

Abstract
The Pathosystems Resource Integration Center (PATRIC, www.patricbrc.org) is designed to provide researchers with the tools and services that they need to perform genomic and other ‘omic’ data analyses. In response to mounting concern over antimicrobial resistance (AMR), the PATRIC team has been developing new tools that help researchers understand AMR and its genetic determinants. To support comparative analyses, we have added AMR phenotype data to over 15 000 genomes in the PATRIC database, often assembling genomes from reads in public archives and collecting their associated AMR panel data from the literature to augment the collection. We have also been using this collection of AMR metadata to build machine learning-based classifiers that can predict the AMR phenotypes and the genomic regions associated with resistance for genomes being submitted to the annotation service. Likewise, we have undertaken a large AMR protein annotation effort by manually curating data from the literature and public repositories. This collection of 7370 AMR reference proteins, which contains many protein annotations (functional roles) that are unique to PATRIC and RAST, has been manually curated so that it projects stably across genomes. The collection currently projects to 1 610 744 proteins in the PATRIC database. Finally, the PATRIC Web site has been expanded to enable AMR-based custom page views so that researchers can easily explore AMR data and design experiments based on whole genomes or individual genes.
Keywords: antimicrobial resistance (AMR), antibiotic, genome annotation, minimum inhibitory concentration, RAST, the SEED
Background
The Pathosystems Resource Integration Center (PATRIC) is one of four bioinformatics resource centers (BRCs) funded by the National Institute of Allergy and Infectious Diseases (NIAID) [1]. The BRC program supports research by providing access to data associated with the NIAID Category A–C pathogenic genera [2], with PATRIC serving as the bacterial database. To provide a rich comparative analysis environment, PATRIC provides access to all publicly available genomes and associated metadata for bacterial and archaeal isolates, which includes >104 000 genomes as of June 2017.
All of the genomes in PATRIC have been consistently annotated using the Rapid Annotation using Subsystems Technology toolkit (RASTtk) [3, 4]. This annotation consistency and subsequent protein family generation [5] serve as the backbone for many of the comparative analysis tools on the Web site [1]. The PATRIC database retains the annotations and identifiers from both GenBank [6, 7] and RefSeq [8] to facilitate side-by-side comparisons across the public data, allowing researchers to quickly find genomes and genes with information that they have gathered from different resources. PATRIC also provides researchers with a private workspace, where they can access bioinformatics services including genome assembly, annotation, RNA sequencing, variation calling, Tn-Seq, similar genome finder, proteome comparison and metabolic model reconstruction. When a user annotates a private genome with the PATRIC annotation service, they can compare their genome with the public collection. This ‘virtual integration’ provides a unique analysis experience that is not available at a similar scale at any other data repository.
Facilitating research on antimicrobial resistance (AMR) has become increasingly important with the recent escalation in resistance and the loss of effectiveness to first-line drugs [9–13]. This resistance has a human cost, with ∼2 million people being sickened and 23 000 dying annually in the United States alone [14]. Here, we describe a set of enhancements introduced to support research on AMR.
AMR strategy
The current strategy for integrating AMR data into PATRIC breaks down roughly into two parts: (1) data collection to support analyses of whole genomes and (2) data collection to support analyses of individual proteins (Figure 1). In both cases, the data are drawn from the literature as well as a number of public resources. Specifics on the data integration, curation and tools are described below.
Figure 1.
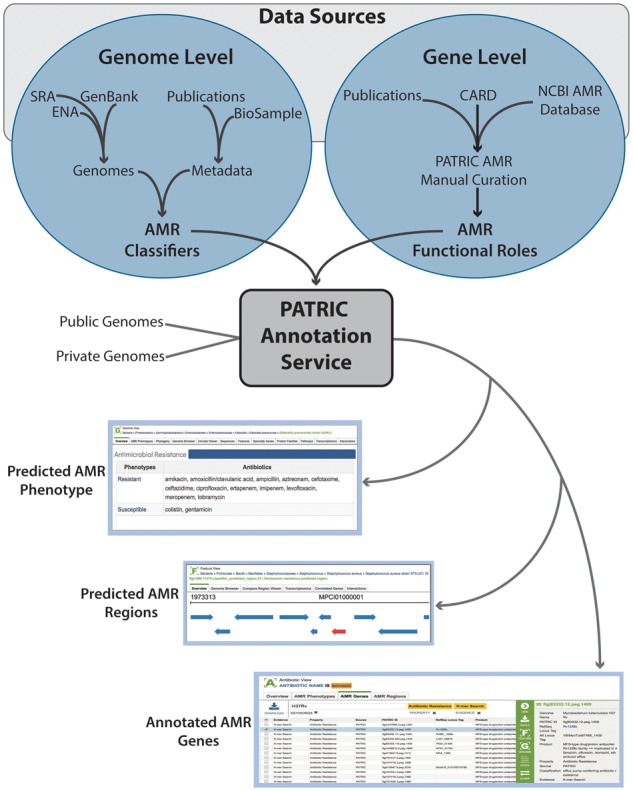
PATRIC annotation process for integrating AMR data in both genomic regions and genes.
