

Abstract
Testicular germ cell tumors (TGCTs) are classified into two main subtypes, seminoma (SE) and non-seminoma (NSE), but their molecular distinctions remain largely unexplored. Here, we used expression data for mRNAs and microRNAs (miRNAs) from The Cancer Genome Atlas (TCGA) to perform a systematic investigation to explain the different telomere length (TL) features between NSE (n = 48) and SE (n = 55). We found that TL elongation was dominant in NSE, whereas TL shortening prevailed in SE. We further showed that both mRNA and miRNA expression profiles could clearly distinguish these two subtypes. Notably, four telomere-related genes (TelGenes) showed significantly higher expression and positively correlated with telomere elongation in NSE than SE: three telomerase activity-related genes (TERT, WRAP53 and MYC) and an independent telomerase activity gene (ZSCAN4). We also found that the expression of genes encoding Yamanaka factors was positively correlated with telomere lengthening in NSE. Among them, SOX2 and MYC were highly expressed in NSE versus SE, while POU5F1 and KLF4 had the opposite patterns. These results suggested that enhanced expression of both TelGenes (TERT, WRAP53, MYC and ZSCAN4) and Yamanaka factors might induce telomere elongation in NSE. Conversely, the relative lack of telomerase activation and low expression of independent telomerase activity pathway during cell division may be contributed to telomere shortening in SE. Taken together, our results revealed the potential molecular profiles and regulatory roles involving the TL difference between NSE and SE, and provided a better molecular understanding of this complex disease.
Keywords: TGCT, seminoma, non-seminoma, telomere length, expression
Introduction
Testicular germ cell tumor (TGCT) is one of the most common malignancies among young men in many countries [1]. Histologically, TGCTs are divided into two major subtypes, seminoma (SE) and non-seminoma (NSE). In fact, 15% of TGCTs are mixed tumors that contain both SE and NSE elements [2]. In terms of cell origin, NSE is considered to originate from earlier gonadal stem cells, and pure SE is derived from later gonadal stem cells [3]. Compared with SE in the same stage, NSE is more likely to metastasize and leads to poorer prognosis [4]. The difference in etiology between SE and NSE has been well studied, and their treatment methods also differ. During the past decade, the survival rate of TGCT patients has increased through treatments such as surgery, radiation therapy and chemotherapy [5–7]. However, ∼20% of the patients show incomplete response or tumor relapse after chemotherapy, and ∼15% of the patients suffer from refractory disease resulting in a poor prognosis [8]. Therefore, a better understanding of the subtype-specific molecular mechanisms will facilitate the development of better treatment options.
Telomeres, which make up the ends of chromosomes, are composed of the repetitive DNA sequence (TTAGGG)n and bound proteins. Telomere is known as playing essential roles for the stable inheritance in eukaryotic chromosomes [9, 10]. Telomeres shorten with each round of cell division. This mechanism limits proliferation of cells to a finite number of cell divisions and induces replicative senescence, differentiation or apoptosis. Telomerase activity is absent in most normal somatic tissues, whereas human cancer cells typically express high level of telomerase [11, 12]. Telomerase is extensively overexpressed in 80–90% of all malignancies, and its activity plays a key role in the maintenance of telomeres (chromosome end structures) and cancer cell immortality in most human malignancies [13]. Telomerase reverse transcriptase (TERT) is a catalytic subunit of the enzyme telomerase. It comprises the most important unit of the telomerase complex with the telomerase RNA component (TERC) [14]. In the differentiated healthy human cells, telomerase is silent because of the transcriptional repression of the TERT, but it is activated during oncogenesis [12]. Approximately 10–15% of human tumors lack telomerase activity, and maintain telomere lengths (TLs) by the telomerase-independent mechanism alternative lengthening of telomeres (ALT) [15]. A recent study has characterized TERT and genomic alterations with TL using The Cancer Genome Atlas (TCGA) samples, and generated TL data for 18 430 samples across 31 cancer types [16]. They reported that a high rate of relatively longer TLs is observed in TGCT (52%) and almost TGCT samples expressed TERT [16]. However, the molecular mechanisms underlying this finding remain unclear, and further it remains unknown whether there is some difference between the two TGCT subtypes regarding the TL-associated features.
In this study, we characterized the distinct TL features of NSE and SE at the molecular level. We observed that TL elongation was dominant in NSE, whereas TL shortening prevailed in SE compared with patient-matched normal samples. We further identified differentially expressed genes (DEGs) and pathways between the two subtypes, identifying contributing TL-associated genes, and analyzed their regulatory networks. Finally, we proposed the potential TL-associated mechanisms. This study represents the systematic investigation of TL-related molecular signatures in TGCT, which provides a better molecular understanding of this disease and facilitates the development of TGCT subtype-specific therapeutic strategy.
Materials and methods
Data sets for TGCT
We downloaded TCGA Level-3 data including clinical data (version 2016.04.27), normalized expression data for mRNA (version 2016.8.16; Platform: IlluminaHiSeq_RNASeqV2) and microRNA (miRNA) (version 2017.07.24; Platform: IlluminaHiSeq_miRNA) expression data from TCGA data portal (https://xenabrowser.net/datapages/). Both the mRNA and miRNA data were generated by Illumina next-generation sequencing platform (HiSeq 2000). Telomere length (TL) information, which calculated by TelSeq software [17] from whole-exome sequencing (WES) data, was downloaded from a previous study [16]. TL ratio was defined as tumor TL/normal TL (tTL/nTL), following Barthel et al. (2017) [16], i.e. the tTL divided by the nTL. Here, the normal samples are from the blood of each patient. Based on clinical data of TGCT and TL information, we excluded the samples without subtype or TL information, and only used primary tumor samples from patients of age between 18 and 45 years to reduce age factor. To reduce molecular noise between NSE and SE, we only used samples from patients diagnosed only with either NSE or SE. As a result, we used 103 of 156 patients in this study, including 48 NSE and 55 SE patients (Supplementary Table S1). In the TL comparison, we used the pair-matched TL information. For molecular comparison in gene and miRNA expression, we only used TGCT tumor samples because of the lack of normal sample data in TCGA. Workflow of the data analysis is illustrated in Supplementary Figure S1.
