

Abstract
Endogenous ions play important roles in the function and pharmacology of G-protein coupled receptors (GPCRs). Historically the evidence for ionic modulation of GPCR function dates to 1973 with studies of opioid receptors, where it was demonstrated that physiologic concentrations of sodium allosterically attenuated agonist binding. This Na+-selective effect was distinct from effects of other monovalent and divalent cations, with the latter usually counteracting sodium’s negative allosteric modulation of binding. Since then, numerous studies documenting the effects of mono- and divalent ions on GPCR function have been published. While ions can act selectively and nonselectively at many sites in different receptors, the discovery of the conserved sodium ion site in class A GPCR structures in 2012 revealed the unique nature of Na+ site, which has emerged as a near-universal site for allosteric modulation of class A GPCR structure and function. In this review, we synthesize and highlight recent advances in the functional, biophysical, and structural characterization of ions bound to GPCRs. Taken together, these findings provide a molecular understanding of the unique roles of Na+ and other ions as GPCR allosteric modulators. We will also discuss how this knowledge can be applied to the redesign of receptors and ligand probes for desired functional and pharmacological profiles.
SIGNIFICANCE STATEMENT
The function and pharmacology of GPCRs strongly depend on the presence of mono and divalent ions in experimental assays and in living organisms. Recent insights into the molecular mechanism of this ion-dependent allosterism from structural, biophysical, biochemical, and computational studies provide quantitative understandings of the pharmacological effects of drugs in vitro and in vivo and open new avenues for the rational design of chemical probes and drug candidates with improved properties.
I. Historical Overview
Endogenous ions are involved in all aspects of human biology, including their key roles in the function and pharmacology of GPCRs, which comprise the largest family of clinically relevant protein targets (Lagerström and Schiöth, 2008; Katritch et al., 2013; Hauser et al., 2017). GPCRs signal both at the plasma membrane and in intracellular membranes, including endosomes and golgi (Calebiro et al., 2010; Irannejad et al., 2013; Vilardaga et al., 2014; Godbole et al., 2017; Eichel and von Zastrow, 2018), and are likely exposed to large spatiotemporal variations in ionic and pH conditions that may affect their function. Thus, for instance, extracellular Na+ is normally maintained in 135–145 mM range, while its intracellular levels are about 10 times lower in most cells (Lodish et al., 2000); intracellular sodium levels rapidly increase during depolarization in neurons. Also, some GPCRs are directly (Wingler et al., 2019) and selectively modulated by inorganic ions as a part of their physiologic function, e.g., CaSR by Ca2+ (Silve et al., 2005) and GPR39 by Zn+ (Sato et al., 2016). Other GPCRs are proton sensing, including GPR68, GPR4, TDAG8, and G2A (Ludwig et al., 2003; Radu et al., 2005; Yang et al., 2007; Liu et al., 2010; Huang et al., 2015b). In this review though, we will mostly focus on the function of endogenous ligands, and therapeutic drugs, being allosterically modulated by ions interacting with GPCRs.
Historically, the first evidence for ionic modulation of GPCRs dates well before they were recognized as a large family of receptors sharing a common seven-transmembrane (7TM) architecture. In 1973, studies of opioid receptors showed that agonist binding is negatively modulated by monovalent cations like Na+ (Pert et al., 1973; Pert and Snyder, 1974), while being positively modulated by divalent cations (Pasternak et al., 1975). Several subsequent studies provided biochemical data suggesting that these effects were mediated by an allosteric mechanism (Simon and Groth, 1975; Horstman et al., 1990). A similar negative allosteric modulation of agonist binding affinity was soon discovered for many other class A GPCRs including adrenergic (Tsai and Lefkowitz, 1978), dopaminergic (Neve, 1991; Neve et al., 1991) and somatostatin (Kong et al., 1993) receptors. Since then, hundreds of papers have appeared documenting the actions of sodium, as well as other cations and anions on the function of many GPCRs [see Katritch et al. (2014) and (Strasser et al., 2015) for review].
Moreover, high-resolution structural information for GPCRs and their complexes, which has emerged in the past few years (Liu et al., 2012b; Fenalti et al., 2014; Miller-Gallacher et al., 2014; Wang et al., 2017) has made it possible to identify a variety of ion binding sites in GPCRs (Fig. 1; Table 1). While some of the ions, like the multiple Zn2+ and Hg2+ ions in rhodopsin structures were introduced to assist crystallization and/or anomalous diffraction phasing (Teller et al., 2001), many other ion binding sites may be relevant for endogenous ligand binding at specific receptors. For example, the crystallographically observed PO43− site in H1 histamine receptor (Shimamura et al., 2011) or Na+ binding in the extracellular loop in the β1AR adrenergic receptor (Miller-Gallacher et al., 2014).
Fig. 1.

Ions identified in GPCR crystal structures. (A) Monovalent ions: Na+ (blue) and Cl− (green). (B) Polyvalent ions Zn2+ (magenta), Hg2+ (cyan), PO43− (phosphate colored green), SO42− (sulfur colored yellow). GPCR structures shown as gray cartoons.
TABLE 1.
All GPCR structures resolved at 3.1 Å or better, with small ions bound to 7TM domain
| Receptor | PDB code | Resolution Range, Å | Ion | Site |
|---|---|---|---|---|
| A2AAR | 4EIY, 5IU4, 5IU7, 5IU8, 5IUA, 5IUB, 5K2A, 5K2B, 5K2C, 5K2D, 5MZJ, 5MZP, 5N2R, 5NLX, 5NM2, 5NM4, 5OLG, 5OLH, 5OLV, 5OLZ, 5OM1, 5OM4, 5VRA | 1.7–2.8 | Na+ | D2.50, conserved, tight bindinga |
| DRD4 | 5WIV | 2.1 | Na+ | D2.50, conserved, tight binding |
| δ-OR | 4N6H, 4RWD | 1.8, 2.7 | Na+ | D2.50, conserved, tight binding |
| PAR1 | 3VW7 | 2.2 | Na+ | D2.50, conserved, tight binding |
| PAR2 | 5NDD | 2.8 | Na+ | D2.50, conserved, tight binding |
| CLTR1 | 6RZ4, 6RZ5 | 2.5–2.7 | Na+ | D2.50, conserved, tight binding |
| β1AR | 3ZPR, 4BVN, 5A8E | 2.1–2.7 | Na+ | D2.50, conserved, tight binding |
| β1AR | 2VT4, 2Y00, 2Y02, 2Y03, 2Y04, 2YCW, 3ZPQ, 3ZPR, 4AMJ, 4BVN, 5A8E, 6H7J, 6H7K, 6H7L, 6H7M, 6H7N, 6H7O | 2.1–3.1 | Na+ | ECL2, backbone, tight binding |
| β2AR | 4LDE, 4LDL | 2.8, 3.1 | Na+ | ECL2, backbone, tight binding |
| SMO, class F | 5L7D | 3.2 | Na+ | Loose binding |
| A2AAR | 5OM1 | 2.1 | Cl− | Helices VI and VIII (H230, R293), loose binding |
| PAR1 | 3VW7 | 2.2 | Cl- | Helices II, III, VI, (K135, R200, K307), loose binding |
| Rhodopsin | 1GZM, 1F88, 1U19, 1L9H, 1HZX, 2HPY, 2G87, 2PED, 3C9L, 3OAX | 2.2–3.0 | Zn2+ | Additive in crystallization protocols. Loose binding in multiple sites. |
| PAF | 5ZKQ | 2.9 | Zn2+ | Helices I, VI, VII (H4, H8, E259, H268), tight binding |
| SMO, class F | 4QIM, 4N4W | 2.6, 2.8 | Zn2+ | Loose binding |
| M2R | 5YC8 | 2.5 | Hg2+ | Used as anomalous scatterers for phasing. Loose binding in multiple sites. |
| Rhodopsin | 1U19, 1L9H, 1HZX, 1F88, 2PED, 2G87, 2HPY, 3OAX | 2.2–3.0 | Hg2+ | |
| 5HT2B | 6DRY, 6DRZ | 2.9, 3.1 | PO43− | Loose binding |
| DRD4 | 5WIU, 5WIV | 2, 2.1 | PO43− | ICL2 and Helix III, tight binding |
| H1R | 3RZE | 3.1 | PO43− | 7TM bundle, ECL, (K179, K191, Y431, H450), tight binding |
| μ-OR | 5C1M | 2.1 | PO43− | Loose binding |
| A2AAR | 3EML | 2.6 | SO42− | Loose binding |
| CB1 | 5U09 | 2.6 | SO42− | Loose binding |
| CCR2 | 5T1A | 2.8 | SO42− | Helix VIII, ICL1, tight binding |
| ETB1 | 5X93, 5GLI | 2.2, 2.5 | SO42− | Inside 7TM, intracellular (R199, R208, K210), tight binding, commonb |
| PD2 | 6D27, 6D26 | 2.7, 2.8 | SO42− | Inside 7TM, intracellular (R143, R2243), tight binding, commonb |
| + Other loose binding sites | ||||
| EP3 | 6M9T | 2.5 | SO42− | Loose binding |
| Rhodopsin | 3PQR, 4J4Q, 4PXF, 5DYS, 5EN0, 5TE3 | 2.3–2.9 | SO42− | Loose binding |
| β2AR | 2RH1, 5D5A | 2.4, 2.5 | SO42− | Inside 7TM, intracellular (T68, R131), tight binding, commonb |
| VI, VII (K270, K273, R328), tight binding | ||||
| μ-OR | 4DKL | 2.8 | SO42− | Inside 7TM, intracellular (T103, R179), tight binding, commonb |
| Other six loose binding sites |
Directly coordinated by two or more ionic interactions with charged residues.
SO42− bound at a common intracellular site in several receptors.
Only the sodium site, however, stands out as highly conserved among most class A GPCRs (Katritch et al., 2014) and potentially tracing its origin to other distant 7TM relatives like prokaryotic channel rhodopsins (Shalaeva et al., 2015). This site binds allosteric sodium in the middle of the 7TM helical bundle of class A GPCRs (Fig. 2, A and B), anchored at the most conserved aspartate residue D2.50 (superscript shows generic numbering of GPCR residues as described in Isberg et al. (2015). Analysis of the structures and sequences of class A GPCRs revealed the highly conserved nature of the sodium pocket, 15 residues of which are conserved exactly in 45 diverse receptors and with minor variations in a vast majority of class A GPCRs families (Fig. 2, C and D) (Katritch et al., 2014). Moreover, those class A receptors that lack the key residues of the sodium site naturally, or via introduced mutations, have their ligand-induced signaling dramatically reduced or completely abolished (Katritch et al., 2014; Massink et al., 2015; White et al., 2018). While our understanding of ion binding sites and their biochemical and physiologic effects on GPCR signaling has greatly expanded in the last few years, we are only beginning to understand the new possibilities for harnessing this knowledge for the discovery of safer and more efficient drugs that have improved subtype and/or functional selectivity (Roth, 2019).
Fig. 2.

Conserved Na+ sites in GPCRs. (A) GPCR superfamily tree with blue circles highlighting receptor structures with Na+ resolved in the conserved pocket and red dots marking GPCR with established allosteric effect of Na+. (B) Overview of the Na+ position in 7TM helical bundle. (C) Type I Na+ site, first discovered in A2AR (green cartoon and sticks) and conserved in majority of Class A GPCRs structures (gray). (D) Distinct coordination of Na+ by two acidic residues D2.50 and D7.49 in Type II sodium site as resolved in PAR1 (cyan), PAR2 and CLTR1 (gray) in δ-branch GPCRs.
II. Structural Data for Conserved and Nonconserved Ion Binding Sites in G-Protein-Coupled Receptors
A. Conserved Sodium Binding Site in Class A G-Protein-Coupled Receptors
1. High-Resolution Structures of Sodium in the Conserved Site
Sodium ion in the conserved sodium pocket was first crystallographically identified in the A2A adenosine receptor (Liu et al., 2012b), quickly followed by PAR1 thrombin (Zhang et al., 2012), β1AR adrenergic (Miller-Gallacher et al., 2014), δ-OR opioid (Fenalti et al., 2014), and D4 dopamine receptors (Wang et al., 2017). Remarkably, despite these structures representing receptors in different major branches of class A GPCRs and having low sequence identity between them (20%–35%), the sodium-binding positions in the structures were found to be almost identical (within 0.5–1.5 Å) with all of them anchored at the negatively charged D2.50 side chain. Moreover, the sequence of all 16 residues lining the sodium binding pocket and their conformations in receptor structures are remarkably conserved either in the whole class A GPCR or its individual branches (Table 2). The unprecedented level of conservation of the Na+ pocket as a structural feature was also emphasized by the fact that the positions of up to 10 water molecules in the pocket comprising Na+/water cluster were found conserved between such distant receptors as A2A, β1AR, and δ-OR. Such a high level of sequence and structural conservation implied a critical functional role of this Na+ site in class A GPCRs (Liu et al., 2012b; Katritch et al., 2014). It is important to note, also, that the sodium pocket lies in close proximity and, in most structures of class A GPCRs, is directly connected to the orthosteric pocket making it potentially accessible to ligand design, as discussed below in section IV.
TABLE 2.
Residue conservation in the sodium pocket for different branches of Class A GPCRs.
Numbers show % conservation, also highlighted by color – from low (white), to medium (yellow), to green (high).
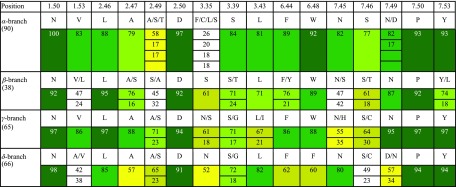 |
A number of high-resolution structures have been more recently obtained (see Table 1), shedding light on the Na+ pocket and revealing new features of the Na+ binding site. Thus, more than 20 additional structures of antagonist-bound complexes for each of the A2A adenosine and β1 adrenergic receptors show that the Na+/water cluster can be reliably resolved in GPCR structures at up to about 2.3 Å resolution. Among them, the X-ray free-electron laser crystal structure of A2AR (1.96 Å resolution) is especially important (Batyuk et al., 2016), because it was determined at room temperature. This structure demonstrated that existence of a well-defined conformation of Na+/water in the pocket detected in crystal structures of many GPCRs was not an artifact of cryo-freezing, but rather a result of the unique stability of the cluster itself.
Key insights were obtained from the antagonist-bond dopamine receptor D4 (DRD4) high-resolution structure (Wang et al., 2017), which was determined both with and without sodium. Importantly, an electron density for sodium was observed only when Na+ (∼200 mM) was added during crystallization, thus providing the most direct structural evidence for Na+ in its binding site. Remarkably, even though another, the sodium-free structure of DRD4, was slightly higher resolution, the electron densities for water molecules forming the Na+/water cluster disappeared, showing that Na+ is critical for the stability of the whole cluster. Indeed, water clusters in polar pockets can usually dynamically form many combinations of their hydrogen bonding network, which compromise detection of individual water molecules. In contrast, the presence of the Na+ with a strong ionic bridge to D2.50 creates a specific configuration of the whole cluster, characterized by well-defined electron densities, as observed previously in A2A, β1AR, and δ-OR high-resolution structures. It is also worth noting that there was no significant difference in the receptor conformation itself between Na+-bound and sodium-free structures of DRD4, even in the sodium-coordinating pocket residues, suggesting that the presence of sodium ion does not “induce” any specific conformational macrostate of the receptor. Instead, the observed stabilizing role of Na+ is manifested in shifting equilibrium toward the same inactive state conformation as observed without sodium.
