

Abstract
For the past 20 years, the Clusters of Orthologous Genes (COG) database had been a popular tool for microbial genome annotation and comparative genomics. Initially created for the purpose of evolutionary classification of protein families, the COG have been used, apart from straightforward functional annotation of sequenced genomes, for such tasks as (i) unification of genome annotation in groups of related organisms; (ii) identification of missing and/or undetected genes in complete microbial genomes; (iii) analysis of genomic neighborhoods, in many cases allowing prediction of novel functional systems; (iv) analysis of metabolic pathways and prediction of alternative forms of enzymes; (v) comparison of organisms by COG functional categories; and (vi) prioritization of targets for structural and functional characterization. Here we review the principles of the COG approach and discuss its key advantages and drawbacks in microbial genome analysis.
Keywords: comparative genomics, genome annotation, enzyme evolution, orthologs, paralogs
Introduction
The success of the entire genomic enterprise critically depends on reliable genome annotation, i.e. correct identification of the genes, which includes accurate determination of gene boundaries and functional annotation of the gene product(s). The Clusters of Orthologous Groups of proteins (COGs) database has been devised as a way to allow phylogenetic classification of proteins from complete microbial genomes [1]. While the COG system has grown over the years (Figure 1), the goal has always been for each COG to represent a family of orthologous protein-coding genes. However, when the compared genomes are separated by long evolutionary distances and possess substantially different numbers of genes, evolutionary relationships between these genes are not accurately captured by the straightforward definition of orthology as a one-to-one relationship because of such evolutionary processes as lineage-specific gene duplication and loss, as well as horizontal gene transfer [7, 8]. Owing to these complexities of the evolutionary relationships among genes, the COGs have become families of co-orthologous genes that embody one-to-many and many-to-many relationships. Hence the term ‘orthologous groups’ (of proteins) that embraces such more complex evolutionary relationships among genes and simplifies the assignment of (general) functions to genes and their products. As the genomic community gradually embraced the notion of co-orthologous relationships between genes [7–9], the COGs have been re-branded Clusters of Orthologous Genes [10].
Figure 1.

Evolution of the COG system. The numbers in parentheses indicate the number of bacterial, archaeal and eukaryotic genomes, respectively, included in the respective COG release [1–6].
During the 20 years since the inception of the COG project, several alternative systems for orthology analysis have been developed [11–20], some of them implementing genome-wide phylogenetic analysis, which, in principle, is supposed to provide robust resolution of evolutionary relationships between orthologs and paralogs. In practice, however, such methods are computationally expensive and fraught with artifacts at different stages, and therefore, simpler approaches such as the COGs continue to be widely used in microbial genomics. The popular EggNOG database (‘Evolutionary genealogy of genes: Non-supervised Orthologous Groups’, http://eggnog.embl.de) applies essentially the same approach as COGs to a much greater number of genomes, but fully relies on automated assignment of orthologs and does not annotate the orthologous gene clusters [21, 22].
Here we briefly review the key principles underlying the COG approach and its applications for genome annotation and comparative analysis. Rather than providing a detailed description of the COG construction methods and the resulting collections of (co)orthologous gene families, our goal here is to highlight the unresolved problems in functional annotation and the possible ways to address them. For a description of the COG database per se, the reader is referred to the previous publications [1, 2–6].
Differences between COGs and other collections of gene and protein families
Functional annotation of proteins encoded in sequenced genomes typically relies on BLASTP [23] or, more recently, HMMer [24] search of protein databases for the most similar sequence, followed by a (semi-)automated transfer of the best hit annotation to the new protein. This approach has a number of well-known drawbacks [25–28]. First, if the sequence similarity is low, there is a distinct possibility that the two proteins have different functions; this problem is exacerbated in cases of transitive annotation of multiple proteins in this manner. Second, the reliance on the best hit often results in a protein ending up being annotated as ‘uncharacterized’ and/or ‘putative’ even when the function of a close homolog is already known. Third, differences in domain architectures of homologous proteins often result in erroneous functional assignment. Given these systematic errors, advanced approaches for functional annotation of proteins increasingly rely on curated databases of protein sequences [29], such as UniProt KnowledgeBase or PANTHER [30, 31], and protein domains, such as Pfam, SMART or SUPERFAMILY [32–34]. Aggregated domain databases InterPro and CDD, which allow an easy comparison of the annotations provided by various databases, often prove to be the most efficient tools [35, 36]. The COG approach shares some features with the curated protein family databases but differs from them in several important aspects.