Telomere-related genes from public resource
A total of 168 unique telomere-related genes (TelGenes) were collected from multiple resources, including MSigDB [18] (version 5.2), TelomeraseDB [19] (April 2013) and published reports (Supplementary Table S2). The TelGenes were manually separated into two groups based on annotation in resource database or published reports: Group-A, TelGenes contained telomerase activity-related genes (68 genes), and Group-B, TelGenes included telomere-related pathways and TL-related genes (100 genes after removing those overlapped with Group-A TelGenes).
mRNA and miRNA analysis between NSE and SE
In the preprocessing step of mRNA analysis, non-expressed genes, which defined as having a log2-transformed RNASeq by expectation maximization (RSEM) expression level of <1 in >50% of samples, were removed [20]. Differential expression was analyzed based on comparing NSE with SE using a linear mixed model and Bayesian t-tests in R package limma. Subtype-differentially expressed genes (subtype-DEGs) were identified with |log2fold change| > 0.585 [i.e. NSE fold change (FC) > 1.5 or SE FC > 1.5] and adjusted P-value (the false discovery rate, FDR) < 0.05.
For the preprocessing of miRNA data, we removed miRNAs with a missing value in > 10% of the samples, and retained miRNAs expression level with > 10 reads per million (RPM) [log2(RPM + 1) > 3.46] in >10% samples [21]. Differentially expressed miRNAs between NSE and SE was identified using limma in the R package, and subtype-differential expression (subtype-DE) miRNAs were identified with |log2FC| > 0.585 and FDR < 0.05.
Gene set enrichment analysis
We used WebGestalt [22] (version 2017.01.27) to examine the enriched KEGG pathways for subtype-DEGs, and required the pathway size with 10–500 genes. The significance level of pathways was set to FDR (Benjamini–Hochberg) < 0.05. For the candidates of TL-associated genes, we executed Reactome Analyze Data tool (http://reactome.org/PathwayBrowser/#TOOL=AT) to define related pathways based on Reactome pathway database (version 61), and pathways with FDR (Benjamini–Hochberg) < 0.05 were considered significant [23].
Correlation analysis between TL ratio and gene expression
To detect TL-associated genes, we used the Spearman's rank correlation coefficient (ρ) to calculate the correlation between TL ratio and gene expression in SE and NSE, respectively. In calculation correlation, genes expression with a value of 0 following log2(RSEM + 1) were set to the missing value. The threshold for the association was set to |ρ| > 0.3 and P-value < 0.05.
Gene regulation network analysis
We built a network using TL-associated genes (|ρ| > 0.3 and P-value < 0.05) and the predicted regulators (TFs and miRNAs), which belonged to subtype-DEGs and subtype-DE miRNAs (|log2FC| > 0.585 and FDR < 0.05). The TF and target information were obtained from TRANSFAC [24] database (version 2016.04) and TRRUST [25] (version 2014.06.24). Based on Spearman's rank correlation coefficient between TFs and target genes, we filtered low correlated TF–target (|ρ| 0.3 and P-value ≥ 0.05; TF–target). To predict miRNAs for target genes, we used TargetScanHuman [26] (version 7.1) and miRBase [27] (release 21) data source, and defined miRNA–target pair with ρ < −0.3 and P-value < 0.05 [28]. In the network, we only used TFs and miRNAs, which, respectively, belonged to subtype-DEGs and subtype-DE miRNAs. Cytoscape [29] (version 3.5.2) was used for network analysis.
Results
Systematic TL difference between NSE and SE
We investigated the TL distribution of 48 NSE and 55 SE patients. The TL information, which was measured by TelSeq from WES data, was obtained from a recent study [16]. For each patient, two TL measures were obtained: one from the normal tissue (referred as nTL), and the other from tumor tissue (referred as tTL) [16] (see ‘Materials and methods’ section; Supplementary Table S1). We further defined the TL ratio as tTL/nTL for each sample. As shown in Figure 1A, we observed no significant difference in nTL between patients with the two subtypes, while tTL and TL ratio were significantly different (Wilcoxon test, P-value < 1.0 × 10−10, Figure 1A and B). In NSE, ∼79% samples exhibited TL elongation (tTL > nTL), while ∼81% of SE patients had TL shortening (tTL < nTL) (Figure 1C). It revealed that the majority of NSE tumors showed TL elongation, whereas TL shortening was dominant in SE tumors. This is consistent with a recent study using fluorescence in situ hybridization [30]. Therefore, the result of TL distribution, which were lengthening in NSE and shortening in SE, is consistent regardless of measurement technique.
Figure 1.

Distinct TL alteration and molecular signatures in two subtypes of TGCT: NSE and SE. (A) Distinct TL between NSE and SE. TL in NSE tumor samples was significantly longer than SE (P-value = 2.42 × 10−13, Wilcoxon test); TL difference is not significant in matched normal samples. (B) TL ratio (tTL/matched nTL) was significantly stronger in NSE than in SE. (C) Distribution of NSE and SE samples by TL. Each dot represents one sample. X-axis indicates the TL in normal tissues from each sample, while Y-axis indicates the TL in tumor tissues from each sample. The samples above the diagonal indicate TL elongation (tTL > nTL), while below the diagonal for TL shortening (tTL < nTL). (D) PCA of mRNA for TGCT samples. The two subtypes were clearly separated. (E) Proportion of subtype-DEGs between the two subtypes. (F) PCA of miRNA for TGCT samples. (G) Proportion of subtype-differential miRNA expression. (H) PCA using mRNA expression of 168 TelGenes (see ‘Material and methods’ section for details) for TGCT samples. (I) Expression of three key TelGenes ATRX, DAXX and TERT in NSE and SE. *: FC > 1.5 (NSE > SE) or FC < 0.67 (1/1.5 = 0.67, NSE < SE) and P-value < 0.05.
To gain clinical insights, we examined the association between TL alteration and clinical stages from I to III. We found Spearman's rank correlation coefficient (ρ) is higher in SE than that in NSE (Spearman’s rank test, |ρ| of SE: 0.28, |ρ| of NSE: 0.01), and TL alteration in SE might be associated with clinical stages. In the comparison between Stages I and III, the TL alteration showed statistically significant difference (t-test, P-value < 0.05) in SE, but not in NSE (Supplementary Figure S2A). For the TERT gene expression, there was no significant difference between clinical stages regarding TL alteration in either NSE or SE (Supplementary Figure S2B).