The recently solved structures of the PAR2 proteinase-activated receptor (Cheng et al., 2017a) and of the cysteinyl leukotriene receptor CLTR1 (Luginina et al., 2019) further confirm the presence of sodium in the δ-branch of GPCRs as was identified previously for PAR1 (Zhang et al., 2012). These receptors provide a distinct structure of the Na+ pocket (Fig. 2D; Table 2), where sodium is coordinated by two acidic residues, D2.50 and D7.49, instead of only D2.50 in most other class A receptors, which have N7.49. This double salt bridge coordination shifts the sodium position about 1.5 Å “down” along the polar channel and changes the overall Na+ coordination and conservation pattern compared with “classical” sodium pocket in α- and γ-branches of GPCRs.
2. Sodium Ion Detection Criteria
While divalent ions in crystal structures are often detected by their anomalous diffraction, monovalent ions including Na+ lack such anomalous diffraction. Reliable detection of monovalent ions like Na+ in the protein crystal structures is based on high resolution (usually <2.3 Å), the strong electron density in a potential Na+ ion position and the unambiguous detection of at least five oxygen (or potentially nitrogen) atoms that comprise the Na+ coordination shell. The sodium ion can be identified then by its 5-atom coordination geometry and short characteristic distances to the coordinating atoms (2.3–2.5 Å), as discussed in Liu et al. (2012b). These criteria help to differentiate Na+ from four-atom tetrahedral coordination of water molecules and characteristic water interaction distances (2.8–3.1 Å). At even higher resolution, e.g., <2.0 Å, the accuracy of measurement may also be sufficient (Cheng et al., 2017a) to differentiate Na+ coordination distances from those of other monovalent ions, e.g., longer distances for K+ (2.6–2.8 Å) and shorter for Li+ (1.9–2.1 Å) (Kuppuraj et al., 2009), thus specifically detecting Na+.
It is important to note that the high structural stability of the protein and of the Na+/water cluster is as important for detection of Na+ as the resolution of the structures. Thus, it was possible to reliably resolve Na+ in PAR2 even at somewhat lower 2.8 Å resolution, because Na+ was coordinated by five oxygen atoms of the protein side chains, including two from the charged carboxy groups of D2.50 and D7.49. At the same time, in some higher resolution structures, for example OX2R (PDB: 5WQC, resolution 1.96 Å), allosteric sodium was apparently absent from the conserved pocket (Suno et al., 2018a). Though some waters of the sodium pocket were resolved, the density for sodium and a neighboring position interacting with D2.50 were not well defined, precluding Na+ detection. This is not surprising, as the Na+ concentration used for this crystallization was 120 mM, which is in the range close to EC50 of Na+ in some receptors (e.g., ∼100 mM for DRD4), and thus may not allow the full saturation required for Na+/water cluster stability and detection. Therefore, the lack of sodium density in this structure does not necessarily mean that the fully conserved Na+ pocket in OX2R does not bind sodium, but rather that it could not be detected crystallographically under the conditions used. Further studies of Na+ in OX2R, including validation of the classic Na+ effect on agonist binding, may be needed to answer this question more definitively (Suno et al., 2018a).
All structures with bound Na+ so far were resolved by crystallography, although it is possible that structural information about sodium pocket may also come in the future from cryo-EM studies. The best GPCR structures by cryo-EM have been solved at ∼3.0 Å resolution (Zhao et al., 2019), but the rapid progress in the cryo-EM field and detection of soluble protein structures at resolutions as high as 1.8 Å (Merk et al., 2016) suggests that this crystal-free technology may ultimately allow deciphering the sodium cluster and other ion binding details as well.
3. Lower Resolution Inactive State Structures of Class A Compatible with Sodium Ion Binding
In many other crystal structures of diverse class A GPCRs where the modest resolution (2.4–2.9 Å) was insufficient reliably to resolve sodium ion, the conserved pocket is still fully compatible with sodium presence in the structure.
In the α-branch of class A GPCRs, this includes A1 adenosine (Cheng et al., 2017b) and most of the aminergic GPCR structures, for example, β2AR (2RH1), D3R (3PBL) that have closely related subtypes with Na+ explicitly determined crystallographically.
In the γ-branch, where sodium was only resolved in high-resolution structure of delta opioid receptor (DOR) so far (Fenalti et al., 2014), the pockets are fully conserved and compatible with Na+ binding in structures of all other opioid receptors including mu (MOR) (Manglik et al., 2012), kappa (KOR) (Wu et al., 2012) and nociceptin (NOP) (Thompson et al., 2012) opioid receptors.
In the β-branch, the NTSR1 neurotensin receptor was previously characterized as having a sodium binding site (White et al., 2012; Krumm et al., 2015), although Na+ was not crystallographically resolved in the NTSR1-agonist complex as it represented a partially active-like state. An interesting observation was made recently for another β-branch GPCR, the ETB endothelin receptor (Shihoya et al., 2017). Though a weak allosteric sodium effect was detected in the ETB receptor, it was above the physiologic sodium concentration (>1 M), probably owing it to the fact that one of the key residues of the otherwise conserved sodium pocket, Y7.53, was replaced by L7.53 in the ETB receptor. Correspondingly, the crystal structure of ETB solved at relatively high resolution 2.2 Å had the cavity filled with electron densities that were more compatible with water molecules than with sodium. Intriguingly, the closely related ETA has much stronger Na+ binding at EC50 = 245 mM, and a high-resolution ETA structure might provide new insights for this family.
4. Structures of Active State G-Protein-Coupled Receptors Are Incompatible with Sodium Binding
Comparison of the sodium pocket conformation in inactive- and active-state structures of A2A, β1AR, muscarinic, 5-HT, and opioid receptors (Fig. 3) reveals that the sodium pocket shape, conformation, and interaction network change dramatically upon activation, resulting in a partially collapsed pocket that is not compatible with high-affinity binding of sodium (Katritch et al., 2014). In general, active state conformations are characterized by an inward movement of the TM7 backbone, which directly clashes with the sodium site and rearranges sodium-coordinating side chains so that they form direct hydrogen bonds instead of Na+ mediated (e.g., between D2.50 and S3.39) that preclude sodium coordination (Fig. 3C). Other major activation related changes, like the outward movement of TM6, also change the shape of the Na+ pocket and disrupt the so-called hydrophobic layer (Yuan et al., 2014). Disruption of the hydrophobic layer, comprising residues 1.53, 2.46, 3.43, 7.53 at the bottom of the Na+ pocket, opens the floodgate for water and sodium ion egress toward the intracellular side.
Fig. 3.

Conformational changes in the sodium pocket upon activation of opioid receptors. (A) Superimposition of high-resolution structures of δ-opioid receptor in inactive state (green, PDB: 4N6H, resolution 1.8 Å) and μ-opioid receptor in active state (orange, PDB: 5C1M, resolution 2.1 Å); conformational changes shown by arrows. (B) Close up of sodium pocket in inactive state. (C) Close up of the pocket in active state. Hydrogen bonds and salt bridges are shown by cyan dotted lines.
Such conformational changes in active-like states were described recently for NTSR1 (White et al., 2012; Krumm et al., 2015) and AT2R (Zhang et al., 2015b, 2017) in structures of complexes with agonists, as well as the structures of fully active MOR (PDB: 5C1M) (Huang et al., 2015a) and KOR (PDB: 6B73) (Che et al., 2018) bond to both agonists and nanobodies. The MOR structure is especially important in this respect because it was solved at a high 2.1 Å resolution. A detailed examination of the pocket structure reveals no electron density suitable for Na+, and in general, the active state conformation is incompatible with Na+ binding (Huang et al., 2015a).
It should be noted, that while the described above general conformational rearrangements are common to active state conformations, the details of newly formed interactions in the pocket can differ between the structures quite dramatically. This contrasts with the very conserved conformation of the residues in the structures of inactive GPCRs. This loss of uniformity can be explained by natural differences in the pocket between receptors, but also by different activation states (intermediate activated to fully active) and a range crystallographic resolution (from 2.1 to 3.5 Å) (Katritch et al., 2014).
5. Allosteric Ligands can Block Sodium Binding
Although the Na+ pocket is small, ∼200 Å3 as estimated in A2A (Liu et al., 2012b), it can bind small molecules like amiloride and its analogs, which have a common positively charged moiety connected to an aromatic ring (Fig. 4) (Liu et al., 2012b; Katritch et al., 2014). The allosteric binding of amiloride has been shown biochemically for several receptors, revealing direct competition with Na+ binding and a strong dependence on mutations in D2.50 and other pocket residues (Howard et al., 1987; Gao and Ijzerman, 2000; Gao et al., 2003a,b; Heitman et al., 2008; Gutiérrez-de-Terán et al., 2013; Massink et al., 2015) (Fig. 4B). Crystallographic observation of ligand binding in the Na+ pocket has been elusive, until recently the structure of leukotriene B4 receptor BLT1 was solved in complex with a ligand reaching into Na+ pocket (Hori et al., 2018). The bitopic ligand BIIL260, spanning the orthosteric pocket and reaching all the way to the sodium binding anchor D2.50, was characterized as an inverse agonist. This is an expected functional effect, as the ligand blocks the sodium site and precludes conformational rearrangements in the pocket, which are required for activation. The bitopic ligand comprised an orthosteric BTL1 selective moiety and a positively charged benzamidine group that forms a salt bridge to D2.50 in a manner similar to amiloride. Interestingly, the study also shows the benzamidine itself has a negative allosteric effect of on BLT1 activation with KB ∼500 μM, which is much weaker than KB values reported for amilorides (Fig. 4B). This allosteric effect of benzamidine was also confirmed in the β2AR, suggesting its potential effect in many other class A GPCRs with a similar Na+ pocket structure. A combination of orthosteric selectivity with a controlled allosteric sodium pocket functionality in bitopic ligands was suggested as a beneficial path for drug discovery, as discussed in section IV below.
Fig. 4.

GPCR pockets for binding allosteric ligands that target conserved sodium binding pocket. Semitransparent surface shows orthosteric pocket (orange) and allosteric conserved sodium pocket (cyan). (A) Overview of the pockets in 7TMD. (B) Amiloride (magenta) bound to KOR in complex with selective antagonist 4-phenylpiperidine derivative JDTic (green) (PDB: 4DJH). (C) Benzamidine (yellow) bound to MOR in complex with irreversible agonist β-FNA (green) (PDB: 4DKL).
6. Mutations Abolishing Sodium Binding
A central role of the Na+ site in activation-related conformational changes suggests that mutation in this site can modulate the stability of specific functional states. Moreover, by removing Na+ as a key “gear” in the transmission mechanism, the conformational space sampled by the receptor along the activation path is modified and can improve the thermostability of the receptor (Katritch et al., 2014). Indeed, several structures of GPCRs have been recently obtained with mutations in the sodium pocket that improved receptor thermostability. Specifically, a mutation in the Na+ anchor residue D7.49N helped to crystallize and solve structures of P2Y1 receptor in complex with antagonists (PDB: 4XNV) (Zhang et al., 2015a), as well as P2Y12 complex with agonist (PDB: 4PXZ) (Zhang et al., 2014). Some of the established sodium-disrupting mutations (D2.50N, S3.39A, and D7.49N) were included as knowledge-based transferrable mutations in the GPCR thermostabilization algorithm (Popov et al., 2018, 2019). Other mutations in the pocket show promise, e.g., introducing Arg in 3.39 position was theoretically predicted as stabilizing mutation (Yasuda et al., 2017) expected to block the Na+ pocket. This mutation recently helped to solve new structures of muscarinic acetylcholine receptor 2 (M2R) (Suno et al., 2018b) and EP4 prostaglandin receptor (Toyoda et al., 2019) in the inactive state.
Because stabilization by sodium pocket destruction usually comes at the expense of losing function, i.e., signaling response to agonists, we specifically studied the structural consequences of some of these mutations in a well-established system such as A2A adenosine receptor (AA2AR) (White et al., 2018). Mutations in sodium-coordinating positions D2.50N and S3.39A were introduced in A2AR and assessed both functionally and structurally. This study demonstrated robust improvement in thermostability for the D2.50N mutation in the apo-, agonist-, and antagonist-bound receptor, supporting its broad importance. Although D2.50N resulted in complete disruption of G-protein signaling mechanism, it retained a full affinity for antagonists, while even improving binding of agonist, as expected for GPCRs with decoupling mutations. Importantly, the crystal structures of A2AR D2.50N and S3.39A mutants in complex with agonist UK432097 were conformationally undistinguishable from the wild-type receptor in the same complex, with only minor local variations in the two to three residues directly interacting with the mutation site. This structural resilience to stabilizing mutations in the sodium pocket again suggests that such mutants can be used to facilitate GPCR crystallization (or improved cryo-EM resolution) with minimal disturbance to the resulting overall structure of the receptor.
B. G-Protein-Coupled Receptors that Lack the Conserved Sodium Site
1. Some Class A G-Protein-Coupled Receptors Lack Specific Sodium Site
A limited number of class A GPCRs lack the key polar residues in the sodium pocket and are apparently not suitable for the selective high-affinity binding of sodium. We estimated about 10%–30% of class A GPCRs lack specific Na+ binding in the conserved pocket, depending on the Na+ affinity cutoff. The most obvious 36 exceptions are listed in Supplemental Table S1 of our previous review (Katritch et al., 2014), including 1) visual rhodopsin and other opsins that lack conservation in the polar pocket, 2) GPCRs lacking D2.50 anchor that are known to lack ligand-induced signaling, some constitutively activated and some acting via dimerization with signaling subtype, 3) some orphan and “putative” GPCR lacking D2.50 anchor where ligand signaling has not been established, or 4) receptors where lack of D2.50 anchor may be compensated by acidic Asp and Glu in positions 7.49 or 3.39 of the sodium pocket.
Several other interesting cases of receptors with rare deviations in the pocket, which result in dramatically reduced or abolished Na+ binding affinity have been studied more recently. Thus, in the NK1 neurokinin receptor (NK1R), the rare E2.50 carboxylic acid side chain, which is longer than the common D2.50, was predicted to occupy the sodium position in this site and make direct interactions with conserved S3.39, T7.46, and N7.49 (Valentin-Hansen et al., 2015). This modeling prediction was recently confirmed by a 2.2 Å resolution structure of NK1R (Schöppe et al., 2019), which also shows that while E2.50 replaces Na+ in the site, the structure of water molecules in the pocket is remarkably conserved as in Na+/water cluster resolved in other GPCRs. It was hypothesized that by replacing mobile Na+ with direct and immobile carboxy side chain interactions of E2.50, the receptor more tightly controls its basal signaling; indeed NK1R lacks appreciable basal activity. Intriguingly, while NK1R lacks Na+ binding and allosteric effects, both can be restored in the NK1R receptor by “reintroducing” D2.50, as it is in other two NK receptors, NK2 and NK3 (Schöppe et al., 2019). The E2.50D and other mutations in the Na+ pocket of NK1R also dramatically change the constitutive and biased signaling profile of NK1R, suggesting that evolution uses deviations from the canonical Na+ site as a way to modulate the functional properties of receptors.
Interestingly, another rare substitution of a small to larger side chain in 7.46 position of the pocket, e.g., S(T,A)7.46N, is found in only two class A GPCRs including angiotensin AT1 receptor. Assessment of the sodium pocket structure of AT1R, including inactive (Zhang et al., 2015b, 2017) and active-like state (Wingler et al., 2019) structures, suggests that N7.46 side chain and its hydrogen bond interactions with N3.35 may interfere with sodium binding, replacing sodium as a conformational stabilizer and making the receptor insensitive to sodium concentration.