Use of complete genomes
A distinct feature of the COG approach is the reliance on complete genome (proteome) sequences, which allows relatively simple and reliable recognition of potential orthologs and paralogs among all proteins encoded in the given genome. With incomplete genomes, there always remains the obvious possibility that the true ortholog of the given gene failed to make it into the final assembly. Like other methods for ortholog identification, the COG approach relies on sequence similarity searches against selected proteomes, aimed at the identification of pairwise best hits. However, instead of imposing predetermined similarity scores for delineation of likely homologs, the COG approach extends the popular concept of two-way (often also called bidirectional, symmetric or reciprocal) best BLAST hits in each particular proteome by adding the more stringent requirement of forming a triangle, or three-way set of best BLAST matches (thus forcing the mathematical property of transitivity [7, 9]) to form a new COG. Owing to the presence of potential paralogs from the same lineage (inparalogs [37]), the original approach [1] only required that at least one such triangle be included that represented symmetrical (bidirectional) matches, with that criteria being imposed by manual supervision of groups initially constructed with an automated method. Later, the process of detection and collapsing such obvious paralogs was performed by an automated method, introduced in the first major update of the COGs [3] and later codified in the EdgeSearch algorithm [38–40]. Proteins from new genomes can be added to the existing COGs by using the new sequences as queries for an RPS-BLAST search of the collection of position-specific scoring matrices generated from COG-specific multiple sequence alignments [41]. The query is assigned to the COG that yields the best score in this search. Technically, this approach is analogous to that used to search domain databases, such as InterPro and CDD, but because the COGs contain previously identified orthologs, in this case, the best hit gives a strong indication of orthology. A detailed discussion of other methods for ortholog identification can be found e.g. in [7, 9, 42–45]. In addition to sequence similarity and phylogenetic proximity, a potentially useful criterion is genomic synteny [39, 40], which, however, in practice is typically used for manual verification of the existing assignments at the quality-control stage.
Flexible similarity cutoffs
The advantage of the triangle-based approach for orthology inference is that it dispenses with artificially imposed sequence similarity cutoffs for different protein families, some of which evolve with dramatically different rates, and permits creation of COGs from proteins that span the entire range of similarity, from barely detectable to extremely high. For example, Na+-binding c subunits (COG0636) of Na+-translocating ATP synthases from bacteria and archaea have low sequence similarity and might not be recognized as orthologs using arbitrarily high BLAST cutoffs; to further complicate the annotation, archaeal protein is often referred to as subunit K [46]. With strict BLAST cutoffs, recognition of orthology becomes particularly complicated for short proteins, including some ribosomal proteins. The COG approach also allows separation of closely related paralogs, such as, for example, 3-isopropylmalate dehydrogenase (LeuB) and isocitrate dehydrogenase (Icd), members of COG0473 and COG0538, respectively, that in most other databases are assigned to the same family (PF00180 in Pfam, SM01329 in SMART, PS00470 in PROSITE, SSF53659 in SUPERFAMILY).
Protein family granularity in COGs
Flexible similarity cutoffs have the built-in advantage of allowing the COGs to be as wide or as narrow as dictated by the evolutionary history of a given gene family. In the above example, the LeuB/Icd family is split into two COGs, which reflects the wide distribution of these enzymes among bacteria and archaea. However, this family also includes even two more closely related enzymes. One of these is tartrate dehydrogenase/decarboxylase that has been characterized in Pseudomonas putida and Agrobacterium vitis [47, 48]. This enzyme is closely related to LeuB, still has the isopropylmalate dehydrogenase activity and has probably evolved from LeuB in the course of the adaptation of the host bacteria to life on tartrate-rich grapevine [47]. The fourth member is homoisocitrate dehydrogenase AksF, which participates in the biosynthesis of the methanoarchaeal coenzyme B [49]. Homoisocitrate dehydrogenase has been described in Methanocaldococcus jannaschii, and a variety of methanogenic archaea encode closely related proteins [49]. At this time, there are too few tartrate dehydrogenases to form a separate COG. As for homoisocitrate dehydrogenase, LeuB and AksF are co-orthologs with respect to the bacterial LeuB enzymes. Accordingly, all members of this family are currently assigned to the same COG0473 (LeuB) and the same arCOG01163 in archaeal COGs [10]. In the future, methanogenic homoisocitrate dehydrogenases might form an archaea-specific COG. For now, however, the split of the family into two COGs appears to represent a reasonable compromise. In contrast, TIGRfams [50] and NCBI Protein Clusters [51] databases divide this family into 6 and 13 clusters, respectively. However, because sequence similarity alone does not allow unequivocal functional assignment, most of these clusters end up with the same functional annotation, either LeuB or Icd.
Phyletic profiles in COGs
An important feature of the COG approach is that a protein (or domain) either belongs or does not belong to it. Accordingly, a genome is either represented in the given COG (by one or more proteins) or it is not. Thus, the COG approach can dispense with the matrix of similarity scores and replace them with the simple yes/no (1 or 0) representation or, alternatively, indicate the number of paralogous members of the given COG in the given genome. Such phyletic patterns, i.e. the patterns of species that are either represented or not represented in the given COGs, are a powerful tool for functional annotation of microbial genomes and evolutionary reconstruction. The most obvious use of phyletic patterns is for identification of supposedly essential genes that are missing in certain genomes [4, 52]. Consistent application of this principle offers an easy way to evaluate genome quality [53, 54], which is why the NCBI’s prokaryotic genome annotation pipeline currently involves routine checking of the submitted genomes for the presence of certain (nearly) universal genes, including those encoding ribosomal proteins and translation system components, as well as RNA polymerase subunits [55, 56]. A conceptually similar application of phyletic patters involves analysis of metabolic pathways and multi-protein functional systems. Obviously, metabolic pathways should not allow accumulation of any intermediate that cannot be further metabolized and represents a dead end: to avoid poisoning the cell, such intermediate would have to be exported into the surrounding milieu. Likewise, an intermediate in the functional metabolic pathway needs to be either imported or synthesized within the cell. Although the possibility of ‘distributed’ pathways cannot be discarded, these simple considerations prove productive when COGs are superimposed on the metabolic map to identify the intermediates that have no known enzymes to produce or metabolize them. Identification of such gaps in pathways often suggests alternative enzymes that can be then identified experimentally [54, 57].
