

Abstract
The biological characteristics and clinical outcomes of gastric cancer (GC) are largely dependent on the histopathological type and degree of differentiation. The identification of the molecular mechanisms underlying the histological grade of GC may provide information about tumorigenesis and tumor progression, and may subsequently be used to develop novel therapeutic agents. The present study obtained the RNA sequencing data and clinical characteristics of patients with GC from The Cancer Genome Atlas. A total of 1,400 differentially expressed genes (DEGs) were screened between two histological grades. Weighted gene co-expression network analysis (WGCNA) was subsequently used to identify nine co-expressed gene modules, and the black module was found to be the most significant for prognosis prediction of tumor. Additionally, the black module was associated with overall survival time, death event, N stage and tumor-node-metastasis (TNM) stage. Functional enrichment analysis revealed that the biological processes of the genes in the black module included ‘Wnt signaling pathway’ and ‘structural molecule activity’. Additionally, 10 network hub genes that were significantly associated with the progression of GC were identified from the black module, and the significance of each hub gene was determined across different TNM stages. Kaplan-Meier survival curves revealed that keratin 40 and glycine decarboxylase were significantly associated with patient prognosis (P<0.05), suggesting that these genes may serve as potential progression and prognosis biomarkers in GC. The present study identified molecular markers that correlated with histological grade in GC. Therefore, the results obtained in the present study may have important clinical implications on treatment selection, risk stratification and prognosis prediction in patients with GC.
Keywords: gastric cancer, histological grade, weighted gene co-expression network analysis, differentially expressed genes, prognostic markers
Introduction
Gastric cancer (GC) is the second most common malignancy in Eastern Asia (particularly in Korea, Mongolia, Japan, and China) and has a five-years survival rate as low as 31.5% (1). The histopathological type is an independent prognostic factor in GC, and a predictor of lymph node metastasis (2–6). Different histopathological types have distinct clinical outcomes and unique biological characteristics. The overall survival (OS) rate for patients with well-differentiated GC is higher than that for patients with poorly differentiated GC. Moreover, different types are associated with specific molecular mechanisms, treatment strategies and prognoses (7–9). However, the exact causes and mechanisms involved in the different types remain unclear. Therefore, it is necessary to further investigate the mechanisms underlying GC differentiation.
Weighted gene co-expression network analysis (WGCNA) has been used to explore the correlations between clinical features of disease and gene clusters (10). WGCNA transforms gene expression data into co-expression modules and provides insights into signaling networks that may be responsible for clinical indicators of tumor progression, including tumor stages, grades and metastasis (11–13). WGCNA is a comprehensive collection of R functions (14), and is widely used to identify candidate biomarkers or therapeutic targets (15,16). In the present study, a co-expression module was constructed using expression data from patients with GC of different histological grades. Gene Ontology (GO) enrichment analysis was subsequently performed on selected modules to identify the hub genes, which may serve as potential therapeutic, diagnostic or prognostic markers.
Materials and methods
Preparation of genetic and clinical data
The workflow for the current study is presented in Fig. 1. The TCGA database included the mRNA sequencing data of 32 normal stomach samples and 376 stomach adenocarcinoma samples, and the clinical data of 408 patients with STAD. Level-3 RNA sequencing data were obtained using an Illumina HiSeq RNAseq v2 RSEM platform. Patients without complete histological grade information were eliminated and 366 patients were available for the next screen. Patients without complete follow-up information or complete clinical information were excluded. A total of 172 patients were included in the WGCNA analysis. Other clinical information, including neoplasm histological grade, American Joint Committee on Cancer pathological tumor-node-metastasis (TNM) stage (17) and anatomic neoplasm subdivision, were retrieved for WGCNA analysis.
Figure 1.

Workflow of data preparation, processing and analysis in the present study.
Screening for differentially expressed genes (DEGs)
R (version 3.4.2) and RStudio (version 1.1.383) (18,19) and two R packages (limma and edgeR, Bioconductor version 3.6; bioconductor.org/), were used to identify DEGs between cancer and normal samples. Additionally, patients were divided into poorly- and moderately/well-differentiated groups to identify the DEGs associated tumor differentiation. The DEG threshold was set at an adjusted P<0.05 and a log2 fold-change >2.
Gene co-expression network construction and module analysis
The gradient method was used to test the independence and average connectivity of different modules with different power values (10). The power values ranged from 1 to 20. The appropriate power value was determined when the degree of independence was 0.9 which was set to eight in the present study, and the module construction was continued using the WGCNA algorithm. In addition, the corresponding gene information for each module was extracted. The WGCNA algorithm may be used to identify co-expression modules (10). WGCNA was implemented in the R package (WGCNA Version 1.68; cloud.r-project.org/) and the heatmaps package (Bioconductor version, 3.6) was used to analyze the strength of interactions.
Identification of the module of interest and functional annotation
The correlation between the modules and the clinical features such as TNM Stage, OS, Division and Grade were assessed by the Pearson's correlation test. Modules with the highest correlation with clinical features were selected as modules of interest. To explore the potential mechanisms by which the genes affect the relevant clinical features, all genes in the module of interest were uploaded to the Database for Annotation, Visualization, and Integrated Discovery (DAVID version 6.8, david.ncifcrf.gov/) (20) and subjected to GO functional (geneontology.org/)and Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.kegg.jp/). The specific cut-off used for terms and pathways was 0.05.
