

Abstract
Background
Synthesis of the hemostatic protein von Willebrand factor (VWF) drives formation of endothelial storage organelles called Weibel‐Palade bodies (WPBs). In the absence of VWF, angiogenic and inflammatory mediators that are costored in WPBs are subject to alternative trafficking routes. In patients with von Willebrand disease (VWD), partial or complete absence of VWF/WPBs may lead to additional bleeding complications, such as angiodysplasia. Studies addressing the role of VWF using VWD patient–derived blood outgrowth endothelial cells (BOECs) have reported conflicting results due to the intrinsic heterogeneity of patient‐derived BOECs.
Objective
To generate a VWF‐deficient endothelial cell model using clustered regularly interspaced short palindromic repeats (CRISPR) genome engineering of blood outgrowth endothelial cells.
Methods
We used CRISPR/CRISPR‐associated protein 9 editing in single‐donor cord blood–derived BOECs (cbBOECs) to generate clonal VWF −/− cbBOECs. Clones were selected using high‐throughput screening, VWF mutations were validated by sequencing, and cells were phenotypically characterized.
Results
Two VWF −/− BOEC clones were obtained and were entirely devoid of WPBs, while their overall cell morphology was unaltered. Several WPB proteins, including CD63, syntaxin‐3 and the cargo proteins angiopoietin (Ang)‐2, interleukin (IL)‐6, and IL‐8 showed alternative trafficking and secretion in the absence of VWF. Interestingly, Ang‐2 was relocated to the cell periphery and colocalized with Tie‐2.
Conclusions
CRISPR editing of VWF provides a robust method to create VWF‐ deficient BOECs that can be directly compared to their wild‐type counterparts. Results obtained with our model system confirmed alternative trafficking of several WPB proteins in the absence of VWF and support the theory that increased Ang‐2/Tie‐2 interaction contributes to angiogenic abnormalities in VWD patients.
Keywords: endothelial cells, gene knockout techniques, protein transport, secretory vesicles, von Willebrand factor
Essentials.
Von Willebrand factor (VWF) synthesis is essential for the formation of the Weibel‐Palade Bodies (WPBs).
Patient‐derived endothelial cell models of von Willebrand disease and VWF deficiency have shown a high degree of phenotypic heterogeneity.
We generated VWF knockout endothelial cells (ECs) using clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR‐associated protein 9 gene editing on cord blood outgrowth ECs (cbBOECs).
WPB‐associated proteins show alternative localization in the absence of VWF, and angiopoietin (Ang)‐2 colocalizes with the receptor Tie‐2.
1. INTRODUCTION
Von Willebrand factor (VWF) is a large multimeric protein that plays a central role in vascular homeostasis. VWF is synthesized by endothelial cells (ECs) and megakaryocytes and secreted VWF mediates adhesion of platelets to sites of vascular damage and acts as a carrier for coagulation factor VIII.1 The importance of synthesis and secretion of VWF is highlighted by the bleeding disorder von Willebrand disease (VWD), which is caused by mutations in the VWF gene that lead to either qualitative or (full or partial) quantitative deficiencies in VWF.2 Elevated levels of VWF, on the other hand, have been associated with increased risk of thrombosis.3
Following its synthesis in ECs, VWF is stored in rod‐shaped storage organelles, called Weibel‐Palade bodies (WPBs), where it is stored together with a wide variety of inflammatory and angiogenic mediators.4, 5, 6 VWF is the driving force behind the formation of its own storage organelle, illustrated by the lack of WPBs in ECs from a patient with severe VWD type 37 or from VWF null animal models8, 9, 10 and the formation of pseudo‐WPBs upon heterologous expression of VWF in other cell types.11, 12, 13 Upon endothelial triggering, that is, after vascular damage, WPBs undergo exocytosis and deliver their cargo to the cell surface or into the vascular lumen, leading to the formation of long VWF strings to which platelets and also leukocytes and erythrocytes can adhere.14, 15, 16, 17, 18
Absence of WPBs not only compromises the hemostatic response of the endothelium but also has consequences for other secretory cargo that relies on WPBs for proper delivery to and across the plasma membrane. Trafficking of several inflammatory mediators, including P‐selectin, CD63, and chemokines such as interleukin (IL)‐6 and IL‐8, is likely to depend on VWF synthesis and WPB formation.19 This is underscored by impaired leukocyte rolling and neutrophil infiltration in wounds in VWF −/− mice, resulting from defective translocation of P‐selectin and/or CD63.8, 20, 21, 22 In addition, storage and secretion of another WPB cargo protein, the proangiogenic mediator angiopoietin (Ang)‐223, 24, 25 is also disturbed when ECs are depleted of VWF.26 Continuous release of Ang‐2 as a consequence of the unavailability of its default storage compartment has been proposed as one of the underlying mechanisms behind angiodysplasia, a clinical complication of VWD that is characterized by recurrent bleeding in the gastrointestinal tract and is associated with vascular malformations of the gut.27, 28 However, studies into angiogenic properties of blood outgrowth endothelial cells (BOECs) derived from patients with VWD, which can be regarded as endothelial models of VWD, have failed to unequivocally support this model.7, 26, 29, 30
Variation in the genetic background between patients as well as controls may be at the basis of the discrepancy between outcomes in BOECs derived from different individuals. This is further confounded by the broad spectrum of VWD‐causing mutations and the residual VWF expression levels that are associated with these mutations.31 To overcome this, there is a need for targeted genetic strategies for long‐term complete ablation of VWF in human primary ECs. Others have previously used the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR‐associated protein 9 (Cas9) system to target gene expression in ECs in vitro.32, 33 In this study, we used CRISPR‐Cas9 genetic engineering to knock out VWF expression using guide RNAs that target the first exon of the VWF gene. For this we used cord blood outgrowth endothelial cells (cbBOECs) from a single donor, with which we created multiple VWF‐deficient and control BOEC clones with an identical genetic background. cbBOECs possess an increased expansion potential compared to other primary ECs,34 which allowed for prolonged culturing and clonal expansion after single‐cell sorting that are necessary to generate genetically homogenous populations of VWF null cells. True VWF −/− cbBOECs were selected using a high‐throughput screen for loss of VWF secretion, and VWF mutations were validated by Sanger and next‐generation sequencing (NGS). Morphologic, functional, and proteomic analyses were used to confirm that CRISPR‐engineered BOECs retained their endothelial properties. We used this model of VWF deficiency to study alternative trafficking of proteins that are normally trafficked via WPBs. Upon the absence of WPBs, we observed an alternative localization pattern for proteins normally associated with the WPB membrane, such as CD63 and the regulator of WPB exocytosis, syntaxin‐3. Alternative targeting was also seen for IL‐6, IL‐8, and Ang‐2, which led to altered storage and secretion of these WPB cargo proteins in VWF −/− BOECs.
2. METHODS
2.1. Antibodies and reagents
Antibodies used in this study are listed in Table S1.
2.2. Isolation of cbBOECs and cell culture
Cord blood was collected from umbilical veins within 48 hours after delivery and was processed for BOEC isolation essentially as described before.35 cbBOECs were cultured in EC growth medium (EGM)‐2 (Lonza, Basel, Switzerland, CC‐3162) supplemented with 18% fetal calf serum (EGM‐18; Bodinco, Alkmaar, The Netherlands).36 Human embryonic kidney 293T (HEK293T) cells were cultured in Gibco Dulbecco's Modified Eagle Medium, high glucose, pyruvate (ThermoFisher, Landsmeer, The Netherlands) for regular passing and seeding and in EGM‐18 for virus production.