Functional categories of genes in COGs
Another widely used feature of the COG system is the assignment of all COGs to one of the 26 functional categories. These categories have evolved over time, with several of them (B, Y, W, Z) describing functions that are found primarily in eukaryotic cells. The recently added V (Defense mechanisms) and X (Mobilome) categories provide for a more detailed description of the dynamics of bacterial and archaeal genomes. Functional categories are assigned in accordance with the cellular roles of the respective COGs, so that, for example, peptide uptake systems are included into category E (Amino acid transport and metabolism), rather than in general ‘Transport’ or other similar categories. Two functional categories of uncharacterized proteins, R (genes with only a generic functional prediction, typically of the biochemical activity) and S (uncharacterized genes), are particularly useful, as they reflect the current level of understanding of protein function on the proteome level and allow tracing the progress in experimental characterization and computational analysis of widespread protein families. The fraction of proteins from a given genome assigned to certain COG functional categories turned out to be a useful whole-genome feature [58] and has been adopted by the Genome Standards Consortium as an essential characteristic of the newly sequenced genomes https://standardsingenomics.biomedcentral.com/submission-guidelines.
Full-length proteins and domains as COG members
Most existing protein family databases include either full-length sequences (NCBI protein database, UniProt, PANTHER, TIGRfams [30, 31]) or separate protein domains (Pfam, SMART, SUPERFAMILY, etc [32–34]). The COG approach allows a degree of flexibility: conserved domain combinations can be included in separate COGs without the need to split them into individual domains. As an example, along with the COG0784 for individual CheY-like receiver (REC) domains of the two-component signal transduction systems (which also includes stand-alone CheY/Spo0F proteins), the current COG collection includes 15 additional REC-domain COGs, such as COG2197 for DNA-binding response regulators of the NarL/FixJ family, containing REC and helix-turn-helix domains; COG0745 for DNA-binding response regulators of the OmpR/PhoB family, which consist of REC and winged-helix domains; COG3279 for DNA-binding response regulators of LytR/AlgR family, containing REC and LytTR domains, and many others [59, 60]. The discrimination between the architectures of proteins that share a common domain provides for a finer granularity of annotation and allows better characterization of the respective proteins. However, non-critical use of COGs for high-throughput domain annotation can result in egregious errors, whereby a multidomain protein receives a misleading annotation of its best COG hit that has a completely different domain architecture. The recent attempts to identify specific domain architectures and limit annotation transfer to proteins with the same domain combination [36] have the potential to resolve this issue.
COG annotation
Functional annotation of COGs, including assignment of COG names, is based on two key principles. First, reliance on orthologous relationships for the COG construction makes it likely, according to the ‘orthology conjecture’, that members of each COG have equivalent functions [7] (with only rare known exceptions [61]). Accordingly, experimentally characterized functions of a single member of a given COG often can be used to assign the functional annotation to the entire COG. Indeed, in most cases, subsequent characterization of additional COG members has confirmed the validity of the initial assignment [6]. Second, all COG names are manually curated with the goal of creating the most appropriate annotation, avoiding the common annotation errors [25], as well as over- and under-predictions. Thus, for those COGs whose members have two or more distinct functions, the annotations (COG names) get expanded to cover the entire range of experimental results. In some cases, the growing number of distinct paralogs justifies splitting a COG into two or more separate COGs with higher sequence conservation and more narrowly defined functional annotation. Many COGs, however, do not include any experimentally characterized members so that their annotation has to rely on computational analyses alone. In such cases, inference of a robust annotation requires careful analysis of their sequences, structures, genomic neighborhoods, phyletic patterns and other cues, which requires a substantial effort that, however, often leads to interesting insights [62, 63]. Such efforts are essential for increasing the fraction of proteins that belong to well-characterized COGs beyond the figure of 60–70% that is currently obtained for most bacterial and archaeal genomes [6]. The overall genome coverage by COGs (including the R- and S-type COGs) has stayed largely the same over the years and currently ranges from ∼65% of the total proteomes in Chlamydiae and Planctomycetes to >80% in Synergistetes and Thermotogae (Figure 2). This stable coverage of bacterial and archaeal genomes by COGs, despite the addition of numerous new genomes, is likely to reflect the open pangenomes of most prokaryotes [65–68] and the extremely rapid turnover of the poorly conserved gene class.
Figure 2.