AMR—integrating data at the genome level
Data collection
To support an environment for comparative analysis, we integrate metadata associated with the public genomes at GenBank [7] into the PATRIC database. This makes it easy to build sets of genomes that are based on collection date, geographic location, host, isolation source, etc. These metadata fields are incorporated both from BioSample [15] and directly from the GenBank file when an assembled genome is added to PATRIC. In some cases, metadata are acquired first hand from the NIAID-funded genome sequencing centers and from collaborators wishing to make their data public. Given the increasing emphasis on research to combat AMR and the decreasing costs of sequencing, we have been able to collect a large number of genomes with AMR panel data in the form of minimum inhibitory concentrations (MICs) or susceptible, intermediate and resistant (SIR) calls [16]. These panel data provide critical context for AMR research by allowing researchers to quickly build data sets for performing protein and gene comparisons, novel gene discovery, whole-genome variation analyses and machine learning (ML) experiments (described below).
To increase the number of genomes with AMR metadata in PATRIC and expand our ability to support AMR-based comparative analyses, we began searching the literature for studies that included sequenced bacterial genomes and AMR panel data. Oftentimes, panel data from these studies were not recorded in the public archives, so PATRIC becomes the only place, where both the assembled genomes and metadata are available in the same place. If a genome was assembled and deposited in GenBank [7], we attach the AMR metadata directly to the corresponding genome in PATRIC. If the reads for a genome were deposited in the Sequence Read Archive (SRA) or the European Nucleotide Archive (ENA) [17, 18], we assemble and annotate the genome using PATRIC services [1, 4, 19]. We then incorporate the genome into the database along with the metadata (Supplementary Document S1).
As laboratory methods for determining MIC values vary, incorporating these data into PATRIC requires a significant manual curation effort. When information is available from the study, we record how the MIC data were generated, including the laboratory method, the units of the measurement and the platform that was used to make the measurements. When an assertion about a phenotype is provided in the form of a SIR call, we record the laboratory standard from the European Committee on Antimicrobial Susceptibility Testing (EUCAST) [20] or the Clinical and Laboratory Standards Institute [21] and the year of the standard. To date, we have attached metadata to PATRIC genomes for ∼9165 genomes and have assembled and annotated ∼6122 genomes from SRA and ENA (Supplementary Table S1). To date, all AMR metadata in PATRIC are phenotypes that are derived from laboratory analyses. Studies often assert the susceptibility or resistance of an organism based on the presence or absence of key AMR genes. We do not currently incorporate data that are only based on genotypic data. The complete collection of AMR data in PATRIC can be downloaded from the PATRIC FTP site: ftp.patricbrc.org/patric2/current_release/RELEASE_NOTES/PATRIC_genomes_AMR.txt.
ML classifiers
As the PATRIC database was rapidly accumulating AMR panel data associated with sequenced genomes, a small number of studies were being published that explored using ML algorithms to study AMR [22–24]. With a sufficient number of genomes and AMR panel data, ML algorithms can be used to predict AMR phenotypes and the genomic regions associated with AMR with no a priori knowledge of the underlying mechanisms. This is an appealing area of exploration for PATRIC because it allows us to leverage our growing metadata collection to predict AMR phenotypes within the annotation service and to identify AMR-associated genomic regions with single-nucleotide polymorphism (SNP)-level resolution, a feature that can be used to inform our ongoing manual protein annotation efforts.
In early 2016, we published a study describing the collection of AMR metadata for genomes and an ML approach that used the AdaBoost algorithm [25, 26] to build classifiers for predicting AMR [16]. At the time, we had sufficient data to make predictions in the species Acinetobacter baumannii, Mycobacterium tuberculosis, Staphylococcus aureus and Streptococcus pneumoniae for nine antibiotics [16] (Table 1). Shortly thereafter, we collaborated with scientists at the Houston Methodist Research Hospital to build classifiers for Klebsiella pneumoniae covering 13 antibiotics using 1777 genomes collected in their hospital system between 2011 and 2015 [27]. Using the same protocol as described in the Davis et al. [16] and Long et al. studies [27], we added 18 additional classifiers to the annotation system that have not been previously reported, including classifiers for M. tuberculosis, Peptoclostridium difficile, Pseudomonas aeruginosa, S. aureus and S. pneumoniae (Table 1). Receiver operating characteristic (ROC) curves for the newly added classifiers are shown in Figure 2.
Table 1.