Distinct gene expression patterns between NSE and SE
To investigate expression differences between NSE and SE, we analyzed subtype-DE between two subtypes of TGCT using collected mRNA and miRNA data from TCGA. As TCGA does not include expression data for normal matched samples for TGCT, we only used tumor samples to perform systematic comparisons between NSE and SE. First, to explore the expression profile of each subtype, we performed principal component analysis (PCA) using expression profiles of mRNAs and miRNAs, respectively. As shown in Figure 1D, both expression of mRNA and miRNA successfully separated the two subtypes based on the first and second principal components (Figure 1D and F). Next, we analyzed subtype-DEGs between NSE and SE. A total of 8263 significant subtype-DEGs were identified (∼47% of 17 635 genes; FDR < 0.05 and |log2FC| > 0.585), of which 4658 genes were highly expressed in NSE and 3605 genes were highly expressed in SE (Figure 1E). Similarly, 311 miRNAs (∼59% of 526 miRNAs; FDR < 0.05 and |log2FC| > 0.585) were differentially expressed between NSE and SE, of which 207 miRNAs were highly expressed in NSE and 104 miRNAs were highly expressed in SE (Figure 1G).
To better understand the expression profile of TelGenes in the two subtypes of TGCT, we collected 168 TelGenes from public resources as described in Supplementary Table S2 (see ‘Material and methods’ section). Initial analysis using the TelGenes in PCA showed that the gene expression of TelGenes also clearly separated NSE and SE samples (Figure 1H). In addition, the TERT and ALT-related genes (ATRX and DAXX) also showed different expression patterns between NSE and SE. Of those, TERT and ATRX showed higher expression in NSE than in SE, while DAXX expression showed a reversed pattern (Figure 1I). This gene expression analysis suggested that the different expression profile might contribute to the TL alteration for the two subtypes of TGCT.
Subtype-DEGs and TelGenes
To understand whether the enriched functions of the subtype-DEGs are related to TL features, we used WebGestalt to find KEGG pathways that enriched with subtype-DEGs (FDR < 0.05). The highly expressed genes in NSE were highly enriched in cellular community-related pathways (e.g. ‘Focal adhesion’, ‘Signaling pathways regulating pluripotency of stem cells’, ‘Gap junction’ and ‘Tight junction’), and signaling and interaction pathways (e.g. ‘ECM-receptor interaction’, ‘Hippo signaling pathway’, ‘PI3K-Akt signaling pathway’, ‘TGF-beta signaling pathway’, ‘Neuroactive ligand-receptor interaction’, ‘Wnt signaling pathway’ and ‘Rap1 signaling pathway’) (Figure 2A and Supplementary Table S3). The Wnt signaling plays crucial roles in stem cell and telomerase activity [31]. Hence, highly expressed genes (e.g. CTNNB1 and MYC) within Wnt signaling pathway might enhance telomerase activity and lead telomere elongation in NSE. In contrast, the highly expressed genes in SE were significantly highly enriched in immune related pathways (e.g. ‘Primary immunodeficiency’, ‘Systemic lupus erythematosus’, ‘T cell receptor signaling pathway’, ‘Natural killer cell mediated cytotoxicity’ and ‘Hematopoietic cell lineage’) (Figure 2B and Supplementary Table S3). This enrichment of the immune pathways in SE is supported by a previous study that a high degree of immune cell infiltration (tumor infiltrating lymphocytes) is often present in SEs [32].
Figure 2.

Gene set enrichment of subtype-DEGs and overlap with TelGenes. (A) Top 10 significantly enriched KEGG pathways in NSE [adjusted P-value (FDR) < 0.05]. (B) Top 10 significantly enriched pathways in SE (FDR < 0.05). Full results are available in Supplementary Table S3. (C) Volcano plot of telomerase activity-related genes (Group-A TelGenes) expression. The Group-A TelGenes, which were also subtype-DEGs, were highlighted in green boxes. Blue dots: highly expressed subtype-DEGs in SE. Red dots: highly expressed subtype-DEGs in NSE. (D) Volcano plot of Group-B TelGene (telomere-related pathways and TL-related genes that removing overlap with Group-A TelGenes) expression. The Group-B TelGenes, which were also subtype-DEGs, were highlighted in white boxes.
We hypothesized that telomerase activity-related genes would highly express in NSE because the enhanced telomerase activity induces TL elongation in cancer. To test our hypothesis, we first separated 168 TelGenes into two groups based on function: Group-A included telomerase activity-related genes (n = 68) based on their function described in relevant articles (Supplementary Table S2); Group-B included telomere-related pathways and TL-related genes (n = 100). Then, the two groups of TelGenes were compared with subtype-DEGs. Among the 68 Group-A TelGenes, 26 were subtype-DEGs, including 17 highly expressed genes in NSE and 9 highly expressed genes in SE (Figure 2C). Especially, four genes (CTNNB1, H19, MYC and WRAP53), which are of well-known importance to increase telomere activity and regulate TERT expression, were highly expressed in NSE. Of them, H19 was expressed extremely higher in NSE than SE (FC = 14.7 and FDR = 1.25 × 10−20). Among the 100 Group-B TelGenes, 37 belonged to subtype-DEGs, including 27 and 10 highly expressed genes in NSE and SE, respectively. Interestingly, we observed 21 histone-related genes (H2A1, H2B1 and H41 family genes, Figure 2D) highly expressed in SE, while only two such genes were found in NSE. Notably, we found a key gene in telomerase-independent telomere elongation, zinc finger and SCAN domain (ZSCAN4) [33], and TERF1 was significantly highly expressed (FC > 3 and FDR < 1 − 10−10) in NSE (Figure 2D). In the cancer cell, ZSCAN4 indirectly interacts with TERF1 and functions in regulation of telomere elongation independent of telomerase activity [34]. These high expressed genes might contribute to the TL elongation feature in NSE.