Hub gene identification and correlation analysis
The modules of interest were visualized using Cytoscape software (version 3.6.0; cytoscape.org/), and the top ten genes with the greatest number of edges were identified as the hub genes (21). A one-way ANOVA with a post-hoc Dunnett's test was used to test for associations between hub genes and the corresponding clinical features (SPSS version 21; IBM Corp.). The patients were subsequently divided into two groups based on the expression of each hub gene (high vs. low; cut-off point, 50%). Survival analysis was performed for all hub genes using the survival package (version 2.42–3, cloud.r-project.org/) in R (22). Kaplan-Meier analysis and a log-rank test were used to assess the effect of hub gene expression on overall survival time. In addition, the online software Kaplan-Meier plotter (kmplot.com/analysis) (23), which performs log-rank tests based on data from Gene Expression Omnibus (GEO) datasets (GSE14210, GSE15459, GSE22377, GSE29272, GSE51105 and GSE62254), was used for further verification.
Results
DEGs screening
DEG analysis was performed on the RNA sequencing data of 376 STAD tissues and 32 adjacent non-tumor tissue samples and a total of 11,031 DEGs were identified using edgeR and limma. The tumor samples were subsequently divided into two groups according to the histological grade: i) The poorly differentiated group (219 samples); ii) and the moderately to well-differentiated group (147 samples). A total of 1,400 DEGs (836 upregulated and 564 downregulated) were identified.
Functional annotation of DEGs between patients with poorly and moderately to well-differentiated GC
To explore the functional significance of DEGs in GC differentiation, the aforementioned 1,400 DEGs were subjected to unbiased GO term and KEGG pathway enrichment analyses. For DEGs associated with GC differentiation, the terms ‘digestion’ [false discovery rate (FDR)=2.08×10−04], ‘extracellular space’ (FDR=4.97×10−21) and ‘structural molecule activity’ (FDR=8.54×10−09) were the most significantly enriched biological process, cellular component and molecular function, respectively, while ‘neuroactive ligand-receptor interaction’ was the most significantly enriched pathway (Table I).
Table I.
List of the top GO terms and KEGG pathways in DEGs.
| A, GO biological process | |||
|---|---|---|---|
| Term | Gene count | P-Value | FDR |
| GO:0007586 digestion | 15 | 1.17×10−07 | 2.08×10−04 |
| GO:0010951 negative regulation of endopeptidase activity | 17 | 2.12×10−05 | 3.70×10−02 |
| GO:0002027 regulation of heart rate | 9 | 3.20×10−05 | 5.70×10−02 |
| GO:1903779 regulation of cardiac conduction | 11 | 5.77×10−05 | 1.02×10−01 |
| GO:0006936 muscle contraction | 15 | 7.72×10−05 | 1.37×10−01 |
| B, Cellular component | |||
| Term | Gene count | P-Value | FDR |
| GO:0005615 extracellular space | 136 | 3.61×10−24 | 4.97×10−21 |
| GO:0005576 extracellular region | 149 | 8.50×10−23 | 1.17×10−19 |
| GO:0030018 Z disc | 23 | 1.44×10−09 | 1.99×10−06 |
| GO:0072562 blood microparticle | 24 | 3.90×10−08 | 5.37×10−05 |
| GO:0043204 perikaryon | 17 | 4.67×10−06 | 6.00×10−03 |
| C, GO molecular function | |||
| Term | Gene count | P-Value | FDR |
| GO:0005198 structural molecule activity | 37 | 5.61×10−12 | 8.54×10−09 |
| GO:0008201 heparin binding | 24 | 5.09×10−08 | 7.74×10−05 |
| GO:0005179 hormone activity | 18 | 9.67×10−08 | 1.47×10−04 |
| GO:0008307 structural constituent of muscle | 9 | 1.71×10−04 | 2.60×10−01 |
| GO:0005102 receptor binding | 29 | 2.54×10−04 | 3.85×10−01 |
| D, KEGG analysis | |||
| Pathway | Gene count | P-Value | FDR |
| hsa04080:Neuroactive ligand-receptor interaction | 28 | 4.48×10−06 | 6.00×10−03 |
| hsa04970:Salivary secretion | 14 | 1.67×10−05 | 2.10×10−02 |
| hsa04971:Gastric acid secretion | 10 | 1.50×10−03 | 1.96 |
| hsa04972:Pancreatic secretion | 11 | 2.50×10−03 | 3.11 |
| hsa04610:Complement and coagulation cascades | 9 | 4.10×10−03 | 5.15 |
GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; FDR, false discovery rate.