AMR classifiers in the PATRIC annotation system
| Species | Antibiotica | Resistant genomesb | Susceptible genomesb | F1 score | Initially described in |
|---|---|---|---|---|---|
| Acinetobacter baumannii | Carbapenem | 122 | 110 | 0.95 | [16] |
| Klebsiella pneumoniae | Amikacin | 1190 | 364 | 0.92 | [27] |
| Klebsiella pneumoniae | Aztreonam | 1377 | 100 | 0.75 | [27] |
| Klebsiella pneumoniae | Cefoxitin | 555 | 976 | 0.80 | [27] |
| Klebsiella pneumoniae | Ciprofloxacin | 119 | 1435 | 0.91 | [27] |
| Klebsiella pneumoniae | Ertapenem | 265 | 178 | 0.96 | [27] |
| Klebsiella pneumoniae | Gentamicin | 786 | 768 | 0.86 | [27] |
| Klebsiella pneumoniae | Imipenem | 1100 | 453 | 0.94 | [27] |
| Klebsiella pneumoniae | Levofloxacin | 246 | 1307 | 0.93 | [27] |
| Klebsiella pneumoniae | Meropenem | 1123 | 430 | 0.92 | [27] |
| Klebsiella pneumoniae | Piperacillin–tazobactam | 322 | 1230 | 0.76 | [27] |
| Klebsiella pneumoniae | Tetracycline | 658 | 896 | 0.79 | [27] |
| Klebsiella pneumoniae | Tobramycin | 501 | 1053 | 0.94 | [27] |
| Klebsiella pneumoniae | Co-trimoxazole | 331 | 1223 | 0.87 | [27] |
| Mycobacterium tuberculosis | Amikacin | 210 | 350 | 0.91 | This study |
| Mycobacterium tuberculosis | Capreomycin | 204 | 350 | 0.83 | This study |
| Mycobacterium tuberculosis | Isoniazid | 250 | 250 | 0.88 | [16] |
| Mycobacterium tuberculosis | Kanamycin | 188 | 250 | 0.87 | [16] |
| Mycobacterium tuberculosis | Ofloxacin | 239 | 250 | 0.79 | [16] |
| Mycobacterium tuberculosis | Rifampicin | 250 | 250 | 0.86 | [16] |
| Mycobacterium tuberculosis | Streptomycin | 250 | 250 | 0.71 | [16] |
| Peptoclostridium difficile | Azithromycin | 213 | 246 | 0.97 | This study |
| Peptoclostridium difficile | Ceftriaxone | 228 | 86 | 0.86 | This study |
| Peptoclostridium difficile | Clarithromycin | 213 | 246 | 0.99 | This study |
| Peptoclostridium difficile | Clindamycin | 310 | 89 | 0.74 | This study |
| Peptoclostridium difficile | Moxifloxacin | 188 | 271 | 0.97 | This study |
| Pseudomonas aeruginosa | Levofloxacin | 192 | 290 | 0.85 | This study |
| Staphylococcus aureus | Ciprofloxacin | 467 | 762 | 0.98 | This study |
| Staphylococcus aureus | Clindamycin | 350 | 274 | 0.97 | This study |
| Staphylococcus aureus | Erythromycin | 484 | 821 | 0.96 | This study |
| Staphylococcus aureus | Gentamicin | 162 | 1144 | 0.98 | This study |
| Staphylococcus aureus | Methicillin | 707 | 886 | 0.99 | [16] |
| Staphylococcus aureus | Penicillin | 886 | 156 | 0.96 | This study |
| Staphylococcus aureus | Tetracycline | 203 | 1029 | 0.97 | This study |
| Staphylococcus aureus | Co-trimoxazole | 142 | 178 | 0.96 | This study |
| Streptococcus pneumoniae | Beta-lactam | 2124 | 584 | 0.90 | [16] |
| Streptococcus pneumoniae | Chloramphenicol | 165 | 289 | 0.94 | This study |
| Streptococcus pneumoniae | Co-trimoxazole | 2124 | 584 | 0.88 | [16] |
| Streptococcus pneumoniae | Erythromycin | 381 | 324 | 0.96 | This study |
| Streptococcus pneumoniae | Tetracycline | 368 | 290 | 0.96 | This study |
aAMR data in PATRIC may be described as individual antibiotics or classes of antibiotics.
bUsed for building the classifiers.
Figure 2.

ROC curves for AdaBoost-based AMR classifiers installed in the annotation service since the publication of the Davis et al. [16] and Long et al. papers [27]. Accuracy and F1 scores are displayed in each inset. ROC curves depict classifiers for (A) P. difficile, (B) S. aureus and (C) K. pneumoniae (Kpn), M. tuberculosis (Mtb), P. aeruginosa (Pae) and S. pneumoniae (Spn). Antibiotic abbreviations are: AZM, azithromycin; CC, clindamycin; CIP, ciprofloxacin; CLR, clarithromycin; CRO, ceftriaxone; E, erythromycin; GM, gentamicin; MFX, moxifloxacin; OX, ofloxacin; P, penicillin; SXT, trimethoprim sulfamethoxazole; TE, tetracycline.
To date, we have maintained a policy of adding classifiers to the annotation system when their accuracies and F1 scores exceed 70% and their top feature k-mers relate to known AMR genes. The classifiers built in this project and described in Table 1 and Figure 2 are integrated into the annotation service and can be accessed through PATRIC and RAST. Phenotype predictions and the associated genomic regions are available for browsing on both Web sites and are described in tutorials at http://tutorial.theseed.org/.
Our AMR metadata collection and classifier building efforts are ongoing at PATRIC. In many cases, the AMR metadata available in published studies report pan-resistant strains, which can be difficult to classify. In an effort to improve the accuracy of the classifiers, we are actively seeking strains with AMR metadata that improve the biological diversity of the collection. This includes collecting strains susceptible to many antibiotics. We are also comparing the results from several ML methods and are in the process of adding classifiers based on these other methods when they outperform AdaBoost [25]. In this manner, an antibiotic and species would be paired with the best ML algorithm in the annotation system.