Another example of a GPCR structure with deviations in the sodium pocket incompatible with selective Na+ binding is the CCR5 chemokine receptor (CCR5R), which lacks two key sodium coordinating side chains in N3.35 and S3.39 positions, which are replaced by Gly instead. Indeed, while the inactive state structure of CCR5R in complex with an antagonist was solved at relatively high (2.2 Å) resolution, no density for Na+ binding was detected (PDB: 5UIW) (Zheng et al., 2017). The structural deviations in the allosteric pocket that compromise Na+ binding appear to be common to a group of other inflammatory chemokine receptors, suggesting that the switch in Na+ pocket played a key evolutionary role in differentiating the chemokine receptor family into homeostatic (CXCR4-like) and inflammatory (CCR5-like) (Taddese et al., 2018) (see more discussion in section V.C). In general, establishing an accurate structure-activity relationship for the Na+ pockets of all class A GPCRs is far from finished and will require a combination of computational modeling and experimental efforts.
2. Non-Class A G-Protein-Coupled Receptors Lack Conserved Sodium Sites in 7-Transmembrane Domains
Potential ion binding sites have been identified or proposed for non-class A GPCRs, both in their 7TM domains and soluble extracellular domains; however, they are structurally distinct from the conserved class A GPCR Na+ site and appear to have different functional and evolutionary roles. In the class B 7TM domain, an allosteric Na+ site was proposed by MD simulations in the glucagon receptor, with the ion coordinated by residues Glu3626.53b, Asn2383.43b, Tyr2393.44b, and Tyr4007.53b, although the predicted ion residence time in the binding site was very short (Selvam et al., 2018). Moreover, the site is conserved only in four class B receptors that have the key acidic Glu residue in 6.53b position, and the biologic significance of ion binding to class B GPCRs remains unclear. None of the class B structures have ions detected in their crystal structures that bind their 7TM or extracellular domains, even though many of the extracellular structures were solved at sub 2.0 Å resolution.
In Class C GPCRs, the internal cavity extends deep in the 7TM bundle reaching approximately the location of the class A Na+ site. There are polar residues like Y6593.40c, T7816.44c, and S8097.45c in this region that create a hydrophilic subpocket, and indeed a water molecule has been resolved in this position in the mGLuR5 metabotropic glutamate receptor structure (mGluR5) solved at 2.2 Å resolution (PDB: 6FFI) (Christopher et al., 2019). This polar triad is conserved in seven of eight mGLuR receptors (but not other class C), suggesting some role for a water binding site. However, the subpocket lacks any acidic residues compatible with specific binding of cations like Na+. None of the currently available class C structures have an ion resolved crystallographically, even at 2.2 Å resolution.
Similarly, in class F GPCRs, exemplified by the smoothened receptor structures (Wang et al., 2013) and the recently solved structure of apo FZD4 receptor (Yang et al., 2018), there is an extended polar channel in the 7TM domain, with water molecules coordinated by the conserved in class F residues Y2622.52a, S3173.40a, and Y4446.41a. Again, none of the residues in the core of the 7TM has an acidic side chain, precluding specific ion binding in this region. Some nonspecific binding ions have been detected in smoothened receptor (SMO) structures, though they are not likely to play a substantial functional role in these receptors.
C. Nonconserved Ion Binding Sites in G-Protein-Coupled Receptors
Sodium ions, as well as other single and polyatomic ions have been found in many crystal structures of GPCRs with sufficient resolution, as listed in Table 1. Thus, for the β1AR and β2AR, in addition to the conserved sodium site, a second Na+ ion was also identified in the ECL2, coordinated by three carboxy groups of the protein backbone and 2 water molecules. This tightly bound Na+, along with a disulfide bond formed by cysteines of the loop, apparently helps to stabilize the α-helix-loop structural motif in the ECL2 of these receptors, and thus apparently serves a structural role. This particular sequence and structural motif, however, can be found in only three receptors of β-AR subfamily and is not conserved in other GPCRs.
Other notable nonconserved sites include phosphate ion PO43− in the ECL region of the H1 histamine (H1R)receptor (Shimamura et al., 2011). As described in this H1R structural paper, the PO43− ion plays an important role in binding and selectivity of some of the ligands (see more discussion in section IV.C), but this site is unique for H1R.
Specific binding of Zn2+ has also been observed in the of PAF1 platelet activating receptor receptor extracellular loops tightly coordinated by three His and one Glu side chains with distances as low as 2.0–2.2 Å (Cao et al., 2018). This can explain Zn2+ induced inhibition of platelet activating factor binding to the receptor and physiologic reduction information of platelets (Nunez et al., 1989). Ions have been described as endogenous ligands to some of the GPCRs. Thus, Ca2+ ion is an endogenous agonist for eponymous CaSR calcium-sensing receptor (Chang et al., 2008; Hannan et al., 2018). CaSR is critical for many of the functions dependent upon the regulation of Ca2+ metabolism, including the parathyroid gland and bone development. The allosteric modulation by extracellular calcium has been studied extensively for another Class C GPCR family, the metabotropic glutamate receptors, where the Ca2+ site adjacent to the Glu site was predicted and biochemically characterized (Jiang et al., 2010, 2014). Most recently, Ca2+ was also revealed as an important allosteric modulator of Class B parathyroid hormone receptor signaling, and the structural determinants of the ion binding were proposed (White et al., 2019). Also, Zn2+ has been described as an endogenous agonist for the GPR39 receptor, for which the activation site and mechanism have been proposed (Storjohann et al., 2008; Sato et al., 2016). It would be very interesting to test these hypotheses to gain a better understanding of the atomistic mechanisms when the high-resolution structures of these receptors are available.
D. Nonspecific Ion Binding in Crystal Structures
Multiple ions have been found crystallographically in the intracellular region of the receptor (Fig. 1; Table 1); however, most of them have loose interactions with the receptor, suggesting nonspecific binding. As the intracellular region is enriched with positively charged Arg and Lys residues, it is not surprising that all of the ions identified in this region are anions, including Cl−, PO43−, and SO42−. In most cases, the ions are coordinated by one or two positively charged side chains, although in general, the binding is rather loose and the ions remain highly exposed to solvent. Note also that these intracellular ion binding sites are not reproduced between receptors and, in most cases, not even between different subunits and different structures of the same receptor, so this binding is probably nonspecific and only identified due to the high concentration of these ions in crystallization conditions.
Some divalent cations like Zn2+ and Hg2+ have been also detected bound at the lipid interface of the 7TM bundle. Interestingly, in almost all these cases these metal cations have been found in structures of rhodopsin, reflecting specific crystallization conditions that used a high concentration of the ions. One other crystal structure with Hg2+ is a M2 muscarinic receptor structure (Suno et al., 2018b), where the ions are bound on the lipid interface of the 7TM domain and do not make any ionic or even substantial polar contacts, making their binding apparently nonspecific.
Many other ions present in high concentrations in crystallographic conditions are likely to be loosely and nonspecifically bound in GPCRs, but were not identified in crystal structures because they lack well-defined electron density. Their physiologic role is usually limited to nonspecific ionic strength effects at high concentrations. For example, some studies report a component of the Na+ allosteric effects that are independent of D2.50 mutation in H1R (Hishinuma et al., 2017). This is not surprising, given numerous charged residues in GPCRs, often in the orthosteric ligand binding pockets, for example, negative anchor residues in all aminergic opioid and positive phosphate binding residues in P2Y purinergic receptors. Nonspecific ion binding in ICL3 region may also directly modulate downstream effector binding and activation. Although most of these effects, including the overall ionic strength of the solvent, can manifest themselves only at high concentration of ions and unlikely involved in GPCR function, they need to be accounted in experiments by using appropriate controls.
III. Functional Role—Why is Sodium So Special for Class A G-Protein-Coupled Receptors?
Sodium is one of the most abundant ions in the human body, essential for cell energetics, homeostasis, neural function, and many other physiologic functions. However, our understanding of Na+ and its role in the physiologic processes involving GPCR signaling is only now starting to unfold.
A. Allosteric Effects of Sodium on Agonist Binding
1. “Classical” Allosteric Effect of Sodium Ion on Agonist Binding
As mentioned above, the selective sodium effect was originally discovered in opioid receptors as a negative allosteric modulation of (NAM) of agonist binding upon increasing sodium concentration (Pert et al., 1973; Pert and Snyder, 1974; Simon and Groth, 1975; Roth et al., 1981). This NAM effect in the μ-opioid receptor (μ-OR) correlated well with ligand efficacy, and for some time was the primary method for differentiating agonists from antagonists; for the latter, the effect was usually neutral or reversed (Pert and Snyder, 1974). This allosteric effect was observed for sodium within the physiologic concentration range of ∼140 mM, and, in some cases, titration of the effect with sodium concentration allowed measurement of KB or EC50 values as a proxy for Na+ binding affinity. Importantly, the described above NAM effect on agonists was found specific for Na+, while showing much less magnitude for Li+ and lacking for K+ and larger monovalent cations (Pert and Snyder, 1974). The divalent cations like Mn2+, Mg2+, and Ca2+ displayed the opposite effect on agonist binding, although the effect was not specific (Pasternak et al., 1975). Since then, studies for numerous other class A GPCRs have demonstrated similar sodium allosteric effect on ligand binding, many of these studies also showing the specificity of Na+ binding by mutations in the pocket and corresponding effects on signaling (for historical data see Table 1 in Katritch et al., 2014).
In the years since the structural detection of Na+ in the highly conserved GPCR site, a resurgence of interest led to further validation and more detailed biochemical characterization of the sodium allosteric effects in these and many other class A GPCRs (Table 3). Thus, for adenosine A2A receptor titration of the NAM effect of Na+ on agonist NECA allowed estimation of its IC50 value at 44 ± 6 mM (Massink et al., 2015; White et al., 2018), while confirming the positive allosteric modulation (PAM) effect on antagonist ZM241385 (4-(2-(7-amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-ylamino)ethyl)phenol). The NAM effect was drastically reduced by S3.39A and W6.48A mutations and completely abolished by D2.50A, N7.45A, and N7.49A mutations in the Na+ pocket.
TABLE 3.
Recent data on allosteric effect of Na+
| Receptor | Allosteric Effect [Na+] | Estimate of Na+ Affinity | PDB ID (Na+ in Structure) | Reference |
|---|---|---|---|---|
| DRD4 | Agonist NAM, reduce basal activity | KB = 98 mM | 5WIV (Na+) | Wang et al. (2017) |
| DRD2 | Agonist NAM, control of other allosteric ligands | KB = 123.1 mM | 6CM4 | Wang et al. (2017), Draper-Joyce et al. (2018) |
| DRD3 | Agonist NAM | KB = 76 mM | 3PBL | Wang et al. (2017) |
| MOR | Agonist NAM, control of other allosteric ligands | KB = 7.3 mM | 4DKL | Wang et al. (2017), Livingston et al. (2018) |
| DOR | Agonist NAM, control of other allosteric ligands | KB = 24.3 mM | 4N6H (Na+) | Wang et al. (2017) |
| A2AR | Agonist NAM | KB = 32.4 mM | 4EIY (Na+) | Wang et al. (2017) |
| CLTR1 | Receptor thermostability | EC50 = 39 mM | 6RZ4, 6RZ5 (Na+) | Luginina et al. (2019) |
| BLT1 | Agonist NAM | EC50 < 200 mMa | 5X33 (Na+ site blocked by ligand) | Hori et al. (2018) |
| V1b | Agonist NAM, required for IP3 signaling | EC50 < 50 mMa | n/a | Koshimizu et al. (2016) |
| OXTR | Agonist NAM | EC50 < 200 mMa | n/a | Schiffmann and Gimpl (2018) |
| H1R | Agonist NAM | EC50 < 100 mMa | 3RZE | Hishinuma et al. (2017) |
| NTSR1 | Agonist NAM | EC50 = 43 mM | 4BUO | White et al. (2018) |
| ETA | Agonist NAM | EC50 = 245 mM | n/a | Shihoya et al. (2017) |
Approximately estimated from Na+ titration curves in the referenced papers.
For dopamine receptors, NAM modulation of agonist binding by Na+ was confirmed for the D4, D2, and D3 dopamine receptors (Michino et al., 2015; Wang et al., 2017), and titration of the effect on Na+ concentration made it possible systematically to compare Na+ affinities for several receptors. Thus, Na+ showed lower affinities to dopamine receptors, KB ∼100 mM (D4), KB ∼123 mM (D2), and KB ∼76 mM (D3) compared with KB as high as 7.3 mM for MOR, 24.3 mM for DOR, and 32.4 mM for A2A. As expected (see functional effect in Section B.1), constitutive activity of the D4 receptor significantly increased at low concentrations of Na+, the effect of which can be blocked by the addition of antagonists (Wang et al., 2017).
For the BLT1 leukotriene receptor, structural studies (Hori et al., 2018) were complemented by biochemical assays that revealed a pronounced negative allosteric effect of Na+ on agonist leukotriene B4 binding. Interestingly, the study also described similar effects for an allosteric small molecule benzamide that competes with Na+ for the sodium pocket binding.
For V1b vasopressin receptors in cell-based assays, a recent study shows that reducing the concentration of external Na+ to below 50 mM dramatically increased cell surface binding of radiolabeled agonist [3H]arginine vasopressin (Koshimizu et al., 2016). Interestingly, though agonist binding was increased, the receptor signaling and internalization were reduced in low Na+ concentrations. This is an important observation, suggesting that the functional effects of Na+ are not limited to NAM effect on agonist binding. Again, the biochemical and functional effects were selective for Na+ compared with Cs+ or NH4+.
For the oxytocin receptor, while the endogenous agonist oxytocin was positively modulated by divalent ions like Mg2+, specific NAM effect of Na+ on oxytocin binding was detected at physiologic Na+ concentration (Schiffmann and Gimpl, 2018). Thus, the increase of Na+ concentration from 0 to 300 mM reduced oxytocin affinity ∼15-fold, while no significant effect was observed for K+ or other monovalent ions.
For the H1 histamine receptor (Hishinuma et al., 2017), all three agonists studied showed expected NAM effect at 100 mM concentration of Na+. While a maximal NAM effect of Na+ was observed for histamine and other two agonists, a set of diverse antagonists showed a whole range of effects from NAM to PAM, with the most pronounced PAM found for the most efficacious second-generation antagonists (antihistamines). While these effects were largely abolished by D2.50N mutation, residual D2.50-independent effects were observed for some of the antagonists like fexofenadine, depending on their physicochemical properties. This observation emphasizes that the observed Na+ effects are often a combination of specific D2.50 pocket Na+ binding and nonspecific effects due to multiple low-affinity binding sites and charge screening effect in ligands and receptors.
For the muscarinic receptor subfamily, early studies suggested a classic Na+ effect in M2 muscarinic receptors (Rosenberger et al., 1980), recently corroborated by mutation studies (Suga and Ehlert, 2013) and molecular dynamics simulations in M3 muscarinic receptors (Miao et al., 2015). However, a strong nonspecific effect of ionic strength (Birdsall et al., 1979) and ionic interactions in the orthosteric pocket may interfere with an accurate assessment of Na+ selective binding in this subfamily.
In general, both the magnitude and affinity (e.g., KB) of the sodium effect can vary dramatically between receptor and between ligands in the same receptor. As the systematic study for six different receptors shows (Wang et al., 2017), the magnitude of agonist potency change by Na+ can exceed 100-fold for some receptors (e.g., MOR), while in other receptors is barely detectable at less than fivefold (DRD4). This may somewhat correlate with the KB of the sodium effect, which is shown to be much higher for MOR than for DRD4.