Co-expression network construction and module analysis
To explore the functional modules in patients with GC with different histological grades, the 1,400 DEGs were subjected to WGCNA. Clinical characters, including neoplasm histological grade, TNM stage and anatomic neoplasm subdivision, were retrieved for WGCNA analysis. A total of 172 patients were included in the WGCNA (Fig. 2). Three outlier samples were discarded. The connectivity between the genes in the gene network satisfied the scale-free network distribution (Fig. S1). Nine co-expressed modules, ranging in size from 34 to 734 genes, were subsequently identified. Each module was assigned a color for reference. The grey module was reserved for genes that had been identified as not co-expressed (Figs. 3 and S2). The genes in each module are listed in Table SI.
Figure 2.
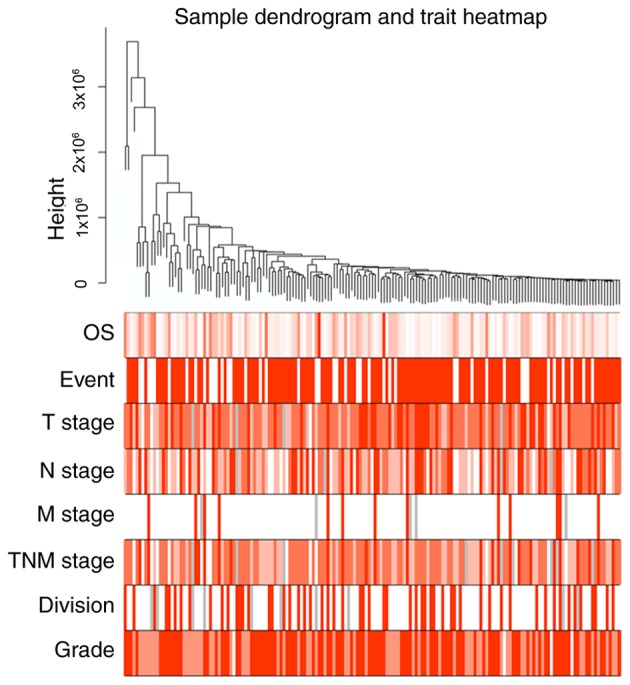
Clustering dendrogram of 169 samples and the associated clinical traits. Three samples were excluded from the original 172 samples. The clustering was based on the expression data of differentially expressed genes between patients with poorly and moderately to well-differentiated gastric cancer. Each clinical trait is depicted with varying color intensity. The darker the color, the stronger the correlation. TNM, tumor-node-metastasis; OS, overall survival.
Figure 3.
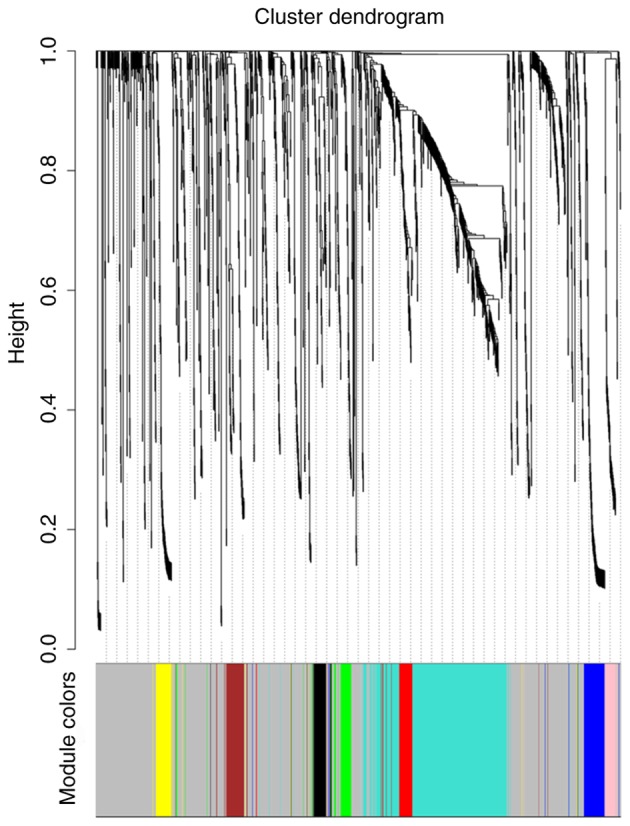
Tree diagram of clustered genes based on the difference in topological overlap and the specified module color. A total of nine co-expression modules were constructed and displayed in different colors.
Identification of key modules and functional annotation
The black module exhibited a greater correlation with OS, event, N stage and TNM stage than the other modules (P<0.05; Fig. 4), and was correlated with the T stage (P=0.08). The genes in the black module may therefore be associated with the survival and prognosis of patients. However, each module was correlated with different clinical features and the red module was correlated with anatomic neoplasm subdivision (P=0.04). Scatterplots of gene significance vs. specific module membership were plotted (Fig. 5). The black module was selected as the module of interest and was subsequently analyzed. To identify the functional involvement of the black module, the 40 genes in the black module were uploaded onto DAVID for KEGG pathway enrichment and GO analyses. Biological processes of the black module included ‘Wnt signaling pathway’ (P=9.80×10−04), ‘structural molecule activity’ (P=0.004) and ‘vitamin transport’ (P=0.005). KEGG pathway analysis revealed that ‘Wnt signaling pathway’ was the only significant pathway (P=0.008; Fig. 6; Table SII).
Figure 4.