AMR—integrating data at the gene level
Data collection
Starting in 2015, the PATRIC annotation team, which also maintains the SEED [28] and RAST projects [3], began a focused effort to incorporate and manually curate protein functions relating to AMR. There are several well-known consortia that strive to provide standardized nomenclature for specific groups of antibiotic resistance genes including tetracycline resistance determinants [29, 30], and different classes of β-lactamases maintained by the Lahey Clinic [31], the University of Stuttgart [32, 33] and the Institute Pasteur [34]. There are also several well-respected databases that provide collections of AMR genes covering broad categories of AMR mechanisms including the Comprehensive Antibiotic Resistance Database (CARD) [35], the Bacterial Antimicrobial Resistance Reference Gene Database [36] hosted by the National Center for Biotechnology Information as part of the National Database of Antibiotic Resistant Organisms (NDARO) and ResFinder [37]. These resources maintain reference sequences for each AMR gene type, providing each with well-curated informative product names (in the case of NDARO) or a specialized Antibiotic Resistance Ontology (ARO, provided by CARD). These collections enable accurate detection and annotation of specific AMR determinates in pathogen isolates by means of supporting the BLAST-based [38, 39] or hidden Markov model (HMM)-based [40] screening of user-submitted sequences against representative sets of AMR sequences. However, in many cases, these AMR annotations project ambiguously because newly discovered proteins can match representative proteins with differing annotations at nearly equal BLAST similarities. For example, a novel CTX-M, SHV or TEM β-lactamase could potentially present the researcher with over a hundred nearly equal BLAST hits against highly homologous but clinically different reference sequence variants, making the choice of the most appropriate product name difficult. In many cases, the best choice would be a novel allele designation, rather than one of the existing curated product names. We believed that a manual curation effort was necessary to integrate AMR sequence variants into distinct functional roles (isofunctional protein families, which are integral for the SEED/PATRIC environment) to ensure that they can be unambiguously projected to the genomes in PATRIC by the annotation service.
As many resources focus more heavily on the horizontally transferred AMR genes, we began our curation effort by building functional roles for AMR-related porin and efflux pump proteins described in the literature that are often chromosomally encoded, reasoning that this would rapidly add new value to the scientific community. Afterward, this naturally led into an effort to incorporate annotations for proteins involved in tetracycline resistance. The proteins involved in efflux pumps are known to play an important role in this type of resistance [41], and there are well-described annotation rules, which have been curated by the community for decades for naming them [30, 42]. More recently, we have been annotating class by class using publicly available resources when possible.
Curation process and k-mer projection
Significant manual curation and modification of the existing RAST/RASTtk automatic annotation pipeline were required to accommodate AMR-related functional roles, as their biology differs significantly from ‘classic’ functional roles encoding prokaryotic enzymatic and nonenzymatic housekeeping functions. The process of creating projectable AMR annotations starts with the incorporation of reference proteins from the literature and public resources. BLAST searches are used to compare reference sequences against the SEED database and PATRIC [1]. The subsequent matching proteins are used to build alignments and trees, which are manually inspected to understand how specific or general an annotation is, and if it will project cleanly in the annotation system. When reference proteins from the literature create ambiguous BLAST matches or split high-similarity clades in the tree, the nomenclature is retained, but then combined into a single annotation that covers the entire clade. The training sets of representative AMR sequence variants from outside sources and the SEED database [28] are then built. They form the basis for each AMR-related functional role. An annotation string for each of the functional roles is assigned, taking into account the SEED database internal nomenclature conventions as well as those developed by the AMR research community and accepted by CARD, ResFinder, NCBI and other resources. Signature k-mers (amino acid 8-mers) are built from these functional roles as described previously [4], and the annotations are then projected to all of the genomes in PATRIC. Trees for the newly annotated AMR proteins are then manually inspected to identify clades that contain multiple annotations, indicating a lack of consistency. Inconsistencies are also identified by comparing the generation of protein families before and after the addition of a new function. The inconsistent proteins are manually re-annotated and this process is iterated until the annotations project stably and accurately across the entire database.
The PATRIC manual curation effort offers a variety of additional benefits to the field of AMR research. For example, this effort is helping to alleviate the well-documented problem of miss-annotation and over prediction of AMR annotations [43, 44]. We are doing this by systematically removing erroneous annotations, which implicate non-AMR-related proteins with antibiotic resistance functions, and by annotating and attaching literature references to these closely related proteins to prevent over-projection of AMR roles, and then curating their projection over the PATRIC collection as described above.
We occasionally discover clades of potential AMR proteins that are surrounded by solid AMR reference sequences, yet have not been described in any reference database. In these cases, we describe the protein as a ‘putative’ AMR protein of a given resistance type, if the sequence identity levels are 50% or better over the entire length of the protein, which enables functional projection. These are obvious targets for characterization in the laboratory. However, if a newly discovered hypothetical clade has a sequence identity that is <50%, we use the less specific annotation string for all its members. In these cases, we use the following annotations: ‘weak similarity to aminoglycoside N(6')-acetyltransferase’ and ‘weak similarity to aminoglycoside N(3)-acetyltransferase’. These are obvious targets for characterization in the laboratory. Finally, having clean sets of AMR-related functional roles facilitates SNP and other comparative analyses at PATRIC and elsewhere by providing relevant sequence peer groups for variation research.
