

Abstract
Factor XII (FXII) becomes a serine protease when blood is exposed to artificial medical surfaces or when pathologic surfaces arise in disease states leading to its autoactivation. Initiation of the blood coagulation cascade was the first recognized activity of FXIIa. Blocking FXIIa activity formed on artificial medical surfaces should reduce induced blood coagulation leading to thrombosis. In contrast to FXII enzymatic activities, less is known about zymogen FXII functions. Studies show that zymogen FXII has biologic activity in various cells in vivo. In endothelium, FXII stimulates cell growth and proliferation and, in vivo, neoangiogenesis after injury. In fibroblasts, transforming growth factor‐β increases FXII expression, which in turn stimulates fibroblast proliferation, contributing to tissue fibrosis. In neutrophils, FXII stimulates Akt2 to initiate neutrophil adhesion, migration, and chemotaxis, priming events leading to NETosis. Factor FXII deficiency leads to decreased neutrophil recruitment and improved wound healing. In dendritic cells, FXII contributes to neuroinflammation, and its deficiency or pharmacologic inhibition renders mice less susceptible to autoimmune encephalomyelitis. These combined studies indicate that FXII also contributes to multiple components of the inflammatory response. In sum, targeting FXII's biologic activities may provide novel approaches to reduce thrombosis and the inflammatory response in various disease states.
Keywords: C1 inhibitor, contact activation, factor XII, high‐molecular‐weight kininogen, prekallikrein, urokinase plasminogen activator receptor
Essentials.
Factor XII (FXII), a zymogen, when activated is a serine protease that initiates blood coagulation.
FXII contact activation on nonphysiologic surfaces is a defense mechanism to foreign bodies.
Zymogen FXII simulates endothelial cell proliferation, neoangiogenesis, and wound repair.
Zymogen FXII stimulates neutrophil adhesion, migration, chemotaxis, and NETosis.
1. INTRODUCTION
Factor XII (FXII) is an 80‐kDa glycoprotein with a plasma concentration of ~40 μg/mL (~500 nmol/L).1, 2 Plasma FXII is mainly synthesized in the liver, but recently a leukocyte form of the protein has been recognized with unique functional activities.3 FXII deficiency, called Hageman trait, was first recognized by Oscar Ratnoff as a defect that led to the prolongation of surface‐activated blood coagulation tests, for example, partial thromboplastin time (PTT).4 Ratnoff performed the PTT assay as part of his routine presurgical evaluation. The PTT was previously invented by Kenneth Brinkhous as an assay to recognize hemophilia, unlike the prothrombin time that was always normal with hemophilia plasma.5, 6 Earl Davie was the first to purify Hageman factor and named it factor XII, according to the developing convention.7 He and Ratnoff first showed that Hageman factor (FXII) activated plasma thromboplastin antecedent (factor XI [FXI) and later that FXIa activated factor IX (FIX).8, 9 These observations were the basis for their formulation of the coagulation cascade hypothesis, simultaneously with R.G. MacFarland, and the contact activation hypothesis for the initiation of blood coagulation.10, 11
The contact activation hypothesis for the initiation of blood coagulation fell from favor within a decade after its proposal for 2 reasons: (1) deficiencies of FXII, prekallikrein (PK), and high‐molecular‐weight kininogen (HK) (the latter 2 proteins were described in the next decade and are cofactors for FXII activation) are not associated with bleeding2, 12, 13; and (2) no convincing physiologic surface was identified as a contact activator. Further, the appreciation of tissue factor and factor VII (FVII) as a physiologic activator of factor X (FX) provided a strong alternative hypothesis for initiation of blood coagulation activation.14, 15, 16 Additionally, the recognition that FXI is activated by thrombin further provided an alternative pathway for FXIa formation, independent of FXII.17, 18
However, FXII‐initiated pathways have found new interest for several reasons. First, FXII, PK, and HK deficiencies in murine models, for the most part, have been shown to reduce thrombosis risk without abnormal bleeding.19, 20, 21 Second, biologic substances such as DNA, RNA, denatured proteins (eg, β‐amyloid), exposed vessel wall collagen, polyphosphate, and neutrophil extracellular traps (NETs) recently have all been shown to serve as platforms for FXII autoactivation in plasma.22, 23, 24, 25, 26 This information is beginning to be translated to understanding disease pathogenesis. For example, in Alzheimer disease, increased pathologic β‐amyloid is associated with evidence of plasma contact activation and the degree of cleaved HK (cHK) correlates with decreased cognitive activity in patients.27, 28 Thus, a new era for FXII and related proteins has emerged.
This review will discuss 2 roles of FXIIa/FXII. First, in vivo FXII contact activation as it occurs in response to injuries (eg, exposure to mechanical devices, bacterial infection, β‐amyloid protein, cancer) that arise in nonphysiologic circumstances. This area, which is classic contact activation, requires the formation of activated FXII (FXIIa) on some kind of surface in medical treatment– or disease‐induced pathologic states. This area is that which is commonly understood about FXII/FXIIa and where most of past and current investigation lies. Second, FXII zymogen also has been recognized to have biologic roles in endothelial cell, fibroblast, neutrophil, and dendritic cell functions. This latter field is based on the emerging recognition of the influence of FXII on cell biology and is the main focus of this review.