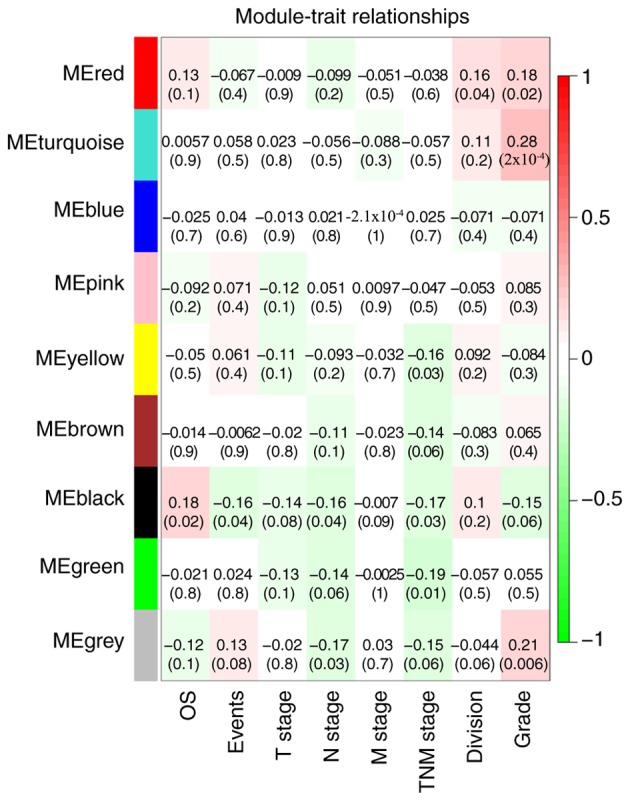
Module-feature association. Each row corresponds to a module eigengene, and the column corresponds to a feature. Each cell contains the corresponding correlation coefficient and P-value. The table was color coded based on correlation according to the color legend. Red represents a positive correlation, green represents a negative correlation, and the darker the color, the greater the significance of the P-value. OS, overall survival; TNM, tumor-node-metastasis.
Figure 5.

Scatterplot of gene significance for (A) T stage, (B) N stage, (C) TNM stage, (D) OS and (E) death events vs. module membership in the black module. (B) Scatterplot of N stage in the black module. (F) Scatterplot of gene significance for anatomic division in the red module. There is a highly significant association between gene significance and module membership in these modules. TNM, tumor-node-metastasis; OS, overall survival; cor, correlation coefficient.
Figure 6.
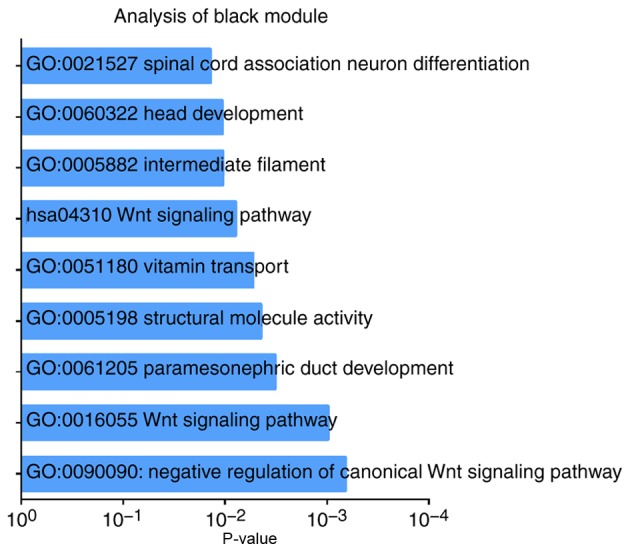
GO functional analysis and KEGG pathway enrichment for genes in the black module. The x-axis shows the-log10 (P-value) and the y-axis shows the GO and KEGG pathway terms. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Hub gene identification and correlation analysis
Cytoscape software was used to construct the co-expression network modules, and the intramodular connectivity was calculated. Genes with high intramodular connectivity were considered as intramodular hub genes (Table SIII). The hub genes in the black module are presented in Fig. 7 (the top ten hub genes, including GLDC, KRT40, GS homeobox 1 (GSX1), keratin associated protein 4–12, distal-less homeobox 4 (DLX4), NFIA antisense RNA 2, Sp7 transcription factor, KRT39 and olfactory receptor family 5 subfamily G member 5 pseudogene). Significant differences (P<0.05) between the hub genes and TNM stages were identified using the one-way ANOVA (Table SIV). The OS analysis of 172 patients divided into two groups according to the median expression of each hub gene (high vs. low) revealed that keratin 40 (KRT40) and glycine decarboxylase (GLDC) were associated with prognosis (P<0.05; Fig. 8A), suggesting that KRT40 and GLDC may act as prognostic biomarkers for GC. Additionally, the effects of KRT40 and GLDC on the OS were validated in datasets obtained from the GEO database (GSE14210, GSE15459, GSE22377, GSE29272, GSE51105 and GSE62254). The significance of the aforementioned genes was further investigated by performing survival analysis (P<0.05; Fig. 8B).
Figure 7.