As of May 2017, the annotation of AMR determinants conferring resistance to tetracycline, β-lactam, aminoglycoside [45, 46], chloramphenicol [47] and MLSKO (macrolides, lincosamides, streptogramins, ketolides and oxazolidinones) [42, 48, 49] antibiotic classes has been completed. These include 450 functional roles for these five major antibiotic classes, as well as 36 roles for closely related non-AMR proteins. This collection comprises a combined set of 7370 reference and SEED proteins with AMR roles and 36 424 proteins with related non-AMR roles. The collection projects consistently to 1 610 744 AMR proteins with AMR roles and 2 518 252 proteins with related non-AMR roles in PATRIC. We have also associated literature references with the majority of the newly curated AMR functional roles in PATRIC, totaling 411 references. The curation effort is ongoing and is focusing on proteins conveying resistance to quinolone, vancomycin, fosfomycin, rifampin/rifamycin, nitroimidazole, bleomycin and other antibiotic classes.
Visualization of AMR data at PATRIC
Several new interfaces have been developed on the PATRIC Web site to allow researchers to fully explore the AMR data available in the resource. These interfaces include information that is summarized across all genomes for the available antibiotics, at the taxon level, and for individual genomes and genes. Details on each of these interfaces are described below.
Antibiotic view
Data from PubChem [50] are now integrated for nearly 100 specific antibiotics that can be viewed on landing pages designed especially to display this information. Each individual antibiotic has a landing page with several tabs that provide a general overview, specific information on the AMR phenotype, the genes associated with that phenotype and the regions within the individual genes or genomes that are linked to resistance or susceptibility to that specific drug (Figure 3).
Figure 3.

Summary information for the antibiotic methicillin at PATRIC. The antibiotic interface provides a summary of the antibiotic, its synonyms and actions, and also provides links via separate tabs for AMR phenotypes, genes and regions across all the data available in PATRIC.
The overview tab includes a general description of the drug, the chemical structure, the mechanism of action, a description of the pharmacological activity and class and known synonyms. The AMR phenotype tab provides a list of all the genomes that have been identified as being susceptible or resistant to that antimicrobial. This tab also includes the laboratory typing method and platform, and the testing standard if that information is available. A third tab, called AMR genes, displays information on the genes associated with resistance. The final tab, AMR regions, includes the location of the specific k-mers that are associated with the genome’s phenotype.
Taxon-level view
PATRIC organizes relevant data for all the available sequenced bacterial and archaeal genomes according to NCBI taxonomy [51]. Data are summarized at each level, from the highest (the Superkingdoms: Bacteria and Archaea) to the strain (or isolate) from which the genome has been sequenced. For each taxonomic level with associated AMR data, PATRIC provides several summaries. A bar graph summarizing the antibiotics, the AMR phenotype (resistant, intermediate or susceptible) and the number of genomes that match that phenotype is available on the overview tab at the top of the main landing page for each taxon (Figure 4A). Clicking on any of the antibiotics displayed in the graph will open a new page that summarizes all the genomes from that taxon level that have the particular AMR phenotype. An alternate tabular view of the data is also available (Figure 4B). The taxon-level summary page also includes an AMR phenotype tab that lists all of the genomes within the selected taxon that have an AMR phenotype, and the data that are associated with it, including specific treatments, phenotypes or laboratory methods. All tables in PATRIC include a dynamic filter for rapid filtering of the genomes based on metadata selections.
Figure 4.

A taxon-level summary on the PATRIC Web site describing AMR phenotype data across all of the genomes that are part of the Staphylococcus genus. (A) A bar graph summarizes the antibiotics, the AMR phenotype (resistant, intermediate or susceptible) and the number of genomes that match that phenotype. (B) The AMR phenotype tabular view, which shows all the genomes that have associated AMR data, includes a dynamic filter for rapid selection of genomes based on the metadata.
Gene view and predicted regions associated with AMR phenotypes
PATRIC provides a summary of data at the gene level, where the physical characteristics of a gene, its functional role(s), available experimental data and associated publications are provided. This view also includes information on homology to genes known to be important in AMR. In addition, PATRIC provides a view for predicted regions within some genes that are associated with AMR phenotypes. The k-mer regions predicted by the ML classifiers are visually indicated and their genomic region can be seen on the genome browser (Figure 5).
Figure 5.

AMR predicted regions, located in the genome of S. aureus strain 08S00974, as visualized in the PATRIC JBrowse viewer [57]. These predicted regions, numbered sequentially by their occurrence in the genome as ‘classifier_predicted_regions 12–15’, were predicted by the ML algorithm that is being used to predict AMR phenotypes. The predicted regions are located in and around a gene (fig|1280.11691.peg.56) that is annotated as ‘Tetracycline resistance, MFS efflux pump = > Tet(K)’. The annotation for this gene came from the focused manual curation effort at PATRIC to incorporate and propagate information for specific genes that were known to play an important role in AMR.