2. FXIIa – A SURFACE‐ACTIVATED DEFENSE MECHANISM INITIATED BY PATHOPHYSIOLOGIC CONTACT ACTIVATION
Robert Colman first called FXII and contact activation a surface‐activated defense mechanism to describe contact activation that occurs with foreign pathogens (bacteria, virus, yeast, parasites) or artificial surfaces (glass, medical apparatus). This review will not go into detail on this content area of FXII because contact activation of FXII has been covered in many reviews by us and others over the past 20 years.29, 30, 31, 32, 33 FXII has the unique property to autoactivate from a zymogen into an active enzyme. The molecular basis of FXII autoactivation is still unknown. The process of autoactivation of FXII is slow (90‐120 minutes) when the zymogen alone is incubated with a negatively charged surface (Figure 1). Autoactivation is greatly accelerated when FXII is incubated with HK and PK or in plasma. FXIIa is a serine protease that has a number of substrates that include PK, FXI, plasminogen, complement component 1r (C1r), C1s, and HK. FXIIa activation of PK to plasma kallikrein (PKa) creates a state where there is reciprocal FXII activation by PKa and formed FXIIa activates more PK, thus amplifying the activity of the system (Figure 1). HK stabilizes PK for optimal activation by FXIIa. Autoactivation, reciprocal activation, and amplification are the essence of the contact activation system (CAS).34, 35 FXIIa activation of FXI initiates the contact activation of blood coagulation,8, 9 and FXIIa activation of plasminogen and C1r and C1s initiates fibrinolysis and classic complement activation, respectively (Figure 1).36, 37, 38
Figure 1.
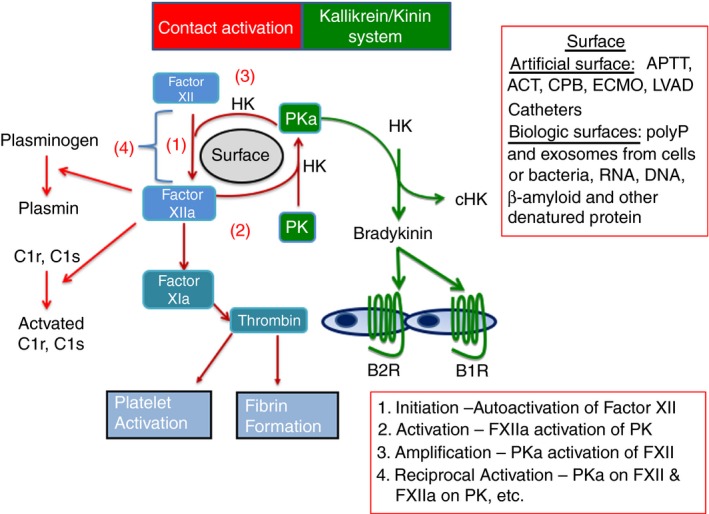
The plasma contact activation and kallikrein/kinin systems. In contact activation, (1) factor XII autoactivates on a surface (artificial or biologic) to form molecules of factor XIIa. Factor XIIa has 5 substrates. (2) It activates PK into plasma kallikrein (PKa). (3) PKa amplifies factor XII activation into factor XIIa. (4) A cycle of reciprocal activation of factor XIIa (FXIIa) on PK and PKa on factor XII is initiated. Factor XIIa also initiates blood coagulation by activating factor XI to factor XIa, contributes to fibrinolysis by activating plasminogen to plasmin, and interacts with the classical complement system by activating C1r and C1s to their active forms. Not shown on the figure, factor XIIa can cleave HK to form cHK and liberate BK. Formed plasma kallikrein also cleaves HK to liberate bradykinin and cHK. Formed bradykinin binds to its 2 G protein–coupled receptors, B2R and B1R to influence vascular homeostasis in health and disease. B2R and B1R, bradykinin B2 and B1 receptors, respectively; HK, intact high‐molecular‐weight kininogen; HKc, cleaved high‐molecular‐weight kininogen; PK, prekallikrein; PKa, plasma kallikrein; polyP, polyphosphate
The formation of PKa by FXIIa activation or any other means initiates what has been called the kallikrein/kinin system (Figure 1). PKa cleaves HK to liberate bradykinin and results in the formation of kininogen fragments (ie, cHK). Bradykinin influences vascular biology by binding to its G protein–coupled receptors, the bradykinin B2 and B1 receptors (Figure 1). The archetype PKa/kinin system activation disorder is acute attacks of hereditary angioedema (HAE).39 HAE syndromes appear to be a group of disorders due to either a deficiency or defect in C1 inhibitor (most commonly recognized [types I and II]) or to a gain of function in FXIIa (Type III), plasmin, or angiopoietin 1.40, 41 In type I HAE that consists of C1 inhibitor deficiency, both PK and HK are consumed, but FXII levels are not altered.39 To date, all these different mechanisms with the exception of the gain‐in‐function angiopoietin I mechanism, result in increased local bradykinin delivery to tissues. Blockade of the bradykinin B2 receptor alleviates symptoms in many patients.42 Importantly, inhibition of active enzymes of the CAS by C1 inhibitor replacement or inhibition of PKa by a monoclonal antibody or nonpeptide mimetic also ameliorates and prevents type I and II HAE in the majority of patients.43, 44, 45 Major questions in understanding the pathogenesis of acute attacks of HAE remain and include the relative roles of FXIIa versus PKa for bradykinin generation and the inciting etiologies for the initiation of acute attacks of the disorder.
Contact activation also is a major participant in gram‐negative sepsis and sepsis from any cause. In Escherichia coli infections in baboons, FXII activation was associated with the hypotension seen in that disorder.46 In this primate animal model, contact activation mainly provoked changes in vascular homeostasis due to bradykinin liberation and not blood coagulation activation through activation of FXI. In disorders such as prostate cancer and Alzheimer disease, there is evidence for a constitutively higher state of contact activation as a result of increased circulating cancer cell exosomes and denatured protein, respectively, in mouse models and human plasma samples.27, 47 The largest single treatment‐related contact activation is the exposure of patients to medical devices such as cardiopulmonary bypass, renal hemodialysis, extracorporeal membrane oxygenation (ECMO), left ventricular assist devices, and indwelling intravenous catheters. In the developed world, several million patients are exposed to these devices annually. Thrombosis in these devices is a major problem, and the present universal means to prevent device thrombosis is the use of unfractionated heparin from natural sources. In a series of investigations, the Colman and Edmund laboratories show that contact activation occurs in a simulated bypass model and that a variety of PKa inhibitors block it as well as platelet and neutrophil activation.48, 49, 50, 51 A recent proof‐of‐concept study shows that a phage display Fab of an antibody to the catalytic region on FXIIa (3F7) reduced contact activation and fibrin deposition on infant membrane oxygenators used in simulated ECMO in rabbits.52 These data suggest that inhibition of contact activation alone may be sufficient to block device surface‐induced blood coagulation without increasing bleeding risk and exposing patients to the deleterious effects of heparin. To date, the only agent in clinical use that is effective to prevent device and catheter thrombosis is unfractionated heparin.53 However, in experimental investigations, heparin along with several FXIIa inhibitors, corn trypsin inhibitor, and antisense oligonucleotides to FXIIa were effective at preventing catheter thrombosis.53, 54 The approach to introduce FXIIa inhibitors as an alternative means to prevent complications of medical devices will be a slow, graduated process in the clinic, as no surgical team with long experience using heparin will immediately eliminate it for a newer agent whose mechanism(s) of action is not fully appreciated. Investigations first will have to show that with contact activation inhibitors, lesser doses of heparin are sufficient to prevent device thrombosis.