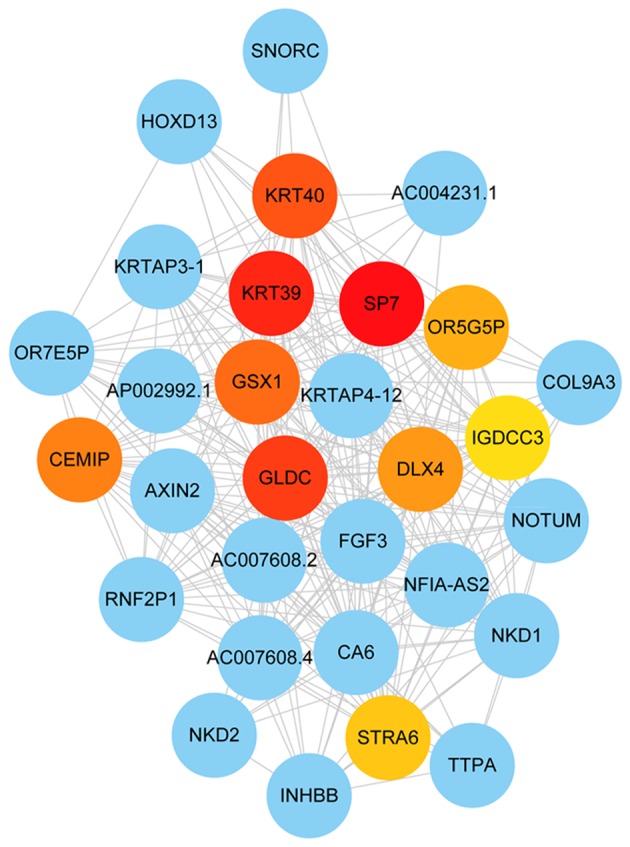
Black module gene network. The top ten hub genes of the black module are presented in red, orange and yellow depending on the gene importance defined as the degree of connectivity. The other genes in the black module are represented in blue.
Figure 8.

(A) Kaplan-Meier curve obtained from the weighted prognostic index classification by KRT40 and GLDC gene expression in patents with gastric cancer from the TCGA database. (B) Kaplan-Meier curve of KRT40 and GLDC expression in patients with gastric cancer in the Gene Expression Omnibus database. The numbers in the brackets represent the confidence intervals. KRT40, keratin 40; GLDC, glycine decarboxylase; HR, hazard ratio.
Discussion
The degree of differentiation of GC is associated with complex gene interactions and often indicates prognosis (24–27). Studying the molecular mechanisms underlying differentiation is important for understanding the pathogenesis and development of GC, and may be helpful for the diagnosis and treatment of GC. However, to the best of our knowledge, there is currently no clinically applicable biomarker for distinguishing between the histological types of GC. The present study used RNA sequencing data and clinical information obtained from 408 GC samples in the TCGA database, 172 of which were included in the final WGCNA to identify robust co-expression modules associated with cancer characteristics.
In cancer studies, candidate molecular biomarkers may be used to distinguish between normal and cancerous tissues (28–32). The present study identified DEGs between GC and paracancerous tissues in 408 samples in the TCGA database. The samples were divided into two groups based on the degree of GC differentiation, and a total of 1,400 DEGs associated with the differentiation of GC were obtained. GO enrichment analysis revealed that the 1,400 DEGs were associated with ‘digestion’, ‘extracellular space’, ‘structural molecule activity’ and ‘neuroactive ligand-receptor interaction’. As the preliminary GO analysis did not clearly explain the role of the DEGs, WGCNA was subsequently used to further analyze the aforementioned DEGs. WGCNA has a number of advantages, as the analysis focuses on the association between clinical features and co-expression, resulting in higher reliability and biological significance (33–35). Therefore, genes in the same module are considered to be functionally associated with each other, and the analysis identifies biologically relevant modules and hub genes that may eventually serve as biomarkers for detection or treatment (10).
The black co-expression module in the current study was correlated with various clinical traits, including OS, death event, N stage and TNM stage. The aforementioned clinical traits were associated with the survival and prognosis of patients. The black module was therefore considered to be a clinically significant gene cluster that required further investigation in the current study.
Functional annotation of the black module revealed that the genes were involved in the ‘Wnt signaling pathway’ and ‘structural molecule activity’, which affect the pathogenesis and development of tumors (36–40). The associations between the TNM stage and the genes in the ‘Wnt signaling pathway’, including NKD inhibitor of WNT signaling pathway 1 and 2 and notum palmitoleoyl-protein carboxylesterase, and the genes involved in ‘structural molecule activity’, including keratin (KRT) 39 and KRT40, were investigated. Furthermore, the black the genes in the black module were analyzed using Cytoscape software and the top ten hub genes, including GLDC, KRT40, GS homeobox 1 (GSX1), keratin associated protein 4–12, distal-less homeobox 4 (DLX4), NFIA antisense RNA 2, Sp7 transcription factor, KRT39 and olfactory receptor family 5 subfamily G member 5 pseudogene, were identified.