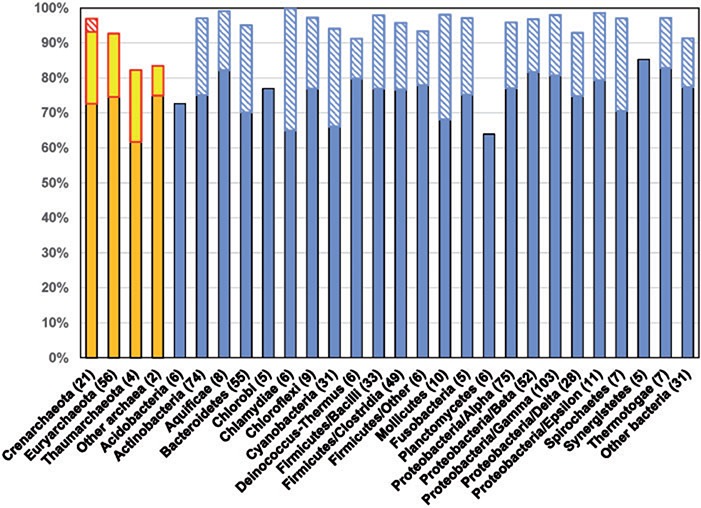
Proteome coverage by the current version of COGs. Archaeal and bacterial phyla and selected classes of Firmicutes and Proteobacteria are listed as in the latest release of the COG database [6]. The orange and blue columns show the fractions of the respective proteomes covered by COGs in each taxonomic group (including R- and S-type COGs that consist of poorly characterized or uncharacterized genes), averaged over the members of that group in the COGs (the respective numbers are shown in parentheses). The ‘Other archaea’ group includes two genomes representing, respectively, Kor- and Nanoarchaeota; the ‘Other bacteria’ group includes members of Deferribacteres, Nitrospirae, Verrucomicrobia and other sparsely sampled phyla, as well as representatives of several candidate phyla. The bright yellow rectangles on top of the archaeal columns indicate the additional coverage of the archaeal proteomes in the latest version of arCOGs [10]. The hatched rectangles indicate the additional coverage of the archaeal and bacterial proteomes in the ATGC-COGs from the latest version of the ATGCs database [64].
Although COG annotations typically describe protein families, in the most recent release of the COG database, owing to the popularity of COG-based annotation, many COG names have been modified to allow functional annotation of individual proteins [6].
Unresolved problems in the COG approach
The wide use of COGs for microbial genome annotation and comparative analysis has illuminated several problems inherent in the COG approach that warrant a brief discussion. These difficulties include, among others, the issues of COG hierarchy, inclusion of paralogs, splitting proteins into separate domains and scalability of the COG approach.
Orthologs, paralogs and xenologs: the missing hierarchy
The very definition of orthology [69] inherently depends on the group of organisms under consideration [7, 9, 37]. For example, in most members of the Crenarchaeota, the family B DNA polymerases are represented by several paralogs which form distinct orthologous families (arCOG00328, arCOG00329, arCOG15272 and others) within this archaeal phylum (all these genes are out-paralogs in Crenarchaeota). In contrast, most of those bacteria that possess the polB gene have a single copy, which is co-orthologous to all archaeal polB genes, so archaea and bacteria share only one orthologous family of polB, COG0417 (all these genes are co-orthologs among prokaryotes with several in-paralogs in archaea). Such complex relationships among homologous genes confound COG analysis because the definition of orthology becomes mutually dependent with the phyletic patterns (the definition of orthology depends on the list of organisms where these genes are present, which itself depends on which of the homologous genes are considered orthologs and which are not). Several formal and informal empirical rules have been proposed to resolve this conundrum [70]. The hierarchical orthologous groups have been implemented in such databases as EggNOG, OMA and OrthoDB [14, 22, 71].
In most of the current COG collections, all COGs are equal, and there is no hierarchical structure; only in arCOGs, an extra level of super-COGs has been introduced to combine paralogous COGs into higher level clusters. Although the non-hierarchical structure of COG collections is convenient for straightforward genome annotation, it has substantial drawbacks. Some COGs include closely related proteins with similar, if not identical, biochemical activities. In such cases, assignment of a protein to a specific COG can be taken, without justification, as an indication that the respective organism possesses one functionality but not the other. A good example is the case of glutamate and glutamine aminoacyl tRNA-synthetases (COG0008). While most bacteria encode two paralogous enzymes that charge the Glu- and Gln-specific tRNAs, archaea (as well as chlamydia, chlorobi, chloroflexi, cyanobacteria and certain members of other bacterial phyla) encode only glutamate-tRNA synthetase and produce glutamyl-tRNA by transamidation of misacylated Glu-tRNAGln [72]. Here, both bacterial paralogs are co-orthologs for the archaeal and chlamydial enzymes, which is why they end up in a single COG. Obviously, splitting COG0008 into two subCOGs would have been a better solution, allowing a precise characterization of the respective enzymes. In some cases, a COG includes a small subgroup with a well-characterized function but the lack of hierarchy results in annotation of generic function only (e.g. an ABC-type transporter).
The single-level definition of orthology can even result in annotations that are largely arbitrary. In some cases (e.g. COG0183, Acetyl-CoA acetyltransferase), COGs are overloaded with paralogs because it is practically impossible to track all extant genes to distinct genes in the common ancestor. On other occasions (COG0050, Translation elongation factor EF-Tu, and COG5256, Translation elongation factor EF-1α), lineage-specific COGs are created for genes that are arguably orthologous because they are sufficiently distinct. The absence of multilevel hierarchy dilutes functional annotation of the characterized members of the COG and weakens the evolutionary reconstructions. Developing and implementing a hierarchical framework is one of the most pressing problems in the COG-based approach to gene classification and genome annotation.