3. FXII ZYMOGEN – A PHYSIOLOGIC EFFECTOR OF CELL GROWTH, PROLIFERATION, AND ANGIOGENESIS
Although the enzymatic and coagulant activities of FXIIa have been known for a long time, the influence of zymogen FXII itself on endothelial cell and neutrophil function were only recently appreciated. The thought that FXII has activities in addition to its proteolytic functions was suggested by the recognition of various consensus sequences in the heavy chain of the protein once it was cloned.55, 56 The heavy chain of FXII from the N‐terminus was noted to have a collagen type II fibronectin binding domain, an epidermal growth factor domain, a second fibronectin type I finger, and a second epidermal growth factor domain. Remarkably, in 1993 when hepatocyte growth factor activator (HGFA) was cloned, it was recognized to have a close sequence (39%) and structural homology to FXII.57 Hepatocyte growth factor is a zymogen that is activated to a serine protease by thrombin, FXIIa, and PKa. It circulates in plasma and has a role in tissue repair and angiogenesis.58 Further, both of these proteins also are structurally homologous to single‐chain urokinase and tissue‐type plasminogen activator (t‐PA).59 Additionally, both HGFA and FXIIa activate hepatocyte growth factor.58, 59
The first functional data that predict FXII's influence on cell biology was the observation by Chien et al60 that FXIIa regulates the expression of monocyte Fc gamma R1 receptors. The authors assumed that this effect was a result of the enzymatic activity of the protein. To their surprise, zymogen FXII also upregulated the receptor's expression.60 Independent studies also indicated that FXII and FXIIa stimulated mitogenesis through mitogen‐activated protein kinase in HepG2 cells.61, 62
4. INFLUENCE OF FXII ON VASCULAR BIOLOGY
Unlike other contact system proteins, FXII is not expressed in cultured human umbilical vein and microvascular endothelial cells. However, labeled FXII specifically binds to cultured endothelial cells (ECs) when grown in monolayers on microtiter plates or when in suspension in the presence of ≥7 to 10 μmol/L free Zn2+.63 Activated platelets release sufficient Zn2+ to support FXII binding to EC.63 Zymogen FXII has a colocalized, multiprotein binding site on cultured ECs that consists of at least urokinase plasminogen activator receptor (uPAR, CD87), gC1qR, and cytokeratin 1.63, 64 These proteins also are the same putative receptors for HK on EC.33, 63, 64 HK, along with FXII, soluble uPAR, cytokeratin 1, peptide Y39HKCTHKGR47 (YHK9) from the collagen type II fibronectin domain on FXII or, to a lesser extent, vitronectin block labeled FXII binding to ECs.64
ECs bind FXII in the presence of Zn2+ to stimulate pERK1/2 and pAktS473. These events are blocked by the mitogen‐activated protein/ERK kinase 1 inhibitor PD98059, the PI3 kinase inhibitor LY294002, antibody 3B10 to uPAR's domain 2, and peptide YHK9 from FXII.65 Peptide FXII signaling in ECs is much faster than bound FXII autoactivation. It occurs within 7 minutes vs autoactivation that takes 60 to 120 minutes. Zymogen FXII and Zn2+ are sufficient for pERK1/2 or pAkt S473 formation. FXII‐induced pAktS473 in ECs is downstream from pERK1/2 because in cells with a subunit deficiency in mTOR, pAktS473 is blocked but pERK1/2 is not.65 In peptide mapping studies, 2 overlapping 20 amino acid peptides from domain 2 of uPAR, L166RG20 and P176GS20, inhibit FXII binding to EC.65 Using other peptides as inhibitors, FXII also interacts with uPAR domains 1 and 3 as well.65 Peptides to the integrin binding site on uPAR domain 2 (NLD9) and uPAR binding site on β‐propeller subunit of the β1 integrin (IQE13) also block FXII signaling.65, 66, 67 Additionally, antibodies to β1 integrin (6S6), an epidermal growth factor antagonist AG1478, and a siRNA to vascular endothelial growth factor (VEGF) R2 (KDR) also interfere with this signaling pathway65 (Figure 2).
Figure 2.