GLDC is involved in glycine metabolism and serves a role in several types of cancer (41–45). KRT39 and KRT40 contribute to the structural integrity of a complex or assembly within or outside a cell (46). GSX1 is among the earliest transcription factors expressed in neuronal progenitors (47) and may be used as a prognostic predictor. DLX4 is a transcription factor encoded by a homeobox gene associated with ovarian cancer (48). The expression value of each hub gene across different TNM stages was significantly different, and KRT40 and GLDC were associated with patient prognosis for 3-year overall survival analysis, suggesting that these hub genes were positively correlated with tumor stage and prognosis of GC.
In summary, the present study established a gene co-expression network to identify network genes associated with the progression of GC, relative to the histological grade. GLDC, KRT40, GSX1 and DLX4 were identified as potential diagnostic and prognostic biomarkers of GC as they showed the highest levels of significance for prognosis. Additional research is required to investigate the roles of the aforementioned genes in in the pathogenesis and progression of GC. The results obtained in the current study may contribute to the improvement of risk stratification, therapeutic decision-making and prognosis prediction for patients with GC.
Supplementary Material
Acknowledgements
Not applicable.
Funding
The current study was funded by the National Natural Science Foundation of China (grant nos. 81472308 and 31470891).
Availability of data and materials
The datasets used and analyzed during the current study are available from TCGA (gdc.cancer.gov/) and Gene Expression Omnibus, (ncbi.nlm.nih.gov/geo/).
Authors' contributions
JC, XX, YC and RW conceived and designed the study. JC and XX acquired and interpreted the data and contributed to the revision of the manuscript. WC and WZ prepared the initial draft of the manuscript and contributed to the acquisition and interpretation of the data. JC, XX, YC and RW analyzed the data. All authors approved the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Folli S, Morgagni P, Roviello F, De Manzoni G, Marrelli D, Saragoni L, Di Leo A, Gaudio M, Nanni O, Carli A, et al. Risk factors for lymph node metastases and their prognostic significance in early gastric cancer (EGC) for the Italian research group for gastric cancer (IRGGC) Jpn J Clin Oncoly. 2001;31:495–499. doi: 10.1093/jjco/hye107. [DOI] [PubMed] [Google Scholar]
- 3.Popiela T, Kulig J, Kolodziejczyk P, Sierzega M, Sierzega M, Polish Gastric Cancer Study Group Long-term results of surgery for early gastric cancer. Brit J Surg. 2002;89:1035–1042. doi: 10.1046/j.1365-2168.2002.02156.x. [DOI] [PubMed] [Google Scholar]
- 4.Adachi Y, Yasuda K, Inomata M, Sato K, Shiraishi N, Kitano S. Pathology and prognosis of gastric carcinoma: Well versus poorly differentiated type. Cancer. 2000;89:1418–1424. doi: 10.1002/1097-0142(20001001)89:7<1418::AID-CNCR2>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 5.Noda S, Soejima K, Inokuchi K. Clinicopathological analysis of the intestinal type and diffuse type of gastric carcinoma. Jpn J Surg. 1980;10:277–283. doi: 10.1007/BF02468788. [DOI] [PubMed] [Google Scholar]
- 6.Ribeiro MM, Sarmento JA, Sobrinho Simões MA, Bastos J. Prognostic significance of Lauren and Ming classifications and other pathologic parameters in gastric carcinoma. Cancer. 1981;47:780–784. doi: 10.1002/1097-0142(19810215)47:4<780::AID-CNCR2820470424>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 7.Kim BS, Oh ST, Yook JH, Kim BS. Signet ring cell type and other histologic types: Differing clinical course and prognosis in T1 gastric cancer. Surgery. 2014;155:1030–1035. doi: 10.1016/j.surg.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 8.Qiu MZ, Cai MY, Zhang DS, Wang ZQ, Wang DS, Li YH, Xu RH. Clinicopathological characteristics and prognostic analysis of Lauren classification in gastric adenocarcinoma in China. J Transl Med. 2013;11:58. doi: 10.1186/1479-5876-11-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flucke U, Mönig SP, Baldus SE, Zirbes TK, Bollschweiler E, Thiele J, Dienes HP, Hölscher AH. Differences between biopsy- or specimen-related Laurén and world health organization classification in gastric cancer. World J Surg. 2002;26:137–140. doi: 10.1007/s00268-001-0195-0. [DOI] [PubMed] [Google Scholar]