Whole proteins versus protein domains
As noted above, COG construction is based on clustering of orthologous domains that are identified as bidirectional best hits in genome-specific BLAST searches. This approach, however, is sensitive to domain rearrangements that occurred after the divergence of the analyzed set of species from their last common ancestor. Particularly severe problems are caused by promiscuous domains, which can attract proteins to spurious COGs through significant but effectively irrelevant sequence similarity to the promiscuous domains. Although this problem can be addressed semi-automatically, e.g. by excluding the hits that cover only a small portion of the protein sequence, precise solutions still require manual intervention. On many occasions, conserved domain architectures allowed construction of consistent COGs that were not substantially affected by the presence of a shared domain (e.g. the widespread helix-turn-helix DNA-binding domain). Conversely, the diversity of domain architectures of proteins involved in microbial signal transduction and containing a number of promiscuous domains (PAS, GAF, CHASE, GGDEF, EAL and others) required splitting some of these proteins into individual domains or domain combinations. As a result, the COGs are a mix of (i) highly specific domain architectures (such as the above-mentioned response regulators), (ii) multiple domain architectures that include a single shared domain and (iii) separate promiscuous domains. To our knowledge, as of this writing, there is no complete, formal solution for optimal dissection of full-length proteins into orthologous domains. At present, for the analysis of multidomain proteins, the best practical approaches are offered by integrated domain identification tools, such as CDD (which includes the COGs) and InterPro.
Scalability of the COG approach and specialized COG collections
The basic COG approach relies first on an exhaustive all-against-all protein comparison that scales as O(n2) with the total number of proteins and then on a search of connected triangles in clusters of reciprocal best hits that scales as O(n3) with the number of proteins in the cluster [38]. Inevitably, the growth of the database outpaces the availability of the computational resources, making regular major updates of the entire COG database impractical. Several divide-and-conquer strategies have been used to circumvent this major difficulty. One approach that has been implemented in several COG updates includes accommodating the new sequences into the existing COGs first, then searching for potential new COGs among the sequences that do not fit the existing ones, and then, moving some sequences from the old COGs to the new ones [10]. The principal direction, however, has involved construction of dedicated COG collections for distinct microbial taxa. In particular, the COGs for archaea (arCOGs) went through several closely curated releases and remain up to date, having become a widely used framework for archaeal genome annotation and analysis [10, 70, 73]. As illustrated in Figure 2, detailed analysis of archaeal protein families increased the coverage of cren-, eury- and thaumarchaeal genomes by 18–20%, so that arCOGs now cover >92% of the proteins encoded in typical genomes of Crenarchaeota and Euryarchaeota. Separate projects have involved construction and analysis of COGs for Cyanobacteria and Gram-positive bacteria of the order Lactobacillales [74, 75].
The COG approach was also implemented in the database of Alignable Tight Genome Clusters (ATGC) that includes closely related bacterial and archaeal genomes [64, 76]. COGs have been constructed separately for each ATGC. These ATGC-COGs largely avoid the problems inherent in the COG analysis at larger evolutionary distances (lineage-specific paralogy, differential gene loss and differences in domain architectures) and have proved an efficient platform for various types of evolutionary reconstructions [77, 78]. In taxa for which ATGCs are available—i.e. those studied in sufficient depth so that multiple closely related genomes are available—the coverage of genomes is again raised so that ATGC-COGs now cover >95% of the proteins encoded in typical genomes (Figure 2).
The COG approach has also been extended beyond cellular organisms to construct COG for viruses that infect bacteria or archaea, and for the large DNA viruses of eukaryotes [79, 80].
The successful application of the early versions of the COGs was to a large extent based on comprehensive manual curation of the COG membership, COG names and supporting information, and a substantial body of computational analysis aimed at predicting functions for poorly characterized COGs. This effort has led to several notable breakthroughs that have been validated by subsequent experiments and opened up new research directions, including the characterization of the CRISPR-Cas system [81, 82], prediction of the archaeal exosome [83], identification of the bacterial c-di-GMP-centered signaling network [84, 85], new bacterial toxin-antitoxin systems [86–88] and archaeal type IV secretion systems [89], and allowing prioritization of uncharacterized proteins (COGs) for further study [90, 91]. However, scaling this labor-consuming approach to accommodate the exponentially growing amount of genomic sequence data is even more challenging than keeping the COGs up to date. That path forward is likely to combine improved automatic approaches to functional annotation with subprojects focusing on specific taxa or functional classes of COGs.
Concluding remarks
The COG approach for identification of orthologous genes was developed as a platform for comparative genomic analysis shortly after the first few microbial genomes have been sequenced. It could have been expected that in 20 years, this simple strategy based on sequence similarity hierarchy would completely give way to more sophisticated, phylogenetic approaches. This, however, is not the case, primarily, because the extended orthology conjecture, according to which bidirectional best hits between genomes correspond to orthologs, and the latter possess equivalent functions, largely holds for prokaryotes given the limited extent of lineage-specific paralogy, differential gene loss and domain shuffling. In contrast, in eukaryotes where all these confounding aspects of genome evolution are pervasive, the COG approach encounters great difficulties, and robust, genome-wide orthology assignment does not seem to be feasible without full-scale phylogenomics. Thus, the COGs are likely to remain an important tool for microbial genome analysis for years to come, so that investment of effort into refinements of this straightforward approach seems to be justified.