2.3. Lentiviral CRISPR‐Cas9 targeting constructs
Guide RNA (gRNA) sequences targeting the first exon of VWF were designed using the MIT CRISPR design tool (http://crispr.mit.edu)37 and the BROAD Institute single guide RNA (sgRNA) designer (http://www.broadinstitute.org/rnai/public/analysis-tools/sgrna-design)38 by submitting the DNA sequence of VWF exon 1 flanked by 100 bp up‐ and downstream (chromosome 12, 6123020‐6123259 positive strand; Figure 1). gRNA sequences were selected that have a high predicted efficiency (BROAD Institute score) and a high inverted off‐target score (MIT CRISPR design tool).37, 38 gRNAs used in this study were gRNA‐1: 5′‐TGGCCCTCATTTTGCCAGGT‐3′ and gRNA‐2: 5′‐AGCACCCCGGCAAATCTGGC‐3′. Complementary oligos were hybridized with BsmBI restriction site compatible overhangs on either side (gRNA‐1: 5′‐CACCGTGGCCCTCATTTTGCCAGGT‐3′ and 5′‐AAACACCTGGCAAAATGAGGGCCAC‐3′, gRNA‐2: 5′‐CACCGAGCACCCCGGCAAATCTGGC‐3′ and 5′‐AAACGCCAGATTTGCCGGGGTGCTC‐3′) to facilitate cloning into BsmBI‐digested LentiCRISPR v2 plasmid39 (a gift from Feng Zhang, Addgene #52961). The resulting constructs LentiCRISPR‐gRNA‐1 and LentiCRISPR‐gRNA‐2 were verified by DNA sequencing analysis.
Figure 1.

Schematic overview of the CRISPR clone generation workflow. Guide RNAs (gRNAs) were designed to target exon 1 of the VWF gene (Step 1) and cloned into a LentiCRISPR V2 vector (Step 2). HEK293T cells were transfected with a vector containing a VWF targeting gRNA (gRNA‐1 or gRNA‐2) or the empty LentiCRISPR vector as a control (CTRL) (Step2). Lentivirus was produced in endothelialcell growth medium 18, and medium was transferred to cord blood outgrowth endothelial cells (cbBOECs) from a single donor for transduction either directly or after combining medium containing 2 gRNAs, sometimes combining virus containing 2 different gRNAs for increased targeting efficiency (Step 4). Transduced cells were selected by puromycin and single‐cell sorted using vascular endothelial (VE)‐cadherin as an endothelial cell surface marker (Step 5). Medium of single‐cell clones was collected for an ELISA‐mediated high‐throughput screen for von Willebrand factor (VWF) deficient clones. Putative knockout clones were expanded to multiple larger culture surfaces for western blot (WB) and sequencing analysis to confirm biallelic VWF knockout (Step 8), after which they were cryopreserved or used for functional assays
2.4. Generation of CRISPR‐edited VWF−/− cbBOECs
Lentivirus production in HEK293T cells and transduction of ECs was performed as described.36 cbBOECs (passage number: 4, confluency: 60%‐80%) were lentivirally transduced with LentiCRISPR v2 as a control (LentiCRISPR‐CTRL), LentiCRISPR‐gRNA‐1, LentiCRISPR‐gRNA‐2 or a combination of LentiCRISPR‐gRNA‐1 and LentiCRISPR‐gRNA‐2 in 6‐well plates (Figure 1). After selection using 1 μg/mL of puromycin for 72 hours, cells were left to recover until confluency. Cells of each condition were single‐cell sorted into gelatin‐coated 96‐well plates by fluorescence‐activated cell sorting (FACS) using a BD FACSAria III cell sorter (BD Biosciences, Breda, The Netherlands) and using anti‐vascular endothelial (VE) cadherin–fluorescein isothiocyanate as an EC marker. EGM‐18 was replaced every 2 to 3 days, and after 7 days colonies started to form in some wells. Conditioned media of wells that were >50% confluent were assayed using VWF ELISA40 to identify candidates no longer secreting VWF. VWF‐deficient wells were passaged and expanded for further analysis including immunoblotting of cell lysates and Sanger sequencing and NGS of the genomic DNA isolated with the DNeasy Blood & Tissue kit (Qiagen, Venlo, The Netherlands). Verified clones were cryopreserved as described.35
2.5. Secretion assay
ECs were seeded in gelatin‐coated 6‐well plates and cultured at full confluency for 4 to 5 days. Twenty‐four hours before the secretion assay, media were replaced for fresh EGM‐18 supplemented with 10 ng/mL of IL‐1β (I9401, Sigma‐Aldrich, St Louis, MO) or vehicle. Twenty‐four‐hour conditioned media were harvested, cells were pretreated for 15 minutes with serum‐free M199 medium (Gibco 22340, ThermoFisher) supplemented with 0.2% (w/v) bovine serum albumin (BSA; Serva, Heidelberg, Germany) and were stimulated with 100 μmol/L of histamine or vehicle for 30 minutes as described previously.41 Releasates were collected, and stimulated and unstimulated ECs were lysed in M199 with 0.2% BSA and 1% Triton X‐100 (Sigma‐Aldrich). Protein secretion and intracellular content were determined using the DuoSet ELISA kit for Ang‐2 (DY623; R&D Systems, Minneapolis, MN), and the Pelikine compact kit for IL‐6 and IL‐8 (M1916 and M1918, respectively; Sanquin, Amsterdam, The Netherlands). Assays were performed according to manufacturer's protocol.
2.6. Statistical analysis
Statistical analyses were performed in Prism 8.1.1 (GraphPad Software, La Jolla, CA). Normal distribution of the data (N = 3) was confirmed using the Shapiro‐Wilk test. In case of 1 dependent and 1 independent variable, a 1‐way analysis of variance (ANOVA) was used with a Tukey's multiple comparisons test and Bonferroni correction method for multiple testing. In case of 2 independent variables, a 2‐way ANOVA was used with Tukey's multiple comparisons test and Bonferroni‐Dunn correction method for multiple testing. Significance values are specified in figure legends.
2.7. Mass spectrometry
Cells were lysed, processed into tryptic peptides and analyzed by mass spectrometry using an Orbitrap Fusion Tribid mass spectrometer (ThermoFisher). Data were processed using the Maxquant computational platform essentially as described.42 The .raw MS files and search/identification files obtained with MaxQuant were deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD013857. A more detailed description of the mass spectrometry sample acquisition and data analysis is provided in Data S1.