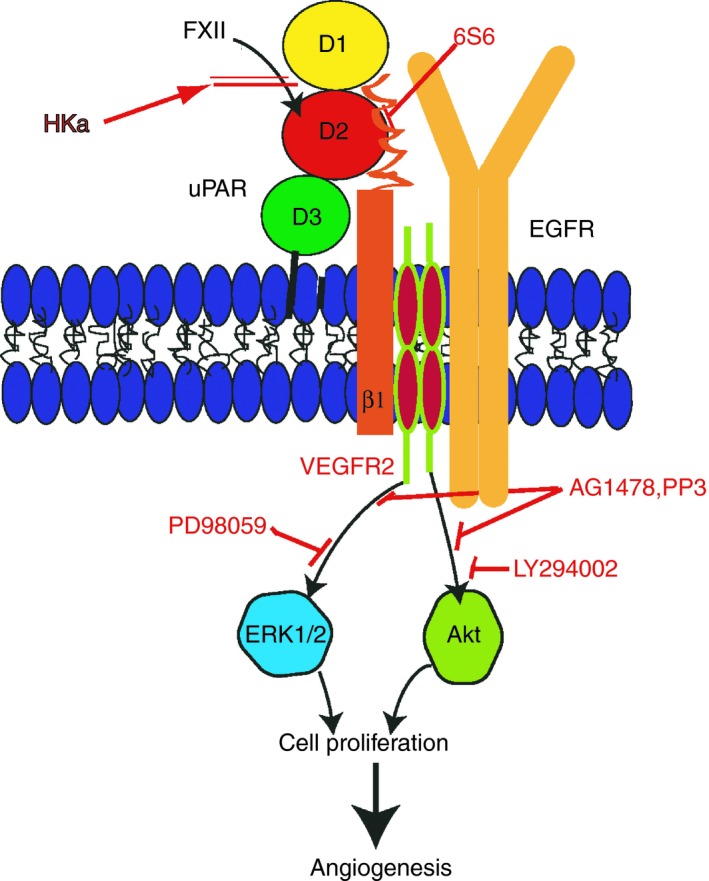
Factor XII signaling pathways leading to angiogenesis. Factor XII (FXII) binds to uPAR domain 2 (D2) to set up a signaling cascade resulting in endothelial cell growth, proliferation, and angiogenesis. High‐molecular‐weight kininogen blocks this pathway by blocking FXII binding to uPAR. uPAR interacts with β1 integrins and this pathway is blocked by mab 6S6. Through mechanisms not completely known, the integrin interacts with endothelial cell growth factor receptor (EGFR). EGFR kinase inhibitors AG1478 and PP3 block this pathway. VEGFR2 also blocks FXII signaling in part, in this pathway. This pathway leads to pERK1/2 and pAktS473 expression. The MEK inhibitor PD98059 and the PI3 kinase inhibitor LY294002 block these phosphoproteins, respectively. Finally, unimpeded this signaling pathway leads to cell proliferation and angiogenesis. Any of the inhibitors enumerated above will block this process. FXII, factor XII; uPAR, urokinase plasminogen activator receptor
The sum of these investigations shows that FXII has a sophisticated signaling system. FXII binding to uPAR signals through β1 integrins and the epidermal growth factor receptor (EGFR) to influence ERK1/2 and Akt phosphorylation. VEGFR2 further modifies this pathway but does not completely regulate FXII‐induced signaling (Figure 2). Our studies also show that this pathway is mediated through β1 integrins, but other integrins may be operative as well.
The consequence of FXII signaling is stimulation of EC growth and proliferation in vitro. EC growth and proliferation are blocked by the same inhibitors to signaling, PD98059, LY294002, LRG20, and 6S6 and analog peptides from HK that compete FXII binding to uPAR.65 This pathway on EC influences neoangiogenesis in vivo. FXII or VEGF stimulates vessel rings to sprout in vitro.65 In matrigel plugs applied to the flank of mice, FXII stimulates new vessel growth in wild‐type (WT) but not in uPAR deleted (plaur −/−) mice.65 Finally, f12 −/− mice have fewer skin vessels constitutively and after 9 days of repair following a skin punch biopsy injury.65 These combined data indicate that FXII has a previously unappreciated role in vessel growth and repair angiogenesis mediated through uPAR or CD87.
5. INFLUENCE OF FXII ON VASCULAR SMOOTH MUSCLE CELLS AND FIBROBLASTS
FXII also stimulates pERK1/2 in cultured vascular smooth muscle cells.68 In human lung fibroblasts, TGF‐β1 induces p‐JNK and translocation of SMAD‐3 to the nucleus to upregulate FXII expression.69 FXII upregulation is associated with increased pERK1/2, pAkt, and p38 since a JNK inhibitor and JNK and SMAD‐3 antisense oligonucleotides did not block them.69 In adult respiratory distress syndrome (ARDS), FXII levels are increased and colocalize with lung fibroblasts.69 In bronchoalveolar lavage fluid from ARDS patients and controls, FXII levels are elevated, and the higher levels correlate with survival outcomes.70 High FXII levels are associated with higher interleukin (IL)‐8, IL‐1β, IL‐6, leukemia inhibitory factor, CXCL5, and tumor necrosis factor‐α levels in human precision cut lung slices (PCLSs).70 FXII also stimulates pERK1/2 and pAkt in PCLSs.70
6. INFLUENCE OF FXII ON NEUTROPHIL FUNCTION
John Rebuck recognized that in skin windows placed on an FXII‐deficient patient, there was less neutrophil migration.71 The Colman laboratory recognized that FXIIa, but not FXII, has the ability to aggregate washed human neutrophils and cause degranulation.72 This function, however, requires an intact heavy chain of FXII because βFXIIa, that is, the form of FXIIa without its heavy chain, does not have the ability to activate neutrophils.72 We examined sterile wound punch biopsy specimens and observed that on days 2 and 5, f12 −/− mice have less neutrophil migration into the healing wounds.3 Similarly, after thioglycolate‐induced sterile peritonitis, f12 −/− mice have reduced peritoneal exudative cells than normal mice.3 Human and mouse neutrophils express FXII, which becomes secreted following cell activation.3
Isolated FXII in the presence of 10 μmol/L Zn2+ binds uPAR on surface plasmon resonance with a KD of 37 ± 29 nmol/L. The major regulator of FXII's interaction with neutrophils, like ECs, is the ambient local zinc ion concentration. Constitutively in the intravascular compartment, there is insufficient free Zn2+ to support FXII binding. Only when the local free Zn2+ concentration becomes 7 to 10 μmol/L, as in the case when there is adjacent platelet activation, can binding occur.63 On WT and f12 −/− neutrophils, zymogen FXII in the presence of 10 μmol/L Zn2+stimulates pAktS473 and pAktS474 (pAkt2), and these reactions are blocked by wortmanin; LY294002; an Akt2 inhibitor (Akti XII); and an intracellular Zn2+ sequestering agent, TPEN.3 FXII Locarno and a double FXII mutant with R353P and S544A mutations equally stimulated neutrophil pAktS474, indicating that all FXII activity on cells does not require its catalytic activity and is a zymogen FXII function. FXII stimulation of neutrophils initiates 3 major events via Akt2: αMβ2 expression, calcium mobilization, and histone H3 citrullination, NET formation over time3 (Figure 3). Murine f12 −/− neutrophils adhere less to fibrinogen and have less F‐Met‐Leu‐Phe–induced migration. FXII itself stimulates migration and chemotaxis and murine f12 −/− neutrophils. In fact, FXII is a more potent chemotaxin than F‐Met‐Leu‐Phe with normal and f12 −/− neutrophils. With plaur −/− neutrophils, FXII does not stimulate chemotaxis.3
Figure 3.