Identification TL-associated candidate genes that related to subtype-DEGs and TelGenes
To define TL-associated genes, we performed a correlation analysis between gene expression and TL using Spearman's rank correlation coefficient (ρ) method. As a result, we found 2185 and 2844 genes associated with TL (|ρ| > 0.3 and P-value < 0.05) in NSE and SE, respectively (Figure 3A and B). Of them, 643 and 519 genes belonged to the subtype-DEGs and TL-associated genes in NSE and SE, respectively. The 643 genes in NSE were further categorized as positively correlated (relative TL elongation, 344 genes) and negatively correlated (relative TL shortening, 299 genes) with TL. Similarly, the 519 genes in SE included 144 genes associated with TL elongation and 375 with TL shortening (see ‘Materials and methods’ section). To study the effect of TelGenes on the candidate genes (643 and 519 genes) in NSE and SE, we overlapped 168 TelGenes with 643 and 519 genes in NSE and SE, respectively. We found that 11 TelGenes overlapped with the candidate genes. Of these, only four TelGenes (AURKB, SSB, TERT and WRAP53) belonged to 344 relative TL elongation genes in NSE (TL-E-NSE genes), while only seven TelGenes (HIST1H2AC, HIST1H2BE, HIST1H2BF, HIST1H2BJ, HIST1H2BK, HIST1H2BO and TCP1) belonged to 375 related TL shortening genes in SE (TL-S-SE genes). In addition, we did not detect any TelGenes within other gene groups in NSE and SE (e.g. related TL shortening in NSE or related TL elongation in SE) (Figure 3C and D).
Figure 3.

Venn diagram and TL-associated genes. (A) Venn diagram of subtype-DEGs and positively correlated with TL genes. SE > NSE: higher expression genes in SE. NSE > SE: higher expression genes in NSE. SE: cor_TL_pos: gene expression positively correlated with TL genes in SE. NSE: cor_TL_pos: gene expression positively correlated with TL genes in NSE. Red and blue boxes refer to the highly expressed subtype-DEGs that positively correlated with TL genes in NSE and in SE, respectively. (B) Venn diagram of subtype-DEGs and negatively correlated with TL genes. SE: cor_TL_neg: gene expression negatively correlated with TL genes in SE. NSE: cor_TL_neg: gene expression negatively correlated with TL genes in NSE. Red and blue boxes refer to the highly expressed subtype-DEGs that negatively correlated with TL genes in NSE and in SE, respectively. (C) Spearman’s correlation (ρ) between subtype-DEGs and TL in NSE. Dots in red indicated the subtype-DEGs that associated with TL in NSE (Spearman's rank correlation coefficient |ρ| > 0.3, P-value < 0.05). Four TelGenes were labeled in green (Group-A TelGenes). X-axis is for gene expression and Y-axis for correlation value. (D) Spearman’s correlation between subtype-DEGs and TL in SE. Dots in blue indicated the subtype-DEGs that associated with TL in SE (|ρ| > 0.3, P-value < 0.05). Seven TelGenes were labeled in green (Group-A TelGenes) or white boxes (Group-B TelGenes).
To understand whether biologically enriched functions relate to TL between TL-E-NSE and TL-S-SE genes, which defined in this study and included 11 TelGenes, we performed functional enrichment analysis using the Reactome database. The TL-E-NSE genes were significantly enriched (FDR < 0.05) with some pathways including ‘Response to metal ions’, ‘Heat shock factor 1 (HSF1)-dependent transactivation’, ‘Pyruvate metabolism’ and ‘The citric acid (TCA) cycle and respiratory electron transport’ (Table 1). In addition, TERT and WRAP53 were included in the ‘Telomere extension by telomerase’ pathway, which was not included in the significantly enriched pathways because only two genes were low power to detect by statistics (data not shown). On the other hand, TL-S-SE genes were significantly enriched with histone-related pathways including ‘DNA methylation’, ‘Packaging of DNA repair’ and ‘Chromatin modifying enzymes’ (Table 1). We found that the histone-related genes also significantly enriched in ‘DNA damage/Telomere stress induced senescence’, ‘DNA double strand break response’, ‘Oxidative stress induced senescence’ and many other pathways (data not shown). We also confirmed enriched pathways using TL shortening associated genes of NSE (299 genes) and TL elongation associated genes of SE (144 genes), but there were no overlapped pathways compared with TL-E-NSE and TL-S-SE genes, respectively. These enriched pathway annotation results may further explain the reason why TL elongation was dominant in NSE than in SE, and TL shortening prevailed in SE.
Table 1.
Pathways enriched with telomere elongation associated genes
| Gene group | Pathway name a | q-value b | # TL-associated genes | # Total genes | TL-associated genes in pathway |
|---|---|---|---|---|---|
| TL elongation- associated genes in NSEc | Response to metal ions | 1.12E-04 | 8 | 21 | MT2A, MT1M, MT1F, MT1G, MT1X, MT1H, MT1E |
| HSF1-dependent transactivation | 0.013 | 9 | 59 | HSPB1, CRYBA4, HSPA1A | |
| Pyruvate metabolism | 0.013 | 8 | 47 | LDHA, PDHA1, GLO1, PDK3, SLC16A3 | |
| TL shortening associated genes in SEd | The TCA cycle and respiratory electron transport | 0.013 | 18 | 219 | NDUFA9, COX7B, PDHA1, NDUFA4, NDUFB11, TACO1, GLO1, ATP5G1, ATP5B, LDHA, NDUFS5, PDK3, SUCLG1, SLC16A3, SLC25A14 |
| DNA methylation | 8.30E-07 | 12 | 37 | HIST1H2BO, HIST1H2BJ, HIST1H2BK, HIST1H2BF, HIST1H2BE, HIST1H2AC, HIST1H3D, HIST1H3E | |
| Packaging of telomere ends | 8.81E-06 | 10 | 34 | HIST1H2BO, HIST1H2BJ, HIST1H2BK, HIST1H2BF, HIST1H2BE, HIST1H2AC | |
| Chromatin modifying enzymes | 0.0024 | 20 | 257 | NCOA2, HIST1H2BO, KDM3B, HIST1H2BJ, HIST1H2BK, SMARCA2, RBBP5, KIAA1267, HIST1H2BF, HIST1H2BE, TBL1X, HIST1H2AC, HIST1H3D, HIST1H3E | |
| Activation of nicotinic acetylcholine receptors | 0.0289 | 4 | 17 | CHRNB4, CHRNA5, CHRNA4, CHRNA7 |
Reactome Analysis Data tool was used for gene set enrichment analysis.
q-value was calculated using the P-value from binomial test after Benjamini–Hochberg’s multiple test correction.
Telomere elongation-associated genes refer to those having positive correlation between TL alteration and gene expression in NSE.
