tsao MS. Targeting the CDK4/6-Rb pathway enhances response to PI3K inhibition in PIK3CA-mutant lung squamous cell carcinoma. Clin Cancer Res. 2018;24:5990–6000. doi: 10.1158/1078-0432.CCR-18-0717. [DOI] [PubMed] [Google Scholar]
- 63.Chen L, Pan J. Dual cyclin-dependent kinase 4/6 inhibition by PD-0332991 induces apoptosis and senescence in oesophageal squamous cell carcinoma cells. Br J Pharmacol. 2017;174:2427–2443. doi: 10.1111/bph.13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma Y, Lin D, Sun W, Xiao T, Yuan J, Han N, Guo S, Feng X, Su K, Mao Y, et al. Expression of targeting protein for xklp2 associated with both malignant transformation of respiratory epithelium and progression of squamous cell lung cancer. Clin Cancer Res. 2006;12:1121–1127. doi: 10.1158/1078-0432.CCR-05-1766. [DOI] [PubMed] [Google Scholar]
- 65.Mollinari C, Kleman JP, Jiang W, Schoehn G, Hunter T, Margolis RL. PRC1 is a microtubule binding and bundling protein essential to maintain the mitotic spindle midzone. J Cell Biol. 2002;157:1175–1186. doi: 10.1083/jcb.200111052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhan P, Xi GM, Liu HB, Liu YF, Xu WJ, Zhu Q, Zhou ZJ, Miao YY, Wang XX, Jin JJ, et al. Protein regulator of cytokinesis-1 expression: Prognostic value in lung squamous cell carcinoma patients. J Thorac Dis. 2017;9:2054–2060. doi: 10.21037/jtd.2017.06.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang J, Zhou Y, Thomas GS, Gu Z, Yang Y, Xu H, Tricot G, Zhan F. NEDD8 inhibition overcomes CKS1B-induced drug resistance by upregulation of p21 in multiple myeloma. Clin Cancer Res. 2015;21:5532–5542. doi: 10.1158/1078-0432.CCR-15-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhan F, Colla S, Wu X, Chen B, Stewart JP, Kuehl WM, Barlogie B, Shaughnessy JD., Jr CKS1B, overexpressed in aggressive disease, regulates multiple myeloma growth and survival through SKP2- and p27Kip1-dependent and -independent mechanisms. Blood. 2007;109:4995–5001. doi: 10.1182/blood-2006-07-038703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Romero Arenas MA, Whitsett TG, Aronova A, Henderson SA, LoBello J, Habra MA, Grubbs EG, Lee JE, Sircar K, Zarnegar R, et al. Protein expression of PTTG1 as a diagnostic biomarker in adrenocortical carcinoma. Ann Surg Oncol. 2018;25:801–807. doi: 10.1245/s10434-017-6297-1. [DOI] [PubMed] [Google Scholar]
- 70.Guo XC, Li L, Gao ZH, Zhou HW, Li J, Wang QQ. The long non-coding RNA PTTG3P promotes growth and metastasis of cervical cancer through PTTG1. Aging (Albany NY) 2019;11:1333–1341. doi: 10.18632/aging.101830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang JL, Cao SW, Ou QS, Yang B, Zheng SH, Tang J, Chen J, Hu YW, Zheng L, Wang Q. The long non-coding RNA PTTG3P promotes cell growth and metastasis via up-regulating PTTG1 and activating PI3K/AKT signaling in hepatocellular carcinoma. Mol Cancer. 2018;17:93. doi: 10.1186/s12943-018-0841-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.You H, Lin H, Zhang Z. CKS2 in human cancers: Clinical roles and current perspectives (Review) Mol Clin Oncol. 2015;3:459–463. doi: 10.3892/mco.2015.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hauge S, Naucke C, Hasvold G, Joel M, Rødland GE, Juzenas P, Stokke T, Syljuåsen RG. Combined inhibition of Wee1 and Chk1 gives synergistic DNA damage in S-phase due to distinct regulation of CDK activity and CDC45 loading. Oncotarget. 2017;8:10966–10979. doi: 10.18632/oncotarget.14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jensen JB, Munksgaard PP, Sørensen CM, Fristrup N, Birkenkamp-Demtroder K, Ulhøi BP, Jensen KM, Ørntoft TF, Dyrskjøt L. High expression of karyopherin-α2 defines poor prognosis in non-muscle-invasive bladder cancer and in patients with invasive bladder cancer undergoing radical cystectomy. Eur Urol. 2011;59:841–848. doi: 10.1016/j.eururo.2011.01.048. [DOI] [PubMed] [Google Scholar]
- 75.Goto Y, Kurozumi A, Arai T, Nohata N, Kojima S, Okato A, Kato M, Yamazaki K, Ishida Y, Naya Y, et al. Impact of novel miR-145-3p regulatory networks on survival in patients with castration-resistant prostate cancer. Br J Cancer. 2017;117:409–420. doi: 10.1038/bjc.2017.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Garnett MJ, Mansfeld J, Godwin C, Matsusaka T, Wu J, Russell P, Pines J, Venkitaraman AR. UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. Nat Cell Biol. 2009;11:1363–1369. doi: 10.1038/ncb1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Akter KA, Hyodo T, Asano E, Sato N, Mansour MA, Ito S, Hamaguchi M, Senga T. Erratum to: UBE2S is associated with malignant characteristics of breast cancer cells. Tumor Biol. 2016;37:6999. doi: 10.1007/s13277-016-4832-5. [DOI] [PubMed] [Google Scholar]
- 78.Ben-Eliezer I, Pomerantz Y, Galiani D, Nevo N, Dekel N. Appropriate expression of Ube2C and Ube2S controls the progression of the first meiotic division. FASEB J. 2015;29:4670–4681. doi: 10.1096/fj.15-274522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed in the present study are available from the corresponding authors on reasonable request.