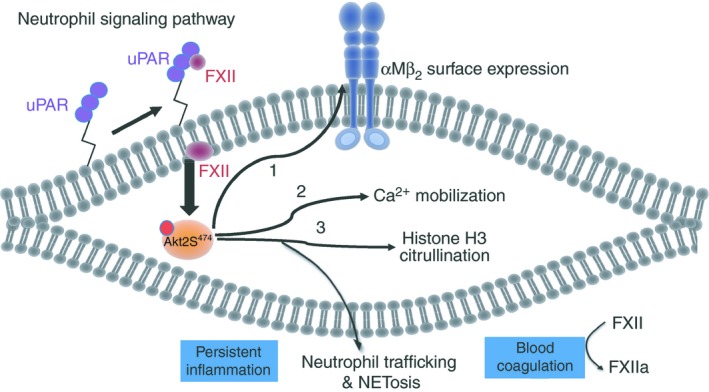
Factor XII signaling pathways in neutrophil activation. The neutrophil FXII‐uPAR interaction leads to phosphorylation of pAkt2 at S474, the major Akt isoform in these cells. pAkt2 phosphorylation has 3 specific effects in neutrophils. First, it promotes the surface expression of αMβ2 integrin that is essential for adhesion, migration and chemotaxis, basic neutrophil functions. Second, it increases intracellular calcium allowing for neutrophil cell‐directed movement. Third, it stimulates citrullination of histone H3 and NETosis. These combined events contribute to neutrophil priming, persistent inflammation at sites of injury, and facilitate further plasma FXIIa generation and blood coagulation. FXII, factor XII; uPAR, urokinase plasminogen activator receptor
These FXII activities translate into in vivo functions. Sterile punch biopsy wounds on f12 −/− mice heal faster than those of WT mice.3 The accelerated wound healing observed correlates with significantly less neutrophil elastase and NETs in the wound milieu.3 When wild‐type bone marrow is transplanted into f12 −/− mice, the degree of peritoneal exudative cells after thioglycolate‐induced inflammation increases to that observed when WT bone marrow is transplanted to WT mice. Likewise, when f12 −/− bone marrow is transplanted into normal mice, there are fewer peritoneal exudative cells after thioglycolate‐induced inflammation like f12 −/− bone marrow transplanted in f12 −/− mice.3 Similar events occur with wound healing after sterile punch biopsies. Bone marrow from f12 −/− mice into normal or f12 −/− murine hosts results in faster wound healing than WT bone marrow transplanted into WT or f12 −/− mice hosts.
These observations have led to the hypothesis that FXII has a complex relationship with neutrophils. Either plasma FXII or released neutrophil FXII has the ability to bind to uPAR on the neutrophil surface to promote neutrophil adhesion, migration, and chemotaxis (Figure 3). FXII also stimulates neutrophil extrusion of NETs that contribute to inflammation and thrombosis in the milieu where these events are taking place.
Recent investigations on dendritic cells also indicate that FXII has a role in neuroinflammation.73 Patients with multiple sclerosis (MS) have been observed to have high levels of circulating FXII during relapse. When a murine model of experimental autoimmune encephalomyelitis, a model of MS, is induced in f12 −/− mice, there is less inflammation, suggesting that FXII mediates the process. These findings are consistent with our neutrophil studies, as dendritic cells and immune activation by FXII mediated through uPAR (CD87) must proceed through a related intracellular signaling pathway.73
7. SUMMARY
Factor XII autoactivates into a pathophysiologic response agent, FXIIa, when it interacts with nonhuman (infectious agents) or human tissue (vessel wall collagen, DNA, RNA, polyphosphate, exosomes). Also, FXIIa is formed when human plasma courses over artificial medical devices. Inhibitors to FXIIa should be useful to ameliorate the effects of these pathophysiologic interactions. Alternatively, zymogen FXII has biologic activity related to cell protection and repair. In endothelium, FXII promotes neoangiogenesis and wound repair via uPAR and β1 integrins. In neutrophils, FXII promotes adhesion, migration, chemotaxis, and NETosis via uPAR and β2 integrins. Considering that FXII has more receptors than uPAR and uPAR interacts with more integrins than β1, additional interactions should be discovered.
8. FUTURE PERSPECTIVES
These data suggest an exciting future for FXII and its related proteins. Because deficiencies of FXII, PK, and HK are not associated with bleeding but each deficiency reduces thrombosis risk, there are good opportunities to use FXIIa inhibitors (intravenous or oral) to prevent contact activation that is associated with all medical devices and acute attacks of HAE. Use of these agents has the potential to reduce thrombosis risk without bleeding on medical devices and in activated FXII states (eg, type III angioedema) associated with HAE. Alternatively, inhibition of FXII, the zymogen, may be useful in sterile inflammatory states such as wound healing in patients with diabetes and other debilitating states.3 In sum, FXIIa and FXII inhibitors in the future may be useful in the management of many disorders such as induced thrombosis on man‐made devices and preventing inflammation in a variety of disorders.