Interestingly, a strong sensitivity to allosteric Na+ was characterized recently for ligands that are allosteric modulators themselves. At the D2 dopamine receptor, the allosteric ligand SB269652 completely loses its modulatory effect in the absence of Na+ ion (Draper-Joyce et al., 2018). Similarly, allosteric ligands effect in MOR was found to be controlled by Na+ presence (Livingston and Traynor, 2014). The effect has been recently observed in other opioid receptors (Livingston et al., 2018), and the authors conclude that disruption of the Na+ ion binding site may represent a common mechanism for allosteric modulation of class A GPCRs.
2. Binding of Sodium Not Always Detected by a “Classical” Allosteric Effect
While the negative allosteric modulation of agonist binding has been long considered a hallmark effect of Na+ in some GPCRs, this effect may be much less pronounced and can easily go undetected in some cases, even when sodium is known to bind in their conserved pocket. Thus, sodium ion anchored by D2.50 has been resolved in high-resolution β1AR structure, revealing the same Na+/water cluster as in A2AR (Miller-Gallacher et al., 2014). But in contrast to A2AR, any attempts to detect this NAM effect in β1AR have failed, suggesting that the β1AR adrenergic receptor lacks any observable dependence of agonist binding on Na+ concentration. In the closely related β2AR (65% sequence identity), mutations D2.50A or D2.50N disrupting Na+ site also failed to detect classic Na+ NAM effect on agonist binding (Strader et al., 1988).
The lack of “classical” NAM sodium effects on GPCRs that actually bind Na+ can arise from combination of several factors, such as 1) weak coupling between allosteric and orthosteric pocket conformations, which reduce the magnitude of the allosteric effect and 2) presence of nonspecific binding effects that can at least partially mask/compensate the specific NAM effect. The detection of the “classical” sodium effect can be further complicated in those cases where affinity of Na+ binding in the specific pocket is low (KB > 100 mM), which makes it harder to differentiate from nonspecific ionic strength effect. Corroborating the first factor above, both β1- and β2-adrenergic receptors are well known to have very weak coupling between extracellular agonist binding and intracellular conformational changes, reflected in their high basal activity (∼20%–40%) and incomplete activation by endogenous ligands (Yao et al., 2009). This weak coupling is reflected also in structural studies, showing the agonist binding per se does not convert β1AR and β2AR to active (R*) state (Rosenbaum et al., 2011; Warne et al., 2011), which needs G protein or arrestin binding for stabilization (Rasmussen et al., 2011). Importantly, this carefully documented case of absence of NAM effect of Na+ on agonist binding in some GPCRs suggest that this “classical” effect, though most easily measurable in vitro, is not, in fact, essential to the functional role of sodium. The NAM effect on agonist binding is only part of the story and probably not the most biologically important part.
B. Evidence for the Functional Importance of the Sodium Ion Site
Although the most commonly documented effect of sodium presence in class A GPCRs is NAM, i.e., reduction of agonist binding and reduction in constitutive activity (Quitterer et al., 1996; Seifert and Wenzel-Seifert, 2001; Wang et al., 2017) there is a substantial evidence that physiologic sodium is actually required for efficient stimulation of the receptors in response to agonists. Indeed, as early as 1982, Cooper et al. (1982) noted the amplification effect of Na+ on agonist-induced cAMP modulation in rat striatal plasma membrane. More recent studies are further corroborating this hypothesis, including both direct dependence of signaling on Na+ concentrations and its displacement by an agonist, as well as indirect effects of mutations in Na+ coordinating residues.
1. Direct Functional Effects of Sodium Ion Presence
Because of the potential interference of changing Na+ concentration with signal transduction downstream from GPCRs, direct measurements of the Na+ concentration effect on GPCR signaling are challenging. Nevertheless, several studies demonstrate that such dependencies can be detected in well-controlled assays. Thus in 1982, Cooper et al. (1982) were the first to note the amplification effect of Na+ on agonist signaling via opioid receptors in rat striatal plasma membrane. Opioid receptors generally signal via the Gi pathway, which inhibits the production of cAMP. The authors found that the presence of Na+ at 80 mM concentrations results in a dramatic increase of the cAMP inhibition effect of the morphine agonist, especially at high GTP concentrations.
Sodium effects on spontaneous opioid receptor GTPase activity and relative agonist efficacy were also studied by Costa et al. (1990, 1992), revealing “paradoxical difference in the way sodium ions affect GTPase activity and ligand binding.” Most importantly, they showed that buffer exchange from K+ to Na+ dramatically reduces basal GTPase activity while maintaining the activity of agonist DADLE, thus selectively amplifying the ligand-induced stimulation (Fig. 5A).
Fig. 5.

Direct measurement of Na+ effect on GPCR signaling. (A) Exchange of K+ to Na+ leads to reduced basal activity, but maintains agonist-induced signal (Costa et al., 1990). (B) Addition of Na+ dramatically enhances differential between agonists and antagonists of G-protein signaling in MOR (Selley et al., 2000). (C) Addition of Na+ reduces basal activity of DRD4 (Wang et al., 2017). This figure reproduced with permission.
Selley et al. (2000) studied the Na+ effect on [35S]-GTPγS binding in CHO cells stably transfected with MOR (mMOR-CHO cells) and in rat thalamus. In both systems, an increase of sodium concentration to physiologic levels (∼140 mM) had a dual effect of 1) reduced basal G-protein signaling and 2) increased receptor stimulation by full agonists, but not partial agonists. In other words, while in the absence of Na+, stimulation by full and partial agonists was almost indistinguishable, increasing Na+ concentrations “magnified relative efficacy differences among agonists” (Fig. 5B).
In the recent study on DRD4 (Wang et al., 2017), which combines structural, biochemical, and functional assessment of the receptor, the authors showed that basal (constitutive) activity dependence on Na+ can be accurately measured. Thus, the study found that constitutive Gαi/o activity at D4 receptor was dramatically (twofold) reduced at physiologic Na+ concentrations. The potentiation of DRD4 constitutive activity in low Na+ concentration can be abolished by selective DRD4 antagonist nemonapride, showing that the effect is entirely D4 receptor-mediated (Fig. 5C).
The aforementioned study of V1b vasopressin receptors (Koshimizu et al., 2016) also shows that agonist binding at high Na+ concentrations was reduced. However, IP3 production assays showed that Na+ in the external buffer was required for signaling. Thus, in the NaCl-containing buffer, the agonist increased the IP3 level from basal 6.6 ± 1.2 to 13.2 ± 0.7 nM in the stimulated receptor. In contrast, in the buffer without NaCl, the agonist-stimulated IP3 levels were below the detection level (<1 nM). These biochemical and functional effects were selective for Na+ compared with Cs+ or NH4+.
In general, all these results suggest that presence of sodium at physiologic concentrations both reduces the basal activity of receptors and enhances the stimulated response to agonist, thus selectively enhancing the overall efficacy of full agonists. The effects were selective to Na+, as shown in K+ replacement experiments. Importantly, when the studies were able to accurately titrate sodium allosteric effects on signaling, whether basal or ligand induced (Costa et al., 1990; Selley et al., 2000; Wang et al., 2017), the Na+ response curves show KB or EC50 values in the range of ∼10–200 mM, approximately corresponding to the affinity of Na+ in the conserved sodium pocket of these receptors.
2. Mutations in Sodium Ion Pocket Reduce or Abolish Receptor Stimulation by Agonists
A substantial body of evidence for the functional role of Na+ comes from indirect studies showing the dramatic impact of mutations in the sodium coordinating residue’s classic allosteric binding effect and on signaling function of at least 20 class A GPCRs, as presented in Table 1 of our previous review (Katritch et al., 2014). Several recent studies corroborate these observations, suggesting that removal of Na+ site via mutations often has similar consequences as removal of Na+ itself from solution. Thus Massink et al. (2015) show that while in A2A adenosine receptor mutations in sodium-coordinating residues S3.39 and N7.45 reduce or abolish classic dependence of agonist binding on Na+, they also increased basal activity and reduced the maximal activity (Emax) of the agonist-induced signal. These mutations result in about a fivefold reduction of total cAMP response to ligand compared with the wild-type signal. Importantly, the mutations did not reduce but even slightly improved EC50 values of agonists in these assays, which is similar to improved agonist affinities in lieu of Na+. In the case of the D2.50N(A) mutations (Massink et al., 2015; White et al., 2018) in adenosine A2AAR, however, any cAMP activity (basal or induced) of the mutants was disrupted, suggesting that in addition to the Na+ anchoring role, D2.50 has other roles in the receptor activation, which may also be related to dynamic change in its protonation state (Vickery et al., 2018). A similar effect was recently observed for GPR3, where D2.50A mutation in a recently characterized sodium site completely abolished signaling (Capaldi et al., 2018).
Interestingly, constitutively active mutants have been also observed in a number of class A GPCRs in position 3.43 at the bottom of the sodium pocket, where conserved hydrophobic residues Leu (∼74% receptors) or Met (∼20%) comprise a hydrophobic layer, keeping the gate closed to the waters and Na+ ion escaping toward the intracellular side (Yuan et al., 2014). One of the studies showed that replacing Leu or Met with any small or polar group in 3.43 position (Arg, Lys, or Ala) all resulted in constitutive activation in thyrotropin receptor (TSHR), but also in β2AR, luteinizing hormone (LHR), and follitropin (FSHR) receptors (Tao et al., 2000). Apparently, mutations breaking the hydrophobic layer facilitate Na+/water cluster disruption and egress into the cytoplasm, activating receptors even without agonist.
3. A Gain of Function by Introducing Acidic Residues in Sodium Ion Pocket
The importance of allosteric Na+ binding itself is further corroborated by gain-of-function effects, when acidic residues in the pocket other than D2.50 were found to restore, at least partially, signaling function of the receptor lacking D2.50. Such gain of function studies performed for the 5-HT2A serotonin receptor and μ-opioid receptor show that whereas the D2.50N mutant abrogated receptor coupling to G-protein, double mutant D2.50N/N7.49D with another Asp in position 7.49 restored Na+ binding and regained most of the functional activity (Sealfon et al., 1995; Xu et al., 1999). Similarly, some GPCRs, for example the sodium-dependent GnRHR, have these residues naturally reversed as N2.50 and D7.49 in the wild-type protein (Flanagan et al., 1999). Another Na+ coordinating position of the pocket, 3.39, also bears Glu in a few olfactory receptors that lack D2.50, which likely helps them to retain their Na+ binding properties and signaling.
4. Disease-Associated Mutations in the Sodium Pocket
Because GPCRs play a critical role in many biologic and pathologic pathways, missense mutations modifying their signaling response underlie many monogenic disorders in retinal, endocrine, metabolic, developmental, and other systems (Spiegel and Weinstein, 2004; Insel et al., 2007; Vassart and Costagliola, 2011). Some of the critical mutations occur in the sodium pocket residues, impacting their functional profile. Thus, a disease-relevant SNP in CLTR2 cysteinyl leukotriene receptor residue L1293.43 has been associated with uveal melanoma and blue nevi (Moore et al., 2016; Moller et al., 2017). Like other mutations in this position described above, the L1293.43N mutant of CLTR2 receptor constitutively activates endogenous Gαq and is unresponsive to stimulation by leukotriene (Moore et al., 2016).
As predicted recently by Hauser et al. (2018), many more GPCR point mutations documented in the Exome Aggregation Consortium database may be pathologically and therapeutically relevant and many of them are located in the sodium pocket. More than 220 potential disease-associated mutations have been suggested in the sodium pocket of more than 80 different clinical targets of class A GPCRs (Hauser et al., 2018). Of these, mutations at D2.50 position were predicted to be deleterious in 24 different class A GPCRs, S3.39 in 14, N7.45 in 13, S7.46 in 15, and Y7.53 in 15 GPCRs. Most of these SNPs are exceedingly rare (rate <10−4) or unique, making their disease association hard to detect and statistically validate. Thus, understanding of their functional role can facilitate full biochemical and in vivo characterization of the mutants, leading to new diagnostics tools for range diseases. Importantly, as the effect of mutations can vary from elevated basal activity to reduced or completely abolished signaling, the same receptor may have several different disease associations.
C. Mechanism of Sodium Ion Functional Involvement
1. Sodium as an Allosteric Cofactor of Class A G-Protein-Coupled Receptor Signaling
The above evidence suggests that along with selective NAM effects on agonist binding, physiologic concentrations of Na+ can reduce the basal activity of receptors and, overall, enhance the magnitude of their stimulation by full agonists. These opposing effects of Na+ on agonist binding and signaling response at GPCRs have been characterized with EC50 or KB values in the same 20–100 mM concentration range, and the effects can be abolished by D2.50N or other mutations in the sodium pocket, suggesting a common functional mechanism that involves Na+ binding in the conserved pocket. In 2014, we (Katritch et al., 2014) proposed a dynamic mechanism of Na+ as an allosteric cofactor in class A GPCR ligand induced signal transduction. It involves Na+ entrance into the conserved pocket from the extracellular side and along the hydrated channel, which is opened in most class A GPCRs. The extracellular entrance of Na+, also observed in all MD simulations (see section V.B below) is also corroborated by the fact that the intracellular side of the pocket of class A GPCRs in an inactive state is sealed by the “hydrophobic layer” right beneath the sodium. Moreover, the intracellular entrance of Na+ is hindered by a major electrostatic barrier due to excess of positive charges (as high as 10–15) found at the cytoplasmic side of receptors. It is well established that the presence of the sodium/water cluster in the conserved pocket stabilizes the receptor in the inactive state, reducing its basal activity and reducing the availability of high-affinity binding sites for agonists (Chung et al., 2011). Unlike agonists, binding of most antagonists is compatible with Na+ binding and therefore a synergistic stabilization of inactive state by Na+ can enhance the affinity of antagonists and inverse agonists.
During activation-related rearrangements in the 7TM bundle and the sodium pocket, Na+ becomes dislodged from its position in the pocket and exits toward cell cytoplasm via the opening formed in the hydrophobic layer upon activation (Yuan et al., 2013). Importantly, the extracellular entrance and intracellular egress of Na+ comprises a transfer of Na+ ion across cell membranes. This transfer goes along with the gradient of Na+ concentration, which is ∼10- to 20-fold higher at the extracellular side, as well as with electrostatic potential on the plasma membrane, and the reverse transfer against the electrochemical gradient is very unlikely. It was estimated that the transmembrane transfer of Na+ along with gradient would result in ∼3 kcal gain in energy, and this transfer can be coupled with signal amplification in class A GPCRs observed in presence of Na+.
One of the more recent studies also pointed to possible protonation of D2.50 upon activation, where increased mobility of Na+ in the pocket results in higher pKa of this acidic side chain (Vickery et al., 2018). Such protonation would result in the total disappearance of the barrier for sodium intracellular egress and thus facilitate activation (Fig. 6).
Fig. 6.

Updated version of the Na+ involvement in GPR activation mechanism. (A) Inactive receptor conformation has an Na+ ion bound to D2.50 in a pocket, which is sealed from the cytosol by a hydrophobic layer around Y7.53. (B) G-protein and agonist bind to the receptor, leading to the formation of a continuous water channel across the GPCR. The increased mobility of the Na+ ion results in a pKa shift and subsequent protonation of D2.50. (C) Neutralization of D2.50 and the presence of the hydrated pathway facilitate transfer of Na+ to the intracellular side, driven by the TM Na+ gradient and the negative cytoplasmic membrane voltage. (D) The expulsion of Na+ toward the cytosol results in a prolonged active state of the receptor. This figure from Vickery et al. (2018) reproduced with permission.