- 10.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Udyavar AR, Hoeksema MD, Clark JE, Zou Y, Tang Z, Li Z, Li M, Chen H, Statnikov A, Shyr Y, et al. Co-expression network analysis identifies spleen tyrosine kinase (SYK) as a candidate oncogenic driver in a subset of small-cell lung cancer. BMC Syst Biol. 2013;7(Suppl 5):S1. doi: 10.1186/1752-0509-7-S5-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, Laurance MF, Zhao W, Qi S, Chen Z, et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci USA. 2006;103:17402–17407. doi: 10.1073/pnas.0608396103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi Z, Derow CK, Zhang B. Co-expression module analysis reveals biological processes, genomic gain, and regulatory mechanisms associated with breast cancer progression. BMC Syst Biol. 2010;4:74. doi: 10.1186/1752-0509-4-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X, Hu AX, Zhao JL, Chen FL. Identification of key gene modules in human osteosarcoma by co-expression analysis weighted gene co-expression network analysis (WGCNA) J Cell Biochem. 2017;118:3953–3959. doi: 10.1002/jcb.26050. [DOI] [PubMed] [Google Scholar]
- 15.Perumal D, Pillai S, Nguyen J, Schaal C, Coppola D, Chellappan SP. Nicotinic acetylcholine receptors induce c-Kit ligand/stem cell factor and promote stemness in an ARRB1/ β-arrestin-1 dependent manner in NSCLC. Oncotarget. 2014;5:10486–10502. doi: 10.18632/oncotarget.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi K, Bing ZT, Cao GQ, Guo L, Cao YN, Jiang HO, Zhang MX. Identify the signature genes for diagnose of uveal melanoma by weight gene co-expression network analysis. Int J Ophthalmol. 2015;8:269–274. doi: 10.3980/j.issn.2222-3959.2015.02.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amin MB, Greene FL, Edge SB, Compton CC, Gershenwald JE, Brookland RK, Meyer L, Gress DM, Byrd DR, Winchester DP. The eighth edition AJCC cancer staging manual: Continuing to build a bridge from a population-based to a more ‘personalized’ approach to cancer staging. CA Cancer J Clin. 2017;67:93–99. doi: 10.3322/caac.21388. [DOI] [PubMed] [Google Scholar]
- 18.Team RC, corp-author. R Foundation for Statistical Computing; Vienna: 2012. R: A language and environment for statistical computing. [Google Scholar]
- 19.RStudio Team, corp-author. RStudio: Integrated Development for R. RStudio, Inc.; Boston, MA: 2015. [Google Scholar]
- 20.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 21.Wisniewski N, Cadeiras M, Bondar G, Cheng RK, Shahzad K, Onat D, Latif F, Korin Y, Reed E, Fakhro R, Deng MC. Weighted gene coexpression network analysis (WGCNA) modeling of multiorgan dysfunction syndrome after mechanical circulatory support therapy. J Heart Lung Transpl. 2013;32(Suppl):S223. doi: 10.1016/j.healun.2013.01.565. [DOI] [Google Scholar]
- 22.Goel MK, Khanna P, Kishore J. Understanding survival analysis: Kaplan-Meier estimate. Int J Ayurveda Res. 2010;1:274–278. doi: 10.4103/0974-7788.76794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lánczky A, Nagy Á, Bottai G, Munkácsy G, Szabó A, Santarpia L, Győrffy B. miRpower: A web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res Treat. 2016;160:439–446. doi: 10.1007/s10549-016-4013-7. [DOI] [PubMed] [Google Scholar]
- 24.Fiocca R, Luinetti O, Villani L, Mastracci L, Quilici P, Grillo F, Ranzani GN. Molecular mechanisms involved in the pathogenesis of gastric carcinoma: Interactions between genetic alterations, cellular phenotype and cancer histotype. Hepatogastroenterology. 2001;48:1523–1530. [PubMed] [Google Scholar]
- 25.Liu W, Rodgers GP. Olfactomedin 4 expression and functions in innate immunity, inflammation, and cancer. Cancer Metastasis Rev. 2016;35:201–212. doi: 10.1007/s10555-016-9624-2. [DOI] [PubMed] [Google Scholar]
- 26.Piao Y, Li Y, Xu Q, Liu JW, Xing CZ, Xie XD, Yuan Y. Association of MTOR and AKT gene polymorphisms with susceptibility and survival of gastric cancer. PLoS One. 2015;10:e0136447. doi: 10.1371/journal.pone.0136447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Streit M, Schmidt R, Hilgenfeld RU, Thiel E, Kreuser ED. Adhesion receptors in malignant transformation and dissemination of gastrointestinal tumors. Recent Results Cancer Res. 1996;142:19–50. doi: 10.1007/978-3-642-80035-1_3. [DOI] [PubMed] [Google Scholar]
- 28.Carlomagno N, Incollingo P, Tammaro V, Peluso G, Rupealta N, Chiacchio G, Sandoval Sotelo ML, Minieri G, Pisani A, Riccio E, et al. Diagnostic, predictive, prognostic, and therapeutic molecular biomarkers in third millennium: A breakthrough in gastric cancer. Biomed Res Int. 2017;2017:7869802. doi: 10.1155/2017/7869802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi YJ, Kim N. Gastric cancer and family history. Korean J Intern Med. 2016;31:1042–1053. doi: 10.3904/kjim.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Panarese I, De Vita F, Ronchi A, Romano M, Alfano R, Di Martino N, Zito Marino F, Ferraraccio F, Franco R. Predictive biomarkers along gastric cancer pathogenetic pathways. Expert Rev Anticancer Ther. 2017;17:417–425. doi: 10.1080/14737140.2017.1301207. [DOI] [PubMed] [Google Scholar]