RELATIONSHIP DISCLOSURES
Dr. Schmaier receives research grants from NIH (HL126645, AI130131; HL144113; HL143402; CA223301), DOD (BC150596P1), Shire, and Cleveland Clinic Foundation Velosano. Dr. Stavrou is supported by NIH (HL137695), the American Heart Association (17GRNT33661005), and the Oscar D. Ratnoff Endowed Professorship.
AUTHOR CONTRIBUTIONS
AHS wrote the first draft; EXS edited all versions of the manuscript, and both authors contributed figures.
Schmaier AH, Stavrou EX. Factor XII – What's important but not commonly thought about. Res Pract Thromb Haemost. 2019;3:599–606. 10.1002/rth2.12235
REFERENCES
- 1. Revak SD, Cochrane CG, Johnston AR, Hugli TE. Structural changes accompanying enzymatic activation of human Hageman factor. J Clin Invest. 1974;54:619–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saito H, Ratnoff OD, Pensky J. Radioimmunoassay of human Hageman factor (factor XII). J Lab Clin Med. 1976;88:506–14. [PubMed] [Google Scholar]
- 3. Stavrou EX, Fang C, Bane KL, Long AT, Naudin C, Kucukal E, et al. Factor XII and uPAR upregulate neutrophil functions to influence wound healing. J Clin Invest. 2018;128:944–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ratnoff OD, Colopy JE. A familial hemorrhagic trait associated with a deficiency of a clot‐promoting fraction of plasma. J Clin Invest. 1955;34:602–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Langdell RD, Wagner RH, Brinkhous KM. Effect of antihemophilic factor on one‐stage clotting tests; a presumptive test for hemophilia and a simple one‐stage antihemophilic factor assy procedure. J Lab Clin Med. 1953;41:637–47. [PubMed] [Google Scholar]
- 6. Quick AJ. The thromboplastin reagent for the determination of prothrombin. Science. 1940;92:113–4. [DOI] [PubMed] [Google Scholar]
- 7. Ratnoff OD, Davie EW. The purification of activated Hageman factor (activated factor XII). Biochemistry. 1962;1:967–75. [DOI] [PubMed] [Google Scholar]
- 8. Ratnoff OD, Davie EW, Mattlett DL. Studies on the action of Hageman factor: evidence that activated Hageman factor in turn activates plasma thromboplastin antecedent. J Clin Invest. 1961;40:803–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ratnoff OD, Davie EW. The activation of Christmas factor (factor IX) by activated plasma thromboplastin antecedent (activated factor XI). Biochemistry. 1962;1:677–85. [Google Scholar]
- 10. Davie EW, Ratnoff OD. Waterfall sequence for intrinsic blood coagulation. Science. 1964;145:1310–2. [DOI] [PubMed] [Google Scholar]
- 11. MacFarland RG. An enzyme cascade in the blood clotting mechanism, and its function as a biochemical amplifier. Nature. 1964;202:498–9. [DOI] [PubMed] [Google Scholar]
- 12. Wuepper KD. Prekallikrein deficiency in man. J Exp Med. 1973;138:1345–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Colman RW, Bagdasarian A, Talamo RC, Scott CF, Seavey M, Guimaraes JA, et al. Williams trait. Human kininogen deficiency with diminished levels of plasminogen proactivator and prekallikrein associated with abnormalities of the Hageman factor‐dependent pathways. J Clin Invest. 1975;56:1650–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nemerson Y, Pitlick FA. Purification and characterization of the protein component of tissue factor. Biochemistry. 1970;9:5100–5. [DOI] [PubMed] [Google Scholar]
- 15. Osterud B, Berre A, Otnass AB, Bjorklid E, Prydz H. Activation of the coagulation factor VII by tissue thromboplastin and calcium. Biochemistry. 1972;11:2853–7. [DOI] [PubMed] [Google Scholar]
- 16. Fair DS, MacDonald MJ. Cooperative interaction between factor VII and cell surface‐expressed tissue factor. J Biol Chem. 1987;262:11692–8. [PubMed] [Google Scholar]
- 17. Naito K, Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J Biol Chem. 1991;266:7353–8. [PubMed] [Google Scholar]
- 18. Gailani D, Broze GJ Jr. Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–12. [DOI] [PubMed] [Google Scholar]
- 19. Renné T, Pozgajová M, Grüner S, Schuh K, Pauer HU, Burfeind P, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202:271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Merkulov S, Zhang WM, Komar AA, Schmaier AH, Barnes E, Zhou Y, et al. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood. 2008;111:1274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stavrou EX, Fang C, Merkulova A, Alhalabi O, Grobe N, Antoniak S, et al. Reduced thrombosis in Klkb1−/− mice is mediated by increased Mas receptor, prostacyclin, Sirt1, and KLF4 and decreased tissue factor. Blood. 2015;125:710–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci U S A. 2007;104:6388–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maas C, Govers‐Riemslag JW, Bouma B, Schiks B, Hazenberg BP, Lokhorst HM, et al. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest. 2008;118:3208–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van der Meijden PE, Munnix IC, Auger JM, Govers‐Riemslag JW, Cosemans JM, Kuijpers MJ, et al. Dual role of collagen in factor XII‐dependent thrombus formation. Blood. 2009;114:881–90. [DOI] [PubMed] [Google Scholar]