D. Other Potential Functional Effects of the Conserved Sodium Ion Binding
1. Voltage Sensing
Selective transfer of Na+ positive charge through the GPCR transmembrane bundle and coupling of this transfer with receptor activation is likely to make GPCRs sensitive to both sodium concentration gradient and the electrostatic potential on the membrane (Ben-Chaim et al., 2006). Several recent studies, indeed, showed that membrane voltage increased the sensitivity of the α2A adrenoreceptor to norepinephrine (Rinne et al., 2013). Activation of another adrenergic receptor, the β1AR, by catecholamine agonists was also shown to be positively modulated by membrane voltage, while depolarization of membrane dramatically reduced signaling (Birk et al., 2015). Similarly, voltage sensitivity of muscarinic acetylcholine receptors to their full agonists was shown for M2, M3, and M5 subtypes (Navarro-Polanco et al., 2011; Rinne et al., 2015). Several studies, including MD-simulations in M2 and the δ-opioid receptor (Vickery et al., 2018), suggested that Na+ binding in the sodium pocket may explain such voltage sensitivity. Limited experimental data from live cell assays, however, have not been conclusive so far. While D2.50 mutations to Ala (Navarro-Polanco et al., 2011) or Asn (Barchad-Avitzur et al., 2016) eliminated gating currents in M2R, voltage sensitivities for agonist binding and conformational changes of the receptor were still present in the mutant (Barchad-Avitzur et al., 2016). This suggests the presence of multiple voltage sensors in muscarinic receptors (Hoppe et al., 2018) and calls for similar assessments of voltage sensitivity in other class A GPCRs, where the effect may be more well defined.
2. pH Dependence
Protonation of D2.50 has been proposed as a facilitator of Na+ egress from class A GPCRs, thus shifting the conformational equilibrium toward their active state and facilitating signaling (Vickery et al., 2018; Hu et al., 2019). This mechanism is consistent with in vitro observations that lower pH increases both basal and ligand-induced activation, for example in the β2AR (Ghanouni et al., 2000). This pH dependence may have important physiologic consequences because, in addition to classic cell membrane signaling, GPCR have been shown to be signaling for an extended period of time from endosomes, where pH is dramatically shifted toward an acidic environment (Calebiro et al., 2010; Irannejad et al., 2013; Vilardaga et al., 2014; Godbole et al., 2017; Eichel and von Zastrow, 2018). The conserved Na+ site protonation would establish a common mechanism for pH dependence for the majority of class A GPCRs; however, more data and further details of the proton transfer need first to be established.
IV. Ion Binding Sites as Ligand Targets—New Approaches to Design Functional Properties
Beyond their physiologic importance, can the ion binding sites in GPCRs be directly exploited for the discovery of new ligands with potentially therapeutically relevant properties? Indeed, structure-based analysis of known GPCRs suggests that ion binding sites can be critical for designing both subtype and functionally selective ligands (Fig. 7).
Fig. 7.

GPCR pockets for binding bitopic ligands that target specific ion binding sites. (A) Overview of the pocket positions in 7TM domain. (B) structure of BTL1 receptor with bitopic ligand, PDB: 5X33 (Hori et al., 2018). (C) Model of bitopic ligands designed for binding to KOR Na+ allosteric site (Zaidi et al., 2019). (D) Structure of complex with orthosteric ligand and PO43− ion, PDB: 3RZE (Shimamura et al., 2011). In all panels, semitransparent surface shows orthosteric pocket (orange), allosteric conserved sodium pocket (cyan), and allosteric site in EC loops (green).
A. Targeting Nonconserved Ion Binding Sites for Subtype Selectivity
Targeting selective ionic interactions can often serve as a beneficial strategy for creating subtype selectivity within closely related subfamily members, with some of such cases being characterized pharmacologically and structurally. One of the examples is the development of highly selective drugs for the H1 histamine receptor (H1R) (Fig. 7D). While the first generation of antihistamines, including doxepin, were not subtype selective, the crystal structure of the H1R-doxepin complex revealed a phosphate ion tightly bound in the extracellular loop (ECL) region and coordinated by nonconserved basic side chains K179ELC2 and K1915.39 (Shimamura et al., 2011). Docking of the second generation antihistamines like acrivastine, levocetirizine, and fexofenadine showed that H1R selectivity and thus improved safety and pharmacological profile of these drugs can be explained by their acidic carboxy groups mimicking the interactions of the PO43− ions.
Interactions with ion binding sites must be taken into consideration in other cases of design of GPCR ligands. The GPR39A, recently identified as Zn2+ modulated receptor, presents a good example of ligand identification for an ion-binding receptor. Ligands were discovered by medium-throughput screening assays, which detected selective modulation of Zn2+ activity on this GPCR39A (Sato et al., 2016). Another interesting example is proton-sensing receptor GPR68, where the proton site has been detected via a combination of molecular modeling and mutagenesis (Huang et al., 2015b). After initial detection of lorazepam as a selective positive allosteric modulator of the proton activation in GPR68, homology modeling and ligand guided optimization approaches were used to develop a model for virtual screening of ∼3M available compounds. The screening yielded several new selective PAMs for GPR68, some of them showing in vivo activity (Huang et al., 2015b). Rapidly improving availability of receptor structures and more relevant templates for structural homology modeling makes such approaches more and more practical in application to other ion-binding GPCRs.
B. Allosteric Ligand Binding in the Conserved Sodium Ion Site
The importance of the conserved Na+ site for the function of class A GPCRs suggests that targeting this site with allosteric or bitopic ligands may be a viable general strategy for modulation of these receptor signaling. The volume of the pocket is usually about 150–250 Å3, thus permitting binding of small fragment-like molecules. Indeed, the Na+ pocket has been characterized as a binding site of an antidiuretic drug, sodium channel blocker amiloride, and its derivatives (Liu et al., 2012b). Amilorides are known as negative allosteric modulators (NAMs) of many class A GPCRs, including adenosine (Howard et al., 1987; Leppik et al., 2000; Gao et al., 2003a,b, 2011; Gutiérrez-de-Terán et al., 2013), dopamine (Neve, 1991; Hoare et al., 2000), muscarinic (Dehaye and Verhasselt, 1995), 5-HT (Pauwels, 1997), GnRHR (Heitman et al., 2008), and potentially many more receptors, as summarized in Katritch et al. (2014). Various affinity estimates for amiloride derivatives show KB raging from ∼1 to 50 μM in their negative allosteric modulation of orthosteric ligand binding. Docking of amiloride and a bulkier derivative HMA (5-(N,N-hexamethylene)amiloride) (Gutiérrez-de-Terán et al., 2013) shows that the positively charged guanidine moiety of the ligand forms a salt bridge to the D2.50 carboxyl, while the bulkier N5 substituents point toward the orthosteric site. The induced docking and conformational modeling also suggested that the fitting of amilorides, especially HMA into the pocket, requires substantial expansion of the pocket, which manifested in adjustments in the N7.45, N7.49, and especially W6.48 side chains. Accordingly, mutations of these residues to alanine in this study only improved affinity of amiloride and HMA severalfold, suggesting that amiloride might not be the optimal chemotype for targeting the pocket. On the chemistry side, a number of additional amiloride derivatives with longer 5N substitutes were characterized in a recent study (Massink et al., 2016), showing that extension of the allosteric ligand into the orthosteric pocket is possible without major reduction of the binding affinity.
Another small molecule characterized recently as an allosteric Na+ pocket binder in the BLT1 receptor is benzamidine (Hori et al., 2018), though its affinity (KB) was estimated much lower than amiloride at ∼500 μM, making it only ∼10 times more potent than Na+ ion itself (based on Fig. 4A in Hori et al., 2018). The study revealed binding of benzamidine and its NAM effect on G-protein activity in two very different receptors, the BLT1 receptor and β1AR, suggesting that it likely binds at the sodium ion binding site in other class A GPCRs as well.
Intriguingly, because Li+ can compete with Na+ in the conserved pocket (Pert et al., 1973), some studies hypothesized that effects of Li+ on functional properties of GPCRs can be implicated in physiologic and psychoactive effects of the Li+ (Dudev et al., 2018). The Li+ effect as a competitor to Na+ binding is especially intriguing because lithium is widely used in treatment of bipolar disorders; however, more evidence is needed to establish the GPCR mode of action of Li+, as this ion can also impart a central nervous system effect via ion channel modulation.
C. Targeting Sodium Ion with Bitopic Ligands
1. Concept of Bitopic Ligands
The highly conserved nature and small size of the sodium pocket itself limit its selectivity, and therefore, the practical utility of small ligands like amilorides and benzamidines as allosteric modulators. On the other hand, a combination of high affinity selective orthosteric moieties with the unique functional properties of the Na+ site allosteric binders could make bitopic ligands an attractive target for ligand design. One recently characterized example of such a bitopic ligand is benzamidine-containing ligand BIIL260 found in the BTL1 receptor structure (Hori et al., 2018). By reaching into the Na+ site and forming a salt bridge with D2.50 carboxyl, as well as hydrogen bonds to S3.39 and S7.45, the positively charged benzamidine moiety is expected to block activation related changes. In agreement with this prediction, BIIL260 was characterized as an inverse agonist, completely blocking the basal activity of the receptor. There are several other benzamidine-containing compounds for BLT1 predicted to bind in a similar manner (Hori et al., 2018).
2. Structure-Based Design of Bitopic Ligands for the Sodium Ion Site
Structure-based rational design of bitopic ligands targeting Na+ site of class A GPCRs was proposed as a potentially broadly applicable mechanism for developing selective ligands with beneficial functional properties, including, e.g., inverse agonism (Katritch et al., 2014). Such design in application to opioid receptors been performed recently (S. Zaidi, T. Che, B. Roth, and V. Katritch, manuscript in preparation). The structure-based design of these ligands (see Fig. 7) is based on the orthosteric agonist nalfurafine, extending its morphinan scaffold at N5 toward the sodium pocket. Both bitopic ligands synthesized and tested, BRI-731 and BRI-751, show high affinities at the three opioid receptors and retain full Gi agonism of nalfurafine. At the same time, the ligands, and especially guanidine-containing BRI-751, effectively block arrestin recruitment, which makes them highly Gi-biased agonists. The ability of bitopic ligands to block arrestin signaling can be explained by the conformational mechanism, where the allosteric moiety (e.g., amiloride) blocks inward movements of TM7 in the Na+ pocket, while still allowing outward movement of TM6. Indeed, several previous studies, e.g., by NMR (Liu et al., 2012a) and fluorescent spectroscopy (Rahmeh et al., 2012), established a direct connection between dynamical changes in TM6 and G-protein biased signaling, while TM7 was associated with arrestin-biased signaling. Like the amiloride derivatives described above, the improved affinities of BRI-751 in the Ala mutants of the pocket side chains suggest that the Na+ site is slightly too small for these moieties and that further optimization could improve the ligand affinities to wild-type receptors.
V. Biophysical and Computational Approaches for Studying Allosteric Ions
As ions impact many aspects of GPCR function, often in subtle dynamic ways, a comprehensive multidisciplinary approach is often needed to fully understand the observed effects. In addition to the key evidence from biochemical and pharmacological studies of allosteric ion effects and structural studies revealing ion binding sites, biophysical and computational approaches make an increasingly important contribution to our understanding of ion dynamics and functional role.
A. Nuclear Magnetic Resonance Spectroscopy
1. Study of G-Protein-Coupled Receptor Dynamics with and without Sodium Ion Site
Nuclear magnetic resonance (NMR) approaches have been widely applied to study conformational dynamics of GPCRs, complementing static the picture produced by crystallography, and the recent examples show that it can greatly contribute to the understanding of the role of ions in GPCR function. Thus, in a recent study (Eddy et al., 2018b) characterized the dynamic behavior of the A2A adenosine receptor (A2AAR) by assessing NMR spectra of Trp and Gly residues in complexes with specific agonists and antagonists. The study showed that the D2.50N mutation disrupting the Na+ site can drastically reduce signaling-related dynamics in the intracellular half of the receptor, while not affecting conformational dynamics in the extracellular ligand binding half. This is in perfect agreement with the “uncoupling” role of D2.50N mutation that abolishes G-protein activation in A2AAR (Eddy et al., 2018a) and validates the dynamic nature of the Na+ allosteric effect.
2. Direct Nuclear Magnetic Resonance Assessment of Allosteric Sodium Ion Binding Kinetics
Recent NMR studies show that the effect of sodium and divalent cations on GPCR signaling dynamics can be assessed directly by NMR (Eddy et al., 2018b; Ye et al., 2018). These studies, also performed with A2AAR, demonstrated that increasing concentrations of Na+ selectively shift the equilibrium toward an inactive ensemble, while the addition of K+ does not have such effect. Moreover, the Ye et al. (2018) study used 23Na NMR binding isotherm to achieve the first direct measurement of the dissociation constant of Na+ in apo A2AR as Kd = 61 ± 27 mM, which is in good agreement with previous indirect estimations of Na+ allosteric effects KB = 32 mM (Massink et al., 2015). Furthermore, relaxation dispersion (CPMG) experiments with 23Na provided a first measure of the bound state lifetime. In the apo state, the specific Na+ bound lifetime was estimated at 480 μs, in agreement with lack of Na+ dissociation in the shorter time scales of <10 μs in molecular dynamic simulations of the inactive state. As expected, binding of the antagonist ZM241385 increased the bound fraction of Na+ by 20% and the average bound state lifetime to 630 μs. Another important observation in this study involves amiloride analog HMA, which was shown by NMR to compete with sodium weakening its effect on titration isotherm.
3. Binding and Effect of Divalent Ions in A2A Adrenergic Receptor is Potentially Nonspecific
In stark contrast with Na+ effects, the same NMR study (Ye et al., 2018) shows that the allosteric effects of divalent cations at A2AAR appear to be nonselective, at least between Ca2+ and Mg2+, and require non-physiologically high concentrations (100–500 mM) of these ions. The divalent ions shifted the equilibrium toward active states of the receptor, and this shift was synergistic with agonist binding, which is similar to observations for divalent ions found in opioid receptors earlier (Pert et al., 1973). The MD simulations in this study suggested that at high cation concentrations they can bind in the extracellular loops of the receptor, bridging the key acidic residues and contracting the pocket, thus facilitating agonist binding and receptor activation.
B. Molecular Dynamic Simulations
Owing to its ability to look into atomistic details and generate hypotheses for experimental validation, molecular dynamics (MD) simulations have become an important tool for deciphering roles of ions in GPCR function. On the other hand, adequate modeling of ions, especially Na+, is becoming a requirement for adequate MD studies of functional effects in most GPCRs, in the same way as ions are absolutely required in the modeling of ion channels.
1. Sodium Access and Universality of Sodium Ion Binding in Class A G-Protein-Coupled Receptors
Starting from the pioneering work on the Na+ access into the allosteric pocket in dopamine receptors (Selent et al., 2010), a number of studies characterized Na+ access kinetics in opioid (Shang et al., 2014) and muscarinic (Miao et al., 2015; Vickery et al., 2016) receptors. Moreover, conventional and accelerated MD simulations were recently performed systematically for 18 different GPCRs with known 3-D structures by Selvam et al. (2018). The studies predict similar sodium entry pathways from the extracellular side into the 2.50 pocket for 15 of these diverse class A receptors. Interestingly, the predicted average mean first passage time for Na+ ranged from about 0.1 to 0.3 μs in opioid and orexin-2 receptors, which have relatively open access to the sodium pocket, to ∼30 μs in sphingosine-1-phosphate receptors, which has the extracellular entrance into the receptor obscured by the extracellular (EC) loops. In two of the receptors assessed, the PAR1 protease activated receptor and the P2Y12 purinergic receptor, occlusion of the EC access blocked the extracellular entrance of Na+ during the modeling time, even though PAR1 and PAR2 protease activated receptor crystal structures have Na+ resolved in the conserved pocket. These observations suggest that either Na+ enters PAR1/PAR2 from the intracellular side against the gradients and electrostatic potential (which is unlikely), or thermal and ligand-induced conformational changes in the EC loops can dynamically open the access for Na+ from the outside of the cell. This study (Selvam et al., 2018) also predicted very long average unbinding times for the D2.50 anchored Na+, ranging from 13 μs in the adenosine receptor to ∼120 μs in the orexin-2 receptor. This estimate is approaching the same order of magnitude (480 μs ) as measured by the NMR study (Ye et al., 2018). While the MD was also used to simulate Na+ binding to a nonconserved site in the class B glucagon receptor, the predicted class B binding was very weak and unstable, with unbinding times much faster (2 μs) than binding times (27 μs), which further contrasts with strong and highly selective Na+ binding in class A receptors.
2. Molecular Dynamic Simulations Corroborate New Functional Hypotheses
Molecular dynamic simulations also suggest some intriguing details of Na+-dependent functions. Thus, Vickery et al. (2016) used the CompEL approach to simulate the Na+ gradient and the voltage across the membrane in MD simulations with δ-OR and M2R. The study suggested that the sodium movement along the transmembrane domain in the water-filled pocket can create a gating charge of 0.5e when the ion travels between the extracellular entry and the allosteric binding site, which would make the receptor highly sensitive to membrane polarization and depolarizations as occurs in neuron signaling. Moreover, another study (Vickery et al., 2018) used comprehensive MD-based pKa predictions to show that D2.50 is likely to become protonated when active-like conformation of the receptors and increased mobility of Na+ can lead to D2.50 protonation, followed by fast and barrier-free egress of Na+ into the cytoplasm.
Another recent MD study on μ-opioid receptor (MOR) used a >11 μs Markov State Model ensemble MD simulation to identify the continuous egression path for Na+ in the MOR active state (Hu et al., 2019). This work draws similar conclusions, showing that protonation of D2.50 can greatly facilitate Na+ egress from the cytosol in an active MOR. The combination of MD with machine learning also provides insight into conformational changes accompanying Na+ movements, predicts energetics, and time scales of Na+ translocation in inactive and active MOR, as well as qualitatively predicts impact of Na+ binding on agonist affinity. Interestingly, the predicted timescales for sodium intracellular relocation on the order of 1 second are comparable to the experimentally derived lifetimes of GPCR/G protein complexes.
Long-time scale accelerated molecular dynamic simulations in the M3 muscarinic receptor in Miao et al. (2015) further corroborated the notion that sodium ion bound to charged D2.50 residue confines the receptor mostly to an inactive state with reduced flexibility. In contrast, the D2.50-protonated receptor, which does not bind Na+ could sample larger conformational space, including large-scale structural rearrangements of the transmembrane helices leading to active-like states. MD simulations in MOR receptor (Yuan et al., 2013) also supported the Na+ trajectory pathway through a receptor entering the D2.50 binding site from the extracellular side and then exiting at the intracellular site upon conformational changes and redistribution of internal water molecules. The simulations suggested that the egress path of Na+ can also help to disrupt the ionic lock between D3.49 and R3.50 in the conserved “DRY” motif and thus can facilitate G-protein activation.
Another recent study performed for CXCR4 chemokine receptor simulations used replica exchange MD with enhanced sampling (Cong and Golebiowski, 2018), showing the special role of Na+ coordinating N3.35 side chain in receptor activation and explaining the constitutive activation role of N3.35A and N3.35S mutations.
For histamine receptors, the MD simulations and the calculation of Gibbs energy of solvation (Wittmann et al., 2014) also show the preference for D2.50 binding of Na+ in human H3-receptor (hH3R), which has an experimentally documented allosteric sensitivity to sodium ions. In contrast, the study suggests that the L7.42Q change between the hH3R and hH4R is responsible for different sodium dynamics in the two closely related receptor subtypes, even though all 16 residues of the sodium pocket are identical. While Q7.42, unique for hH4R in not directly involved in sodium binding, it can disrupt the water-filled channel connecting orthosteric and allosteric sodium pocket in hH4R and kinetically block the sodium access, which may explain hH4R insensitivity to sodium effects.
MD simulations are now routinely used in many GPCR structural studies to decipher the functional effects of the Na+ ion, e.g., for D4 dopamine receptor (Wang et al., 2017). The MD simulations of the DRD4 inactive state suggest that sodium and the antagonist nemonapride can mutually stabilize each other’s binding, in agreement with the increased affinity of nemonapride-like antagonists in the presence of sodium.
C. Phylogenetic Analysis
While the exceptional level of sequence and structure conservation of the sodium pocket in class A GPCRs has been characterized (Katritch et al., 2014), detailed study of variations in the sequence and structure of the pocket can be used to decipher interesting trends in the evolution of GPCR function. Thus, lack of conservation in the residues corresponding to sodium pocket of opsin receptors suggests different functional features of these receptors. Indeed, driven by much higher energies of photochemical switches in the covalent retinal ligand and the requirement for fast on-off switching, opsins have apparently lost their Na+ binding and functional dependence. For GPCRs with dissolvable ligands, however, loss of Na+ anchor residues in the receptor pocket of GPCR subtypes leads to loss of ligand-induced signaling, e.g., NTSR2 neurotensin receptor (NTSR2) is nonresponsive to neurotensin and presumably acts via dimerization with its fully functional and sodium-dependent NTSR1 neurotensin receptor subtype (Katritch et al., 2014).
Phylogenetic analysis also has been applied to make new insights into evolution of function in whole GPCR families, such as chemokine receptors (Taddese et al., 2018). The study used principal component analysis of sequence covariations in nested GPCR sequence sets to decipher evolutionary determinants of chemokine receptors. This approach identified residue positions 2.49, 3.35, and 7.45 as the hallmark positions that define the emergence of chemokine receptors and their subsequent divergence into homeostatic (e.g., CXCR4) and inflammatory (e.g., CCR5) receptors. Polar N3.35 and S3.39 in these key residue positions define high stability of the Na+ binding in homeostatic receptors like CXCR4, while glycines G3.35 and G3.39 in these positions result in loose and weak Na+ binding in CCR5 and other inflammatory receptors. Further analysis of chemokines in species also suggested that evolution of chemokine receptors might be driven, at least in part, by dramatic changes in the sodium binding mode. Thus, the ancient jawless fishes had a highly conserved sodium binding site, while this conservation was loosened during the divergence of the chemokine family in modern species.
VI. Unanswered Questions and Future Perspectives
Despite dramatic progress in the understanding of the role of ions in GPCR function, allosteric regulation and fine tuning, numerous questions and venues of inquiry remain wide open.
First, it is still unclear how, with so many potential binding sites in GPCRs for various cations and anions (Strasser et al., 2015), the conserved Na+ site became an almost universal site across the most populous and highly diverse class of class A GPCRs. Some hints were proposed in a recent study (Shalaeva et al., 2015) tracing sodium site evolution to the much more ancient 7TM family of prokaryotic sodium-translocating rhodopsins, though more comprehensive research is needed to clarify the origins and evolution of the Na+ site in GPCRs, which may be facilitated by the flow of sequencing data from diverse prokaryotic and eukaryotic species.
Second, ascribing a precise functional role of sodium in the activation mechanism remains challenging. While its role as an NAM for agonist binding is most commonly studied, for the super-conserved site it absolutely is also required for effective agonist-induced signal transduction in class A GPCRs. The definitive experimental proof that not just the residues of the pocket, but the presence of Na+ in the pocket itself, is critical for the function remains to be obtained. Because Na+ is critical for the function of many cell components and has an obviously major impact on ion channels involved in GPCR signaling, such ultimate experimental proof requires very accurate and well-controlled experiments where removal of Na+ and abrogation of its transmembrane gradient does not impact the assay itself.
Third, while the bulk of the structural and molecular modeling studies predict an extracellular entrance of Na+ into the allosteric pocket and subsequent intracellular egress upon receptor activation, a definitive electrochemical or biochemical experiment directly demonstrating the transmembrane transfer of sodium by GPCRs across plasma membrane remains to be done. The key challenge for the electrophysiological assessment is an ultra-low current in this system, where a single charge transfer is occurring only once per GPCR activation event. Such currents are below the sensitivity thresholds of most methods conventionally used to characterize ion channels, pumps, and transporters and require either supersensitive electrophysiology or other approaches, e.g., using 22Na+ radioisotope.
Fourth, new approaches are needed to further investigate physiologic relevance of Na+ modulation in some GPCRs and cell types. This includes further studies of gating potential effects in GPCR (Vickery et al., 2018), which may play a role in GPCR response to depolarization in neurons. Another interesting aspect is GPCR signaling in endosomes, where acidic environments can contribute to D2.50 protonation and explain prolonged, pH-dependent signaling observed in this system. In general, the allosteric and the functional effects of sodium need to be incorporated as part of allosteric models and signaling pathway analysis, quantitatively explaining often unusual pharmacology of GPCR signaling in different cells and environments.
Finally, while the first few examples of direct targeting Na+ site by bitopic ligands have been emerging (Hori et al., 2018; S. Zaidi, T. Che, B. Roth, and V. Katritch, manuscript in preparation), there are myriad possibilities for using the Na+ pocket in many GPCR targets to rationally design chemical probes with new properties and pharmacological profiles.
Acknowledgments
We would like to thank Drs. Ulrich Zachariae, Vadim Cherezov, Raymond C. Stevens, and Enrique Abola for helpful discussions.
Abbreviations
- A2AR
A2A adenosine receptor
- BLT1
leukotriene B4 receptor 1
- cAMP
cyclic adenosine monophosphate
- CaSR
calcium sensing receptor
- CB1
cannabinoid receptor type 1
- CCR2
C-C chemokine receptor type 2
- CLTR1
cysteinyl leukotriene receptor 1
- CPMG
Carr Purcell Meiboom and Gill NMR
- DOR
delta opioid receptor
- DRD2
dopamine receptor D2
- DRD3
dopamine receptor D3
- DRD4
dopamine receptor D4
- EC
extracellular
- ECL
extracellular loop
- ECL2
extracellular loop 2
- EM
electron microscopy
- EP3
prostaglandin E receptor subtype EP3
- ETA
endothelin ETA receptor
- ETB1
endothelin ETB1 receptor
- GPCR
G-protein-coupled receptors
- HMA
5-(N,N-hexamethylene)amiloride
- MD
molecular dynamics
- M2R
muscarinic acetylcholine receptor 2
- MOR
μ-opioid receptor
- Na+
sodium ion
- NAM
negative allosteric modulation
- NMR
nuclear magnetic resonance
- NTS
neurotensin receptor
- NTSR1
neurotensin receptor type 1
- NTSR2
neurotensin receptor type 2
- PAM
positive allosteric modulation
- PAR1
protease-activated receptor 1
- PAR2
protease-activated receptor 2
- µ-OR
µ-opioid receptor
- 5HT2B
5-hydroxytryptamine receptor 2B
- 7TM
7 transmembrane
Authorship Contributions
Participated in research design: Katritch, Roth.
Performed data analysis: Katritch, Zarzycka, Zaidi.
Wrote or contributed to the writing of the manuscript: Katritch, Roth, Zarzycka, Zaidi.
Footnotes
The research was supported by National Institutes of Health National Institute on Drug Abuse Grants [DA038858, P01DA035764, R37DA035764]; National Institute of Mental Health Grant [R01MH112205]; the National Institute of Mental Health Psychoactive Drug Screening Program; and the Michael Hooker Distinguished Professorship to B.L.R. and The Netherlands Association for Scientific Research Rubicon fellowship [019.161LW.035].