3. RESULTS
3.1. Generation of CRISPR/Cas9 mediated VWF knockout cord blood outgrowth endothelial cells
Two gRNAs (gRNA‐1 and gRNA‐2) were designed to target the first exon of VWF and were cloned into the lentiviral LentiCRISPR expression vector (Figure 1), which simultaneously expresses gRNAs and the double strand break inducing Cas9 endonuclease. cbBOECs were transduced with LentiCRISPR without targeting gRNA (CTRL), gRNA‐1, gRNA‐2, or a combination and were then selected for effective virus transduction by puromycin. Introduction of double strand breaks using CRISPR/Cas9 gene editing generally results in a heterogeneous pattern of insertions and deletions, which may not all lead to knockouts. To enable separation and isolation of biallelic knockouts from the pool of cells that most likely also contains nonedited and monoallelic knockouts, we single‐cell sorted the transduced cells and grew them up in 96‐well plate format. We performed a first screen for VWF‐deficient clones by measuring secreted VWF in conditioned media (Figures 1 and 2A). From this, we selected 10 clones that lost the capacity to secrete VWF, as well as 4 control clones, and expanded those to 6‐well plates. It must be noted that it is important to initially select multiple clones, as viability of both control and knockout cells appeared to be decreased in some clones after virus transduction, single‐cell sorting, and repeated passaging. Immunoblotting was used to confirm absence of VWF in cell lysates (Figure 2B). We selected 2 knockout (VWF −/− A and VWF −/− B) and 2 control clones (CTRL A and CTRL B) with comparable growth characteristics and expansion potential, which were then further expanded for cryopreservation.
Figure 2.

Screening and mutation analysis of VWF knockout clones. (A) Conditioned medium of single‐cell clones that had reached >50% confluence was collected, and an ELISA for (secreted) von Willebrand factor (VWF) was performed as a first screen for VWF‐deficient clones. Arrows indicate control (CTRL) and gRNA targeted clones that were selected for further screening. (B) After selected clones had been expanded to 6‐well plates and reached confluence, cells were lysed and VWF deficiency was assayed using immunoblotting with polyclonal anti‐VWF and anti‐β‐actin as a loading control. Two control clones (CTRL A and CTRL B) and two VWF −/− clones (VWF −/− A and VWF −/− B) were selected, as indicated by the arrows. (C) Sanger sequencing and next‐generation sequencing on exon 1 of VWF were used to identify CRISPR/Cas9‐induced mutations. Sanger sequence traces are shown for all clones, and major mutations in both VWF −/− clones are indicated in the figure
Sanger sequencing and NGS of genomic DNA of the 2 selected clones revealed a number of CRISPR‐induced mutations, which were found directly adjacent to the gRNA hybridization sites (Figure 2C, Table S2). One allele of clone VWF −/− A contained a single nucleotide insertion (c.13insA), causing a frameshift that leads to a premature stop codon in the second exon (p.R5KfsX41). The other allele contained a 184‐bp deletion starting at position −169 until position 15 in exon 1, which led to the removal of its start codon. In line with this large deletion, we observed a smaller PCR fragment next to the normal‐sized fragment when amplifying VWF exon 1 from the genomic DNA of this clone (Figure S1). Clone VWF −/− B contained the same c.13insA mutation as VWF −/− A on both alleles and possibly a c.55insA mutation on a single allele, which combined with the c.13insA mutation would cause a frameshift causing a premature stop codon in exon 3 (Figure 2C, Table S2). Other low‐frequency variants that were picked up by NGS were most probably the result of sequence read errors (Table S2). These results show that the loss of VWF expression is the direct result of targeted mutations brought about by CRISPR/Cas9‐induced double strand breaks in the first exon.
We next sought to confirm that our CRISPR‐edited BOEC clones had retained their EC characteristics. CTRL and VWF −/− BOEC clones all formed confluent monolayers with a typical cobblestone morphology (not shown). All clones expressed endothelial markers VE‐cadherin (CD144; Figure 3) and platelet EC adhesion molecule‐1 (CD31; Figure S2) at cell‐cell junctions with no obvious differences between VWF −/− and CTRL clones. Immunostainings using a selection of monoclonal and polyclonal antibodies directed against VWF showed normal distribution and morphology of WPBs in CTRL clones A and B, whereas VWF−/− clones were completely devoid of WPBs or remaining VWF immunoreactivity (Figure 3). Finally, all clones were able to form networks in a Matrigel‐based morphogenesis assay in the presence of vascular endothelial growth factor (VEGF; Figure S3A) and maintained LDL uptake capacity (Figure S3B). These results show that we have generated VWF null BOEC clones that have otherwise preserved their normal endothelial properties.
Figure 3.

Morphology of VWF −/− cbBOECs is similar to controls and Weibel‐Palade bodies are lost. Two control clones (CTRL A and B) and two VWF −/− clones (VWF −/− A and B) were fixed after 5 to 7 days of confluence and immunostained with anti‐vascular endothelial (VE) ‐cadherin (magenta), mouse monoclonal anti‐human VWF (cyan) and rabbit polyclonal anti‐hVWF (green). Boxed areas are magnified on the right. Scale bars represent 20 μm
3.2. Whole‐proteome analysis of VWF‐deficient BOEC
To determine whether, apart from morphologic or functional differences, loss of VWF induces changes in the overall protein expression profile of ECs. We compared expression profiles of 2 independent CTRL and 2 independent VWF −/− BOEC clones using label‐free mass spectrometry–based protein quantification. From this, we quantified the abundance of 4371 proteins, of which only a limited number (17) was significantly up‐ or downregulated in VWF −/− BOECs (Figure 4A; a separate, side‐by‐side comparison of the individual clones is shown in Figure S4). As expected, the largest difference was seen for VWF, further confirming that we knocked out its expression in our CRISPR‐engineered BOECs. Proteins with statistically significant changes in expression are shown in Figure 4B, which includes among others retinal dehydrogenase 1 (ALDH1A1), nucleoredoxin (NXN), and collagen XII alpha chain (COL12A1) (Figure 4B). In contrast, expression of the WPB‐associated proteins CD63 and syntaxin‐3 (STX3), which were represented in our data set, remained unchanged (Figure 4C). This suggests that these proteins are still present, although they may display alternative localizations in the absence of WPBs. The expression profiles of a select array of WPB and endothelial markers43 presented no differences between CTRL and VWF −/− clones (Figure 4C), establishing that apart from lacking VWF, these cells have retained their endothelial characteristics.
Figure 4.