- 25. Müller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139:1143–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zamolodchikov D, Chen ZL, Conti BA, Renné T, Strickland S. Activation of the factor XII‐driven contact system in Alzheimer's disease patient and mouse model plasma. Proc Natl Acad Sci U S A. 2015;112:4068–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yamamoto‐Imoto H, Zamolodchikov D, Chen ZL, Bourne SL, Rizvi S, Singh P, et al. A novel detection method of cleaved plasma high‐molecular‐weight kininogen reveals its correlation with Alzheimer's pathology and cognitive impairment. Alzheimers Dement (Amst). 2018;10:480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Colman RW, Schmaier AH. Contact activation: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood. 1997;90:3819–43. [PubMed] [Google Scholar]
- 30. Schmaier AH, McCrae KR. The plasma kallikrein‐kinin system: its evolution from contact activation. J Thromb Haemost. 2007;5:2323–9. [DOI] [PubMed] [Google Scholar]
- 31. Stavrou E, Schmaier AH. Factor XII: what does it contribute to our understanding of the physiology and pathophysiology of hemostasis & thrombosis. Thromb Res. 2010;125:210–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Renné T, Schmaier AH, Nickel KF, Blombäck M, Maas C. In vivo roles of factor XII. Blood. 2012;120:4296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost. 2016;14:28–39. [DOI] [PubMed] [Google Scholar]
- 34. Tankersley DL, Finlayson JS. Kinetics of activation and autoactivation of human factor XII. Biochemistry. 1984;23:273–9. [DOI] [PubMed] [Google Scholar]
- 35. Schmaier AH. Contact activation In: Losalzo J, Schafer AI, editors Thrombosis and hemorrhage. 2nd ed Baltimore: Williams & Wilkins, 1998; p. 105–27. [Google Scholar]
- 36. Goldsmith GH Jr, Saito H, Ratnoff OD. The activation of plasminogen by Hageman factor (factor XII) and Hageman factor fragments. J Clin Invest. 1978;62:54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ghebrehiwet B, Silverberg M, Kaplan AP. Activation of the classical pathway of complement by Hageman factor fragment. J Exp Med. 1981;153:665–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ghebrehiwet B, Randazzo BP, Dunn JT, Silverberg M, Kaplan AP. Mechanisms of activation of the classical pathway of complement by Hageman factor fragment. J Clin Invest. 1983;71:1450–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schapira M, Silver LD, Scott CF, Schmaier AH, Prograis LJ Jr, Curd JG, et al. Prekallikrein activation and high‐molecular‐weight kininogen consumption in hereditary angioedema. N Engl J Med. 1983;308:1050–3. [DOI] [PubMed] [Google Scholar]
- 40. Schmaier AH. The hereditary angioedema syndromes. J Clin Invest. 2018;129:66–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bafunno V, Firinu D, D'Apolito M, Cordisco G, Loffredo S, Leccese A, et al. Mutation of the angiopoietin‐1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol. 2018;141:1009–17. [DOI] [PubMed] [Google Scholar]
- 42. Cicardi M, Banerji A, Bracho F, Malbrán A, Rosenkranz B, Riedl M, et al. Icatibant, a new bradykinin‐receptor antagonist, in hereditary angioedema. N Engl J Med. 2010;363:532–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Banerji A, Riedl MA, Bernstein JA, Cicardi M, Longhurst HJ, Zuraw BL, et al.; HELP Investigators . Effect of lanadelumab compared with placebo on prevention of hereditary angioedema attacks: a randomized clinical trial. JAMA. 2018;320:2108–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Banerji A, Busse P, Shennak M, Lumry W, Davis‐Lorton M, Wedner HJ, et al. Inhibiting plasma kallikrein for hereditary angioedema prophylaxis. N Engl J Med. 2017;376:717–28. [DOI] [PubMed] [Google Scholar]
- 45. Aygören‐Pürsün E, Bygum A, Grivcheva‐Panovska V, Magerl M, Graff J, Steiner UC, et al. Oral plasma kallikrein inhibitor for prophylaxis in hereditary angioedema. N Engl J Med. 2018;379:352–62. [DOI] [PubMed] [Google Scholar]
- 46. Pixley RA, De La Cadena R, Page JD, Kaufman N, Wyshock EG, Chang A, et al. The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia. In vivo use of a monoclonal anti‐factor XII antibody to block contact activation in baboons. J Clin Invest. 1993;91:61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nickel KF, Ronquist G, Langer F, Labberton L, Fuchs TA, Bokemeyer C, et al. The polyphosphate‐factor XII pathway drives coagulation in prostate cancer‐associated thrombosis. Blood. 2015;126:1379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wachtfogel YT, Harpel PC, Edmunds LH Jr, Colman RW. Formation of C1s‐C1‐inhibitor, kallikrein‐C1‐inhibitor, and plasmin‐alpha 2‐plasmin‐inhibitor complexes during cardiopulmonary bypass. Blood. 1989;73:468–71. [PubMed] [Google Scholar]
- 49. Wachtfogel YT, Kucich U, Hack CE, Gluszko P, Niewiarowski S, Colman RW, et al. Aprotinin inhibits the contact, neutrophil, and platelet activation systems during simulated extracorporeal perfusion. J Thorac Cardiovasc Surg. 1993;106:1–9; discussion 9–10. [PubMed] [Google Scholar]
- 50. Wachtfogel YT, Bischoff R, Bauer R, Hack CE, Nuijens JH, Kucich U, et al. Alpha 1‐antitrypsin Pittsburgh (Met358–>Arg) inhibits the contact pathway of intrinsic coagulation and alters the release of human neutrophil elastase during simulated extracorporeal circulation. Thromb Haemost. 1994;72:843–7. [PubMed] [Google Scholar]
- 51. Wachtfogel YT, Kettner C, Hack CE, Nuijens JH, Reilly TM, Knabb RM, et al. Thrombin and human plasma kallikrein inhibition during simulated extracorporeal circulation block platelet and neutrophil activation. Thromb Haemost. 1998;80:686–91. [PubMed] [Google Scholar]
