

Abstract
Laboratory tests of platelet function are instrumental in studying platelet physiology and inherited or acquired platelet abnormalities. Light transmission aggregometry, developed in the early 1960s, is still considered the gold standard for the identification and diagnosis of platelet function disorders. Since then, novel techniques have been developed, including flow‐based assays and flow cytometry. In this tutorial, we describe the basic methodologies for the preparation of citrated platelet‐rich plasma and washed platelet suspensions and discuss their respective advantages and limitations as well as important factors to consider to perform high‐quality tests of platelet function. In addition, the methodologies of the main platelet function tests (light transmission aggregation, flow‐based assays, and flow cytometric assays) are described, and their respective strengths and limitations are discussed to assess various aspects of platelet biology.
Keywords: flow cytometry, hemostasis, platelet aggregation, platelet function tests, platelet‐rich plasma
Essentials.
High‐quality tests of platelet function are required to study platelet physiology.
Preparation of citrated platelet‐rich plasma and washed platelets is described.
The main platelet function tests are described and discussed.
These tests are essential to identify platelet defects responsible for bleeding syndromes.
1. INTRODUCTION
Hemostatic properties were first ascribed to blood platelets by the Italian pathologist Giulio Bizzozero more than 100 years ago, who observed that distinct cells in human blood were able to form thrombi within damaged areas of the vessel wall.1 Following this seminal work, the key role of platelets in hemostasis and thrombosis was progressively elucidated during the first part of the 20th century with the description of many bleeding disorders due to platelet dysfunction, such as May‐Hegglin anomaly, Glanzmann thrombasthenia and Bernard‐Soulier syndrome.2, 3, 4 It is noteworthy that the only tests available at this time to assess platelet function were a manual platelet count, inspection of a peripheral blood smear, and measurement of the bleeding time, introduced by Duke in 1910.5 A new era in platelet research commenced in the early 1960s with the introduction of the platelet aggregometer by Gustav Born, which revolutionized the study of platelet function, providing fundamental insights into the way platelets behave and respond to various stimuli.6 In the 1970s, flow‐based assays were developed, reproducing a very wide range of physiological and pathological blood flow conditions to better reflect the in vivo situation. Later, in the 1980s, the development of monoclonal antibodies and their use in flow cytometric assays allowed both the phenotypic characterization of blood platelets and the introduction of new functional tests dedicated to platelets.7 More recently, flow cytometry has enabled the assessment of platelet aggregation in whole blood,8, 9 which can also be studied using the impedance measurement technique.10 However, these 2 methods are still not widely used and not yet recommended for the exploration of functional platelet defects. The combined use of these various tools developed since the 1960s now provides research teams and physicians with a valuable set of tests to study platelet physiology and identify functional defects of platelets responsible for bleeding syndromes.
The purpose of this tutorial is to describe the basic methodologies for the preparation of citrated platelet‐rich plasma (cPRP) and washed platelet suspensions, to discuss their respective advantages and limitations as well as important factors to consider to perform high‐quality tests of platelet function. Among these functional tests, we describe the methodologies of light transmission aggregation, flow‐based assays, and flow cytometric assays, with particular emphasis on their respective advantages and limits.
2. PLATELET FUNCTION TESTING
2.1. In vivo test of platelet function: the bleeding time
The bleeding time assay was the first screening test in humans and is still the only one used to assess platelet function in vivo. It consists of measuring the time required for bleeding to stop after making a superficial incision in the patient's skin using Duke's method (standardized puncture of the ear lobe with a lancet)5 or later a more reliable method developed by Ivy, using a blood pressure cuff on the upper arm to maintain constant pressure and an incision on the anterior surface of the forearm.11 Until the early 1990s, this test was still considered to be useful to identify congenital and acquired platelet disorders. However, due to its poor reproducibility, low sensitivity and specificity, and lack of predictive value,12 this assay is no longer used in routine practice and has been replaced by alternative techniques to assess platelet function in vitro as detailed below.
2.2. Light transmission aggregometry
First described in 1962 by Born and O'Brien,6, 13 an aggregometer is a photometer that measures the transmission of light through a thermoregulated vial containing a platelet suspension maintained under constant agitation. This turbidimetric technique is used to measure in vitro the ability of platelets to aggregate with each other in response to various activating agents. The progressive formation of platelet aggregates leads to an increase in light transmission that is characteristic of the agonist used, provided the targeted signaling pathway is functional. Shortly after the introduction of this method, tests with platelets from patients with Glanzmann thrombasthenia showed that these cells were unable to aggregate, regardless of the agonist employed. We now know that this is due to the complete deficiency or absence of functional αIIbβ3, the integrin required for platelet aggregation.14 Since the 1960s, the general principle of this test has not changed, although improvements have been made mostly in regulating the agitation and temperature, or in the light transmission recording device, which are now mainly controlled by specialized electronic components and software. In the late 1970s, a test was developed to simultaneously measure platelet aggregation and quantify the ATP secreted by platelets into the extracellular medium throughout the aggregation process.15, 16 It is based on a bioluminescent quantification of ATP, whereby the latter reacts with luciferin and firefly luciferase to cause light emission. This technique allows the detection of secretion anomalies but does not differentiate between a primary secretion defect and dense granule deficiencies.
Specialized clinical hemostasis laboratories evaluate platelet function in cPRP using light transmission aggregometry (LTA), which is still considered to be the gold standard for the identification and diagnosis of platelet function disorders. Although the preparation of cPRP is rapid and provides a high yield of platelets, the investigation of platelet functions in cPRP harbors certain limitations. The preparation contains an anticoagulant, which modifies the pH and lowers the extracellular calcium concentration to the micromolar range, while the plasma proteins present can play a role in platelet functions. Furthermore, cPRP is stable for no more than 2 hours. To overcome these limitations, platelets can be isolated from blood anticoagulated with acid‐citrate‐dextrose (ACD) using a series of centrifugations and washing steps with suspension in a physiological buffer under well‐defined conditions, as initially described by Mustard's group.17 In the following section, we outline the basic methodologies for the preparation of cPRP and washed platelet suspensions.
2.2.1. Platelet aggregation using human cPRP
Trisodium citrate, which chelates positively charged calcium ions and thereby blocks calcium‐dependent clotting factor reactions, is the standard anticoagulant for platelet aggregometry in cPRP. At a citrate concentration of 0.38% (13 mmol/L, pH 7.5), the calcium level is sufficiently reduced to prevent coagulation, with enough calcium remaining to allow platelet aggregation; 0.32% instead of 0.38% citrate is commonly used in many laboratories. Of note, maximal platelet aggregation is slightly higher in 0.32% compared to 0.38% citrate probably due to the higher concentration of calcium in PRP anticoagulated with 0.32% citrate.18 Therefore, either 0.32% or 0.38% sodium citrate concentration is acceptable, as long as its use is consistent in each center, as recommended by the Platelet Physiology Subcommittee of the Scientific and Standardization Committee of the ISTH.19
The reduced calcium concentration of the plasma is responsible for the occurrence of an artifact called the second wave of aggregation after addition of a critical amount of ADP (Figure 1). This second wave of aggregation occurs not only in cPRP but also in an artificial medium with a low calcium concentration, as shown by Mustard et al.17 It is secondary to activation of the arachidonic acid pathway and the formation of thromboxane A2 (TxA2) and is accompanied by secretion of the contents of both dense and alpha granules. On the other hand, it is blocked by aspirin and other cyclooxygenase‐inhibiting drugs. The occurrence of this second wave of aggregation induced by high concentrations of ADP allows the detection of secretion anomalies and aspirin or nonsteroidal anti‐inflammatory drug (NSAID) intake. In contrast, in a physiological calcium environment (2 mmol/L), that is, in PRP anticoagulated with hirudin or when platelets are suspended in a physiological buffer containing 2 mmol/L calcium, ADP induces only reversible platelet aggregation without secretion, whatever its concentration (Figure 1).