Key Points.
Robust orthology identification is essential for accurate genome annotation.
Reconstructions of genome evolution are based on orthology and paralogy.
COGs are an essential tool in microbial genomics.
Several specialized COG projects have been developed.
Acknowledgments
The authors would like to thank all former members of the COG team for their contributions to the project.
Biographies
Michael Y. Galperin is a Lead Scientist at the NCBI’s (NIH) Computational Biology Branch. He uses comparative genomics to study evolution of membrane energetics and bacterial metabolic and signaling pathways.
David M. Kristensen is an Assistant Professor at the University of Iowa’s Department of Biomedical Engineering. He uses tools of comparative genomics, bioinformatics and systems biology to study evolution of genes in viruses and microbes.
Kira S. Makarova is a Staff Scientist at the NCBI’s Computational Biology Branch. Her area of expertise is comparative genomics and sequence analysis of microbial genomes.
Yuri I. Wolf is a Lead Scientist at the National Center for Biotechnology Information in Bethesda, Maryland. His research is focused on quantitative aspects of evolutionary and comparative genomics.
Eugene V. Koonin is a Senior Investigator and Leader of the Evolutionary Genomics Group at the National Center for Biotechnology Information at the NIH. He studies various aspects of genome evolution.
Funding
The authors are supported by Intramural Research Program of the US National Institutes of Health at the National Library of Medicine. D.M.K. acknowledges the support of the Department of Biomedical Engineering at the University of Iowa (Iowa City, USA).
References
- 1. Tatusov RL, Koonin EV, Lipman DJ.. A genomic perspective on protein families. Science 1997;278:631–7. [DOI] [PubMed] [Google Scholar]
- 2. Koonin EV, Tatusov RL, Galperin MY.. Beyond complete genomes: from sequence to structure and function. Curr Opin Struct Biol 1998;8:355–63. [DOI] [PubMed] [Google Scholar]
- 3. Tatusov RL, Galperin MY, Natale DA, et al. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 2000;28:33–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tatusov RL, Natale DA, Garkavtsev IV, et al. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 2001;29:22–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tatusov RL, Fedorova ND, Jackson JD, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 2003;4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Galperin MY, Makarova KS, Wolf YI, et al. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res 2015;43:D261–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koonin EV. Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet 2005;39:309–38. [DOI] [PubMed] [Google Scholar]
- 8. Gabaldon T, Koonin EV.. Functional and evolutionary implications of gene orthology. Nat Rev Genet 2013;14:360–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kristensen DM, Wolf YI, Mushegian AR, et al. Computational methods for gene orthology inference. Brief Bioinform 2011;12:379–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Makarova KS, Wolf YI, Koonin EV.. Archaeal Clusters of Orthologous Genes (arCOGs): an update and application for analysis of shared features between Thermococcales, Methanococcales, and Methanobacteriales. Life 2015;5:818–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kanehisa M, Sato Y, Kawashima M, et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2016;44:D457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen F, Mackey AJ, Stoeckert CJ Jr, et al. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res 2006;34:D363–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Uchiyama I, Mihara M, Nishide H, et al. MBGD update 2015: microbial genome database for flexible ortholog analysis utilizing a diverse set of genomic data. Nucleic Acids Res 2015;43:D270–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Altenhoff AM, Skunca N, Glover N, et al. The OMA orthology database in 2015: function predictions, better plant support, synteny view and other improvements. Nucleic Acids Res 2015;43:D240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heinicke S, Livstone MS, Lu C, et al. The Princeton Protein Orthology Database (P-POD): a comparative genomics analysis tool for biologists. PLoS One 2007;2:e766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huerta-Cepas J, Capella-Gutierrez S, Pryszcz LP, et al. PhylomeDB v4: zooming into the plurality of evolutionary histories of a genome. Nucleic Acids Res 2014;42:D897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kriventseva EV, Tegenfeldt F, Petty TJ, et al. OrthoDB v8: update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res 2015;43:D250–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Powell S, Forslund K, Szklarczyk D, et al. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res 2014;42:D231–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sonnhammer EL, Ostlund G.. InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res 2015;43:D234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaduk M, Riegler C, Lemp O, et al. HieranoiDB: a database of orthologs inferred by Hieranoid. Nucleic Acids Res 2017;45:D687–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jensen LJ, Julien P, Kuhn M, et al. eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res 2008;36:D250–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huerta-Cepas J, Szklarczyk D, Forslund K, et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 2016;44:D286–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Altschul SF, Madden TL, Schaffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25:3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol 2011;7:e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Galperin MY, Koonin EV.. Sources of systematic error in functional annotation of genomes: domain rearrangement, non-orthologous gene displacement and operon disruption. In Silico Biol 1998;1:55–67. [PubMed] [Google Scholar]
- 26. Schnoes AM, Brown SD, Dodevski I, et al. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol 2009;5:e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gilks WR, Audit B, De Angelis D, et al. Modeling the percolation of annotation errors in a database of protein sequences. Bioinformatics 2002;18:1641–9. [DOI] [PubMed] [Google Scholar]
- 28. Valencia A. Automatic annotation of protein function. Curr Opin Struct Biol 2005;15:267–74. [DOI] [PubMed] [Google Scholar]
- 29. Gaudet P, Livstone MS, Lewis SE, et al. Phylogenetic-based propagation of functional annotations within the Gene Ontology Consortium. Brief Bioinform 2011;12:449–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mi H, Huang X, Muruganujan A, et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res 2017;45:D183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res 2017;45:D158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Letunic I, Doerks T, Bork P.. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res 2015;43:D257–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oates ME, Stahlhacke J, Vavoulis DV, et al. The SUPERFAMILY 1.75 database in 2014: a doubling of data. Nucleic Acids Res 2015;43:D227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Finn RD, Coggill P, Eberhardt RY, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 2016;44:D279–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Finn RD, Attwood TK, Babbitt PC, et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res 2017;45:D190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marchler-Bauer A, Bo Y, Han L, et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res 2017;45:D200–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sonnhammer EL, Koonin EV.. Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet 2002;18:619–20. [DOI] [PubMed] [Google Scholar]
- 38. Kristensen DM, Kannan L, Coleman MK, et al. A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 2010;26:1481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lechner M, Hernandez-Rosales M, Doerr D, et al. Orthology detection combining clustering and synteny for very large datasets. PLoS One 2014;9:e105015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dewey CN. Positional orthology: putting genomic evolutionary relationships into context. Brief Bioinform 2011;12:401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marchler-Bauer A, Zheng C, Chitsaz F, et al. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res 2013;41:D348–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alexeyenko A, Tamas I, Liu G, et al. Automatic clustering of orthologs and inparalogs shared by multiple proteomes. Bioinformatics 2006;22:e9–15. [DOI] [PubMed] [Google Scholar]
- 43. Chen F, Mackey AJ, Vermunt JK, et al. Assessing performance of orthology detection strategies applied to eukaryotic genomes. PLoS One 2007;2:e383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Altenhoff AM, Dessimoz C.. Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Comput Biol 2009;5:e1000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Altenhoff AM, Dessimoz C.. Inferring orthology and paralogy. Methods Mol Biol 2012;855:259–79. [DOI] [PubMed] [Google Scholar]
- 46. Mulkidjanian AY, Galperin MY, Makarova KS, et al. Evolutionary primacy of sodium bioenergetics. Biol Direct 2008;3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tipton PA, Beecher BS.. Tartrate dehydrogenase, a new member of the family of metal-dependent decarboxylating R-hydroxyacid dehydrogenases. Arch Biochem Biophys 1994;313:15–21. [DOI] [PubMed] [Google Scholar]
- 48. Salomone JY, Crouzet P, De Ruffray P, et al. Characterization and distribution of tartrate utilization genes in the grapevine pathogen Agrobacterium vitis. Mol Plant Microbe Interact 1996;9:401–8. [DOI] [PubMed] [Google Scholar]
- 49. Howell DM, Graupner M, Xu H, et al. Identification of enzymes homologous to isocitrate dehydrogenase that are involved in coenzyme B and leucine biosynthesis in methanoarchaea. J Bacteriol 2000;182:5013–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Haft DH, Selengut JD, Richter RA, et al. TIGRFAMs and genome properties in 2013. Nucleic Acids Res 2013;41:D387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Klimke W, Agarwala R, Badretdin A, et al. The national center for biotechnology information's protein clusters database. Nucleic Acids Res 2009;37:D216–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yutin N, Puigbo P, Koonin EV, et al. Phylogenomics of prokaryotic ribosomal proteins. PLoS One 2012;7:e36972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Natale DA, Galperin MY, Tatusov RL, et al. Using the COG database to improve gene recognition in complete genomes. Genetica 2000;108:9–17. [DOI] [PubMed] [Google Scholar]
- 54. Koonin EV, Galperin MY (2003) Sequence—Evolution—Function: Computational Approaches in Comparative Genomics. Boston: Kluwer Academic. [PubMed] [Google Scholar]
- 55. Tatusova T, Ciufo S, Fedorov B, et al. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res 2014;42:D553–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tatusova T, DiCuccio M, Badretdin A, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 2016;44:6614–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Galperin MY, Koonin EV.. Functional genomics and enzyme evolution. Homologous and analogous enzymes encoded in microbial genomes. Genetica 1999;106:159–70. [DOI] [PubMed] [Google Scholar]
- 58. Galperin MY, Kolker E.. New metrics for comparative genomics. Curr Opin Biotechnol 2006;17:440–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Galperin MY. Structural classification of bacterial response regulators: diversity of output domains and domain combinations. J Bacteriol 2006;188:4169–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Galperin MY. Diversity of structure and function of response regulator output domains. Curr Opin Microbiol 2010;13:150–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Diaz R, Vargas-Lagunas C, Villalobos MA, et al. argC orthologs from Rhizobiales show diverse profiles of transcriptional efficiency and functionality in Sinorhizobium meliloti. J Bacteriol 2011;193:460–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Prunetti L, El Yacoubi B, Schiavon CR, et al. Evidence that COG0325 proteins are involved in PLP homeostasis. Microbiology 2016;162:694–706. [DOI] [PubMed] [Google Scholar]
- 63. Zallot R, Yuan Y, de Crecy-Lagard V.. The Escherichia coli COG1738 member YhhQ is involved in 7-cyanodeazaguanine (preQ0) transport. Biomolecules 2017;7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kristensen DM, Wolf YI, Koonin EV.. ATGC database and ATGC-COGs: an updated resource for micro- and macro-evolutionary studies of prokaryotic genomes and protein family annotation. Nucleic Acids Res 2017;45:D210–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tettelin H, Masignani V, Cieslewicz MJ, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci USA 2005;102:13950–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tettelin H, Riley D, Cattuto C, et al. Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol 2008;11:472–7. [DOI] [PubMed] [Google Scholar]
- 67. Puigbo P, Lobkovsky AE, Kristensen DM, et al. Genomes in turmoil: quantification of genome dynamics in prokaryote supergenomes. BMC Biol 2014;12:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wolf YI, Makarova KS, Lobkovsky AE, et al. Two fundamentally different classes of microbial genes. Nat Microbiol 2016;2:16208. [DOI] [PubMed] [Google Scholar]
- 69. Fitch WM. Distinguishing homologous from analogous proteins. Syst Zool 1970;19:99–113. [PubMed] [Google Scholar]
- 70. Makarova KS, Sorokin AV, Novichkov PS, et al. Clusters of orthologous genes for 41 archaeal genomes and implications for evolutionary genomics of archaea. Biol Direct 2007;2:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zdobnov EM, Tegenfeldt F, Kuznetsov D, et al. OrthoDB v9.1: cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res 2017;45:D744–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Curnow AW, Hong K, Yuan R, et al. Glu-tRNAGln amidotransferase: a novel heterotrimeric enzyme required for correct decoding of glutamine codons during translation. Proc Natl Acad Sci USA 1997;94:11819–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wolf YI, Makarova KS, Yutin N, et al. Updated clusters of orthologous genes for Archaea: a complex ancestor of the Archaea and the byways of horizontal gene transfer. Biol Direct 2012;7:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mulkidjanian AY, Koonin EV, Makarova KS, et al. The cyanobacterial genome core and the origin of photosynthesis. Proc Natl Acad Sci USA 2006;103:13126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Makarova KS, Koonin EV.. Evolutionary genomics of lactic acid bacteria. J Bacteriol 2007;189:1199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Novichkov PS, Ratnere I, Wolf YI, et al. ATGC: a database of orthologous genes from closely related prokaryotic genomes and a research platform for microevolution of prokaryotes. Nucleic Acids Res 2009;37:D448–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Novichkov PS, Wolf YI, Dubchak I, et al. Trends in prokaryotic evolution revealed by comparison of closely related bacterial and archaeal genomes. J Bacteriol 2009;191:65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ran W, Kristensen DM, Koonin EV.. Coupling between protein level selection and codon usage optimization in the evolution of bacteria and archaea. MBio 2014;5:e00956-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yutin N, Colson P, Raoult D, et al. Mimiviridae: clusters of orthologous genes, reconstruction of gene repertoire evolution and proposed expansion of the giant virus family. Virol J 2013;10:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Grazziotin AL, Koonin EV, Kristensen DM.. Prokaryotic Virus Orthologous Groups (pVOGs): a resource for comparative genomics and protein family annotation. Nucleic Acids Res 2017;45:D491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Makarova KS, Aravind L, Grishin NV, et al. A DNA repair system specific for thermophilic Archaea and bacteria predicted by genomic context analysis. Nucleic Acids Res 2002;30:482–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Makarova KS, Grishin NV, Shabalina SA, et al. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 2006;1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Koonin EV, Wolf YI, Aravind L.. Prediction of the archaeal exosome and its connections with the proteasome and the translation and transcription machineries by a comparative-genomic approach. Genome Res 2001;11:240–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Galperin MY, Nikolskaya AN, Koonin EV.. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol Lett 2001;203:11–21. [DOI] [PubMed] [Google Scholar]
- 85. Amikam D, Galperin MY.. PilZ domain is part of the bacterial c-di-GMP binding protein. Bioinformatics 2006;22:3–6. [DOI] [PubMed] [Google Scholar]
- 86. Makarova KS, Wolf YI, Koonin EV.. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol Direct 2009;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Fozo EM, Makarova KS, Shabalina SA, et al. Abundance of type I toxin-antitoxin systems in bacteria: searches for new candidates and discovery of novel families. Nucleic Acids Res 2010;38:3743–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Makarova KS, Wolf YI, Snir S, et al. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J Bacteriol 2011;193:6039–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Makarova KS, Koonin EV, Albers SV.. Diversity and evolution of type IV pili systems in Archaea. Front Microbiol 2016;7:667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Galperin MY, Koonin EV.. ′Conserved hypothetical′ proteins: prioritization of targets for experimental study. Nucleic Acids Res 2004;32:5452–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Galperin MY, Koonin EV.. From complete genome sequence to ′complete′ understanding? Trends Biotechnol 2010;28:398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]