- 52. Larsson M, Rayzman V, Nolte MW, Nickel KF, Björkqvist J, Jämsä A, et al. A factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Sci Transl Med. 2014;6(222):222ra17. [DOI] [PubMed] [Google Scholar]
- 53. Yau JW, Stafford AR, Liao P, Fredenburgh JC, Roberts R, Weitz JI. Mechanism of catheter thrombosis: comparison of the antithrombotic activities of fondaparinux, enoxaparin, and heparin in vitro and in vivo. Blood. 2011;118:6667–74. [DOI] [PubMed] [Google Scholar]
- 54. Yau JW, Liao P, Fredenburgh JC, Stafford AR, Revenko AS, Monia BP, et al. Selective depletion of factor XI or factor XII with antisense oligonucleotides attenuates catheter thrombosis in rabbits. Blood. 2014;123:2102–7. [DOI] [PubMed] [Google Scholar]
- 55. Cool DE, Edgell CJ, Louie GV, Zoller MJ, Brayer GD, MacGillivray RT. Characterization of human blood coagulation factor XII cDNA. Prediction of the primary structure of factor XII and the tertiary structure of beta‐factor XIIa. J Biol Chem. 1985;260:13666–76. [PubMed] [Google Scholar]
- 56. Cool DE, MacGillivray RT. Characterization of the human blood coagulation factor XII gene. Intron/exon gene organization and analysis of the 5′‐flanking region. J Biol Chem. 1987;262:13662–73. [PubMed] [Google Scholar]
- 57. Miyazawa K, Shimomura T, Kitamura A, Kondo J, Morimoto Y, Kitamura N. Molecular cloning and sequence analysis of the cDNA for a human serine protease responsible for activation of hepatocyte growth factor. Structural similarity of the protease precursor to blood coagulation factor XII. J Biol Chem. 1993;268:10024–8. [PubMed] [Google Scholar]
- 58. Fukushima T, Uchiyama S, Tanaka H, Kataoka H. Hepatocyte growth factor activator: a proteinase linking tissue injury with repair. Int J Mol Sci. 2018;19:3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shimomura T, Miyazawa K, Komiyama Y, Hiraoka H, Naka D, Morimoto Y, et al. Activation of hepatocyte growth factor by two homologous proteases, blood‐coagulation factor XIIa and hepatocyte growth factor activator. Eur J Biochem. 1995;229:257–61. [DOI] [PubMed] [Google Scholar]
- 60. Chien P, Pixley RA, Stumpo LG, Colman RW, Schreiber AD. Modulation of the human monocyte binding site for monomeric immunoglobulin G by activated Hageman factor. J Clin Invest. 1988;82:1554–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schmeidler‐Sapiro KT, Ratnoff OD, Gordon EM. Mitogenic effects of coagulation factor XII and factor XIIa on HepG2 cells. Proc Natl Acad Sci U S A. 1991;88:4382–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gordon EM, Venkatesan N, Salazar R, Tang H, Schmeidler‐Sapiro K, Buckley S, et al. Factor XII‐induced mitogenesis is mediated via a distinct signal transduction pathway that activates a mitogen‐activated protein kinase. Proc Natl Acad Sci U S A. 1996;93:2174–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mahdi F, Madar ZS, Figueroa CD, Schmaier AH. Factor XII interacts with the multiprotein assembly of urokinase plasminogen activator receptor, gC1qR, and cytokeratin 1 on endothelial cell membranes. Blood. 2002;99:3585–96. [DOI] [PubMed] [Google Scholar]
- 64. Mahdi F, Shariat‐Madar Z, Todd RF 3rd, Figueroa CD, Schmaier AH. Expression and colocalization of cytokeratin 1 and urokinase plasminogen activator receptor on endothelial cells. Blood. 2001;97:2342–50. [DOI] [PubMed] [Google Scholar]
- 65. LaRusch GA, Mahdi F, Shariat‐Madar Z, Adams G, Sitrin RG, Zhang WM, et al. Factor XII stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFR to initiate angiogenesis. Blood. 2010;115:5111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Degryse B, Resnati M, Czekay RP, Loskutoff DJ, Blasi F. Domain 2 of the urokinase receptor contains an integrin‐interacting epitope with intrinsic signaling activity: generation of a new integrin inhibitor. J Biol Chem. 2005;280:24792–803. [DOI] [PubMed] [Google Scholar]
- 67. Wei Y, Tang CH, Kim Y, Robillard L, Zhang F, Kugler MC, et al. Urokinase receptors are required for alpha 5 beta 1 integrin‐mediated signaling in tumor cells. J Biol Chem. 2007;282:3929–39. [DOI] [PubMed] [Google Scholar]
- 68. Fernando AN, Fernando LP, Fukuda Y, Kaplan AP. Assembly, activation, and signaling by kinin‐forming proteins on human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2005;289:H251–7. [DOI] [PubMed] [Google Scholar]
- 69. Jablonska E, Markart P, Zakrzewicz D, Preissner KT, Wygrecka M. Transforming growth factor‐β1 induces expression of human coagulation factor XII via Smad3 and JNK signaling pathways in human lung fibroblasts. J Biol Chem. 2010;285:11638–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hess R, Wujak L, Hesse C, Sewald K, Jonigk D, Warnecke G, et al. Coagulation factor XII regulates inflammatory responses in human lungs. Thromb Haemost. 2017;117:1896–907. [DOI] [PubMed] [Google Scholar]
- 71. Rebuck JW. The skin window as a monitor of leukocytic functions in contact activation factor deficiencies in man. Am J Clin Pathol. 1983;79:405–13. [DOI] [PubMed] [Google Scholar]
- 72. Wachtfogel YT, Pixley RA, Kucich U, Abrams W, Weinbaum G, Schapira M, et al. Purified plasma factor XIIa aggregates human neutrophils and causes degranulation. Blood. 1986;67:1731–7. [PubMed] [Google Scholar]
- 73. Göbel K, Pankratz S, Asaridou CM, Herrmann AM, Bittner S, Merker M, et al. Blood coagulation factor XII drives adaptive immunity during neuroinflammation via CD87‐mediated modulation of dendritic cells. Nat Commun. 2016;7:11626. [DOI] [PMC free article] [PubMed] [Google Scholar]