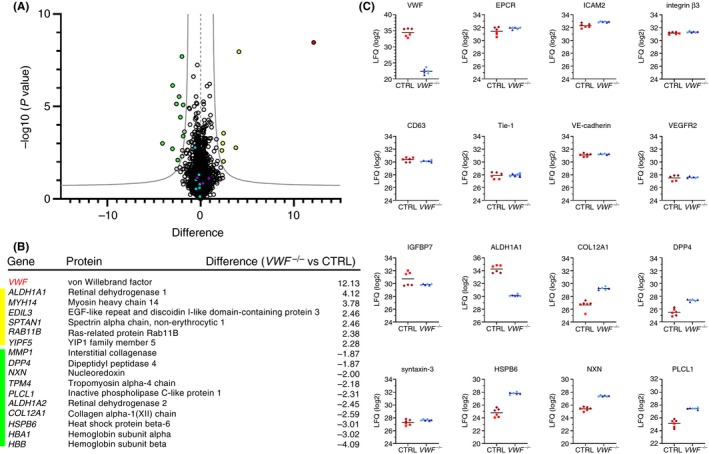
Whole proteome analysis of control and VWF −/− cord blood outgrowth endothelial cells (cbBOECs) reveals limited number of differentially expressed proteins. (A) Volcano plot showing differentially expressed proteins in 2 independent CTRL clones (CTRL I and II) and 2 independent VWF −/− clones (VWF −/− I and II), determined in triplicate. Difference in expression is shown on the x‐axis and the logarithmic P value (−log10 [P value]) is shown on the y‐axis. Significance cutoff line is based on false discovery rate (FDR) = 0.05 and S0 = 1.0. Highlighted data points represent VWF (red), Weibel‐Palade body (WPB) proteins (purple), proteins downregulated in VWF −/− (yellow), proteins upregulated in VWF −/− (green), and several unchanged endothelial markers (blue). (B) Table listing proteins that were significantly differentially expressed in VWF −/− BOECs compared to CTRL BOECs, and their corresponding difference in expression, shown in the same color coding as (A). (C) Expression profile plots showing label‐free quantification (LFQ) (log2) values of VWF, WPB‐associated proteins CD63, IGFBP7, and syntaxin‐3 (STX3) and several well‐established endothelial markers: endothelial protein C receptor (EPCR), intracellular adhesion molecule 2 (ICAM2), vascular endothelial growth factor receptor 2 (VEGFR2), integrin β3, Tie‐1, and vascular endothelial (VE)‐cadherin. LFQ plots of a selection of the hits in 4B are also shown: retinal dehydrogenase (ALDH1A1), collagen XII alpha (COL12A1), dipeptidyl peptidase 4 (DPP4), heat shock protein beta 6 (HSP6B), nucleoredoxin (NXN) and inactive phospholipase C‐like protein (PLCL1). Triplicates of individual clones are represented as CTRL I (brown diamonds) and CTRL II (red squares), VWF −/− I (dark‐blue triangles), VWF −/− II (light‐blue circles)
3.3. Alternative routing of Weibel‐Palade body cargo proteins
As genetic ablation of VWF also leads to the absence of WPBs, we hypothesized that proteins that are normally stored in or associated with these granules would now be subject to alternative trafficking pathways, resulting in a change in cellular expression. To test this, we investigated the localization of a number of WPB (cargo) proteins that are involved in inflammation, angiogenesis, and WPB trafficking using immunocytochemistry. In VWF‐deficient ECs, 2 chemokines that are normally stored in WPBs upon upregulation with IL‐1β, IL‐6, and IL‐8, were now primarily found in small punctate structures, which may represent small constitutive secretory vesicles and a ribbon‐like pattern consistent with their accumulation in the Golgi (Figure 5A,C).44, 45, 46, 47 Because only a small portion of IL‐6 and IL‐8 is stored inside WPBs,19, 46 we addressed whether loss of WPBs has an effect on functional release of these cytokines. Therefore, we stimulated CTRL and VWF −/− for 24 hours with IL‐1β and assayed their unstimulated and histamine‐evoked release by ELISA (Figure 5B and D). Overall, no significant differences were found in terms of intracellular content or absolute release between control and knockout ECs. In VWF −/−, B cytokine release relative to intracellular content appears to be increased, but not in VWF −/− A. However, the production of IL‐6 and IL‐8 is dramatically increased in all clones upon IL‐1β, and release over 24 hours is clearly considerably higher than the stored fraction. Another trend was visible, which showed that an induction of cytokine release upon histamine stimulation that was seen in CTRL clones was less pronounced in VWF −/− clones, although the data showed limited statistically significant differences due to variation between experiments.
Figure 5.
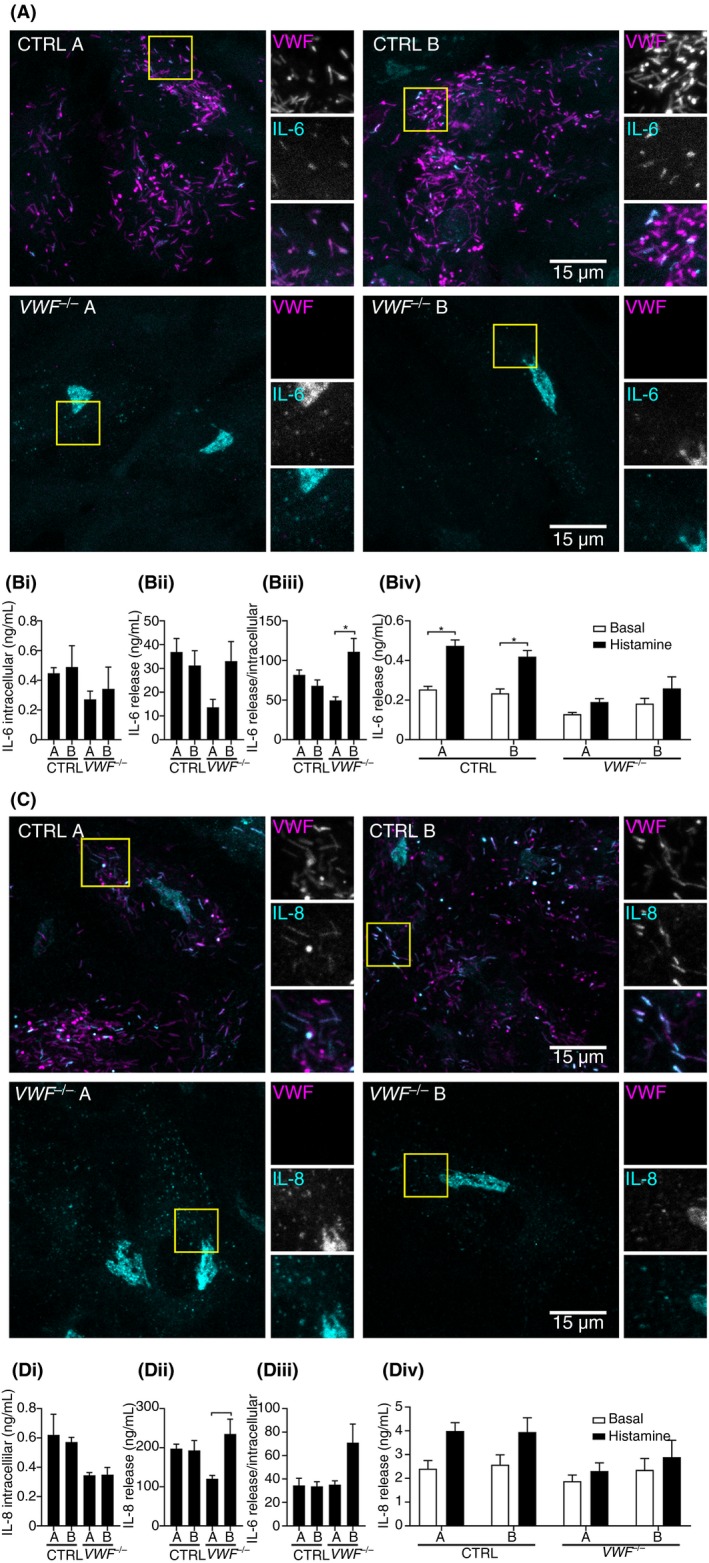
Interleukin (IL)‐6 and IL‐8 show alternative localization in VWF −/− cord blood outgrowth endothelial cells (cbBOECs). (A and C) Two control clones (CTRL A and B) and 2 VWF −/− clones (VWF −/− A and B) were cultured for 5 to 7 days after reaching confluence, the last 24 hours including 10 ng/mL IL‐1β. Immunostainings were performed with mouse monoclonal anti‐human VWF (magenta) and (A) anti‐IL‐6 (cyan) or (C) anti‐IL‐8 (cyan). Boxed areas are magnified on the right. Scale bars represent 15 μm. (B and D) Cytokine storage and secretion in IL‐1β–treated CTRL and VWF −/− BOECs. IL‐6 (Bi) and IL‐8 (Di) intracellular content were measured by ELISA of lysates. (Bii‐iii) Steady‐state release of IL‐6 (Bii‐Biii) and IL‐8 (Dii‐Diii) was measured by ELISA in 24‐hour unstimulated release samples and was expressed as absolute levels (Bii and Dii) or normalized to intracellular content (Biii and Diii). (Biv and Div) Stimulus‐induced Ang‐2 release was assayed using 30 minutes of unstimulated vs. histamine‐treated CTRL and VWF −/− BOECs. Data are shown as mean ± standard error of the mean of 3 independent biological replicates, performed in triplo. Statistical analysis was 1‐way analysis of variance with a significance level of *P < 0.05, **P < 0.01, ***P < 0.001
The angiogenesis mediator Ang‐2 is another protein that, like IL‐8, is thought to be coackaged in the WPB during its formation at the trans‐Golgi network (TGN), possibly through noncovalent interaction with VWF.48 Also for Ang‐2, we observed a punctate pattern in the absence of WPBs, as well as an overall decrease in Ang‐2 signal (Figure 6). However, whereas the punctae of IL‐6 and IL‐8 were diffusely distributed throughout the cell, Ang‐2 shows enrichment at cell‐cell junctions (Figure 6 see also Figure S5). It has been suggested previously that altered angiogenic properties of ECs lacking VWF may be caused by increased constitutive release of Ang‐2, which would then lead to autocrine/paracrine regulation of Tie‐2 signaling.28 In line with this, we observed that Ang‐2, which in CTRL cells primarily localizes to WPBs and shows minimal overlap with Tie‐2, is primarily found on Tie‐2 positive structures that are enriched at cell‐cell junctions of VWF−/− cells (Figure S5). This suggests that in the absence of its storage compartment, constitutively released Ang‐2 associates with Tie‐2 on the plasma membrane. Next, we performed secretion assays with the CTRL and VWF −/− clones and measured the Ang‐2 concentration in the releasates vs. the lysates by ELISA. Strikingly, lysates of VWF −/− clones contained less Ang‐2 than CTRL clones (Figure 6Bi), while the basal release in 24 hours was increased in VWF −/− B (Figure 6Bii), substantiating that in the absence of WPBs Ang‐2 is not stored intracellularly but is continuously released. Both VWF −/− clones exhibited higher Ang‐2 release relative to intracellular content compared to CTRL clones (Figure 6Biii). In response to histamine, CTRL clones showed a strong increase of Ang‐2 release, whereas in the VWF −/− clones, Ang‐2 release remained unchanged to unstimulated cells, suggesting that no Ang‐2 is stored in histamine‐responsive vesicles. Absolute Ang‐2 release in response to histamine was indeed decreased in VWF‐deficient clones.