Telomere shortening associated genes refer to those having negative correlation between TL alteration and gene expression in SE.
Regulation network analysis and identification key miRNAs from TL-associated genes
Among the TL-E-NSE and TL-S-SE, 11 TelGenes have been reported and showed the related telomere stability or TL in different studies. To better illustrate the TL alteration patterns and difference between NSE and SE within the 11 TelGenes, we drew scatter plots based on the TL and gene expression value, and observed different patterns between NSE and SE (Figure 4A). TERT and AURKB had positive correlation both of NSE and SE; however, almost nine TelGenes showed opposite patterns.
Figure 4.

Comparison of correlation and regulator networks between NSE and SE within 11 TelGenes. (A) Spearman's rank correlation coefficient between TL alteration and expression of 11 TelGenes in NSE and SE. Four highly expressed TelGenes in NSE versus SE, and seven highly expressed TelGenes in SE versus NSE. X-axis is for gene expression and Y-axis for log(TL ratio). TL ratio: tTL/nTL. For each gene, a linear regression model was fitted using log(TL ratio) versus its gene expression in NSE (red) and SE (blue). P: P-value. For each gene in each subtype, the fitted lines are displayed in NSE (red) and SE (blue), and the shaded area indicates 95% confidence interval. (B) Network of TFs and miRNAs for TelGenes between NSE and SE. Defined the TFs–target (Spearman's rank correlation coefficient |ρ| > 0.3) and miRNA–target (ρ < −0.3) pairs with P-value < 0.05, |log2FC| > 0.585 and FDR < 0.05. Edges in green: negative correlation; edges in orange: positive correlation; and edge thickness is proportional to the correlation value. Box in orange: higher expression genes or miRNAs in NSE or SE. Box in green: low expression genes or miRNAs in NSE or SE.
To understand the regulation of 11 TelGenes by transcription factors (TFs) or miRNAs, we constructed the TF–target and miRNA–target regulatory networks using the integration of information from three databases, TRANSFAC [24], TRRUST [25] and TargetScan [26]. To select subtype-DEG-related TFs and subtype-DE miRNAs in each subtype, we filtered out the TFs and miRNAs, which were neither subtype-DEGs nor subtype-DE miRNAs. We also filtered out TF–targets and miRNA–targets with low correlation coefficient (see ‘Materials and methods’ section). With this approach, we constructed a TF–target and miRNA–target network, and presented the 11 TelGenes, which displayed genes in Figure 4A. Among the 11 TelGenes, we found that only 6 genes (AURKB, TERT, HIST1H2BE, HIST1H2BF, HIST1H2BO and TCP1) had inferred regulators (Figure 4B). Furthermore, we observed that the regulators for the same target genes showed different quantity and correlation between NSE and SE. For example, TERT was regulated by 13 TFs and 4 miRNAs in NSE, while only 3 TFs regulated TERT in SE; MYC was positively correlated with TERT expression in NSE, but it was negatively correlated in SE; 61 miRNAs showed negative correlation with three target genes (HIST1H2BE, HIST1H2BO and TCP1) in NSE, but 22 miRNAs were related with these three genes in SE.
To identify potential key miRNAs within telomere lengthening and telomere shortening in NSE and SE, respectively, we sorted miRNAs by quantity of target genes and selected the top one as key miRNA in TL-E-NSE and TL-S-SE genes, respectively. We found that miR-181c and miR-24 were key miRNA candidates, and they may be important for posttranscriptional regulation of gene expression in NSE and SE, respectively (Supplementary Figures S3 and S4). In addition, we observed that the quantity of miRNAs for the same target genes was especially different between NSE and SE in the miRNA–target network (Supplementary Figures S3 and S4), which may partly explain the different TLs between these two subtypes.
A proposed model for TL difference between NSE and SE
From our study results and the evidence by the previous studies, we proposed a mechanistic model for TL elongation and shortening in NSE and SE, respectively (Figure 5). In our analysis, highly expressed genes in NSE were significantly enriched (FDR < 0.05) in ‘Signaling pathways regulating pluripotency of stem cells’ pathways among the KEGG pathways (Supplementary Table S3). Moreover, gene expression of TERT and WRAP53, which is included in the ‘Telomere extension by telomerase’ pathway, had a positive correlation with TL in NSE. Previous reports showed that TERT expression correlated with telomerase activity [35], and WRAP53 controlled telomerase [36]. Specifically, ZSCAN4 promotes genomic stability during reprogramming in induced pluripotent stem (iPS) cells [37], and indirectly interacts with TERF1 and functions in regulation of telomere elongation independent of telomerase activity in cancer cells [34]. In this study, we found that the expression of ZSCAN4 and TERF1 had high positive correlation (ρ > 0.6 and P-value < 0.05) with TERT expression, and had low positive correlation (0.1 < ρ < 0.3) with TL only in NSE. Based on ZSCAN4 function, we also confirmed reprogramming factors (Supplementary Figure S5). Among the four known reprogramming factors [Yamanaka factors: OCT4 (POU5F1), SOX2, KLF4 and MYC], which function in reprogramming of pluripotent stem cells [38], SOX2 and MYC were highly expressed (FC > 1.5 and FDR < 0.05) in NSE compared with SE. Of these, MYC can induce telomerase activation through TERT expression [35]. In contrast, POU5F1 and KLF4 were highly expressed in SE versus NSE (Supplementary Figure S5). In the TL association with gene expression, four reprogramming genes had positive correlation with TL elongation in NSE, while only SOX2 and MYC had positive correlation in SE (Supplementary Figure S5). This result suggests that enhanced expression of reprogramming genes has more contribution in NSE than in SE during TL elongation. Thus, synergetic high expression pattern of ZSCAN4, reprogramming factors and telomerase may be important for influencing TL elongation in NSE.
Figure 5.
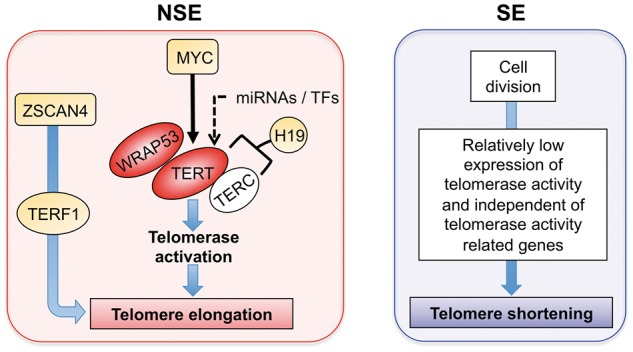
A proposed model for explaining the potential molecular mechanisms of TL regulation in NSE and SE. Red oval: DEG that was positively correlated with TL (ρ > 0.3). Orange: subtype-DEG.
Discussion
Our results showed that TL and gene expression features were significantly different between NSE and SE. As expected, telomerase activity-related genes were highly expressed in NSE compared with SE because TL elongation dominated in NSE. Interestingly, we revealed that immune-related pathways were significantly enriched and histone-related genes were highly expressed in SE. Histone-related genes feature prominently in many pathways such as DNA damage, cell senescence and DNA methylation. These results might suggest that telomeres were unstable in a number of patients with SE. In the regulation network, we found the regulators were different between the two subtypes, especially a number of miRNAs. This different regulation may in part explain the different TL distribution between NSE and SE.
Telomerase is active in germ cell cancers [39]. Telomerase activity and telomere maintenance are correlated with the immortality of cancer cells, germ cells and embryonic stem cells (ES cells) [40]. In our analysis, the expression levels of two genes that encode the essential components of active telomerase, TERT and WRAP53 (also known as TCAB1), were more highly expressed in NSE than in SE, and their expression was positively correlated with TL in NSE as well. WRAP53 controls telomerase trafficking and is required for telomere synthesis in human cancer cells [36]. Thus, the highly expressed TERT and WRAP53 may be an important factor for telomerase activation and TL lengthening in NSE. Recent report showed that a high rate of relatively longer TLs in TGCT (52%), and TERT was expressed in >90% samples in TGCT [16]. These results implied that TERT expression might be not a deciding factor for TL elongation in TGCT.
NSE is thought to originate from earlier gonadal stem cells, and tends to metastasize to various tissue sites [3]. Gene set enrichment analysis showed that highly expressed genes in NSE were enriched in genes that are involved in stem cell and Wnt signaling pathways (Supplementary Table S3). Moreover, Wnt/β-catenin signaling can lead to an enhanced TERT expression in stem cells and cancer cells, which results in the stabilization of telomeres [31]. In NSE, CTNNB1 encoding β-catenin was highly expressed compared with SE (FC = 2.3 and FDR < 6.4 × 10−15), which implies that the high expression of CTNNB1 can lead to an enhanced TERT expression in NSE. Our hypothesis that more stem cell properties may result in the stabilization of telomeres by telomere elongation in NSE.
On the other hand, the elevated expression of MYC was another important clue to TL lengthening in NSE. TERT is a direct target of MYC, and upregulation of MYC is closely correlated with activation of TERT and telomerase [41]. In our study, the expression of MYC was positively correlated with TERT in NSE. In addition, our analysis revealed that other telomerase activity- or TL-related genes were also significantly highly expressed and might contribute TL elongation in NSE, i.e. H19, ZSCAN4 and TERF1. Overexpression of H19 has been shown to increase the binding of TERT to TERC, thus leading to enhanced telomerase activity and telomere lengthening [42]. The expression of H19 was significantly high in NSE, showing >8-fold higher expression than in SE. ZSCAN4 regulates telomere elongation and genomic stability during reprogramming in ES cell. However, it is not associated with increased telomerase activity [33, 37]. Furthermore, ZSCAN4 indirectly interacts with TERF1 and functions in regulation of telomere elongation independent of telomerase activity in cancer cells [34].
MiRNAs function as key regulators of gene expression in various cancers. In the miRNA–target network analysis, miR-181c was the most important for regulating TL elongation in NSE based on the number of target genes. The previous functional analysis showed that miR-181c inhibition increased cell proliferation, migration and invasion [43]. Therefore, a low level of miR-181c expression may be one of the reasons for TL elongation and cell proliferation in NSE.
Shortening of telomeres is associated with each round of cell division because of the inability of conventional DNA polymerases to replicate the ends of chromosomes [44]. TERF1 exerts a protective function against the DNA damage response at telomeres, and that facilitates the replication of telomeric DNA [45]. Histones function as chaperones during DNA replication and repair [46]. Our study showed that histone-related genes were significantly highly expressed in SE, and these genes are related with cellular senescence pathways, including ‘DNA Damage/Telomere Stress Induced Senescence’, ‘Oxidative Stress Induced Senescence’ and ‘Cellular Senescence’ in Reactome pathways. Moreover, the expression of TL elongation-related genes (e.g. TERT, MYC, TERF1 and ZSCAN4) was lower in SE than in NSE. Therefore, telomere shortening in SE may be associated with low expression of TL elongation-related genes during continual proliferation of cancer cells.
Previous studies revealed that high TERT expression, high telomerase activity or both had been correlated with poor prognosis in several cancer types such as colorectal cancer [47], non-small cell lung cancer [48, 49] and breast cancer [50]. Specifically, telomere shortening is associated with poor prognosis, and telomerase activity correlates with DNA repair impairment in non-small cell lung cancer [49]. Furthermore, abnormal telomere elongation is common and also correlates with advanced stages and/or poor survival in some cancers [51–53]. Our study found significant significance of TL alteration between the Stages I and III in SE. However, difference in TERT expression was not significant between the stages of SE (Supplementary Figure S2). In NSE, neither TL alteration nor TERT expression was significantly different between stages.
This study has some limitations. First, we used molecular data only from tumor samples because omics data of normal samples are not available in the public TCGA data resource. Second, our analysis only focused on the two main subtypes of TGCT because sample size in NSE subtypes is too small to further compare. Third, the overall sample size (n = 103) was not large enough to investigate the confounding effect of age in TL. Thus, further studies with a larger number of samples are required to compare tumor tissue with normal tissue using omics approaches and elucidate the effects of various factors on TL alteration, such as age, tumor stage, clinical outcome and more detailed subtypes of TGCT. Fourth, our analysis is primary based on correlation analysis, but correlations do not imply causality. Finally, the observed difference in mRNA and miRNA expression, the gene signatures, as well as their pathways and networks, tended to be false positives because such results were based on genome-wide data analyses. The experimental verification and further functional studies should be performed to make the findings more reliable and their mechanisms more insightful. These results are not immediately useful for clinical practice either. Hence, the results in this study should be interpreted with caution.
In conclusion, our work demonstrates distinct TL pattern and molecular difference between NSE and SE using expression data set, and different expression of TelGenes might be attributed to the significantly different TL features. This study revealed that enhanced expression of both telomere-related genes (e.g. TERT, WRAP53, MYC and ZSCAN4), and Yamanaka factors might induce telomere elongation in NSE. Additionally, the miR-181c might be an important regulator of expression of TL elongation-associated genes in NSE. On the other hand, the relatively low expression of telomerase and other genes that are independent of telomerase activity during continual cell division may lead to TL shortening in SE. Our approach and findings may be helpful to explain the characterization of metastasis and poor prognosis in NSE, and may provide a basis for better prognosis and treatment of TGCT.
Key Points.
We observed that TL elongation was dominant in NSE, whereas TL shortening prevailed in SE when compared with patient-matched normal samples.
Near half of mRNAs and micoRNAs showed significantly differential expression between the two subtypes of TGCT, and telomerase activity-related genes were significantly highly expressed in NSE than in SE.
In the pathway enrichment, stem cell pathway and metabolism-related pathways were enriched in NSE, while immune-related pathways were significantly enriched in SE.
NSE showed significantly higher expression of ZSCAN4 and telomerase activity-related genes (TERT, WRAP53 and MYC) than SE, and these genes were positively correlated with TL elongation in NSE. In addition, the expression of Yamanaka factors was positively correlated with telomere elongation in NSE.
miR-181c might be an important regulator of expression of TL elongation-associated genes in NSE.
Supplementary Material
Acknowledgements
The authors thank Dr Guangchun Han for technical support in R package and Dr Mingyu Shao for downloading TCGA data. The authors also thank Dr Irmgard Willcockson for proofing English of an earlier version of the manuscript. The authors also thank the Data Science and Informatics Core for Cancer Research supported by CPRIT (grant number RP170668).
Hua Sun is a postdoctoral fellow in the Bioinformatics and Systems Medicine Laboratory (BSML), Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston.
Pora Kim is a postdoctoral fellow in the Bioinformatics and Systems Medicine Laboratory (BSML), Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston.
Peilin Jia is an assistant professor in Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston.
Ae Kyung Park is a visiting scientist in the Bioinformatics and Systems Medicine Laboratory (BSML), Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston. She is an Associate Professor in College of Pharmacy, Sunchon National University, Republic of Korea.
Han Liang is an associate professor in Department of Bioinformatics and Computational Biology and Department of Systems Biology, the University of Texas MD Anderson Cancer Center.
Zhongming Zhao is a professor in the Center for Precision Health, School of Biomedical Informatics, the University of Texas Health Science Center at Houston. He directs the Bioinformatics and Systems Medicine Laboratory (BSML).
Funding
This work was partially supported by National Institutes of Health (grant numbers R01LM012806, R01LM011177, R21CA196508, R01A174586 and U24A209851).
References
- 1. Valberg M, Grotmol T, Tretli S, et al. . A hierarchical frailty model for familial testicular germ-cell tumors. Am J Epidemiol 2014;179(4):499–506. [DOI] [PubMed] [Google Scholar]
- 2. Horwich A, Shipley J, Huddart R.. Testicular germ-cell cancer. Lancet 2006;367(9512):754–65. [DOI] [PubMed] [Google Scholar]
- 3. Tu SM, Lin SH, Logothetis CJ.. Stem-cell origin of metastasis and heterogeneity in solid tumours. Lancet Oncol 2002;3(8):508–13. [DOI] [PubMed] [Google Scholar]
- 4. Kobayashi K, Saito T, Kitamura Y, et al. . Oncological outcomes in patients with stage I testicular seminoma and nonseminoma: pathological risk factors for relapse and feasibility of surveillance after orchiectomy. Diagn Pathol 2013;8(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Houldsworth J, Korkola JE, Bosl GJ, et al. . Biology and genetics of adult male germ cell tumors. J Clin Oncol 2006;24(35):5512–8. [DOI] [PubMed] [Google Scholar]
- 6. Lopes LF, Macedo CR, Pontes EM, et al. . Cisplatin and etoposide in childhood germ cell tumor: brazilian pediatric oncology society protocol GCT-91. J Clin Oncol 2009;27(8):1297–303. [DOI] [PubMed] [Google Scholar]
- 7. Lopes LF, Macedo CR, Aguiar Sdos S, et al. . Lowered cisplatin dose and no bleomycin in the treatment of pediatric germ cell tumors: results of the GCT-99 protocol from the Brazilian germ cell pediatric oncology cooperative group. J Clin Oncol 2016;34(6):603–10. [DOI] [PubMed] [Google Scholar]
- 8. International Prognostic Factors Study Group, Lorch A, Beyer J, et al. . Prognostic factors in patients with metastatic germ cell tumors who experienced treatment failure with cisplatin-based first-line chemotherapy. J Clin Oncol 2010;28:4906–11. [DOI] [PubMed] [Google Scholar]
- 9. Blackburn EH. The molecular structure of centromeres and telomeres. Annu Rev Biochem 1984;53:163–94. [DOI] [PubMed] [Google Scholar]
- 10. O'Sullivan RJ, Karlseder J.. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol 2010;11:171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shay JW, Wright WE.. Role of telomeres and telomerase in cancer. Semin Cancer Biol 2011;21(6):349–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim NW, Piatyszek MA, Prowse KR, et al. . Specific association of human telomerase activity with immortal cells and cancer. Science 1994;266(5193):2011–5. [DOI] [PubMed] [Google Scholar]
- 13. Shay JW, Bacchetti S.. A survey of telomerase activity in human cancer. Eur J Cancer 1997;33(5):787–91. [DOI] [PubMed] [Google Scholar]
- 14. Weinrich SL, Pruzan R, Ma L, et al. . Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat Genet 1997;17(4):498–502. [DOI] [PubMed] [Google Scholar]
- 15. Heaphy CM, Subhawong AP, Hong SM, et al. . Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol 2011;179(4):1608–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barthel FP, Wei W, Tang M, et al. . Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet 2017;49(3):349–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ding Z, Mangino M, Aviv A, et al. . Estimating telomere length from whole genome sequence data. Nucleic Acids Res 2014;42(9):e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Subramanian A, Tamayo P, Mootha VK, et al. . Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Podlevsky JD, Bley CJ, Omana RV, et al. . The telomerase database. Nucleic Acids Res 2008;36:D339–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Werling DM, Parikshak NN, Geschwind DH.. Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nat Commun 2016;7:10717.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lim EL, Trinh DL, Scott DW, et al. . Comprehensive miRNA sequence analysis reveals survival differences in diffuse large B-cell lymphoma patients. Genome Biol 2015;16(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang B, Kirov S, Snoddy J.. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 2005;33:W741–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao J, Cheng F, Zhao Z.. Tissue-specific signaling networks rewired by major somatic mutations in human cancer revealed by proteome-wide discovery. Cancer Res 2017;77(11):2810–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wingender E, Dietze P, Karas H, et al. . TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res 1996;24(1):238–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Han H, Shim H, Shin D, et al. . TRRUST: a reference database of human transcriptional regulatory interactions. Sci Rep 2015;5(1):11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Agarwal V, Bell GW, Nam JW, et al. . Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015;4:e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kozomara A, Griffiths-Jones S.. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 2014;42(D1):D68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang W, Jia P, Hutchinson KE, et al. . Clinically relevant genes and regulatory pathways associated with NRASQ61 mutations in melanoma through an integrative genomics approach. Oncotarget 2015;6:2496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shannon P, Markiel A, Ozier O, et al. . Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13(11):2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shekhani MT, Barber JR, Bezerra SM, et al. . High-resolution telomere fluorescence in situ hybridization reveals intriguing anomalies in germ cell tumors. Hum Pathol 2016;54:106–12. [DOI] [PubMed] [Google Scholar]
- 31. Hoffmeyer K, Raggioli A, Rudloff S, et al. . Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science 2012;336(6088):1549–54. [DOI] [PubMed] [Google Scholar]
- 32. Boublikova L, Buchler T, Stary J, et al. . Molecular biology of testicular germ cell tumors: unique features awaiting clinical application. Crit Rev Oncol Hematol 2014;89(3):366–85. [DOI] [PubMed] [Google Scholar]
- 33. Zalzman M, Falco G, Sharova LV, et al. . Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature 2010;464(7290):858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee K, Gollahon LS.. ZSCAN4 and TRF1: a functionally indirect interaction in cancer cells independent of telomerase activity. Biochem Biophys Res Commun 2015;466(4):644–9. [DOI] [PubMed] [Google Scholar]
- 35. Fernandez PC, Frank SR, Wang L, et al. . Genomic targets of the human c-Myc protein. Genes Dev 2003;17(9):1115–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Venteicher AS, Abreu EB, Meng Z, et al. . A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 2009;323(5914):644–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang J, Lv W, Ye X, et al. . Zscan4 promotes genomic stability during reprogramming and dramatically improves the quality of iPS cells as demonstrated by tetraploid complementation. Cell Res 2013;23(1):92–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takahashi K, Tanabe K, Ohnuki M, et al. . Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131(5):861–72. [DOI] [PubMed] [Google Scholar]
- 39. Albanell J, Bosl GJ, Reuter VE, et al. . Telomerase activity in germ cell cancers and mature teratomas. J Natl Cancer Inst 1999;91(15):1321–6. [DOI] [PubMed] [Google Scholar]
- 40. Hiyama E, Hiyama K.. Telomere and telomerase in stem cells. Br J Cancer 2007;96(7):1020–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sagawa Y, Nishi H, Isaka K, et al. . The correlation of TERT expression with c-myc expression in cervical cancer. Cancer Lett 2001;168(1):45–50. [DOI] [PubMed] [Google Scholar]
- 42. Pu H, Zheng Q, Li H, et al. . CUDR promotes liver cancer stem cell growth through upregulating TERT and C-Myc. Oncotarget 2015;6(38):40775–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li Y, Wang H, Li J, et al. . MiR-181c modulates the proliferation, migration, and invasion of neuroblastoma cells by targeting Smad7. Acta Biochim Biophys Sin 2014;46(1):48–55. [DOI] [PubMed] [Google Scholar]
- 44. Greider CW, Blackburn EH.. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985;43(2 Pt 1):405–13. [DOI] [PubMed] [Google Scholar]
- 45. Donate LE, Blasco MA.. Telomeres in cancer and ageing. Philos Trans R Soc Lond B Biol Sci 2011;366(1561):76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ransom M, Dennehey BK, Tyler JK.. Chaperoning histones during DNA replication and repair. Cell 2010;140(2):183–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tatsumoto N, Hiyama E, Murakami Y, et al. . High telomerase activity is an independent prognostic indicator of poor outcome in colorectal cancer. Clin Cancer Res 2000;6:2696–701. [PubMed] [Google Scholar]
- 48. Wang L, Soria JC, Kemp BL, et al. . hTERT expression is a prognostic factor of survival in patients with stage I non-small cell lung cancer. Clin Cancer Res 2002;8:2883–9. [PubMed] [Google Scholar]
- 49. Frias C, Garcia-Aranda C, De Juan C, et al. . Telomere shortening is associated with poor prognosis and telomerase activity correlates with DNA repair impairment in non-small cell lung cancer. Lung Cancer 2008;60(3):416–25. [DOI] [PubMed] [Google Scholar]
- 50. Kirkpatrick KL, Clark G, Ghilchick M, et al. . hTERT mRNA expression correlates with telomerase activity in human breast cancer. Eur J Surg Oncol 2003;29(4):321–6. [DOI] [PubMed] [Google Scholar]
- 51. Oh BK, Kim H, Park YN, et al. . High telomerase activity and long telomeres in advanced hepatocellular carcinomas with poor prognosis. Lab Invest 2008;88(2):144–52. [DOI] [PubMed] [Google Scholar]
- 52. Gertler R, Doll D, Maak M, et al. . Telomere length and telomerase subunits as diagnostic and prognostic biomarkers in Barrett carcinoma. Cancer 2008;112(10):2173–80. [DOI] [PubMed] [Google Scholar]
- 53. Gertler R, Rosenberg R, Stricker D, et al. . Telomere length and human telomerase reverse transcriptase expression as markers for progression and prognosis of colorectal carcinoma. J Clin Oncol 2004;22(10):1807–14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.