Figure 1.
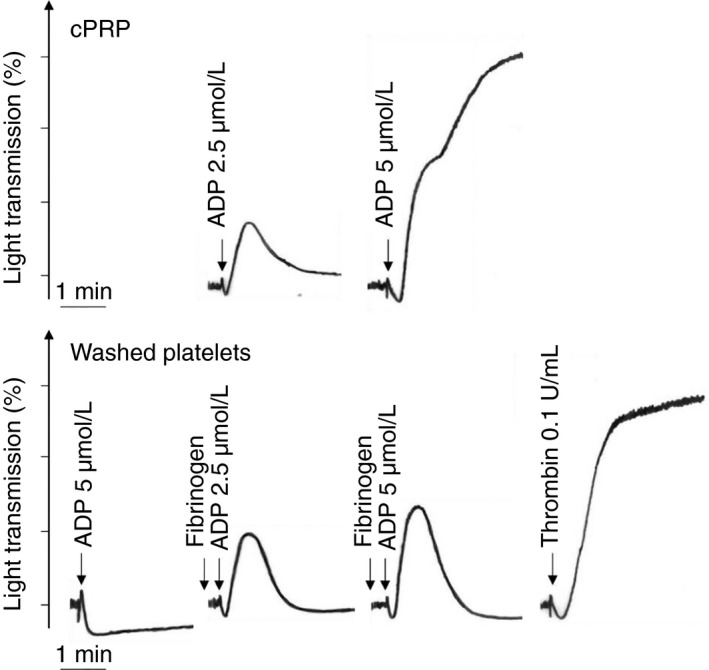
Platelet aggregation profiles to various agonists in citrated PRP or washed platelet suspensions. Citrated PRP: aggregation responses to ADP (2.5 or 5 μmol/L). Washed platelets: the platelets were resuspended in Tyrode's albumin buffer containing calcium (2 mmol/L). Responses to ADP (2.5 or 5 μmol/L) in the absence or presence of fibrinogen (0.25 mg/mL), or thrombin (0.1 U/mL). ADP is provided by Chrono‐Log (Havertown, PA) and bovine thrombin by Sigma. cPRP, citrated platelet‐rich plasma
Preparation of human cPRP
Blood (15 mL) is collected from an antecubital vein, using three 4.5 mL Vacutainer (Becton Dickinson, Franklin Lakes, NJ) tubes (clinical‐grade nonpyrogenic, noncytotoxic, and nuclease‐free borosilicate glass) containing sodium citrate anticoagulant (1 volume of trisodium citrate [3.8%] for 9 volumes of blood). The blood in the 3 Vacutainer tubes is then pooled in a 15‐mL plastic tube (polypropylene or polystyrene), gently mixed, and centrifuged at 250 g in a Sorvall centrifuge (Sorvall RC3BP swinging‐bucket centrifuge with an H6000A‐HBB6 rotor, Kendro Laboratory Products, Les Ulis, France) at room temperature, for a period of time depending on the quantity of blood (Table 1) to obtain cPRP. To maximize the efficiency of the platelet recovery in the upper layer, without contamination due to red blood cells rising from the bottom layer, the centrifugal slow brake must be applied during rotor deceleration. After this centrifugation step, the upper layer containing cPRP is carefully collected with a 10‐mL syringe without disturbing the middle layer containing white blood cells (also called the “buffy coat”). The cPRP is transferred into a new 15‐mL plastic tube, which is capped and allowed to “rest” at room temperature for 15 minutes before platelet testing. The platelet count in cPRP is not adjusted to a standardized level using autologous platelet‐poor plasma (PPP). LTA in cPRP should be completed within a maximum of 4 hours after blood sampling, as recommended by the ISTH guidelines.19
Table 1.
Centrifugation times and relative centrifugal forces (RCFs) for platelet isolation and washing steps
| Volume of fluid (mL) | RCF (g) | RPM | Time (min) | Brake | |
|---|---|---|---|---|---|
| 15‐mL tube | 50‐mL tube | ||||
| Human blood | |||||
| 15 | 50 | 250 | 926 | 16 | No |
| 10 | 40 | 250 | 926 | 15 | No |
| 6 | 25 | 250 | 926 | 13 | No |
| Human PRP | |||||
| 10 | 40 | 2200 | 2749 | 16 | Yes |
| 9 | 35 | 2200 | 2749 | 15 | Yes |
| 8 | 30 | 2200 | 2749 | 14 | Yes |
| 6.5 | 25 | 2200 | 2749 | 13 | Yes |
| 5 | 20 | 2200 | 2749 | 12 | Yes |
| 4.5 | 15 | 2200 | 2749 | 10 | Yes |
| Washing steps | |||||
| 0 | 40 | 1900 | 2254 | 8 | Yes |
| 8 | 30 | 1900 | 2254 | 8 | Yes |
Centrifugation times depend on the volume of fluid; the time and relative centrifugal force (g) are given here for the Sorvall RC3BP centrifuge with an H6000A‐HBB6/HLR6 rotor (diameter: 55.4 cm).
Abbreviations: PRP, platelet‐rich plasma; RCF, relative centrifugal forces.
2.2.2. Platelet aggregation using washed human platelets
To study the intrinsic properties of human platelets, use of washed platelets in suspension in a physiological buffer is essential to isolate the cells from the anticoagulant and their plasma environment, in particular the thrombin, which can be generated during blood collection and the preparation of PRP. Furthermore, suspending platelets in an artificial medium allows one to manipulate the inorganic ions, proteins, and other constituents in the suspension. Human platelets are isolated by sequential centrifugation and resuspension in Tyrode's albumin buffer, an iso‐osmotic phosphate buffer at pH 7.35 containing calcium (2 mmol/L), magnesium (1 mmol/L), and glucose (0.1%, w/v) and supplemented with human serum albumin (HSA) (0.35%, w/v), using the technique developed by Mustard's group17 and slightly modified by Cazenave's group,20 which replaced prostaglandin E1 by prostaglandin I2 (PGI2) and used human instead of bovine albumin.