Figure 6.

Angiopoietin‐2 (Ang‐2) localizes to the cell‐cell junctions in VWF −/− cord blood outgrowth endothelial cells (cbBOECs). (A) Two control clones (CTRL A and B) and 2 VWF −/− clones (VWF −/− A and B) were fixed after 5 to 7 days of confluence and immunostained with mouse monoclonal anti‐human VWF (cyan), anti‐Ang‐2 (green) and anti‐β‐catenin (magenta). Boxed areas are magnified on the right. Scale bars represent 10 μm. (B) Dysregulation of Ang‐2 storage and secretion in the absence of von Willebrand factor (VWF). (Bi) Ang‐2 intracellular content was measured by ELISA of lysates. (Bii‐iii) Steady‐state release of Ang‐2 was measured by ELISA in 24‐hour unstimulated release samples and was expressed as absolute levels (Bii) or normalized to intracellular content (Biii). (Biv) Stimulus‐induced Ang‐2 release was assayed using 30 minutes of unstimulated vs. histamine‐treated CTRL and VWF −/− BOECs. Data are shown as mean ± SEM of 3 independent biological replicates, performed in triplo. Statistical analysis was one‐way analysis of variance with a significance level of *P < 0.05, **P < 0.01, ***P < 0.001
A different localization pattern was observed for syntaxin‐3 and CD63, 2 proteins that are normally found both on WPBs and on endosomes.40, 49 It has previously been shown that CD63 is trafficked to the WPBs via the endosomal system in an annexin A8‐ and AP‐3–dependent manner.21, 50, 51, 52 As expected, whereas CD63 localization to the elongated WPBs is lost in VWF −/− clones, its localization to spherical vesicles that are most likely endosomes remains similar (Figure 7). Interestingly, syntaxin‐3 shows a similar pattern, suggesting that this protein may normally follow a similar route as CD63 to arrive at a WPB.
Figure 7.

CD63 and syntaxin‐3 show loss of localization to elongated vesicles but unaltered pattern on rounded vesicles in VWF −/− cord blood outgrowth endothelial cells (cbBOECs). Two control clones (CTRL A and B) and 2 VWF −/− clones (VWF −/− A and B) were fixed after 5 to 7 days of confluence and immunostained with mouse monoclonal anti‐human VWF (magenta) and (A) anti‐CD63 (cyan) or (B) anti‐syntaxin‐3 (STX3, cyan). Boxed areas are magnified on the right. Scale bars represent 10 μm
4. DISCUSSION
In this study, we generated stable VWF knockout ECs using CRISPR/Cas9 gene editing, resulting in ECs entirely devoid of WPBs. To our knowledge, this is the first study to report the ablation of an entire organelle using CRISPR/Cas9. A number of studies have previously employed CRISPR/Cas9 to knock out expression of targets in ECs. In most of those studies, no single‐cell selection and clonal expansion was performed, but a bulk population was used in which a proportion of the cells may still express the targeted protein.33, 53 Single‐cell cloning is needed to arrive at genetically homogeneous populations of ECs that have lost expression of the target gene, but the extensive passaging involved places high demands on the proliferative capacity of modified endothelial cells. Our approach was similar to a methodological study in which clonal endothelial lines were generated after single‐cell sorting of CRISPR‐edited cbBOECs,32 primary ECs that have increased expansive capacity compared other primary ECs such as human umbilical vein ECs or peripheral BOECs.34, 54 Similar to what we find in our study, clonal CRISPR‐engineered BOEC lines had retained sufficient expansion capacity for downstream experimental applications. As our target of interest is a secreted rather than a cell surface protein, we used an assay for secreted protein as our initial clone selection followed by cell expansion for further clone selection through analysis of intracellular protein. For knockout of intracellular, nonsecreted proteins of interest, an extra expansion step would be required for immediate clone selection through western blotting, as we have previously described.52
To ensure that phenotypic differences that we observed are not the result of (1) variation in genetic background between donors, (2) clonal effects that are not directly linked to the inactivation of the target, or (3) clonal expansion of single cells, we used BOECs from a single donor, and we selected multiple clones from CRISPR‐edited cells as well as control cells that were subjected to the same single‐cell sorting and expansion procedure in parallel. Even with these precautions, we observed that a substantial number of clonal candidates were generated that ceased to expand shortly after the clonal selection procedure or that suffered from severely reduced proliferative capacity. Whether this is the result of lentiviral transduction and/or expression of Cas9 protein or whether this is an inherent problem of a primary cell system being pushed to the limit of its proliferative capacity by clonal expansion is at this point unclear. Careful surveying of a large number of candidates for their proliferative capacity may therefore be necessary to generate enough clones for the intended experimental analysis. This also complicates the use of (CRISPR‐edited) BOECs in assays that model angiogenesis, which are highly dependent on proliferation of ECs, and their outcomes would be influenced by a rapid drift in proliferative capacity between individual clones. For that particular purpose, it may be more suited to use ECs derived from induced pluripotent stem cells (iPSCs). CRISPR/Cas9 gene editing in iPSCs as well as their differentiation into ECs has been well established.55, 56
Genetic removal of WPBs revealed alternative trafficking pathways for WPB cargo and associated proteins. Contrary to most WPB cargo proteins, which are copackaged with VWF in newly forming WPBs, CD63 first traffics via the endosomal system, and that is why it is not observed in immature WPBs.50, 51 We have recently shown that CD63, but also the WPB v‐SNARE protein vesicle‐associated membrane protein 8 (VAMP8), depend on the AP‐3 complex for their targeting to the WPBs.52 The WPB regulating t‐SNARE protein syntaxin‐3, one of the cognate SNARE partners of VAMP8, has also been observed both on WPBs and on round vesicles that are most likely endosomes, but how it is trafficked to WPBs remains unknown. Interestingly, we observe a similar localization pattern for CD63 and syntaxin‐3, with localization to WPBs and endosomes in control cells and only to endosomes in VWF −/− cells. This shows that syntaxin‐3 targeting to endosomes is not dependent on WPBs and is therefore most likely not initiated by a journey through the secretory pathway. Instead, it may be directly incorporated into the endosomes from the Golgi, from which it will then commence its cycling between WPBs, plasma membrane, and endosomes in a similar manner as CD63.