References
- Barchad-Avitzur O, Priest Michael F, Dekel N, Bezanilla F, Parnas H, Ben-Chaim Y. (2016) A novel voltage sensor in the orthosteric binding site of the M2 muscarinic receptor. Biophys J 111:1396–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batyuk A, Galli L, Ishchenko A, Han GW, Gati C, Popov PA, Lee MY, Stauch B, White TA, Barty A, et al. (2016) Native phasing of x-ray free-electron laser data for a G protein-coupled receptor. Sci Adv 2:e1600292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Chaim Y, Chanda B, Dascal N, Bezanilla F, Parnas I, Parnas H. (2006) Movement of ‘gating charge’ is coupled to ligand binding in a G-protein-coupled receptor. Nature 444:106–109. [DOI] [PubMed] [Google Scholar]
- Birdsall NJ, Burgen AS, Hulme EC, Wells JW. (1979) The effects of ions on the binding of agonists and antagonists to muscarinic receptors. Br J Pharmacol 67:371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk A, Rinne A, Bunemann M. (2015) Membrane potential controls the efficacy of catecholamine-induced beta1-adrenoceptor activity. J Biol Chem 290:27311–27320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calebiro D, Nikolaev VO, Persani L, Lohse MJ. (2010) Signaling by internalized G-protein-coupled receptors. Trends Pharmacol Sci 31:221–228. [DOI] [PubMed] [Google Scholar]
- Cao C, Tan Q, Xu C, He L, Yang L, Zhou Y, Zhou Y, Qiao A, Lu M, Yi C, et al. (2018) Structural basis for signal recognition and transduction by platelet-activating-factor receptor. Nat Struct Mol Biol 25:488–495. [DOI] [PubMed] [Google Scholar]
- Capaldi S, Suku E, Antolini M, Di Giacobbe M, Giorgetti A, Buffelli M. (2018) Allosteric sodium binding cavity in GPR3: a novel player in modulation of Aβ production. Sci Rep 8:11102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W, Tu C, Chen TH, Bikle D, Shoback D. (2008) The extracellular calcium-sensing receptor (CaSR) is a critical modulator of skeletal development. Sci Signal 1:ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che T, Majumdar S, Zaidi SA, Ondachi P, McCorvy JD, Wang S, Mosier PD, Uprety R, Vardy E, Krumm BE, et al. (2018) Structure of the nanobody-stabilized active state of the kappa opioid receptor. Cell 172:55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng RKY, Fiez-Vandal C, Schlenker O, Edman K, Aggeler B, Brown DG, Brown GA, Cooke RM, Dumelin CE, Dore AS, et al. (2017a) Structural insight into allosteric modulation of protease-activated receptor 2. Nature 545:112–115. [DOI] [PubMed] [Google Scholar]
- Cheng RKY, Segala E, Robertson N, Deflorian F, Dore AS, Errey JC, Fiez-Vandal C, Marshall FH, Cooke RM. (2017b) Structures of human A1 and A2A adenosine receptors with xanthines reveal determinants of selectivity. Structure 25:1275–1285. [DOI] [PubMed] [Google Scholar]
- Christopher JA, Orgován Z, Congreve M, Doré AS, Errey JC, Marshall FH, Mason JS, Okrasa K, Rucktooa P, Serrano-Vega MJ, et al. (2019) Structure-based optimization strategies for G protein-coupled receptor (GPCR) allosteric modulators: a case study from analyses of new metabotropic glutamate receptor 5 (mGlu5) X-ray structures. J Med Chem 62:207–222. [DOI] [PubMed] [Google Scholar]
- Chung KY, Rasmussen SG, Liu T, Li S, DeVree BT, Chae PS, Calinski D, Kobilka BK, Woods VL, Jr, Sunahara RK. (2011) Conformational changes in the G protein Gs induced by the beta2 adrenergic receptor. Nature 477:611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong X, Golebiowski J. (2018) Allosteric Na+-binding site modulates CXCR4 activation. Phys Chem Chem Phys 20:24915–24920. [DOI] [PubMed] [Google Scholar]
- Cooper DM, Londos C, Gill DL, Rodbell M. (1982) Opiate receptor-mediated inhibition of adenylate cyclase in rat striatal plasma membranes. J Neurochem 38:1164–1167. [DOI] [PubMed] [Google Scholar]
- Costa T, Lang J, Gless C, Herz A. (1990) Spontaneous association between opioid receptors and GTP-binding regulatory proteins in native membranes: specific regulation by antagonists and sodium ions. Mol Pharmacol 37:383–394. [PubMed] [Google Scholar]
- Costa T, Ogino Y, Munson PJ, Onaran HO, Rodbard D. (1992) Drug efficacy at guanine nucleotide-binding regulatory protein-linked receptors: thermodynamic interpretation of negative antagonism and of receptor activity in the absence of ligand. Mol Pharmacol 41:549–560. [PubMed] [Google Scholar]
- Dehaye JP, Verhasselt V. (1995) Interaction of amiloride with rat parotid muscarinic and alpha-adrenergic receptors. Gen Pharmacol 26:155–159. [DOI] [PubMed] [Google Scholar]
- Draper-Joyce CJ, Verma RK, Michino M, Shonberg J, Kopinathan A, Herenbrink CK, Scammells PJ, Capuano B, Abramyan AM, Thal DM, et al. (2018) The action of a negative allosteric modulator at the dopamine D2 receptor is dependent upon sodium ions. Sci Rep 8:1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudev T, Mazmanian K, Lim C. (2018) Competition between Li+ and Na+ in sodium transporters and receptors: which Na+-binding sites are “therapeutic” Li+ targets? Chem Sci 9:4093–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy MT, Gao ZG, Mannes P, Patel N, Jacobson KA, Katritch V, Stevens RC, Wuthrich K. (2018a) Extrinsic tryptophans as NMR probes of allosteric coupling in membrane proteins: application to the A2A adenosine receptor. J Am Chem Soc 140:8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy MT, Lee MY, Gao ZG, White KL, Didenko T, Horst R, Audet M, Stanczak P, McClary KM, Han GW, et al. (2018b) Allosteric coupling of drug binding and intracellular signaling in the A2A adenosine receptor. Cell 172:68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichel K, von Zastrow M. (2018) Subcellular organization of GPCR signaling. Trends Pharmacol Sci 39:200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, Roth BL, Stevens RC. (2014) Molecular control of delta-opioid receptor signalling. Nature 506:191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan CA, Zhou W, Chi L, Yuen T, Rodic V, Robertson D, Johnson M, Holland P, Millar RP, Weinstein H, et al. (1999) The functional microdomain in transmembrane helices 2 and 7 regulates expression, activation, and coupling pathways of the gonadotropin-releasing hormone receptor. J Biol Chem 274:28880–28886. [DOI] [PubMed] [Google Scholar]
- Gao ZG, Ijzerman AP. (2000) Allosteric modulation of A(2A) adenosine receptors by amiloride analogues and sodium ions. Biochem Pharmacol 60:669–676. [DOI] [PubMed] [Google Scholar]
- Gao ZG, Kim SK, Gross AS, Chen A, Blaustein JB, Jacobson KA. (2003a) Identification of essential residues involved in the allosteric modulation of the human A(3) adenosine receptor. Mol Pharmacol 63:1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Melman N, Erdmann A, Kim SG, Muller CE, IJzerman AP, Jacobson KA. (2003b) Differential allosteric modulation by amiloride analogues of agonist and antagonist binding at A(1) and A(3) adenosine receptors. Biochem Pharmacol 65:525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Verzijl D, Zweemer A, Ye K, Goblyos A, Ijzerman AP, Jacobson KA. (2011) Functionally biased modulation of A(3) adenosine receptor agonist efficacy and potency by imidazoquinolinamine allosteric enhancers. Biochem Pharmacol 82:658–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanouni P, Schambye H, Seifert R, Lee TW, Rasmussen SG, Gether U, Kobilka BK. (2000) The effect of pH on beta(2) adrenoceptor function. Evidence for protonation-dependent activation. J Biol Chem 275:3121–3127. [DOI] [PubMed] [Google Scholar]
- Godbole A, Lyga S, Lohse MJ, Calebiro D. (2017) Internalized TSH receptors en route to the TGN induce local Gs-protein signaling and gene transcription. Nat Commun 8:443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez-de-Terán H, Massink A, Rodriguez D, Liu W, Han GW, Joseph JS, Katritch I, Heitman LH, Xia L, Ijzerman AP, et al. (2013) The role of a sodium ion binding site in the allosteric modulation of the A(2A) adenosine G protein-coupled receptor. Structure 21:2175–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan FM, Kallay E, Chang W, Brandi ML, Thakker RV. (2018) The calcium-sensing receptor in physiology and in calcitropic and noncalcitropic diseases. Nat Rev Endocrinol 15:33–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, Gloriam DE. (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov 16:829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser AS, Chavali S, Masuho I, Jahn LJ, Martemyanov KA, Gloriam DE, Babu MM. (2018) Pharmacogenomics of GPCR drug targets. Cell 172:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman LH, Ye K, Oosterom J, Ijzerman AP. (2008) Amiloride derivatives and a nonpeptidic antagonist bind at two distinct allosteric sites in the human gonadotropin-releasing hormone receptor. Mol Pharmacol 73:1808–1815. [DOI] [PubMed] [Google Scholar]
- Hishinuma S, Kosaka K, Akatsu C, Uesawa Y, Fukui H, Shoji M. (2017) Asp73-dependent and-independent regulation of the affinity of ligands for human histamine H-1 receptors by Na+. Biochem Pharmacol 128:46–54. [DOI] [PubMed] [Google Scholar]
- Hoare SR, Coldwell MC, Armstrong D, Strange PG. (2000) Regulation of human D(1), d(2(long)), d(2(short)), D(3) and D(4) dopamine receptors by amiloride and amiloride analogues. Br J Pharmacol 130:1045–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe A, Marti-Solano M, Drabek M, Bunemann M, Kolb P, Rinne A. (2018) The allosteric site regulates the voltage sensitivity of muscarinic receptors. Cell Signal 42:114–126. [DOI] [PubMed] [Google Scholar]
- Hori T, Okuno T, Hirata K, Yamashita K, Kawano Y, Yamamoto M, Hato M, Nakamura M, Shimizu T, Yokomizo T, et al. (2018) Na(+)-mimicking ligands stabilize the inactive state of leukotriene B4 receptor BLT1. Nat Chem Biol 14:262–269. [DOI] [PubMed] [Google Scholar]
- Horstman DA, Brandon S, Wilson AL, Guyer CA, Cragoe EJ, Jr, Limbird LE. (1990) An aspartate conserved among G-protein receptors confers allosteric regulation of alpha 2-adrenergic receptors by sodium. J Biol Chem 265:21590–21595. [PubMed] [Google Scholar]
- Howard MJ, Hughes RJ, Motulsky HJ, Mullen MD, Insel PA. (1987) Interactions of amiloride with alpha- and beta-adrenergic receptors: amiloride reveals an allosteric site on alpha 2-adrenergic receptors. Mol Pharmacol 32:53–58. [PubMed] [Google Scholar]
- Hu X, Wang Y, Hunkele A, Provasi D, Pasternak GW, Filizola M. (2019) Kinetic and thermodynamic insights into sodium ion translocation through the μ-opioid receptor from molecular dynamics and machine learning analysis. PLoS Comput Biol 15:e1006689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, et al. (2015a) Structural insights into mu-opioid receptor activation. Nature 524:315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XP, Karpiak J, Kroeze WK, Zhu H, Chen X, Moy SS, Saddoris KA, Nikolova VD, Farrell MS, Wang S, et al. (2015b) Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature 527:477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel PA, Tang C-M, Hahntow I, Michel MC. (2007) Impact of GPCRs in clinical medicine: monogenic diseases, genetic variants and drug targets. Biochim Biophys Acta 1768:994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, et al. (2013) Conformational biosensors reveal GPCR signalling from endosomes. Nature 495:534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isberg V, de Graaf C, Bortolato A, Cherezov V, Katritch V, Marshall FH, Mordalski S, Pin JP, Stevens RC, Vriend G, et al. (2015) Generic GPCR residue numbers - aligning topology maps while minding the gaps. Trends Pharmacol Sci 36:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JY, Nagaraju M, Meyer RC, Zhang L, Hamelberg D, Hall RA, Brown EM, Conn PJ, Yang JJ. (2014) Extracellular calcium modulates actions of orthosteric and allosteric ligands on metabotropic glutamate receptor 1alpha. J Biol Chem 289:1649–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Huang Y, Wong HC, Zhou Y, Wang X, Yang J, Hall RA, Brown EM, Yang JJ. (2010) Elucidation of a novel extracellular calcium-binding site on metabotropic glutamate receptor 1{alpha} (mGluR1{alpha}) that controls receptor activation. J Biol Chem 285:33463–33474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. (2013) Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol 53:531–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC. (2014) Allosteric sodium in class A GPCR signaling. Trends Biochem Sci 39:233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong H, Raynor K, Yasuda K, Bell GI, Reisine T. (1993) Mutation of an aspartate at residue 89 in somatostatin receptor subtype 2 prevents Na+ regulation of agonist binding but does not alter receptor-G protein association. Mol Pharmacol 44:380–384. [PubMed] [Google Scholar]
- Koshimizu TA, Kashiwazaki A, Taniguchi J. (2016) Combined sodium ion sensitivity in agonist binding and internalization of vasopressin V1b receptors. Sci Rep 6:25327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm BE, White JF, Shah P, Grisshammer R. (2015) Structural prerequisites for G-protein activation by the neurotensin receptor. Nat Commun 6:7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppuraj G, Dudev M, Lim C. (2009) Factors governing metal-ligand distances and coordination geometries of metal complexes. J Phys Chem B 113:2952–2960. [DOI] [PubMed] [Google Scholar]
- Lagerström MC, Schiöth HB. (2008) Structural diversity of G protein-coupled receptors and significance for drug discovery [published correction appears in Nat Rev Drug Discov (2008) 7:542]. Nat Rev Drug Discov 7:339–357. [DOI] [PubMed] [Google Scholar]
- Leppik RA, Mynett A, Lazareno S, Birdsall NJ. (2000) Allosteric interactions between the antagonist prazosin and amiloride analogs at the human alpha(1A)-adrenergic receptor. Mol Pharmacol 57:436–445. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. (2012a) Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 335:1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J-P, Nakakura T, Tomura H, Tobo M, Mogi C, Wang J-Q, He X-D, Takano M, Damirin A, Komachi M, et al. (2010) Each one of certain histidine residues in G-protein-coupled receptor GPR4 is critical for extracellular proton-induced stimulation of multiple G-protein-signaling pathways. Pharmacol Res 61:499–505. [DOI] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, AP IJ, et al. (2012b) Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston KE, Stanczyk MA, Burford NT, Alt A, Canals M, Traynor JR. (2018) Pharmacologic evidence for a putative conserved allosteric site on opioid receptors. Mol Pharmacol 93:157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston KE, Traynor JR. (2014) Disruption of the Na+ ion binding site as a mechanism for positive allosteric modulation of the mu-opioid receptor. Proc Natl Acad Sci USA 111:18369–18374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish H, Berk A, Zipursky S. (2000) Molecular Cell Biology, W. H. Freeman, New York. [Google Scholar]
- Ludwig MG, Vanek M, Guerini D, Gasser JA, Jones CE, Junker U, Hofstetter H, Wolf RM, Seuwen K. (2003) Proton-sensing G-protein-coupled receptors. Nature 425:93–98. [DOI] [PubMed] [Google Scholar]
- Luginina A, Gusach A, Marin E, Mishin A, Brouillette R, Popov P, Shiriaeva A, Besserer-Offroy E, Longpré J, Lyapina E, et al. (2019) Structure-based mechanism of cysteinyl leukotriene receptor inhibition by antiasthmatic drugs. Sci Adv in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. (2012) Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature 485:321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massink A, Gutiérrez-de-Terán H, Lenselink EB, Ortiz Zacarías NV, Xia L, Heitman LH, Katritch V, Stevens RC, IJzerman AP. (2015) Sodium ion binding pocket mutations and adenosine A2A receptor function. Mol Pharmacol 87:305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massink A, Louvel J, Adlere I, van Veen C, Huisman BJ, Dijksteel GS, Guo D, Lenselink EB, Buckley BJ, Matthews H, et al. (2016) 5′-Substituted amiloride derivatives as allosteric modulators binding in the sodium ion pocket of the adenosine A2A receptor. J Med Chem 59:4769–4777. [DOI] [PubMed] [Google Scholar]
- Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis MI, Pragani R, Boxer MB, Earl LA, Milne JLS, et al. (2016) Breaking cryo-EM resolution barriers to facilitate drug discovery. Cell 165:1698–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y, Caliman AD, McCammon JA. (2015) Allosteric effects of sodium ion binding on activation of the m3 muscarinic g-protein-coupled receptor. Biophys J 108:1796–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michino M, Free RB, Doyle TB, Sibley DR, Shi L. (2015) Structural basis for Na(+)-sensitivity in dopamine D2 and D3 receptors. Chem Commun (Camb) 51:8618–8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Gallacher JL, Nehme R, Warne T, Edwards PC, Schertler GF, Leslie AG, Tate CG. (2014) The 2.1 Å resolution structure of cyanopindolol-bound β1-adrenoceptor identifies an intramembrane Na+ ion that stabilises the ligand-free receptor. PLoS One 9:e92727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller I, Murali R, Müller H, Wiesner T, Jackett LA, Scholz SL, Cosgarea I, van de Nes JA, Sucker A, Hillen U, et al. (2017) Activating cysteinyl leukotriene receptor 2 (CYSLTR2) mutations in blue nevi. Mod Pathol 30:350–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AR, Ceraudo E, Sher JJ, Guan Y, Shoushtari AN, Chang MT, Zhang JQ, Walczak EG, Kazmi MA, Taylor BS, et al. (2016) Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat Genet 48:675–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Polanco RA, Moreno Galindo EG, Ferrer-Villada T, Arias M, Rigby JR, Sanchez-Chapula JA, Tristani-Firouzi M. (2011) Conformational changes in the M2 muscarinic receptor induced by membrane voltage and agonist binding. J Physiol 589:1741–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve KA. (1991) Regulation of dopamine D2 receptors by sodium and pH. Mol Pharmacol 39:570–578. [PubMed] [Google Scholar]