Future improvements
We continue to peruse resources and publications to identify new genomes and AMR genes to incorporate into PATRIC. These will be used to expand the AMR phenotype predictions and AMR gene analysis to new genera and new antibiotics. We plan to map AMR properties to the genus-specific families (PLfams) to support comparative analysis of AMR genes, incorporate new AMR gene trees and allow users to build nucleotide-based multiple sequence alignments to identify SNPs and their association with AMR phenotypes. We are acutely aware that several important types of AMR determinants are not amenable to being encoded and automatically propagated via the automated annotation propagation strategy described above. These include antibiotic targets, which are largely cellular proteins performing essential household cellular functions, and such proteins are grouped into ‘classic’ functional roles in SEED/PATRIC. They carry functional annotations that are unrelated to AMR. Antibiotic susceptibility in these target proteins is determined by a few, or even a single, non-synonymous mutation in the corresponding gene [52–54]. Likewise, single mutations in noncoding DNA regions, including promoters, operators and attenuators, can lead to dramatic increase in MIC, or an increase in resistance levels to particular antimicrobials [55, 56]. These cases will be treated separately in PATRIC. We are in the process of designing tools specific for SNP detection and analysis targeted at the gene level. While PATRIC does not currently enable examining AMR data from metagenomes or from population-based studies, this is something that we plan to provide in future releases.
Key Points.
PATRIC includes AMR information at both the genome and gene level, and uses manual curation and ML to integrate these data into the annotation service. A large collection of AMR-specific functional roles has been manually curated, and this information is propagated by the annotation service. With summaries of the available data across all taxonomic levels and new interfaces, researchers can quickly locate and examine these data in their private genomes and compare with the PATRIC collection.
Supplementary Material
Biographies
Dionysios A. Antonopoulos is a Microbiologist who is a staff scientist in the Biosciences Division at Argonne National Laboratory and an Assistant Professor in the University of Chicago Department of Medicine in Illinois, USA.
Rida Assaf is a PhD student in the Department of Computer Science at the University of Chicago in Illinois, USA.
Ramy Karam Aziz is a Professor and Acting Chair at the Department of Microbiology and Immunology, Faculty of Pharmacy, Cairo University, Cairo Egypt. His research focuses on microbial and viral genomics and metagenomics.
Thomas S. Brettin is a Strategic Program Manager for Computing and Life Sciences within the Computing, Environmental and Biological Sciences Directorate at Argonne National Laboratory in Illinois, USA.
Christopher Bun has a PhD degree in Computational Biology in the Department of Computer Science, University of Chicago in Illinois, USA.
Neal Conrad is a Software Engineering Associate at Argonne National Laboratory and the University of Chicago Computation Institute who specializes in Web application development and user experience for bioinformatics.
James J. Davis is a Computational Biologist at Argonne National Laboratory and the University of Chicago Computation Institute in Illinois, USA.
Emily M. Dietrich is a Coordinating Writer/Editor at Argonne National Laboratory and a joint appointment at the University of Chicago Computation Institute in Illinois, USA.
Terrence Disz, PhD, is a Bioinformatics Software Specialist at the Fellowship for Interpretation of Genomes in Illinois, USA.
Svetlana Gerdes, PhD, is a Comparative Genomics Specialist at the Fellowship for Interpretation of Genomes in Illinois, USA.
Ron Kenyon is a Project Director at the Biocomplexity Institute of Virginia Tech, Blacksburg, Virginia, USA.
Dustin Machi is a Senior Software Architect at the Biocomplexity Institute of Virginia Tech, Blacksburg, Virginia, USA.
Chunhong Mao is a Research Assistant Professor at the Biocomplexity Institute of Virginia Tech, Blacksburg, Virginia, USA.
Daniel E. Murphy-Olson is a Cloud Services Team Lead at Argonne National Laboratory and Joint Staff at the University of Chicago Computation Institute in Illinois, USA.
Eric K. Nordberg is a Research Scientist and Software Engineer with the Biocomplexity Institute of Virginia Tech, Blacksburg, Virginia, USA.
Gary J. Olsen is a Microbiologist with a particular interest in comparative genome analysis at the University of Illinois at Urbana-Champaign in Illinois, USA.
Robert Olson is a Senior Software Engineer in the Computing, Environment and Life Sciences Directorate of Argonne National Laboratory and the Computation Institute at the University of Chicago, in Illinois, USA.
Ross Overbeek is a Founding Fellow of the Fellowship to Interpret Genomes, as well as Senior Computational Scientist at the Computation Institute, University of Chicago, in Illinois, USA.
Bruce Parrello is a Research Professional in the Computing, Environment, and Life Sciences Division at Argonne National Laboratory in Illinois, USA.
Gordon D. Pusch has a PhD degree in Physics. He is a member of the Fellowship for Interpretation of Genomes, and is a codeveloper and co-maintainer of the SEED and RAST genome annotation systems.
John Santerre is a PhD candidate in Machine Learning in the Department of Computer Science, University of Chicago in Illinois, USA.