Trafficking of the chemokines IL‐6 and IL‐8, which are incorporated directly into WPBs during their formation at the TGN, is also substantially altered in the absence of WPBs. This was mainly reflected in a loss of the stimulus‐sensitive pool of these chemokines in response to agonist triggering (Figure 5B and D). The lack of stimulus‐induced release of IL‐6 and particularly IL‐8, which is a potent neutrophil chemoattractant, that we observe in VWF −/− BOECs suggests that apart from the direct57 and indirect (via P‐selectin surface presentation8) role that VWF plays in adhesion of leukocytes, impaired regulated secretion of chemokines could further contribute to defects in neutrophil recruitment in the absence of VWF. Many interleukins, including IL‐6 and IL‐8, have been implicated in cardiovascular disease such as atherosclerosis (reviewed in Apostolakis et al58). This raises the question of whether dysregulated endothelial chemokine release as a result of VWF deficiency may also affect atherogenesis. Several studies in murine models have indeed shown that VWF‐deficient mice are protected from experimental atherosclerosis.59, 60, 61 However, firm evidence for a similar protective effect in patients with VWD has so far not been found (reviewed in62). In vitro IL‐6 and IL‐8 constitutive secretion did not show large differences between CTRL and VWF −/− BOECs (Figure 5), which is consistent with previous observations that the bulk of newly synthesized IL‐6 and IL‐8 does not enter WPBs but is released via the constitutive secretion pathway.19, 46 As far as we are aware, no quantitative analyses have been performed on IL‐6 and/or IL‐8 levels in VWD patients, but based on our in vitro observations, the likelihood of these deviating significantly from healthy subjects is limited, given that most EC‐derived IL‐6 or IL‐8 is not released via the WPBs.
WPBs also facilitate the long‐term storage of the angiogenesis mediator Ang‐2 and ensure its on‐demand availability during proangiogenic conditions, for instance, in response to triggering of WPB release by VEGF.28 Previous studies have also shown altered levels of continuous Ang‐2 release from ECs from patients with VWD or after small interfering RNA–mediated VWF knockdown, which has been suggested to be in part the underlying cause of altered angiogenic properties of VWD patient–derived BOECs and the angiodysplasia that is observed in some patients with VWD.26, 30 This hypothesis is supported by the current finding that when Ang‐2 cannot be stored in the WPBs, it no longer appears to localize to storage vesicles but is released constitutively (Figure 6). However, a recent study on circulating levels of angiogenic mediators in patients with moderate and severe VWD did not find (statistically significant) differences in circulating levels of Ang‐2 in patients with normal and impaired VWF synthesis.63 Secreted Ang‐2 takes part in the complex regulation of angiogenesis through the interaction with a number of EC surface receptor proteins, including Tie‐2 and several integrins.64, 65 Notably, we observed increased colocalization of Ang‐2 with Tie‐2 at cell‐cell junctions, which we speculate represents extracellular binding of Ang‐2 to its receptor. This suggests that the absence of WPBs may lead to increased interactions of Ang‐2 with its cell surface receptors and thereby influence the angiogenic activity of ECs in an autocrine/paracrine manner. Future studies are needed to further unravel how the endothelium dynamically regulates angiogenesis through release of angiogenic factors.
5. RELATIONSHIP DISCLOSURE
The authors report no conflicts of interest.
AUTHOR CONTRIBUTIONS
MS, MK, JW, AG, MH, BN, FPJvA, and MdB performed research and analyzed data; MvdB, CM, and JV contributed vital expertise; MS, MK, and RB designed the research and wrote the paper.
Supporting information
ACKNOWLEDGMENTS
We thank Karin van Leeuwen for her help with the NGS analysis. This study was supported by grants from the Landsteiner Foundation for Blood Transfusion Research (LSBR‐1244 and LSBR‐1707), Sanquin (PPOC‐2018‐21) and the Dutch Thrombosis Foundation (TSN 56‐2015 and 2017‐01). RB is supported by a European Hematology Association Research Fellowship.
Schillemans M, Kat M, Westeneng J, et al. Alternative trafficking of Weibel‐Palade body proteins in CRISPR/Cas9‐engineered von Willebrand factor–deficient blood outgrowth endothelial cells. Res Pract Thromb Haemost. 2019;3:718–732. 10.1002/rth2.12242
Maaike Schillemans and Marije Kat contributed equally.