- 31.Shin VY, Chu KM. MiRNA as potential biomarkers and therapeutic targets for gastric cancer. World J Gastroenterol. 2014;20:10432–10439. doi: 10.3748/wjg.v20.i30.10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tahara T, Arisawa T. DNA methylation as a molecular biomarker in gastric cancer. Epigenomics. 2015;7:475–486. doi: 10.2217/epi.15.4. [DOI] [PubMed] [Google Scholar]
- 33.Chou WC, Cheng AL, Brotto M, Chuang CY. Visual gene-network analysis reveals the cancer gene co-expression in human endometrial cancer. BMC Genomics. 2014;15:300. doi: 10.1186/1471-2164-15-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo Y, Coskun V, Liang A, Yu J, Cheng L, Ge W, Shi Z, Zhang K, Li C, Cui Y, et al. Single-cell transcriptome analyses reveal signals to activate dormant neural stem cells. Cell. 2015;161:1175–1186. doi: 10.1016/j.cell.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kunowska N, Rotival M, Yu L, Choudhary J, Dillon N. Identification of protein complexes that bind to histone H3 combinatorial modifications using super-SILAC and weighted correlation network analysis. Nucleic Acids Res. 2015;43:1418–1832. doi: 10.1093/nar/gku1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang H, Chen Z, Ni X. Tissue transglutaminase-1 promotes stemness and chemoresistance in gastric cancer cells by regulating Wnt/β-catenin signaling. Exp Biol Med (Maywood) 2017;242:194–202. doi: 10.1177/1535370216670541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review) Int J Oncol. 2017;51:1357–1369. doi: 10.3892/ijo.2017.4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song Y, Li ZX, Liu X, Wang R, Li LW, Zhang Q. The Wnt/β-catenin and PI3K/Akt signaling pathways promote EMT in gastric cancer by epigenetic regulation via H3 lysine 27 acetylation. Tumour Biol. 2017;39:1010428317712617. doi: 10.1177/1010428317712617. [DOI] [PubMed] [Google Scholar]
- 39.Wang H, Duan XL, Qi XL, Meng L, Xu YS, Wu T, Dai PG. Concurrent hypermethylation of SFRP2 and DKK2 activates the Wnt/β-catenin pathway and is associated with poor prognosis in patients with gastric cancer. Mol Cells. 2017;40:45–53. doi: 10.14348/molcells.2017.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woo DK, Kim HS, Lee HS, Kang YH, Yang HK, Kim WH. Altered expression and mutation of beta-catenin gene in gastric carcinomas and cell lines. Int J Cancer. 2001;95:108–113. doi: 10.1002/1097-0215(20010320)95:2<108::AID-IJC1019>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 41.Kim D, Fiske BP, Birsoy K, Freinkman E, Kami K, Possemato RL, Chudnovsky Y, Pacold ME, Chen WW, Cantor JR, et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature. 2015;520:363–367. doi: 10.1038/nature14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Min HL, Kim J, Kim WH, Jang BG, Kim MA. Epigenetic silencing of the putative tumor suppressor gene GLDC (Glycine Dehydrogenase) in gastric carcinoma. Anticancer Res. 2016;36:179–187. [PubMed] [Google Scholar]
- 43.Zhuang H, Li Q, Zhang X, Ma X, Wang Z, Liu Y, Yi X, Chen R, Han F, Zhang N, Li Y. Downregulation of glycine decarboxylase enhanced cofilin-mediated migration in hepatocellular carcinoma cells. Free Radic Biol Med. 2018;120:1–12. doi: 10.1016/j.freeradbiomed.2018.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Berezowska S, Galvan JA, Langer R, Bubendorf L, Savic S, Gugger M, Schmid RA, Marti TM. Glycine decarboxylase and HIF-1α expression are negative prognostic factors in primary resected early-stage non-small cell lung cancer. Virchows Arch. 2017;470:323–330. doi: 10.1007/s00428-016-2057-z. [DOI] [PubMed] [Google Scholar]
- 45.Zhang WC, Shyh-Chang N, Yang H, Rai A, Umashankar S, Ma S, Soh BS, Sun LL, Tai BC, Nga ME, et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell. 2012;148:259–272. doi: 10.1016/j.cell.2011.11.050. [DOI] [PubMed] [Google Scholar]
- 46.Yu Z, Wildermoth JE, Wallace OA, Gordon SW, Maqbool NJ, Maclean PH, Nixon AJ, Pearson AJ. Annotation of sheep keratin intermediate filament genes and their patterns of expression. Exp Dermatol. 2011;20:582–588. doi: 10.1111/j.1600-0625.2011.01274.x. [DOI] [PubMed] [Google Scholar]
- 47.Pei Z, Wang B, Chen G, Nagao M, Nakafuku M, Campbell K. Homeobox genes Gsx1 and Gsx2 differentially regulate telencephalic progenitor maturation. Proc Natl Acad Sci USA. 2011;108:1675–1680. doi: 10.1073/pnas.1008824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haria D, Trinh BQ, Ko SY, Barengo N, Liu J, Naora H. The homeoprotein DLX4 stimulates NF-κB activation and CD44-mediated tumor-mesothelial cell interactions in ovarian cancer. Am J Pathol. 2015;185:2298–2308. doi: 10.1016/j.ajpath.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and analyzed during the current study are available from TCGA (gdc.cancer.gov/) and Gene Expression Omnibus, (ncbi.nlm.nih.gov/geo/).