- Neve KA, Cox BA, Henningsen RA, Spanoyannis A, Neve RL. (1991) Pivotal role for aspartate-80 in the regulation of dopamine D2 receptor affinity for drugs and inhibition of adenylyl cyclase. Mol Pharmacol 39:733–739. [PubMed] [Google Scholar]
- Nunez D, Kumar R, Hanahan DJ. (1989) Inhibition of [3H]platelet activating factor (PAF) binding by Zn2+: a possible explanation for its specific PAF antiaggregating effects in human platelets. Arch Biochem Biophys 272:466–475. [DOI] [PubMed] [Google Scholar]
- Pasternak GW, Snowman AM, Snyder SH. (1975) Selective enhancement of [3H]opiate agonist binding by divalent cations. Mol Pharmacol 11:735–744. [PubMed] [Google Scholar]
- Pauwels PJ. (1997) Competitive and silent antagonism of recombinant 5-HT1B receptors by amiloride. Gen Pharmacol 29:749–751. [DOI] [PubMed] [Google Scholar]
- Pert CB, Pasternak G, Snyder SH. (1973) Opiate agonists and antagonists discriminated by receptor binding in brain. Science 182:1359–1361. [DOI] [PubMed] [Google Scholar]
- Pert CB, Snyder SH. (1974) Opiate receptor binding of agonists and antagonists affected differentially by sodium. Mol Pharmacol 10:868–879. [Google Scholar]
- Popov P, Kozlovskii I, Katritch V. (2019) Computational design for thermostabilization of GPCRs. Curr Opin Struct Biol 55:25–33. [DOI] [PubMed] [Google Scholar]
- Popov P, Peng Y, Shen L, Stevens RC, Cherezov V, Liu ZJ, Katritch V. (2018) Computational design of thermostabilizing point mutations for G protein-coupled receptors. eLife 7:e34729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quitterer U, AbdAlla S, Jarnagin K, Müller-Esterl W. (1996) Na+ ions binding to the bradykinin B2 receptor suppress agonist-independent receptor activation. Biochemistry 35:13368–13377. [DOI] [PubMed] [Google Scholar]
- Radu CG, Nijagal A, McLaughlin J, Wang L, Witte ON. (2005) Differential proton sensitivity of related G protein-coupled receptors T cell death-associated gene 8 and G2A expressed in immune cells. Proc Natl Acad Sci USA 102:1632–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmeh R, Damian M, Cottet M, Orcel H, Mendre C, Durroux T, Sharma KS, Durand G, Pucci B, Trinquet E, et al. (2012) Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc Natl Acad Sci USA 109:6733–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011) Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne A, Birk A, Bunemann M. (2013) Voltage regulates adrenergic receptor function. Proc Natl Acad Sci USA 110:1536–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne A, Mobarec JC, Mahaut-Smith M, Kolb P, Bunemann M. (2015) The mode of agonist binding to a G protein-coupled receptor switches the effect that voltage changes have on signaling. Sci Signal 8:ra110. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, Devree BT, Sunahara RK, et al. (2011) Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature 469:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger LB, Yamamura HI, Roeske WR. (1980) Cardiac muscarinic cholinergic receptor binding is regulated by Na+ and guanyl nucleotides. J Biol Chem 255:820–823. [PubMed] [Google Scholar]
- Roth BL. (2019) Molecular pharmacology of metabotropic receptors targeted by neuropsychiatric drugs. Nat Struct Mol Biol 26:535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Laskowski MB, Coscia CJ. (1981) Evidence for distinct subcellular sites of opiate receptors. Demonstration of opiate receptors in smooth microsomal fractions isolated from rat brain. J Biol Chem 256:10017–10023. [PubMed] [Google Scholar]
- Sato S, Huang XP, Kroeze WK, Roth BL. (2016) Discovery and characterization of novel GPR39 agonists allosterically modulated by zinc. Mol Pharmacol 90:726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann A, Gimpl G. (2018) Sodium functions as a negative allosteric modulator of the oxytocin receptor. Biochim Biophys Acta Biomembr 1860:1301–1308. [DOI] [PubMed] [Google Scholar]
- Schöppe J, Ehrenmann J, Klenk C, Rucktooa P, Schütz M, Doré AS, Plückthun A. (2019) Crystal structures of the human neurokinin 1 receptor in complex with clinically used antagonists. Nat Commun 10:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sealfon SC, Chi L, Ebersole BJ, Rodic V, Zhang D, Ballesteros JA, Weinstein H. (1995) Related contribution of specific helix 2 and 7 residues to conformational activation of the serotonin 5-HT2A receptor. J Biol Chem 270:16683–16688. [DOI] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K. (2001) Unmasking different constitutive activity of four chemoattractant receptors using Na+ as universal stabilizer of the inactive (R) state. Receptors Channels 7:357–369. [PubMed] [Google Scholar]
- Selent J, Sanz F, Pastor M, De Fabritiis G. (2010) Induced effects of sodium ions on dopaminergic G-protein coupled receptors. PLoS Comput Biol 6:e1000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selley DE, Cao CC, Liu Q, Childers SR. (2000) Effects of sodium on agonist efficacy for G-protein activation in mu-opioid receptor-transfected CHO cells and rat thalamus. Br J Pharmacol 130:987–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvam B, Shamsi Z, Shukla D. (2018) Universality of the sodium ion binding mechanism in class A G-protein-coupled receptors, Angew Chem Int Ed Engl 57, pp 3048–3053. [DOI] [PubMed] [Google Scholar]
- Shalaeva DN, Galperin MY, Mulkidjanian AY. (2015) Eukaryotic G protein-coupled receptors as descendants of prokaryotic sodium-translocating rhodopsins. Biol Direct 10:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, LeRouzic V, Schneider S, Bisignano P, Pasternak GW, Filizola M. (2014) Mechanistic insights into the allosteric modulation of opioid receptors by sodium ions. Biochemistry 53:5140–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihoya W, Nishizawa T, Yamashita K, Inoue A, Hirata K, Kadji FMN, Okuta A, Tani K, Aoki J, Fujiyoshi Y, et al. (2017) X-ray structures of endothelin ETB receptor bound to clinical antagonist bosentan and its analog. Nat Struct Mol Biol 24:758–764. [DOI] [PubMed] [Google Scholar]
- Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, et al. (2011) Structure of the human histamine H1 receptor complex with doxepin. Nature 475:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silve C, Petrel C, Leroy C, Bruel H, Mallet E, Rognan D, Ruat M. (2005) Delineating a Ca2+ binding pocket within the venus flytrap module of the human calcium-sensing receptor. J Biol Chem 280:37917–37923. [DOI] [PubMed] [Google Scholar]
- Simon EJ, Groth J. (1975) Kinetics of opiate receptor inactivation by sulfhydryl reagents: evidence for conformational change in presence of sodium ions. Proc Natl Acad Sci USA 72:2404–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel AM, Weinstein LS. (2004) Inherited diseases involving G proteins and G protein–coupled receptors. Annu Rev Med 55:27–39. [DOI] [PubMed] [Google Scholar]
- Storjohann L, Holst B, Schwartz TW. (2008) Molecular mechanism of Zn2+ agonism in the extracellular domain of GPR39. FEBS Lett 582:2583–2588. [DOI] [PubMed] [Google Scholar]
- Strader CD, Sigal IS, Candelore MR, Rands E, Hill WS, Dixon RA. (1988) Conserved aspartic acid residues 79 and 113 of the beta-adrenergic receptor have different roles in receptor function. J Biol Chem 263:10267–10271. [PubMed] [Google Scholar]
- Strasser A, Wittmann HJ, Schneider EH, Seifert R. (2015) Modulation of GPCRs by monovalent cations and anions. Naunyn Schmiedebergs Arch Pharmacol 388:363–380. [DOI] [PubMed] [Google Scholar]
- Suga H, Ehlert FJ. (2013) Effects of asparagine mutagenesis of conserved aspartic acids in helix 2 (D2.50) and 3 (D3.32) of M1-M4 muscarinic receptors on the irreversible binding of nitrogen mustard analogs of acetylcholine and McN-A-343. Biochemistry 52:4914–4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suno R, Kimura KT, Nakane T, Yamashita K, Wang J, Fujiwara T, Yamanaka Y, Im D, Horita S, Tsujimoto H, et al. (2018a) Crystal structures of human orexin 2 receptor bound to the subtype-selective antagonist EMPA. Structure 26:7–19. [DOI] [PubMed] [Google Scholar]
- Suno R, Lee S, Maeda S, Yasuda S, Yamashita K, Hirata K, Horita S, Tawaramoto MS, Tsujimoto H, Murata T, et al. (2018b) Structural insights into the subtype-selective antagonist binding to the M2 muscarinic receptor. Nat Chem Biol 14:1150–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddese B, Deniaud M, Garnier A, Tiss A, Guissouma H, Abdi H, Henrion D, Chabbert M. (2018) Evolution of chemokine receptors is driven by mutations in the sodium binding site. PLoS Comput Biol 14:e1006209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao YX, Abell AN, Liu X, Nakamura K, Segaloff DL. (2000) Constitutive activation of g protein-coupled receptors as a result of selective substitution of a conserved leucine residue in transmembrane helix iii. Mol Endocrinol 14:1272–1282. [DOI] [PubMed] [Google Scholar]
- Teller DC, Okada T, Behnke CA, Palczewski K, Stenkamp RE. (2001) Advances in determination of a high-resolution three-dimensional structure of rhodopsin, a model of G-protein-coupled receptors (GPCRs). Biochemistry 40:7761–7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, Huang XP, Trapella C, Guerrini R, Calo G, et al. (2012) Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 485:395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda Y, Morimoto K, Suno R, Horita S, Yamashita K, Hirata K, Sekiguchi Y, Yasuda S, Shiroishi M, Shimizu T, et al. (2019) Ligand binding to human prostaglandin E receptor EP4 at the lipid-bilayer interface. Nat Chem Biol 15:18–26. [DOI] [PubMed] [Google Scholar]
- Tsai BS, Lefkowitz RJ. (1978) Agonist-specific effects of monovalent and divalent cations on adenylate cyclase-coupled alpha adrenergic receptors in rabbit platelets. Mol Pharmacol 14:540–548. [PubMed] [Google Scholar]
- Valentin-Hansen L, Frimurer TM, Mokrosinski J, Holliday ND, Schwartz TW. (2015) Biased Gs versus Gq proteins and beta-arrestin signaling in the NK1 receptor determined by interactions in the water hydrogen bond network. J Biol Chem 290:24495–24508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassart G, Costagliola S. (2011) G protein-coupled receptors: mutations and endocrine diseases. Nat Rev Endocrinol 7:362–372. [DOI] [PubMed] [Google Scholar]
- Vickery ON, Carvalheda CA, Zaidi SA, Pisliakov AV, Katritch V, Zachariae U. (2018) Intracellular transfer of Na(+) in an active-state G-protein-coupled receptor. Structure 26:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickery ON, Machtens JP, Tamburrino G, Seeliger D, Zachariae U. (2016) Structural mechanisms of voltage sensing in G protein-coupled receptors. Structure 24:997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardaga JP, Jean-Alphonse FG, Gardella TJ. (2014) Endosomal generation of cAMP in GPCR signaling. Nat Chem Biol 10:700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wu H, Katritch V, Han GW, Huang XP, Liu W, Siu FY, Roth BL, Cherezov V, Stevens RC. (2013) Structure of the human smoothened receptor bound to an antitumour agent. Nature 497:338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wacker D, Levit A, Che T, Betz RM, McCorvy JD, Venkatakrishnan AJ, Huang XP, Dror RO, Shoichet BK, et al. (2017) D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 358:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AG, Schertler GF, Tate CG. (2011) The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature 469:241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AD, Fang F, Jean-Alphonse FG, Clark LJ, An H-J, Liu H, Zhao Y, Reynolds SL, Lee S, Xiao K, et al. (2019) Ca2+ allostery in PTH-receptor signaling. Proc Natl Acad Sci USA 116:3294–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JF, Noinaj N, Shibata Y, Love J, Kloss B, Xu F, Gvozdenovic-Jeremic J, Shah P, Shiloach J, Tate CG, et al. (2012) Structure of the agonist-bound neurotensin receptor. Nature 490:508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KL, Eddy MT, Gao ZG, Han GW, Lian T, Deary A, Patel N, Jacobson KA, Katritch V, Stevens RC. (2018) Structural connection between activation microswitch and allosteric sodium site in GPCR signaling. Structure 26:259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingler LM, McMahon C, Staus DP, Lefkowitz RJ, Kruse AC. (2019) Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell 176:479–490.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann HJ, Seifert R, Strasser A. (2014) Sodium binding to hH3R and hH 4R–a molecular modeling study. J Mol Model 20:2394. [DOI] [PubMed] [Google Scholar]
- Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, et al. (2012) Structure of the human kappa-opioid receptor in complex with JDTic. Nature 485:327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Ozdener F, Li JG, Chen C, de Riel JK, Weinstein H, Liu-Chen LY. (1999) Functional role of the spatial proximity of Asp114(2.50) in TMH 2 and Asn332(7.49) in TMH 7 of the mu opioid receptor. FEBS Lett 447:318–324. [DOI] [PubMed] [Google Scholar]
- Yang LV, Radu CG, Roy M, Lee S, McLaughlin J, Teitell MA, Iruela-Arispe ML, Witte ON. (2007) Vascular abnormalities in mice deficient for the G protein-coupled receptor GPR4 that functions as a pH sensor. Mol Cell Biol 27:1334–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wu Y, Xu TH, de Waal PW, He Y, Pu M, Chen Y, DeBruine ZJ, Zhang B, Zaidi SA, et al. (2018) Crystal structure of the Frizzled 4 receptor in a ligand-free state. Nature 560:666–670. [DOI] [PubMed] [Google Scholar]
- Yao XJ, Velez Ruiz G, Whorton MR, Rasmussen SG, DeVree BT, Deupi X, Sunahara RK, Kobilka B. (2009) The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc Natl Acad Sci USA 106:9501–9506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda S, Kajiwara Y, Toyoda Y, Morimoto K, Suno R, Iwata S, Kobayashi T, Murata T, Kinoshita M. (2017) Hot-spot residues to be mutated common in G protein-coupled receptors of class A: identification of thermostabilizing mutations followed by determination of three-dimensional structures for two example receptors. J Phys Chem B 121:6341–6350. [DOI] [PubMed] [Google Scholar]
- Ye L, Neale C, Sljoka A, Lyda B, Pichugin D, Tsuchimura N, Larda ST, Pomes R, Garcia AE, Ernst OP, et al. (2018) Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat Commun 9:1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Filipek S, Palczewski K, Vogel H. (2014) Activation of G-protein-coupled receptors correlates with the formation of a continuous internal water pathway. Nat Commun 5:4733. [DOI] [PubMed] [Google Scholar]
- Yuan S, Vogel H, Filipek S. (2013) The role of water and sodium ions in the activation of the μ-opioid receptor, Angew Chem Int Ed Engl 52, pp 10112–10115. [DOI] [PubMed] [Google Scholar]
- Zhang C, Srinivasan Y, Arlow DH, Fung JJ, Palmer D, Zheng Y, Green HF, Pandey A, Dror RO, Shaw DE, et al. (2012) High-resolution crystal structure of human protease-activated receptor 1. Nature 492:387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Gao ZG, Zhang K, Kiselev E, Crane S, Wang J, Paoletta S, Yi C, Ma L, Zhang W, et al. (2015a) Two disparate ligand-binding sites in the human P2Y1 receptor. Nature 520:317–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Han GW, Batyuk A, Ishchenko A, White KL, Patel N, Sadybekov A, Zamlynny B, Rudd MT, Hollenstein K, et al. (2017) Structural basis for selectivity and diversity in angiotensin II receptors. Nature 544:327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Unal H, Gati C, Han GW, Liu W, Zatsepin NA, James D, Wang D, Nelson G, Weierstall U, et al. (2015b) Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell 161:833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang K, Gao ZG, Paoletta S, Zhang D, Han GW, Li T, Ma L, Zhang W, Muller CE, et al. (2014) Agonist-bound structure of the human P2Y12 receptor. Nature 509:119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao LH, Ma S, Sutkeviciute I, Shen DD, Zhou XE, de Waal PW, Li CY, Kang Y, Clark LJ, Jean-Alphonse FG, et al. (2019) Structure and dynamics of the active human parathyroid hormone receptor-1. Science 364:148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Han GW, Abagyan R, Wu B, Stevens RC, Cherezov V, Kufareva I, Handel TM. (2017) Structure of CC chemokine receptor 5 with a potent chemokine antagonist reveals mechanisms of chemokine recognition and molecular mimicry by HIV. Immunity 46:1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]