REFERENCES
- 1. Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424. [DOI] [PubMed] [Google Scholar]
- 2. Leebeek FWG, Eikenboom JCJ. Von Willebrand's disease. N Engl J Med. 2016;375:2067–80. [DOI] [PubMed] [Google Scholar]
- 3. Sadler JE. von Willebrand factor: two sides of a coin. J Thromb Haemost. 2005;3:1702–9. [DOI] [PubMed] [Google Scholar]
- 4. Rondaij MG, Bierings R, Kragt A, van Mourik JA, Voorberg J. Dynamics and plasticity of Weibel‐Palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:1002–7. [DOI] [PubMed] [Google Scholar]
- 5. Metcalf DJ, Nightingale TD, Zenner HL, Lui‐Roberts WW, Cutler DF. Formation and function of Weibel‐Palade bodies. J Cell Sci. 2008;121:19–27. [DOI] [PubMed] [Google Scholar]
- 6. Schillemans M, Karampini E, Kat M, Bierings R. Exocytosis of Weibel‐Palade bodies: how to unpack a vascular emergency kit. J Thromb Haemost. 2019;17:6–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Groeneveld DJ, van Bekkum T, Dirven RJ, Wang J‐W, Voorberg J, Reitsma PH, et al. Angiogenic characteristics of blood outgrowth endothelial cells from patients with von Willebrand disease. J Thromb Haemost. 2015;13:1854–66. [DOI] [PubMed] [Google Scholar]
- 8. Denis CV, André P, Saffaripour S, Wagner DD. Defect in regulated secretion of P‐selectin affects leukocyte recruitment in von Willebrand factor‐deficient mice. Proc Natl Acad Sci USA. 2001;98:4072–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haberichter SL, Merricks EP, Fahs SA, Christopherson PA, Nichols TC, Montgomery RR. Re‐establishment of VWF‐dependent Weibel‐Palade bodies in VWD endothelial cells. Blood. 2005;105:145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Meyer SF, Vanhoorelbeke K, Chuah MK, Pareyn I, Gillijns V, Hebbel RP, et al. Phenotypic correction of von Willebrand disease type 3 blood‐derived endothelial cells with lentiviral vectors expressing von Willebrand factor. Blood. 2006;107:4728–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wagner DD, Saffaripour S, Bonfanti R, Sadler JE, Cramer EM, Chapman B, et al. Induction of specific storage organelles by von Willebrand factor propolypeptide. Cell. 1991;64:403–13. [DOI] [PubMed] [Google Scholar]
- 12. Voorberg J, Fontijn R, Calafat J, Janssen H, van Mourik JA, Pannekoek H. Biogenesis of von Willebrand factor–containing organelles in heterologous transfected CV‐1 cells. EMBO J. 1993;12:749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Michaux G, Abbitt KB, Collinson LM, Haberichter SL, Norman KE, Cutler DF. The physiological function of von Willebrand's factor depends on its tubular storage in endothelial Weibel‐Palade bodies. Dev Cell. 2006;10:223–32. [DOI] [PubMed] [Google Scholar]
- 14. André P, Denis CV, Ware J, Saffaripour S, Hynes RO, Ruggeri ZM, et al. Platelets adhere to and translocate on von Willebrand factor presented by endothelium in stimulated veins. Blood. 2000;96:3322–8. [PubMed] [Google Scholar]
- 15. Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, et al. ADAMTS‐13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–9. [DOI] [PubMed] [Google Scholar]
- 16. Zheng Y, Chen J, López JA. Flow‐driven assembly of VWF fibres and webs in in vitro microvessels. Nat Commun. 2015;6:7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bridges DJ, Bunn J, van Mourik JA, Grau G, Preston RJS, Molyneux M, et al. Rapid activation of endothelial cells enables Plasmodium falciparum adhesion to platelet‐decorated von Willebrand factor strings. Blood. 2010;115:1472–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smeets MWJ, Bierings R, Meems H, Mul FPJ, Geerts D, Vlaar APJ, et al. Platelet‐independent adhesion of calcium‐loaded erythrocytes to von Willebrand factor. PLoS One. 2017;12:e0173077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Knipe L, Meli A, Hewlett L, Bierings R, Dempster J, Skehel P, et al. A revised model for the secretion of tPA and cytokines from cultured endothelial cells. Blood. 2010;116:2183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Doyle EL, Ridger V, Ferraro F, Turmaine M, Saftig P, Cutler DF. CD63 is an essential cofactor to leukocyte recruitment by endothelial P‐selectin. Blood. 2011;118:4265–73. [DOI] [PubMed] [Google Scholar]
- 21. Poeter M, Brandherm I, Rossaint J, Rosso G, Shahin V, Skryabin BV, et al. Annexin A8 controls leukocyte recruitment to activated endothelial cells via cell surface delivery of CD63. Nat Commun. 2014;5:3738. [DOI] [PubMed] [Google Scholar]
- 22. Denis C, Methia N, Frenette PS, Rayburn H, Ullman‐Culleré M, Hynes RO, et al. A mouse model of severe von Willebrand disease: defects in hemostasis and thrombosis. Proc Natl Acad Sci U S A. 1998;95:9524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fiedler U, Scharpfenecker M, Koidl S, Hegen A, Grunow V, Schmidt JM, et al. The Tie‐2 ligand angiopoietin‐2 is stored in and rapidly released upon stimulation from endothelial cell Weibel‐Palade bodies. Blood. 2004;103:4150–6. [DOI] [PubMed] [Google Scholar]
- 24. van Breevoort D, van Agtmaal EL, Dragt BS, Gebbinck JK, Dienava‐Verdoold I, Kragt A, et al. Proteomic screen identifies IGFBP7 as a novel component of endothelial cell–specific Weibel‐Palade bodies. J Proteome Res. 2012;11:2925–36. [DOI] [PubMed] [Google Scholar]
- 25. van Agtmaal EL, Bierings R, Dragt BS, Leyen TA, Fernandez‐Borja M, Horrevoets AJG, et al. The shear stress‐induced transcription factor KLF2 affects dynamics and angiopoietin‐2 content of Weibel‐Palade bodies. PLoS One. 2012;7:e38399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Starke RD, Ferraro F, Paschalaki KE, Dryden NH, McKinnon TA, Sutton RE, et al. Endothelial von Willebrand factor regulates angiogenesis. Blood. 2011;117:1071–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Franchini M, Mannucci PM. Von Willebrand disease‐associated angiodysplasia: a few answers, still many questions. Br J Haematol. 2013;161:177–82. [DOI] [PubMed] [Google Scholar]
- 28. Randi AM, Smith KE, Castaman G. Von Willebrand factor regulation of blood vessel formation. Blood. 2018;132:132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Starke RD, Paschalaki KE, Dyer CEF, Harrison‐Lavoie KJ, Cutler JA, McKinnon TA, et al. Cellular and molecular basis of von Willebrand disease: studies on blood outgrowth endothelial cells. Blood. 2013;121:2773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Selvam SN, Casey LJ, Bowman ML, Hawke LG, Longmore AJ, Mewburn J, et al. Abnormal angiogenesis in blood outgrowth endothelial cells derived from von Willebrand disease patients. Blood Coagul Fibrinolysis. 2017;28:521–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lillicrap D. von Willebrand disease: advances in pathogenetic understanding, diagnosis, and therapy. Hematology Am Soc Hematol Educ Program. 2013;2013:254–60. [DOI] [PubMed] [Google Scholar]
- 32. Abrahimi P, Chang WG, Kluger MS, Qyang Y, Tellides G, Saltzman WM, et al. Efficient gene disruption in cultured primary human endothelial cells by CRISPR/Cas9. Circ Res. 2015;117:121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gong H, Liu M, Klomp J, Merrill BJ, Rehman J, Malik AB. Method for dual viral vector mediated CRISPR‐Cas9 gene disruption in primary human endothelial cells. Sci Rep. 2017;7:42127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van Beem RT, Verloop RE, Kleijer M, Noort WA, Loof N, Koolwijk P, et al. Blood outgrowth endothelial cells from cord blood and peripheral blood: angiogenesis‐related characteristics in vitro. J Thromb Haemost. 2009;7:217–26. [DOI] [PubMed] [Google Scholar]