Sodium citrate (0.38%, 13 mmol/L, pH7.5)–anticoagulated blood cannot be used to prepare washed platelets, as there remains sufficient residual calcium to allow thrombin generation and platelet activation during centrifugation of cPRP. ACD (22 mmol/L trisodium citrate, pH 6.5) is the anticoagulant of choice for blood collection to prepare washed platelets. ACD and 3.8% trisodium citrate solutions differ markedly in pH, ACD‐anticoagulated blood having a pH of 6.5 and sodium citrate–anticoagulated blood a pH of 7.5. They also differ in the citrate concentration, which is higher in ACD‐anticoagulated (22 mmol/L) as compared to citrate‐anticoagulated (13 mmol/L) blood. At this acidic pH and with a lower extracellular calcium concentration, platelet aggregation is totally abolished in ACD‐anticoagulated blood.21
To isolate the platelets without activating them, PGI2, also called prostacyclin, is added during the preparation steps to transiently prevent any platelet activation. Addition of heparin further prevents the generation of traces of thrombin. At the end of the washing procedure, apyrase (adenosine 5′‐triphosphate diphosphohydrolase, EC 3.6.1.5) is added to the platelet suspension. Apyrase, at a concentration of 0.02 U/mL, degrades any traces of ATP or ADP released by the platelets, avoiding the development of refractoriness to ADP and maintaining their discoid shape.22 These preparation conditions provide a suspension of human platelets that is stable for 5 to 8 hours at 37°C.20
Washed platelets prepared under these conditions aggregate after addition of collagen, thrombin, or arachidonic acid and release the contents of their granules. Adrenaline is not itself an aggregating agent but potentiates the aggregation induced by other agonists through activation of α2A‐adrenergic receptors coupled to the inhibition of adenylyl cyclase activity.23, 24 Importantly, the adrenaline‐induced aggregation observed in cPRP is an artifact due to the presence of traces of ADP and/or thrombin.24 In this situation, in the presence of fibrinogen and 2 mmol/L calcium, the platelets aggregate in response to ADP and then disaggregate, without the occurrence of a second wave of aggregation accompanied by the secretion of granule contents (Figure 1).
Preparation of washed human platelets
Blood collection
The blood donors should not have taken drugs affecting platelet responses, such as aspirin and other NSAIDs or thienopyridines like ticlopidine and clopidogrel, during the previous 2 weeks. Blood is collected from a forearm vein through a gauge 16 thin‐walled needle mounted on a short length (10‐20 cm) of plastic tubing. The generation of traces of thrombin during the collection of blood for the preparation of washed platelets must be avoided. Therefore, it is particularly important to perform a nontraumatic venipuncture and to discard the first few milliliters of blood, which are contaminated with tissue factor and contain trace amounts of thrombin. For similar reasons, Vacutainer blood collection should be avoided. Instead, the blood is drawn with a rapid flow directly into a conical 50‐mL plastic (polypropylene or polystyrene) centrifuge tube containing 1 volume of ACD anticoagulant for 6 volumes of blood. Immediately afterwards, the tubes are placed in a water bath at 37°C for a maximum storage period of 15 minutes.
Washed platelet preparation
All operations performed during isolation and washing of the platelets are carried out at 37°C. The compositions of the washing and suspending solutions are given in Table 2.
The anticoagulated blood is centrifuged at 250 g in a Sorvall centrifuge (Sorvall RC3BP swinging‐bucket centrifuge with an H6000A‐HBB6/HLR6 rotor) prewarmed to 37°C, for a period of time depending on the quantity of blood (Table 1) to obtain PRP. To maximize the efficiency of the platelet recovery in the upper layer, without contamination due to red blood cells rising from the bottom layer, the centrifugal slow brake must be applied during rotor deceleration.
The PRP is carefully collected with a 10‐mL syringe and transferred into a new conical 50‐mL centrifuge tube.
After 10 minutes’ incubation at 37°C, the PRP is centrifuged at 2200 g for a period of time depending on the quantity of plasma (Table 1).
The supernatant consisting of PPP is discarded using a Pasteur pipette connected to a vacuum pump, taking care to remove all traces of plasma from the tube walls or near the platelet pellet to avoid any generation of thrombin during the subsequent washing steps. Platelets stimulated by exposure to low concentrations of thrombin may have reduced granule contents or be more sensitive to ADP‐ or adrenaline‐induced aggregation.
The platelet pellet is resuspended in Tyrode's albumin buffer containing 10 U/mL heparin and 0.5 μmol/L PGI2, using a wide‐bore plastic Pasteur pipette (Pastettes, Dutscher, Brumath, France), to prevent platelet activation and plasma coagulation. A 10‐mL volume of buffer is required to resuspend the platelet pellet from 50 to 100 mL of blood. To stabilize the pH of the platelet suspension at about 7.3, it is recommended that the operator exhale air (containing elevated CO2 levels as compared to atmospheric air) into the tube before closing it at each step of the washing procedure.21
After 10 minutes’ incubation at 37°C, 0.5 μmol/L PGI2 is added to this “first wash” platelet suspension, and it is immediately centrifuged at 1900 g for 8 minutes (for 30‐40 mL of suspension).
The platelet pellet is resuspended in Tyrode's albumin buffer containing 0.5 μmol/L PGI2. A platelet count is performed using an automatic blood cell counter.
After 10 minutes’ incubation at 37°C, 0.5 μmol/L PGI2 is added to this “second wash” suspension and it is immediately centrifuged at 1900 g for 8 minutes (for 30‐40 mL of suspension).
The platelet pellet is resuspended in Tyrode's albumin buffer containing 0.02 U/mL apyrase, and the cell count is adjusted to 300 000/μL with the same buffer. The washed platelet preparation is kept at 37°C in a closed tube under a 5% CO2/95% air atmosphere and under these conditions is stable for 5 to 8 hours.
Table 2.
Preparation of Tyrode's albumin buffer 0.35%, pH 7.3
| 100 mL | 200 mL | 300 mL | 400 mL | 500 mL | 1000 mL | |
|---|---|---|---|---|---|---|
| Stock I (mL) | 5 | 10 | 15 | 20 | 25 | 50 |
| Stock II (mL) | 1 | 2 | 3 | 4 | 5 | 10 |
| Stock III (mL) | 2 | 4 | 6 | 8 | 10 | 20 |
| HEPES 0.5 mol/L (mL) | 1 | 2 | 3 | 4 | 5 | 10 |
| Human serum albumin 20% (mL) | 1.8 | 3.6 | 5.4 | 7.2 | 9 | 18 |
| Glucose (g) | 0.1 | 0.2 | 0.3 | 0.4 | 0.5 | 1 |
Reagents for blood collection and washed platelet preparation
ACD anticoagulant solution: 25 g of trisodium citrate dihydrate, 14 g of citric acid monohydrate and 20 g of anhydrous D(+) glucose are dissolved in a final volume of 1 L of distilled water. The final concentrations are 85, 66.6, and 111 mmol/L, respectively; the solution has an osmolarity of 450 mOsm/L and a pH of about 4.5. One volume of anticoagulant is required for 6 volumes of blood.
-
Tyrode's albumin buffer: 137 mmol/L NaCl, 2 mmol/L KCl, 12 mmol/L NaHCO3, 0.3 mmol/L NaH2PO4, 1 mmol/L MgCl2, 2 mmol/L CaCl2, 5.5 mmol/L glucose, and 5 mmol/L HEPES, pH 7.3, containing 0.35% human serum albumin (Table 1). The stock solutions for Tyrode's buffer are as follows:
Stock I: 160 g (2.73 mol/L) NaCl, 4 g (53.6 mmol/L) KCl, 20 g (238 mmol/L) NaHCO3, and 1.16 g (8.6 mmol/L) NaH2PO4.H2O are dissolved in 1 L of distilled water and stored at 4°C. To facilitate the dissolution of the different salts, it is recommended to add the NaCl, KCl, and NaH2PO4 together into 600 mL and the NaHCO3 separately into 200 mL, which must be warmed to 80°C to obtain dissolution. When all the salts have dissolved, the 2 solutions are mixed and the volume is made up to 1 L.
Stock II: 20.33 g (0.1 mol/L) MgCl2.6H2O in 1 L of distilled water.
Stock III: 21.9 g (0.1 mol/L) CaCl2.6H2O in 1 L of distilled water.
HEPES stock: 119 g (0.5 mol/L) N‐[2‐hydroxyethyl]piperazine‐N′‐[2‐ethanesulfonic acid] sodium salt in 1 L of distilled water. HEPES is used as the pH buffer in Tyrode's solution. pH buffers such as Tris (tris[hydroxymethyl]aminomethane) should be avoided because, like other amines, they inhibit some platelet responses and potentiate others. The stock solutions are stable at 4°C for 1 year.
Tyrode's albumin buffer (0.35% albumin): Before mixing the different stock solutions, 50 mL of distilled water should be poured into the beaker to prevent the precipitation of calcium phosphate or calcium carbonate when adding stock I and stock III. All the washing solutions are kept at 37°C throughout the platelet preparation. Table 2: 5 mL stock I, 1 mL stock II, 2 mL stock III, 1 mL HEPES stock, 1.75 mL HSA stock (20%) and 0.1 g anhydrous D(+) glucose made up to a final volume of 100 mL with distilled water. The pH is adjusted to 7.3 with HCl and the osmolarity to 295 mOsm/L with 30% NaCl or distilled water.
Tyrode's buffer: 50 mL stock I and 950 mL distilled water; the pH is adjusted to 7.3 with HCl.
Apyrase preparation: apyrase (adenosine 5′‐triphosphate diphosphohydrolase, EC 3.6.1.5) is an ADP scavenger that is added to the final platelet suspension at 0.02 U/mL, a concentration sufficient to prevent the desensitization of platelet ADP receptors during storage at 37°C. High‐grade apyrase is commercially available from Sigma (St. Quentin Fallavier, France) (type VII apyrase) or Agro‐Bio (La Ferté‐Saint‐Aubin, France) (Apyrase ADP premium). However, we employ apyrase prepared in a 2‐stage procedure using a modification of the method of Molnar and Lorand.25 Some commercially available preparations are not suitable because they contain impurities (eg, potato lectin, a platelet agglutinating agent) and 5′‐nucleotidase activity, which hydrolyses AMP to adenosine. In the final suspending medium, the concentration of apyrase is sufficient to convert 0.25 μmol/L ATP to AMP in 120 seconds at 37°C. Alternatively, the apyrase concentration can be determined to maintain platelet sensitivity to ADP, without having an appreciable inhibitory effect on ADP‐induced aggregation (tested in the presence of fibrinogen). If too much apyrase is used, ADP‐induced aggregation will be decreased and, conversely, if too little is used, the platelets will become refractory to ADP (desensitization of platelet ADP receptors). The ratio of ADPase to ATPase activity is highly variable, depending on the origin of the potatoes, and can change during storage.
PGI2: Prostacyclin (sodium salt, ref P‐6188, Sigma) is prepared as a 1 mmol/L stock solution in ice‐cold buffer (50 mmol/L Tris‐HCl, pH 8.8) and stored in 100‐μL aliquots at −20°C. PGI2 is added at each step of the platelet washing procedure, at a final concentration of 0.5 μmol/L (0.5 μL of the stock solution for 1 mL of platelet suspension), to inhibit transient platelet activation during the preparation.20 Because the half‐life of PGI2 at 37°C is short (a few minutes), it must be added to the washing solution just before centrifugation or platelet resuspension. The PGI2 stock solution should be put on ice immediately after thawing and kept at 4°C and should not be frozen again.
Human fibrinogen (4%, w/v): the lyophilized powder (Grade L, Kabi, Stockholm, Sweden) is dissolved in 0.9% NaCl (4 g in 100 mL) and treated with diisopropylfluorophosphate (DiFP, Sigma‐Aldrich‐Fluka) to inactivate traces of contaminant plasma serine proteases.20 Aliquots (1 mL at about 10 mg/mL) are stored at −20°C and thawed and warmed to 37°C before use. Note that DiFP is highly toxic and should only be used under appropriate conditions (gloves, mask, fume hood).
2.2.3. Platelet aggregation tests using LTA
Platelet aggregometry can be performed using commercially available multichannel aggregometers. The platelet suspension (either cPRP or washed platelets) is stirred at 900 to 1200 rpm in disposable cuvettes maintained at 37°C. PPP and Tyrode's albumin buffer, respectively, are used to establish the maximum (100%) of light transmission. Addition of an agonist (eg, collagen, ADP, arachidonic acid, etc) to the suspension induces platelet aggregation, resulting in an increase in light transmission through the cuvette. The quality of a washed platelet suspension is verified by testing the aggregation response to the weak platelet agonist ADP. A reversible wave of platelet aggregation should be observed following stimulation of washed platelets with ADP (5 μmol/L) in the presence of added fibrinogen (Figure 1). Classical platelet responses to a panel of agonists can then be monitored, including the maximal extent of aggregation (%), the slope of the curve and the latency time (lag phase), the shape change, and primary and secondary aggregation.
2.3. Whole blood aggregometry
Platelet aggregation studies can be performed in whole blood, most commonly anticoagulated with sodium citrate, using an impedance technique. This test measures the change in electrical resistance or impedance between 2 electrodes placed at a fixed distance within the blood sample.10, 26 Adhesion of platelets to the electrodes is followed by the aggregation of other platelets, leading to an increase in impedance. As it is carried out in whole blood, this assay obviates the need to prepare a platelet suspension and allows one to measure platelet functions under more physiological conditions, taking into account the contributions of other blood components that may affect these functions. Nevertheless, this method is not widely used because it provides no information on shape change, the kinetics of platelet aggregation, the 2‐phase response, or platelet disaggregation,27 parameters that are essential to accurately assess platelet function defects. Furthermore, impedance aggregometry cannot be used in thrombocytopenic patients because the extent of platelet aggregation induced by most agonists decreases linearly with the decrease in platelet count.28, 29
2.4. Flow cytometric platelet analyses
Regardless of the LTA test used, whether it is performed with cPRP or washed platelets, the preanalytical steps remain long and require a significant amount of blood. Therefore, the use of flow cytometry, requiring only a few microliters of whole blood, is a valuable alternative for platelet function testing. Flow cytometric analysis, which is based on the detection of cell surface proteins with fluorescently labeled antibodies, offers several advantages for the evaluation of platelet functions.30 The fluorescence of a large number of platelets (10 000) is measured in a small volume and within a limited time interval. Platelets can be analyzed in their physiological environment (ie, whole blood) in the presence of erythrocytes and leukocytes, as well as in platelet suspensions. Due to the requirement for only a small amount of blood, the technique is also suitable for neonatal studies or patients with thrombocytopenia.
Quantification of platelet membrane glycoproteins (GPs) is widely used in diagnosis and research laboratories.31, 32 Commercial kits are now available to quantify the major platelet GPs, including αIIbβ3, the GPIb‐V‐IX complex, and α2β1. Other commercial kits can be used to quantify any platelet receptor (Platelet Calibrator, BioCytex, Marseille, France), for example, GPVI, provided a monoclonal antibody against the target protein is available.33 Flow cytometry can also be employed to determine the activation state of circulating platelets in whole blood, using antibodies directed against proteins newly expressed on the platelet surface during activation. In addition, the reactivity of platelets in whole blood can be evaluated following addition of an agonist. Platelet activation markers include the fibrinogen‐binding site exposed by a conformational change in αIIbβ3 on activated platelets, recognized by the monoclonal antibody PAC‐1, thereby allowing the detection of activated platelets only.34 Granule secretion is revealed by the exposure of P‐selectin, a component of the granule membrane of resting platelets, which is expressed on the platelet surface only after alpha granule release and may be quantified using a monoclonal antibody against P‐selectin.35 The development of a phosphatidylserine‐exposing procoagulant surface on activated platelets can also be detected by monitoring the binding of labeled annexin V.36
Practical issues regarding the use of flow cytometry for platelet function testing have been discussed in comprehensive reviews.30, 36 Briefly, in the case of in vitro stimulation, anticoagulated whole blood is generally diluted 10‐fold before incubation with agonists. Antibodies are then added for 15 minutes. Incubation is performed at room temperature and not on ice, as low temperature can affect platelet activation. The samples are then diluted (usually 20‐fold) to stop the reaction and analyzed by flow cytometry without further washing. If the flow cytometric analysis is to be performed later, the samples can be fixed with paraformaldehyde (0.5%‐1%). However, fixation should not be performed in the presence of unbound antibodies, as this can cause nonspecific binding of antibodies to platelets due to cross linking. Although the small size of platelets makes them detectable through their forward/side‐scattering characteristics alone, it is recommended to include a platelet identification marker using an antibody targeting a platelet‐specific protein such as GPIb or GPIX.
2.5. Platelet reactivity in flowing blood
Platelet adhesion, activation, and aggregation occurs in vivo under a wide spectrum of blood flow conditions ranging from 10 to 5000 per second under normal physiological circumstances and reaching 40 000 per second in the apex of stenotic vessels.37, 38 Such rheological conditions cannot be obtained in classical platelet function testing systems like LTA, where the stirring generates only very low shear rates (<100 per second). Therefore, the development of flow‐based assays in the 1970s, reproducing a wide range of physiological and pathological blood flows, enabled the study of platelet aggregation under conditions that better mimic the in vivo environment.39 These studies have significantly improved our understanding of platelet aggregation as it occurs in vivo. For instance, in LTA the mechanism of aggregation depends primarily on interplatelet bonds supported by αIIbβ3, with the GPIb‐V‐IX complex playing no role at all. In sharp contrast, it has been shown that this complex plays a key role in thrombus growth, for example, in platelet‐platelet interactions, by mediating the recruitment of circulating platelets to von Willebrand factor (VWF) exposed at the surface of activated platelets within the thrombus.40 Moreover, we recently reported that platelet aggregation under flow also involves GPVI, whereas this receptor does not participate in platelet‐platelet interactions under the low shear conditions found in LTA.41 In addition, the part played by soluble agonists such as thrombin, ADP, or TxA2 might be overestimated in suspension assays like LTA where they are not washed away by the flow.
Another advantage of flow systems is that they allow one to explore the initial steps of platelet recruitment to a surface, which cannot be evaluated in suspension assays. To do so, the flow devices are coated with purified proteins binding to 1 or 2 platelet receptors. Perfusion of whole blood over these proteins at various wall shear rates gives insight into the functions of adhesion receptors in platelet tethering, activation, or aggregation. In flow devices it is also easy to image adherent platelets, which is not feasible in suspensions. This enables one to visualize the steps of platelet adhesion and activation in real time, while simultaneously looking at P‐selectin exposure or αIIbβ3 activation or measuring intraplatelet calcium signals. Thrombus formation is likewise easily quantified by measuring the surface coverage in 2‐dimensional or the thrombus volume in 3‐dimensional high‐speed confocal microscopy. Real‐time experiments provide insight into the kinetics of the various steps of thrombus formation. Because flow‐based assays are technically challenging, they can be carried out only in specialized laboratories.
The following section describes the basic methodology required to perform blood perfusion assays under laminar flow as they are routinely carried out in our laboratory. Such techniques permit one to study various platelet functions, including adhesion, activation, and aggregation, under a wide range of hemodynamic conditions found in physiological and pathological situations. We also provide information on quantification methods.
2.6. Methodology for blood perfusion experiments
2.6.1. Coating of the substrate
Flow experiments with human blood are generally performed using rectangular glass capillary microslides (height/width: 0.2/2 mm or 0.1/1 mm). Solutions containing various adhesive proteins are allowed to fill the microslides by capillary action. The main adhesive proteins employed in our laboratory to study platelet adhesion, activation, and aggregation are Horm Collagen (200 μg/mL) (Takeda, Konstanz, Germany), VWF (10 μg/mL) (purified in‐house from factor VIII concentrates) and fibrinogen (100 μg/mL) (Sigma‐Aldrich), diluted in phosphate buffered saline (PBS). The microslides are stored in a humid chamber overnight at 4°C. The next morning, they are rinsed with PBS, passivated by filling for 1 hour with PBS containing 1% human serum albumin, and then rinsed again with PBS before being inserted in the flow system.
2.6.2. Assembly of the flow‐based system
Double‐sided sticky tape (3M) is wrapped around each end of the capillary microslide (3‐4 times) to hermetically seal a 10‐mm length of tubing on each side (Figure 2A: Tubing 1). Truncated plastic micropipette tips of various sizes (Figure 2A: Tip) are inserted into tubing 1 to connect to (1) a Vacutainer blood collection system (Becton Dickinson) on the right side and (2) a 10‐mm length of wide tubing (Figure 2A: Tubing 2) leading to a thinner but much longer piece of tubing (Figure 2A: Tubing 3) on the left side. The Vacutainer blood collection system is inserted into a combination of 2 interconnected 3‐way taps linked to the reservoirs: (1) a 3‐ to 15‐mL syringe without plunger containing the anticoagulated blood, (2) a 30‐mL syringe without plunger containing the buffer (PBS or Tyrode's buffer), and (3) a 10‐mL syringe used to clean the system and remove any air bubbles (Figure 2B). On the other side, tubing 3 is connected through type 2 tubing and a 3‐way tap to (1) a glass syringe and a pump (Standard Infuse/Withdraw PHD 2000, Harvard Apparatus, Hollinston, MA) used to draw the fluids through the flow device and (2) the waste (Figure 2C). References for the microslides, tubings, micropipettes, and syringes are provided in Table 3.
Figure 2.
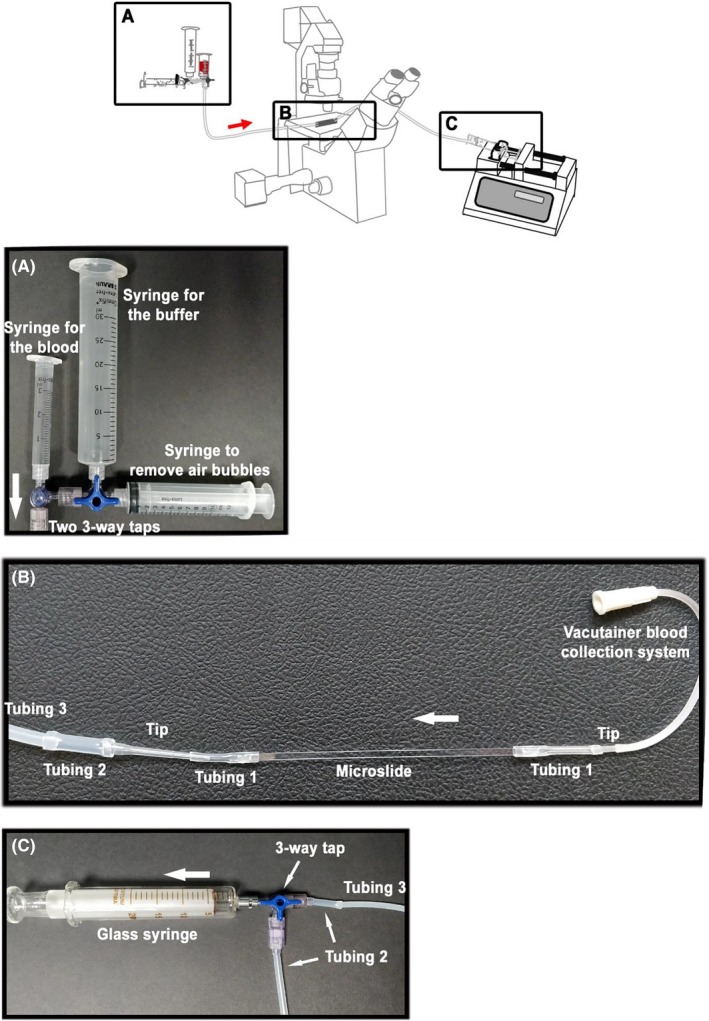
Assembly of the flow‐based system
Table 3.
References of the capillary microslides, tubings, micropipettes, and syringes for blood flow experiments
| Microslides | Tubing 1 | Tubing 2 | Tubing 3 | Micropipette tips | Syringe |
|---|---|---|---|---|---|
| CMC Scientific (Wuxi City, China) | Watson Marlow (La Queue Lez Yvelines, France) | VWR (Radnor, PA) | Watson Marlow | Sorenson(Salt Lake City, UT) | Fortuna Optima all‐glass syringe 30 mL |
| Ref 3520‐100 | Ref 990.0015.005 | Ref 228‐0706 | Ref 913.AJ08.016 | P10 | |
| Size 0.2 × 2 mm | Internal diameter 1.5 mm | Internal diameter 0.8 mm | Internal diameter 0.8 mm | ||
| CamLab | Tygon 3350 | ||||
| Ref 1152980 | Ref T330A‐053 | ||||
| Size 0.1 × 1 mm | Internal diameter 1 mm |
2.6.3. Flow experiments
Once the system has been closed, it is of critical importance to remove any air bubbles. To obtain a given wall shear rate in the capillary, the flow rate needs to be set up according to the formula: S = (6Q)/(Wh2), where S is the shear rate (s‐1), Q is the flow rate (mL/s), W is the width (m), and h the height (m) of the capillary microslide. Experiments are performed for 3 to 15 minutes at wall shear rates no lower than 100 per second to avoid any blood cell sedimentation. Platelet adhesion is monitored in real time using differential interference contrast technique on an inverted microscope and thrombus formation is quantified by confocal microscopy after fluorescent labeling blood with 0.5 μmol/L of DIOC6 (3,3′‐dihexyloxacarbocyanine iodide, ThermoFisher Scientific).
3. APPLICATIONS
3.1. In research
Suspensions of washed platelets in a well‐defined medium under physiological conditions of pH, temperature, osmolarity, and inorganic ions permit study of the intrinsic properties of platelets, without interference from the plasma environment or anticoagulant. Such suspensions are usually stable for 5 to 8 hours at 37°C, as compared to no more than 2 hours for citrated PRP. However, as washed platelet suspensions are more difficult to prepare than cPRP, they are available only in specialized research laboratories. In these suspensions, addition of apyrase is absolutely necessary because in its absence, washed platelets do not aggregate or change shape in response to ADP. This so‐called refractory state is due to desensitization of the P2Y1 receptor for ADP.42 When studying the platelet P2X1 receptor for ATP, a high concentration of apyrase is required.43, 44 Indeed, this nonselective cation channel rapidly desensitizes and hence apyrase must be added, not only to the final resuspension medium but also at each step of the washing procedure. Under these conditions, the selective P2X1 receptor agonist alpha, beta‐methylene‐ATP (αβMeATP) induces a rapid calcium influx in human platelets, associated with a transient shape change and centralization of the secretory granules, although dense granule release has not been detected.45 Nonetheless, in the presence of such high concentrations of apyrase, the aggregation response induced by strong agonists is diminished due to the degradation of nucleotides released by the platelets.
Washed platelets aggregate and secrete their granule contents in response to strong activators (collagen, thrombin, arachidonic acid). ADP triggers a reversible wave of aggregation without release of the platelet granule contents, even at high concentrations. As ADP does not induce granule secretion, fibrinogen must be added before stimulation of the cells with this agent.
Washed platelet suspensions are thus ideal for investigations using LTA, flow cytometry, electron microscopy, western blotting of platelet lysates, or radioligand binding, to mention only a few applications.41, 46, 47, 48
3.2. In the clinic
LTA and flow cytometry are currently the principal assays used in biological laboratories to diagnose thrombopathies in patients presenting mucocutaneous hemorrhagic syndromes. Given the complexity of some of the assays, these explorations are carried out in most cases in specialized clinical hemostasis laboratories. Several experts, including the Subcommittee on Platelet Physiology of the ISTH, have recently proposed a prioritization of the tests required to accurately diagnose a platelet function defect.19, 49, 50 These experts recommend as the first line of approach platelet aggregation tests in cPRP together with quantification of the main platelet GPs by flow cytometry and some functional assays such as determination of the activation state of αIIbβ3. As they are easier to perform, platelet aggregation studies in cPRP are generally preferred to tests needing washed platelets, which are still rarely used in diagnostic laboratories. However, in the event of thrombocytopenia, functional studies will necessarily require a washed platelet suspension, the only method currently available enabling one to adjust the final platelet count without inducing functional artifacts.
A combination of LTA and flow cytometry thus makes it possible to quickly identify the most well‐known hemorrhagic platelet disorders such as Glanzmann thrombasthenia and Bernard‐Soulier syndrome, as well as several moderate thrombopathies like GPVI deficiencies or anomalies of the TxA2 pathway. This combination of tests also allows one to detect less obvious platelet function defects, which will then require further investigation using electron microscopy, protein biochemistry, receptor/ligand affinity studies in radioimmunoassay and/or molecular biology. Such is the case in particular for qualitative or quantitative deficiencies of the alpha (gray platelet syndrome) or delta granules (storage pool diseases), platelet cytoskeleton abnormalities (MYH9, TUBB1, FLNA, WAS) and P2Y12 deficiencies. Nevertheless, despite all the tools now at our disposal, some authors consider that 30% to 50% of platelet function disorders remain undefined, which motivates the increasingly widespread use of high‐throughput sequencing techniques to identify new forms of thrombopathies.51, 52
4. CONCLUSION
The combined use of the various tools described here allows identification and classification of inherited or acquired abnormalities in platelet function, to study the effect of drugs that inhibit platelet aggregation and to understand their pharmacological mechanism of action. The use of rigorously standardized methods for platelet preparation, reagents, and functional tests is of primary importance for achieving reproducible results.
RELATIONSHIP DISCLOSURE
The authors have no competing financial and/or non‐financial interests in relation to the work described.
Hechler B, Dupuis A, Mangin PH, Gachet C. Platelet preparation for function testing in the laboratory and clinic: Historical and practical aspects. Res Pract Thromb Haemost. 2019;3:615–625. 10.1002/rth2.12240
REFERENCES
- 1. de Gaetano G. A new blood corpuscle: an impossible interview with Giulio Bizzozero. Thromb Haemost. 2001;86:973–9. [PubMed] [Google Scholar]
- 2. Coller BS. Historical perspective and future directions in platelet research. J Thromb Haemost. 2011;9(Suppl 1):374–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cattaneo M. Inherited platelet‐based bleeding disorders. J Thromb Haemost. 2003;1:1628–36. [DOI] [PubMed] [Google Scholar]
- 4. Cox K, Price V, Kahr WH. Inherited platelet disorders: a clinical approach to diagnosis and management. Expert Rev Hematol. 2011;4:455–72. [DOI] [PubMed] [Google Scholar]
- 5. Duke W. The relation of blood platelets to hemorrhagic disease: description of a method for determining the bleeding time and coagulation time and report of three cases of hemorrhagic diseases relieved by transfusion. JAMA. 1910;55:1185–92. [Google Scholar]
- 6. Born GV. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature. 1962;194:927–9. [DOI] [PubMed] [Google Scholar]
- 7. Adelman B, Michelson AD, Handin RI, Ault KA. Evaluation of platelet glycoprotein Ib by fluorescence flow cytometry. Blood. 1985;66:423–7. [PubMed] [Google Scholar]
- 8. Fox SC, Sasae R, Janson S, May JA, Heptinstall S. Quantitation of platelet aggregation and microaggregate formation in whole blood by flow cytometry. Platelets. 2004;15:85–93. [DOI] [PubMed] [Google Scholar]
- 9. De Cuyper IM, Meinders M, van de Vijver E, de Korte D, Porcelijn L, de Haas M, et al. A novel flow cytometry‐based platelet aggregation assay. Blood. 2013;121:e70–80. [DOI] [PubMed] [Google Scholar]
- 10. Cardinal DC, Flower RJ. The electronic aggregometer: a novel device for assessing platelet behavior in blood. J Pharmacol Methods. 1980;3:135–58. [DOI] [PubMed] [Google Scholar]
- 11. Quick AJ. The bleeding time as a test of hemostatic function. Am J Clin Pathol. 1975;64:87–94. [DOI] [PubMed] [Google Scholar]
- 12. Rodgers RP, Levin J. A critical reappraisal of the bleeding time. Semin Thromb Hemost. 1990;16:1–20. [DOI] [PubMed] [Google Scholar]
- 13. O'Brien JR. Platelet aggregation: part I some effects of the adenosine phosphates, thrombin, and cocaine upon platelet adhesiveness. J Clin Pathol. 1962;15:446–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nurden AT, Fiore M, Nurden P, Pillois X. Glanzmann thrombasthenia: a review of ITGA2b and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood. 2011;118:5996–6005. [DOI] [PubMed] [Google Scholar]
- 15. Cattaneo M. Light transmission aggregometry and ATP release for the diagnostic assessment of platelet function. Semin Thromb Hemost. 2009;35:158–67. [DOI] [PubMed] [Google Scholar]
- 16. Feinman RD, Lubowsky J, Charo I, Zabinski MP. The lumi‐aggregometer: a new instrument for simultaneous measurement of secretion and aggregation by platelets. J Lab Clin Med. 1977;90:125–9. [PubMed] [Google Scholar]
- 17. Mustard JF, Perry DW, Ardlie NG, Packham MA. Preparation of suspensions of washed platelets from humans. Br J Haematol. 1972;22:193–204. [DOI] [PubMed] [Google Scholar]
- 18. Germanovich K, Femia EA, Cheng CY, Dovlatova N, Cattaneo M. Effects of pH and concentration of sodium citrate anticoagulant on platelet aggregation measured by light transmission aggregometry induced by adenosine diphosphate. Platelets. 2018;29:21–6. [DOI] [PubMed] [Google Scholar]
- 19. Cattaneo M, Cerletti C, Harrison P, Hayward CP, Kenny D, Nugent D, et al. Recommendations for the standardization of light transmission aggregometry: a consensus of the working party from the platelet physiology subcommittee of SSC/ISTH. J Thromb Haemost. 2013. [DOI] [PubMed] [Google Scholar]
- 20. Cazenave JP, Hemmendinger S, Beretz A, Sutter‐Bay A, Launay J. Platelet aggregation: a tool for clinical investigation and pharmacological study. Methodology. Ann Biol Clin (Paris). 1983;41:167–79. [PubMed] [Google Scholar]
- 21. Han P, Ardlie NG. The influence of pH, temperature, and calcium on platelet aggregation: maintenance of environmental pH and platelet function for in vitro studies in plasma stored at 37 degrees C. Br J Haematol. 1974;26:373–89. [DOI] [PubMed] [Google Scholar]
- 22. Ardlie NG, Perry DW, Packham MA, Mustard JF. Influence of apyrase on stability of suspensions of washed rabbit platelets. Proc Soc Exp Biol Med. 1971;136:1021–3. [DOI] [PubMed] [Google Scholar]
- 23. Ardlie NG, Glew G, Schwartz CJ. Influence of catecholamines on nucleotide‐induced platelet aggregation. Nature. 1966;212:415–7. [DOI] [PubMed] [Google Scholar]
- 24. Lanza F, Beretz A, Stierle A, Hanau D, Kubina M, Cazenave JP. Epinephrine potentiates human platelet activation but is not an aggregating agent. Am J Physiol. 1988;255:H1276–88. [DOI] [PubMed] [Google Scholar]
- 25. Molnar J, Lorand L. Studies on apyrases. Arch Biochem Biophys. 1961;93:353–63. [DOI] [PubMed] [Google Scholar]
- 26. Mackie IJ, Jones R, Machin SJ. Platelet impedance aggregation in whole blood and its inhibition by antiplatelet drugs. J Clin Pathol. 1984;37:874–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Toth O, Calatzis A, Penz S, Losonczy H, Siess W. Multiple electrode aggregometry: a new device to measure platelet aggregation in whole blood. Thromb Haemost. 2006;96:781–8. [PubMed] [Google Scholar]
- 28. Femia EA, Scavone M, Lecchi A, Cattaneo M. Effect of platelet count on platelet aggregation measured with impedance aggregometry (multiplate analyzer) and with light transmission aggregometry. J Thromb Haemost. 2013;11:2193–6. [DOI] [PubMed] [Google Scholar]
- 29. Hanke AA, Roberg K, Monaca E, Sellmann T, Weber CF, Rahe‐Meyer N, et al. Impact of platelet count on results obtained from multiple electrode platelet aggregometry (multiplate). Eur J Med Res. 2010;15:214–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Michelson AD. Evaluation of platelet function by flow cytometry. Pathophysiol Haemost Thromb. 2006;35:67–82. [DOI] [PubMed] [Google Scholar]
- 31. Cohn RJ, Sherman GG, Glencross DK. Flow cytometric analysis of platelet surface glycoproteins in the diagnosis of Bernard‐Soulier syndrome. Pediatr Hematol Oncol. 1997;14:43–50. [DOI] [PubMed] [Google Scholar]
- 32. Lindahl TL, Festin R, Larsson A. Studies of fibrinogen binding to platelets by flow cytometry: an improved method for studies of platelet activation. Thromb Haemost. 1992;68:221–5. [PubMed] [Google Scholar]
- 33. Best D, Senis YA, Jarvis GE, Eagleton HJ, Roberts DJ, Saito T, et al. GPVI levels in platelets: relationship to platelet function at high shear. Blood. 2003;102:2811–8. [DOI] [PubMed] [Google Scholar]
- 34. Shattil SJ, Hoxie JA, Cunningham M, Brass LF. Changes in the platelet membrane glycoprotein IIb.IIIa complex during platelet activation. J Biol Chem. 1985;260:11107–14. [PubMed] [Google Scholar]
- 35. Saboor M, Moinuddin M, Ilyas S. New horizons in platelets flow cytometry. Malays J Med Sci. 2013;20:62–6. [PMC free article] [PubMed] [Google Scholar]
- 36. Ramstrom S, Sodergren AL, Tynngard N, Lindahl TL. Platelet function determined by flow cytometry: new perspectives? Semin Thromb Hemost. 2016;42:268–81. [DOI] [PubMed] [Google Scholar]
- 37. Kroll MH, Hellums JD, McIntire LV, Schafer AI, Moake JL. Platelets and shear stress. Blood. 1996;88:1525–41. [PubMed] [Google Scholar]
- 38. Nesbitt WS, Giuliano S, Kulkarni S, Dopheide SM, Harper IS, Jackson SP. Intercellular calcium communication regulates platelet aggregation and thrombus growth. J Cell Biol. 2003;160:1151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jackson SP. The growing complexity of platelet aggregation. Blood. 2007;109:5087–95. [DOI] [PubMed] [Google Scholar]
- 40. Kulkarni S, Dopheide SM, Yap CL, Ravanat C, Freund M, Mangin P, et al. A revised model of platelet aggregation. J Clin Invest. 2000;105:783–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mangin PH, Onselaer MB, Receveur N, Le Lay N, Hardy AT, Wilson C, et al. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica. 2018;103:898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Baurand A, Eckly A, Bari N, Leon C, Hechler B, Cazenave JP, et al. Desensitization of the platelet aggregation response to ADP: differential down‐regulation of the P2Y1 and P2CYC receptors. Thromb Haemost. 2000;84:484–91. [PubMed] [Google Scholar]
- 43. Hechler B, Lenain N, Marchese P, Vial C, Heim V, Freund M, et al. A role of the fast ATP‐gated P2X1 cation channel in thrombosis of small arteries in vivo. J Exp Med. 2003;198:661–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vial C, Hechler B, Leon C, Cazenave JP, Gachet C. Presence of P2X1 purinoceptors in human platelets and megakaryoblastic cell lines. Thromb Haemost. 1997;78:1500–4. [PubMed] [Google Scholar]
- 45. Hechler B, Cattaneo M, Gachet C. The P2 receptors in platelet function. Semin Thromb Hemost. 2005;31:150–61. [DOI] [PubMed] [Google Scholar]
- 46. Gachet C, Cattaneo M, Ohlmann P, Hechler B, Lecchi A, Chevalier J, et al. Purinoceptors on blood platelets: further pharmacological and clinical evidence to suggest the presence of two ADP receptors. Br J Haematol. 1995;91:434–44. [DOI] [PubMed] [Google Scholar]
- 47. Hechler B, Leon C, Vial C, Vigne P, Frelin C, Cazenave JP, et al. The P2Y1 receptor is necessary for adenosine 5’‐diphosphate‐induced platelet aggregation. Blood. 1998;92:152–9. [PubMed] [Google Scholar]
- 48. Hechler B, Ohlmann P, Chafey P, Ravanat C, Eckly A, Maurer E, et al. Preserved functional and biochemical characteristics of platelet components prepared with amotosalen and ultraviolet a for pathogen inactivation. Transfusion. 2013;53:1187–200. [DOI] [PubMed] [Google Scholar]
- 49. Carubbi C, Masselli E, Nouvenne A, Russo D, Galli D, Mirandola P, et al. Laboratory diagnostics of inherited platelet disorders. Clin Chem Lab Med. 2014;52:1091–106. [DOI] [PubMed] [Google Scholar]
- 50. Gresele P; Subcommittee on Platelet Physiology of the International Society on Thrombosis and Hemostasis . Diagnosis of inherited platelet function disorders: guidance from the SSC of the ISTH. J Thromb Haemost. 2015;13:314–22. [DOI] [PubMed] [Google Scholar]
- 51. Balduini CL, Melazzini F, Pecci A. Inherited thrombocytopenias—recent advances in clinical and molecular aspects. Platelets. 2017;28:3–13. [DOI] [PubMed] [Google Scholar]
- 52. Gresele P, Harrison P, Bury L, Falcinelli E, Gachet C, Hayward CP, et al. Diagnosis of suspected inherited platelet function disorders: results of a worldwide survey. J Thromb Haemost. 2014;12:1562–9. [DOI] [PubMed] [Google Scholar]