Maulik Shukla is a Senior Software Engineer, Computing in the Environment and Life Sciences, Argonne National Laboratory in Illinois, USA.
Rick L. Stevens is the Associate Laboratory Director for Computing, Environment and Life Sciences Directorate at Argonne National Laboratory and Professor of Computer Science in the Computation Institute at the University of Chicago in Illinois, USA.
Margo Van Oeffelen is a Technical Assistant at the Fellowship for Interpretation of Genomes.
Veronika Vonstein, PhD, is a Founding Fellow and President of the Fellowship for Interpretation of Genomes.
Andrew S. Warren is a Senior Software Architect at the Biocomplexity Institute of Virginia Tech, Blacksburg, Virginia, USA.
Alice R. Wattam is a Research Assistant Professor at the Biocomplexity Institute of Virginia Tech, Blacksburg, Virginia, USA.
Fangfang Xia is a Computer Scientist in the Computing, Environment and Life Sciences Directorate of Argonne National Laboratory and a Research Fellow at Computation Institute of the University of Chicago in Illinois, USA.
Hyunseung Yoo is a Software Engineer at Argonne National Laboratory and the University of Chicago Computation Institute in Illinois, USA.
Funding
The NIAID, National Institutes of Health, Department of Health and Human Services (grant number HHSN272201400027C to R.L.S.).
References
- 1. Wattam AR, Davis JJ, Assaf R, et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res 2017;45:D535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Greene JM, Collins F, Lefkowitz EJ, et al. National Institute of Allergy and Infectious Diseases bioinformatics resource centers: new assets for pathogen informatics. Infect Immun 2007;75:3212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aziz RK, Bartels D, Best AA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 2008;9:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brettin T, Davis JJ, Disz T, et al. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep 2015;5:8365.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davis JJ, Gerdes S, Olsen GJ, et al. PATtyFams: protein families for the microbial genomes in the PATRIC database. Front Microbiol 2016;7:118.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clark K, Karsch-Mizrachi I, Lipman DJ, et al. GenBank. Nucleic Acids Res 2016;44:D67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benson DA, Cavanaugh M, Clark K, et al. GenBank. Nucleic Acids Res 2017;45:D37.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. O'Leary NA, Wright MW, Brister JR, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 2016;44:D733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. World Health Organization. Antimicrobial Resistance. Draft Global Action Plan on Antimicrobial Resistance. Geneva: WHO, 2015. [Google Scholar]
- 10. Eurosurveillance Editorial Team. WHO member states adopt global action plan on antimicrobial resistance. Euro Surveill 2015;20. [PubMed] [Google Scholar]
- 11. Fauci AS, Collins FS. New strategies in battle against antibiotic resistance. https://directorsblog.nih.gov/2014/09/18/new-strategies-in-battle-against-antibiotic-resistance/.
- 12. Roca I, Akova M, Baquero F, et al. The global threat of antimicrobial resistance: science for intervention. New Microbes New Infect 2015;6:22–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen L. Notes from the field: pan-resistant New Delhi metallo-beta-lactamase-producing Klebsiella pneumoniae—Washoe County, Nevada, 2016. MMWR Morb Mortal Wkly Rep 2017;66:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Centers for Disease Control and Prevention. Antibiotic resistance threats in the United States, 2013. Centres for Disease Control and Prevention, US Department of Health and Human Services, 2013.
- 15. Barrett T, Clark K, Gevorgyan R, et al. BioProject and BioSample databases at NCBI: facilitating capture and organization of metadata. Nucleic Acids Res 2012;40:D57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davis JJ, Boisvert S, Brettin T, et al. Antimicrobial resistance prediction in PATRIC and RAST. Sci Rep 2016;6:27930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kodama Y, Shumway M, Leinonen R.. The Sequence Read Archive: explosive growth of sequencing data. Nucleic Acids Res 2012;40:D54–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leinonen R, Akhtar R, Birney E, et al. The European Nucleotide Archive. Nucleic Acids Res 2010;gkq967.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012;19:455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. European Committee on Antimicrobial Susceptibility Testing. EUCAST guidelines for detection of resistance mechanisms and specific resistances of clinical and/or epidemiological importance. EUCAST, Basel, Switzerland, 2013. http://www/eucast. org/clinical_breakpoints.
- 21. Patel J, Cockerill F, Alder J, et al. Performance standards for antimicrobial susceptibility testing; twenty-fourth informational supplement In: CLSI Standards for Antimicrobial Susceptibility Testing. Clinical and Laboratory Standards Institute, Wayne, PA, vol. 34, 2014, 1–226. [Google Scholar]
- 22. Bradley P, Gordon NC, Walker TM, et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat Commun 2015;6:10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Drouin A, Giguère S, Sagatovich V, et al. Learning interpretable models of phenotypes from whole genome sequences with the Set Covering Machine. arXiv, preprint arXiv:1412.1074 [q-bio.GN], 2014. [Google Scholar]
- 24. Santerre JW, Davis JJ, Xia F, et al. Machine learning for antimicrobial resistance. arXiv:1607.01224 [stat.ML], 2016. [Google Scholar]
- 25. Freund Y, Schapire RE, A decision-theoretic generalization of on-line learning and an application to boosting. In: European Conference on Computational Learning Theory Springer, 1995, 23–37.
- 26. Freund Y, Schapire R, Abe N.. A short introduction to boosting. J Jpn Soc Artif Intell 1999;14:1612. [Google Scholar]
- 27. Long SW, Olsen RJ, Eager TN, et al. Population genomic analysis of 1,777 extended-spectrum beta-lactamase producing Klebsiella pneumoniae, Houston, Texas: unexpected abundance of clonal group 307. mBio, vol. 8, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Overbeek R, Olson R, Pusch GD, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 2014;42:D206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Levy SB, McMurry LM, Barbosa TM, et al. Nomenclature for new tetracycline resistance determinants. Antimicrob Agents Chemother 1999;43:1523–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chopra I, Roberts M.. Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev 2001;65:232–60. second page, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bush K, Pazkill T, Jacoby J. ß-lactamase classification and amino acid sequences for TEM, SHV and OXA extended-spectrum and inhibitor resistant enzymes. http://www.lahey.org/Studies/.
- 32. Thai QK, Bös F, Pleiss J.. The lactamase engineering database: a critical survey of TEM sequences in public databases. BMC Genomics 2009;10:390.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fischer M, Thai QK, Grieb M, et al. DWARF–a data warehouse system for analyzing protein families. BMC Bioinformatics 2006;7:495.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pasteur I. Klebsiella sequence typing. http://bigsdb.pasteur.fr/klebsiella/klebsiella.html.
- 35. McArthur AG, Waglechner N, Nizam F, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother 2013;57:3348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.NCBI. Bacterial antimicrobial resistance reference gene database, 2017. https://www.ncbi.nlm.nih.gov/bioproject/?term=3130472017.
- 37. Zankari E, Hasman H, Cosentino S, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 2012;67:2640–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boratyn GM, Camacho C, Cooper PS, et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res 2013;41:W29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Madden T. The BLAST sequence analysis tool. In: The NCBI Handbook [Internet], 2nd ed., National Center for Biotechnology Information, Bethesda, MD, 2013. [Google Scholar]
- 40. Haft DH, Selengut JD, Richter RA, et al. TIGRFAMs and genome properties in 2013. Nucleic Acids Res 2013;41:D387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun J, Deng Z, Yan A.. Bacterial multidrug efflux pumps: mechanisms, physiology and pharmacological exploitations. Biochem Biophys Res Commun 2014;453:254–67. [DOI] [PubMed] [Google Scholar]
- 42. Roberts MC, Sutcliffe J, Courvalin P, et al. Nomenclature for macrolide and macrolide-lincosamide-streptogramin B resistance determinants. Antimicrob Agents Chemother 1999;43:2823–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Furnham N, Garavelli JS, Apweiler R, et al. Missing in action: enzyme functional annotations in biological databases. Nat Chem Biol 2009;5:521–5. [DOI] [PubMed] [Google Scholar]
- 44. Schnoes AM, Brown SD, Dodevski I, et al. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol 2009;5:e1000605.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ramirez MS, Tolmasky ME.. Aminoglycoside modifying enzymes. Drug Resist Updat 2010;13:151–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shaw KJ, Rather PN, Hare RS, et al. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol Rev 1993;57:138–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van Hoek AH, Mevius D, Guerra B, et al. Acquired antibiotic resistance genes: an overview. Front Microbiol 2011;2:203.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Roberts MC. Update on macrolide-lincosamide-streptogramin, ketolide, and oxazolidinone resistance genes. FEMS Microbiol Lett 2008;282:147–59. [DOI] [PubMed] [Google Scholar]
- 49. Roberts MC. Nomenclature for tetracycline genes/nomenclature center for MLS genes, 2017. http://faculty.washington.edu/marilynr/2017.
- 50. Kim S, Thiessen PA, Bolton EE, et al. PubChem substance and compound databases. Nucleic Acids Res 2015;4:44.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Federhen S. Type material in the NCBI taxonomy database. Nucleic Acids Res 2015;43:D1086–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maness MJ, Sparling PF.. Multiple antibiotic resistance due to a single mutation in Neisseria gonorrhoeae. J Infect Dis 1973;128:321–30. [DOI] [PubMed] [Google Scholar]
- 53. Mac Aogain M, Kilkenny S, Walsh C, et al. Identification of a novel mutation at the primary dimer interface of GyrA conferring fluoroquinolone resistance in Clostridium difficile. J Glob Antimicrob Resist 2015;3:295–9. [DOI] [PubMed] [Google Scholar]
- 54. Santos-Lopez A, Bernabe-Balas C, Ares-Arroyo M, et al. A naturally occurring single nucleotide polymorphism in a multicopy plasmid produces a reversible increase in antibiotic resistance. Antimicrob Agents Chemother 2017;61:e01735–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Martinez J, Baquero F.. Mutation frequencies and antibiotic resistance. Antimicrob Agents Chemother 2000;44:1771–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Suzuki S, Horinouchi T, Furusawa C.. Prediction of antibiotic resistance by gene expression profiles. Nat Commun 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Buels R, Yao E, Diesh CM, et al. JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol 2016;17:66.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.