- 35. Martin‐Ramirez J, Hofman M, van den Biggelaar M, Hebbel RP, Voorberg J. Establishment of outgrowth endothelial cells from peripheral blood. Nat Protoc. 2012;7:1709–15. [DOI] [PubMed] [Google Scholar]
- 36. van Breevoort D, Snijders AP, Hellen N, Weckhuysen S, van Hooren KWEM, Eikenboom J, et al. STXBP1 promotes Weibel‐Palade body exocytosis through its interaction with the Rab27A effector Slp4‐a. Blood. 2014;123:3185–94. [DOI] [PubMed] [Google Scholar]
- 37. Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA targeting specificity of RNA‐guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off‐target effects of CRISPR‐Cas9. Nat Biotechnol. 2016;34:184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sanjana NE, Shalem O, Zhang F. Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods. 2014;11:783–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schillemans M, Karampini E, van den Eshof BL, Gangaev A, Hofman M, van Breevoort D, et al. Weibel‐Palade body localized syntaxin‐3 modulates von Willebrand factor secretion from endothelial cells. Arterioscler Thromb Vasc Biol. 2018;38:1549–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van Hooren KWEM, van Breevoort D, Fernandez‐Borja M, Meijer AB, Eikenboom J, Bierings R, et al. Phosphatidylinositol‐3,4,5‐triphosphate‐dependent Rac exchange factor 1 regulates epinephrine‐induced exocytosis of Weibel‐Palade bodies. J Thromb Haemost. 2014;12:273–81. [DOI] [PubMed] [Google Scholar]
- 42. Salerno F, Engels S, van den Biggelaar M, van Alphen FPJ, Guislain A, Zhao W, et al. Translational repression of pre‐formed cytokine‐encoding mRNA prevents chronic activation of memory T cells. Nat Immunol. 2018;19:828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goncharov NV, Nadeev AD, Jenkins RO, Avdonin PV. Markers and biomarkers of endothelium: when something is rotten in the state. Oxid Med Cell Longev. 2017;2017:9759735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Utgaard JO, Jahnsen FL, Bakka A, Brandtzaeg P, Haraldsen G. Rapid secretion of prestored interleukin 8 from Weibel‐Palade bodies of microvascular endothelial cells. J Exp Med. 1998;188:1751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wolff B, Burns AR, Middleton J, Rot A. Endothelial cell “memory” of inflammatory stimulation: human venular endothelial cells store interleukin 8 in Weibel‐Palade bodies. J Exp Med. 1998;188:1757–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bierings R, van den Biggelaar M, Kragt A, Mertens K, Voorberg J, van Mourik JA. Efficiency of von Willebrand factor–mediated targeting of interleukin‐8 into Weibel‐Palade bodies. J Thromb Haemost. 2007;5:2512–9. [DOI] [PubMed] [Google Scholar]
- 47. Kiskin NI, Hellen N, Babich V, Hewlett L, Knipe L, Hannah MJ, et al. Protein mobilities and P‐selectin storage in Weibel‐Palade bodies. J Cell Sci. 2010;123:2964–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McKinnon TA, Starke RD, Ediriwickrema K, Randi AM, Laffan M. Von Willebrand factor binds to the endothelial growth factor angiopoietin‐2 both within endothelial cells and upon release from Weibel Palade bodies. Blood. 2011;118:698 LP – 698. [DOI] [PubMed] [Google Scholar]
- 49. Vischer UM, Wagner DD. CD63 is a component of Weibel‐Palade bodies of human endothelial cells. Blood. 1993;82:1184–91. [PubMed] [Google Scholar]
- 50. Kobayashi T, Vischer UM, Rosnoblet C, Lebrand C, Lindsay M, Parton RG, et al. The tetraspanin CD63/lamp3 cycles between endocytic and secretory compartments in human endothelial cells. Mol Biol Cell. 2000;11:1829–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harrison‐Lavoie KJ, Michaux G, Hewlett L, Kaur J, Hannah MJ, Lui‐Roberts WW, et al. P‐selectin and CD63 use different mechanisms for delivery to Weibel‐Palade bodies. Traffic. 2006;7:647–62. [DOI] [PubMed] [Google Scholar]
- 52. Karampini E, Schillemans M, Hofman M, van Alphen F, de Boer M, Kuijpers TW, et al. Defective AP‐3‐dependent VAMP8 trafficking impairs Weibel‐Palade body exocytosis in Hermansky‐Pudlak syndrome type 2 blood outgrowth endothelial cells. Haematologica. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cai M, Li S, Shuai Y, Li J, Tan J, Zeng Q. Genome‐wide CRISPR‐Cas9 viability screen reveals genes involved in TNF‐α‐induced apoptosis of human umbilical vein endothelial cells. J Cell Physiol. 2019;234:9184–93. [DOI] [PubMed] [Google Scholar]
- 54. Ingram DA, Mead LE, Tanaka H, Meade V, Fenoglio A, Mortell K, et al. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004;104:2752–60. [DOI] [PubMed] [Google Scholar]
- 55. Lin Y, Gil CH, Yoder MC. Differentiation, evaluation, and application of human induced pluripotent stem cell‐derived endothelial cells. Arterioscler Thromb Vasc Biol. 2017;37:2014–25. [DOI] [PubMed] [Google Scholar]
- 56. Cheng L‐T, Sun L‐T, Tada T. Genome editing in induced pluripotent stem cells. Genes Cells Stem Cell Rev Rep. 2012;17:431–8. [DOI] [PubMed] [Google Scholar]
- 57. Petri B, Broermann A, Li H, Khandoga AG, Zarbock A, Krombach F, et al. von Willebrand factor promotes leukocyte extravasation. Blood. 2010;116:4712–9. [DOI] [PubMed] [Google Scholar]
- 58. Apostolakis S, Vogiatzi K, Amanatidou V, Spandidos DA. Interleukin 8 and cardiovascular disease. Cardiovasc Res. 2009;84:353–60. [DOI] [PubMed] [Google Scholar]
- 59. Methia N, André P, Denis CV, Economopoulos M, Wagner DD. Localized reduction of atherosclerosis in von Willebrand factor–deficient mice. Blood. 2001;98:1424–8. [DOI] [PubMed] [Google Scholar]
- 60. Gandhi C, Ahmad A, Wilson KM, Chauhan AK. ADAMTS13 modulates atherosclerotic plaque progression in mice via a VWF‐dependent mechanism. J Thromb Haemost. 2014;12:255–60. [DOI] [PubMed] [Google Scholar]
- 61. Doddapattar P, Dhanesha N, Chorawala MR, Tinsman C, Jain M, Nayak MK, et al. Endothelial cell–derived von Willebrand factor, but not platelet‐derived, promotes atherosclerosis in apolipoprotein E–deficient mice. Arterioscler Thromb Vasc Biol. 2018;38:520–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. van Galen KPM, Tuinenburg A, Smeets EM, Schutgens REG. Von Willebrand factor deficiency and atherosclerosis. Blood Rev. 2012;26:189–96. [DOI] [PubMed] [Google Scholar]
- 63. Groeneveld DJ, Sanders YV, Adelmeijer J, Mauser‐Bunschoten EP, van der Bom JG, Cnossen MH, et al. Circulating angiogenic mediators in patients with moderate and severe von Willebrand disease: a multicentre cross‐sectional study. Thromb Haemost. 2018;118:152–60. [DOI] [PubMed] [Google Scholar]
- 64. Felcht M, Luck R, Schering A, Seidel P, Srivastava K, Hu J, et al. Angiopoietin‐2 differentially regulates angiogenesis through TIE2 and integrin signaling. J Clin Invest. 2012;122:1991–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Thomas M, Felcht M, Kruse K, Kretschmer S, Deppermann C, Biesdorf A, et al. Angiopoietin‐2 stimulation of endothelial cells induces alphavbeta3 integrin internalization and degradation. J Biol Chem. 2010;285:23842–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials