

Abstract
mPer1, a mouse gene, is a homolog of theDrosophila clock gene period and has been shown to be closely associated with the light-induced resetting of a mammalian circadian clock. To investigate whether the rapid induction of mPer1 after light exposure is necessary for light-induced phase shifting, we injected an antisense phosphotioate oligonucleotide (ODN) to mPer1 mRNA into the cerebral ventricle. Light-induced phase delay of locomotor activity at CT16 was significantly inhibited when the mice were pretreated withmPer1 antisense ODN 1 hr before light exposure.mPer1 sense ODN or random ODN treatment had little effect on phase delay induced by light pulses. In addition, glutamate-induced phase delay of suprachiasmatic nucleus (SCN) firing rhythm was attenuated by pretreatment with mPer1antisense ODN, but not by random ODN. The present results demonstrate that induction of mPer1 mRNA is required for light- or glutamate-induced phase shifting, suggesting that the acute induction of mPer1 mRNA in the SCN after light exposure is involved in light-induced phase shifting of the overt rhythm.
Keywords: antisense oligonucleotide, circadian rhythm, firing rhythm, mPer1, phase shift, suprachiasmatic nucleus
Circadian rhythms, which persist in the absence of environmental cues, are observed in a wide variety of organisms (Edmunds, 1988). To maintain synchrony with the daily environmental cycle, organisms responded to environmental cues, especially light, to reset or entrain their circadian rhythms. In mammals, the suprachiasmatic nucleus (SCN) of hypothalamus has been shown to be a primary circadian pacemaker of locomotor activity and various physiological phenomena (Hastings, 1997). The genetic and molecular mechanisms that control circadian rhythms were initiated by studies of Drosophila rhythms (Konopka and Benzer, 1971). The circadian rhythms evident in the locomotor activity of adult flies and in gating of eclosion were altered by mutations in two genes, period (dPer) and timeless(tim) (Hall, 1998; Young, 1998). Protein levels and mRNA levels of these genes undergo robust circadian oscillation, and both proteins co-regulate their own regulation by negative feedback loops. In mammals, previous studies have demonstrated that mRNAs of immediately early genes (IEGs), including c-fos and junB, are markedly induced by light in the SCN (Rusak et al., 1990;Morris et al., 1998). However, the molecular component of the circadian clock and the mechanism by which the light entrains the circadian clock have only been recently elucidated. The recent isolation of dPer homologous genes, Per1 (Sun et al., 1997; Tei et al., 1997), Per2 (Albrecht et al., 1997; Shearman et al., 1997; Takumi et al., 1998b), and Per3(Zylka et al., 1998; Takumi et al., 1998a) from human and mouse have significantly clarified the molecular mechanisms of the circadian clock in mammals. These genes are rhythmically expressed in the SCN. We showed that brief exposure to light during subjective night results in a large and rapid induction of mPer1 expression (Shigeyoshi et al., 1997). The induction of mPer1 (<20 min) by light is more rapid than the accumulation of c-fos protein (Shigeyoshi et al., 1997). This suggests that c-fos protein is not directly involved in the rapid induction of the mPer1 gene.
To investigate whether induction of mPer1 transcripts by light exposure is necessary for light-induced phase shifts, we injected an antisense phosphotioate oligonucleotide (ODN) to mPer1mRNA intracerebroventricularly 1 hr before light exposure. We found that inhibition of light-induced mPer1 expression by antisense oligonucleotide in vivo significantly represses light-induced phase shifts of the mouse circadian locomotor rhythm. We have reported that treatments with glutamate, NMDA, or substance P, or stimulation of the optic chiasm produce changes in the phase of the firing rhythm of SCN neurons in vitro with a phase–response curve similar to that induced by light exposurein vivo (Shibata et al., 1992, 1994; Shibata and Moore, 1993). Direct application of mPer1 antisense ODN to the SCN in hypothalamic slices in vitro produced an attenuation of the glutamate-induced phase shift in a manner similar to the reduction of the light-induced phase shifts observed in vivo. These results suggest that acute induction ofmPer1 mRNA in the SCN after light exposure is involved in light-induced phase shifts of overt rhythms.
MATERIALS AND METHODS
Phosphotioate ODNs. The published sequence ofmPer1 was used to design an antisense ODN targeted to the region of the mRNA containing the initiation ATG. The sequences of themPer1 antisense and sense ODNs (18-mer) were 5′-TAG GGG ACC ACT CAT GTC T-3′ and 5′-A GAC ATG AGT GGT CCC CTA-3′, respectively. The sequences of random ODN (18-mer) and vasopressin precursor gene (AVP) antisense ODN (18-mer) were 5′-CCG TTA GTA CTG AGC TGA C-3′ and 5′-CAT CCT GGC GAG CAT AGG T-3′, respectively. The random ODN contained an equivalent GC content as the antisense and sense ODNs ofmPer1. All ODNs were purified by HPLC to reduce the possible toxicity of phosphotioate ODNs.
Animals and surgery. Male Balb/c mice (Takasugi Saitama, Japan) purchased 6 weeks postpartum were maintained on a 12 hr light/dark cycle with light on at 8:30 A.M. Animals were given food and water ad libitum. Stainless steel guide cannulas (6.0 mm, 23 gauge) were implanted bilaterally intracerebroventricularly (4.5 mm anterior and 1.1 mm lateral to lambda and depth of 2.1 mm below the skull) using a stereotaxic frame (Narishige, Tokyo, Japan). After 2 d recovery, animals were moved to continuous darkness for at least 2 weeks before ODN administration. For assessment of the locomotor activity, mice were housed individually, and their locomotor activity rhythm was measured by area sensors (model FA-05 F5B; Omron, Tokyo, Japan) with a thermal radiation detector system, and data were stored on a personal computer.
After free-running for 14–20 d in constant darkness, mice were randomly assigned to mPer1 antisense ODN, mPer1sense ODN, mPer1 random ODN, AVP antisense ODN, or vehicle (sterilized saline). A 5 μl aliquot of each ODN (2–6 nmol) was unilaterally injected into the lateral ventricle via an injection cannula (external diameter, 0.35 mm) extending 0.5 mm below the tip of the guide cannula at a rate of 1 μl min−1 using a 10 μl Hamilton syringe. Injection was performed at circadian time 1 (CT1; CT12, onset time of locomotor activity), CT4, CT8, CT15, or CT21, then animals were returned to their individual cages. For light exposure experiments, implanted mice were again randomly assigned an ODN, and 60 min after the injection, each animal was exposed to a light pulse lasting 15 min at CT16. Light (20 lux) was administered while the mice were in a Plexiglas cylinder. After treatment, animals were returned to constant darkness. Some mice were first exposed to the light, and then ODNs were administered 0 or 120 min after light exposure. Each group received a repeated intracerebroventricular injection (3 or 4 times for each animal) after at least 14 d. Injections were randomly administered into the right or left ventricle. To verify that ODNs were administered into the cerebral ventricle, mice were injected with 5 μl of saturated fast green, and their brains were examined macroscopically after sectioning. Anisomycin (50 μg in 5 μl of saline) and MK-801 (10 μg in 5 μl of saline) were also intracerebroventricularly injected in the same manner. After treatment, animals were returned to constant darkness. The phase of the rhythm was assessed visually by applying a straight edge to the onset of activity on successive days before the light pulse and again beginning ∼3 d after a light treatment and determining the difference in phases on the day of the light exposure (Daan and Pittendrigh, 1976). At least four independent experiments using different mice were done at each group.
Slice culture and measurement of neural activity rhythm. On the first day, coronal hypothalamic slices (400 μm thickness) including SCN were obtained between zeitgeber time 9–11 (ZT9–11) from male Balb/c mice (10–14 weeks). Then, slices were preincubated and treated with vehicle or mPer1antisense, sense, or random ODNs (each 20 μm) in Krebs’ Ringer’s solution (in mm: NaCl 129, KCl 4.2, MgSO4 1.3, KH2PO4 1.2, CaCl2 1.5, glucose 25, NaHCO3 22.4, and HEPES 25, with gentamycin 0.5 mg/ml, pH 7.3–7.4) for 4 hr (ZT12–16). At ZT16, the slices were removed into the buffer containing glutamate (10 μm) for 15 min. After drug treatment, perfusion with normal medium was reinstated. The spontaneous action potentials of single SCN cells were recorded extracellularly through glass electrodes filled with 3 m NaCl during the second day in vitro. Stable single unit activity was recorded over 5 min intervals. The activities of all cells recorded during a single experiment were averaged into 2 hr intervals using 1 hr lags. Previous studies have shown that this procedure yields a pattern of electrical activity for the population of SCN neurons that varies little between animals, and that the time of peak electrical activity is a reliable marker of the phase of the SCN pacemaker (Shibata and Moore, 1993;Shibata et al., 1994).
Biochemical analysis. To detect injected biotinylated ODN, anesthetized mice were perfused intracardially with ice-cold saline and 4% paraformamide in 0.1 m phosphate buffer (PB), pH 7.4, and then their brains were removed, post-fixed for 24 hr at 4°C and placed in 0.1 m PB with 20% sucrose for 24 hr. The distributions of ODN in the serial cryostat sections (30 μm) containing SCN were determined using a Vecstatain ABC Elite kit (Vector Laboratories, Burlingame, CA).
RT-PCR analysis. The effect of mPer1 antisense ODN on mPer1 expression in the SCN was examined by RT-PCR. Mice were entrained to light/dark cycle for 2 weeks. Mice were transferred to constant darkness for one extra daily cycle, and at ZT15, mice were administered antisense ODN (2, 4, and 6 nmol) and vehicle. Half of both groups received light treatment (20 lux, 15 min) at ZT16. At ZT17.5, brains (n = 4 for each group) were removed and placed in ice-cold saline. Slices (0.5-mm-thick) of mice brain that contained SCN were frozen on dry ice, and the SCNs were punched out with a 26 gauge needle. Total RNA from the SCN (n = 4) was extracted in each group by Trizol solution (BRL, Bethesda, MD). A one-step RT-PCR system (BRL) was used for reverse transcription of ∼100 ng of RNA, and mPer1,mPer2, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA were amplified by PCR. RT-PCR reactions were performed for 21 cycles with mPer1, mPer2, and GAPDH primers in a single tube. The primer pairs used for the amplification of each product are as follows: 5′-CCA GGC CCG GAG AAC CTT TTT-3′ and 5′-CGA AGT TTG AGC TCC CGA AGT G-3′ (mPer1); 5′-ACA CCA CCC CTT ACA AGC TTC-3′ and CGC TGG ATG ATG TCT GGC TC-3′ (mPer2); and 5′-GAC CTC AAC TAC ATG GTC TAC A-3′ and TGG CCG TGA TGG CAT GGA CT-3′ (GAPDH). The sizes of the PCR products of mPer1,mPer2, and GAPDH were 402, 779, and 436 bp, respectively. PCR products were run on 3% agarose gels, and DNA in the appropriate bands were detected with an EDAS-120 system (Eastman Kodak, Rochester, NY).
RESULTS
Phase shift effects of mPer1 antisense ODN on various CTs
Administration of 6 nmol of mPer1 antisense ODN at various CTs (CT1, 8, 15, 21) were compared with vehicle administration (Fig. 1B). Significant phase delays were observed when mPer1 antisense ODN was administrated at CT1. There were no significant differences betweenmPer1 antisense ODN administrated at other CTs. To examine whether this ODN shifting effect is specific to mPer1antisense ODN, we examined the effects of intracerebroventricular administration of four different ODNs and anisomycin on behavioral rhythms (Fig. 1A,C). Administration of anisomycin, which inhibits protein synthesis, has been shown to induce phase shifts when it was injected into the SCN region (Inouye et al., 1988). Two ODNs, mPer1 antisense ODN and AVP antisense ODN, had specific mRNA targets, whereas the other two ODNs, sense and random ODNs, lacked specific mRNA targets. We found that phase delays were observed when anisomycin (50 μg) or mPer1 antisense (6 nmol) ODN was administered (p < 0.01; Student’s t test). No significant phase shifts were observed after injection of the other ODNs or vehicle (Fig.1C). The magnitude of the phase shifts by mPer1antisense ODN were dose-related, with injection of 4 nmol of themPer1 antisense ODN producing a phase shift approximately half the size of the one at a 6 nmol dose. No phase delays were observed at 2 nmol doses.
Fig. 1.
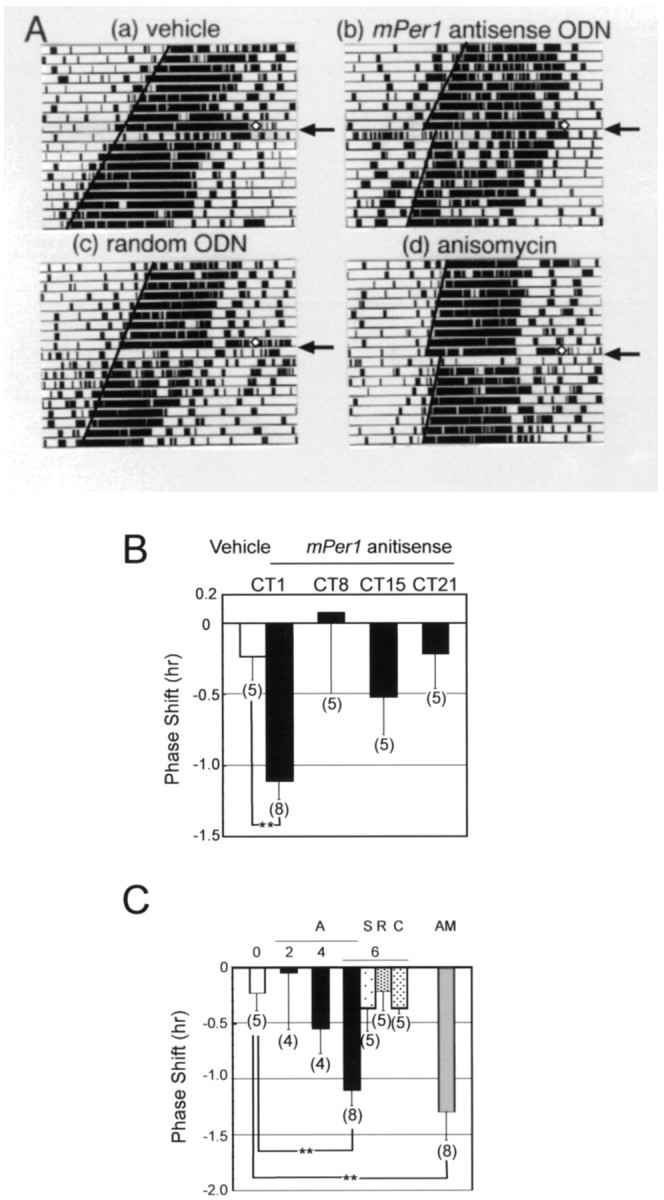
Effect of ODN administration at CT1 on the mouse circadian locomotor rhythm. A, Locomotor activity records of vehicle (a), mPer1antisense ODN (b), random ODN (c), and anisomycin (d)-injected mice. Each animal was injected at CT1 (⋄ in the figure) intracerebroventricularly (5 μl; 1 μl min−1) and returned to constant darkness.B, Phase–response curve for mPer1antisense ODN administration at CT1, CT8, CT15, and CT21.Numbers in parentheses indicate the number of experiments. Injection of mPer1 antisense ODN at CT1 induced a significant phase delay (**p < 0.01; Student’s t test). C, Phase shifts of mouse locomotor rhythm by various ODNs or anisomycin injection at CT1.0, Vehicle; A, mPer1antisense ODN; S, sense ODN; R, random ODN; C, AVP antisense ODN. The number in the figure indicates the amount (in nanomoles) of injected ODN.Numbers in parentheses indicate the number of experiments. Injection of mPer1 antisense ODN and anisomycin significantly phase delayed locomotor rhythm (**p < 0.01; Student’s ttest).
Effect of ODN on light-induced phase shifts
We previously demonstrated that mPer1 induction by light is strongly correlated with phase shifts in behavioral rhythms (Shigeyoshi et al., 1997). Thus, we examined the effect ofmPer1 antisense ODN on light-induced phase shifts (Fig.2A,B). Mice injected with vehicle at CT15 followed by exposure to a light pulse for 15 min at CT16 had a marked phase delay in the circadian rhythm of locomotor activity of ∼2 hr. MK-801, an NMDA receptor antagonist, injected intracerebroventricularly at CT15 markedly depressed the light-induced phase delay at CT16, as previously reported (p < 0.01; Student’s t test) (Colwell et al., 1990; Shibata et al., 1994). Injection ofmPer1 antisense ODN at CT15 attenuated the light-induced phase delay at CT16 in a dose-dependent manner [phase shift, −0.480 ± 0.194 hr (6 nmol injection of antisense ODN) vs −2.204 ± 0.141 hr (vehicle injection); p = 0.0001]. However, injection of mPer1 antisense alone at CT15 did not alter locomotor activity (Fig. 2B).mPer1 antisense administration immediately after light exposure (CT16.3) also inhibited the light-induced phase shift, but less efficiently, and administration of it 2 hr after light exposure (CT18) did not inhibit the phase delay (Fig. 2B). The other ODNs injected at CT15 did not affect the light-induced phase delay at CT16.
Fig. 2.
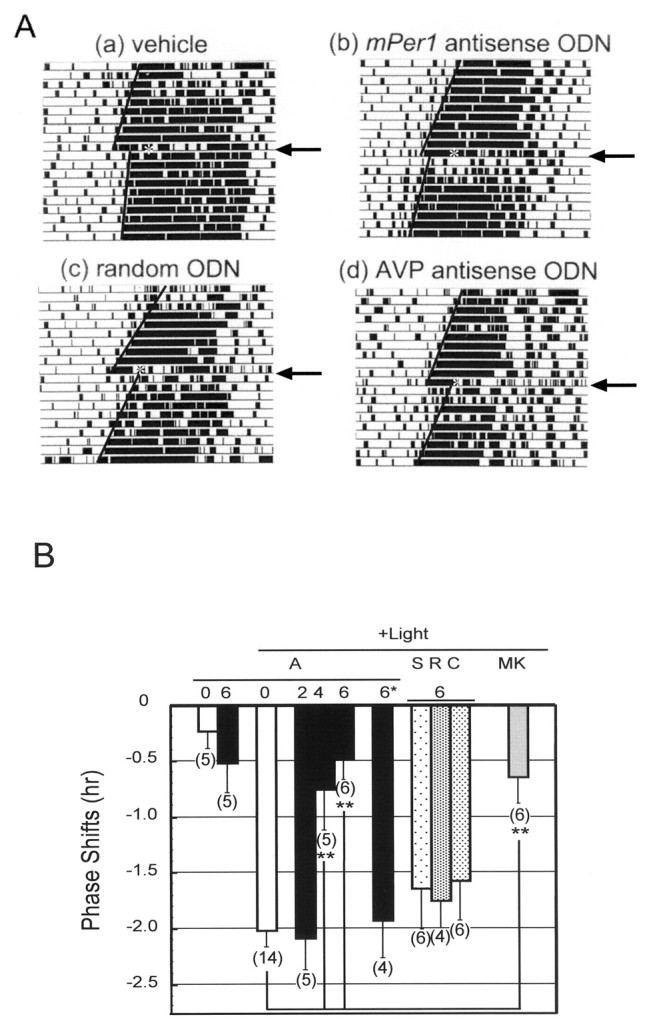
Effect of ODN injection on light-induced phase delay of locomotor activity rhythm. Mice were injected with ODNs at CT15 under the safety light, 1 hr after injection, mice were exposed to light (20 lux) for 15 min and returned to constant darkness.A, Locomotor activity records of vehicle (a), mPer1 antisense (b), random ODN (c), and AVP antisense ODN (d)-injected mouse.B, Light-induced phase shifts in mPer1antisense ODN (A), sense ODN (S), random ODN (R), AVP antisense ODN (C), and MKC-801 (MK)-injected mouse. * indicates that antisense ODN was administered 2 hr after the light pulse. The number in the figure indicates the amount (nanomoles) of injected ODN. 0 indicates the vehicle administration. Numbers in parentheses indicate the number of experiments. Preinjection of mPer1antisense ODN (4 and 6 nmol) and MK-801 significantly reduced light-induced phase shift (**p < 0.01; Student’st test). Injection of mPer1 antisense ODN 2 hr after light exposure did not have any effects.
Effect of antisense ODN on in vitro SCN neural activity rhythm
We and other researchers have reported that treatments with glutamate produce changes in the phase of the firing rhythm of SCN neurons in vitro with a phase–response curve similar to that induced by light exposure in vivo (Shibata et al., 1994; Shirakawa and Moore, 1994). Thus, we examined the effects ofmPer1 antisense ODN in vitro. In control experiments, coronal hypothalamic slices containing whole SCN were treated in vitro for 4 hr on day 1 between ZT12 and ZT16 with drug-free perfusion medium. In these experiments, the mean peak of electrical activity on the subsequent day occurred at ZT6–7 (ZT6.0 ± 0.5; n = 4) (Fig.3A,B). For slices treated with glutamate in vitro at ZT16 on day 1, the peak was around ZT9 on day 2 (Fig. 3). Glutamate-induced phase delay at ZT16 was significantly blocked by 4 hr pretreatment withmPer1 antisense ODN (ZT12–16) but not by pretreatment with random ODN. However, mPer1 antisense ODN did not produce phase changes when applied alone for 4 hr (ZT12–16).
Fig. 3.
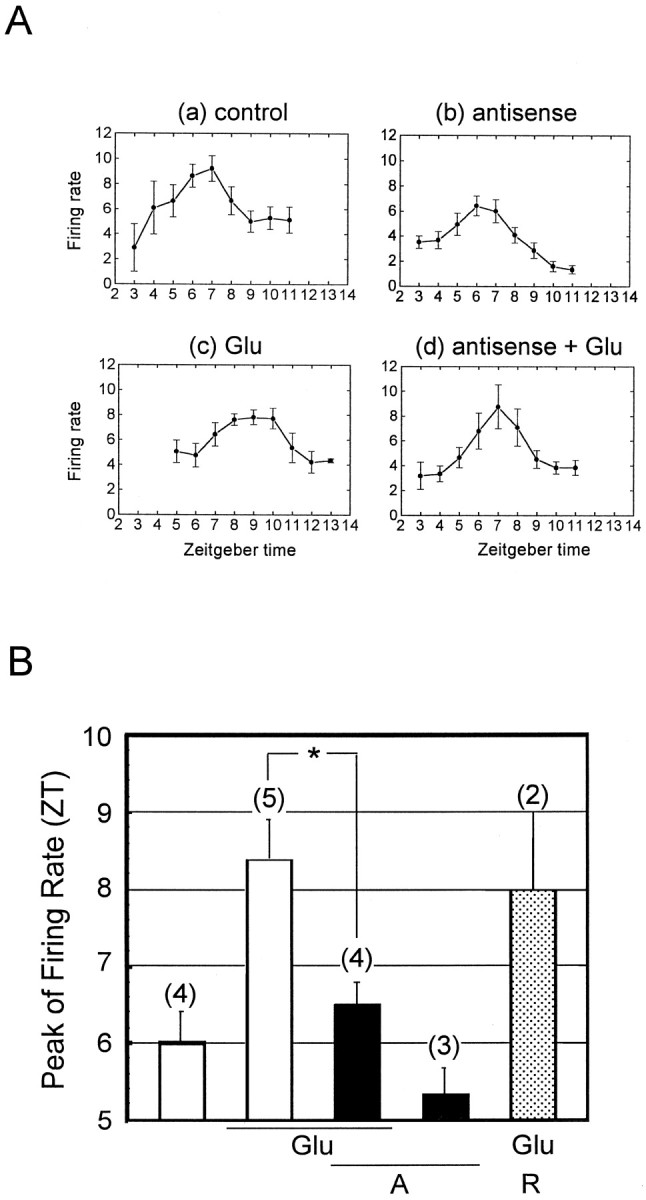
Effect of mPer1 antisense ODN on glutamate–induced phase delay of SCN firing rhythm in vitro. A, The average neuronal activity rhythms in the SCN recorded from mice slice on day 2. Each pointindicates the 2 hr means ± SEM of firing rate of single SCN cells from ZT2–14. B, Average phase shifts induced by glutamate and glutamate plus mPer1 antisense ODN. Eachbar indicates the peak of firing rate (mean ± SEM). Numbers in parentheses indicate the number of slices. Preincubation of mPer1 antisense ODN significantly reduced glutamate-induced phase shift (*p < 0.05 vs glutamate alone; Student’st test). Glu, Glutamate;A, mPer1 antisense ODN; R,mPer1 random ODN.
Distribution of ODN in the brain and effect of antisense ODN on mRNA
Distribution of antisense ODN was examined by injection and staining of biotinylated ODN in the fixed slice section of the brain. The ODN were most extensively distributed around the third ventricle, including the SCN (Fig.4A). Inhibition ofmPer1 induction by mPer1 antisense ODN in the SCN 1.5 hr after light exposure at CT16 was examined by RT-PCR. Before amplification, RNA were preliminarily tested for possible genomic DNA contamination. Gel analysis showed bands of expected lengths. Light exposure at CT16 induced expression of mPer1 mRNA (180 ± 24% of nonlight group; n = 4) and mPer2mRNA (160 ± 25%; n = 4) 90 min after light pulse. Light induction of mPer1 mRNA was considerably inhibited when 4 or 6 nmol of mPer1 antisense ODN was administrated (Fig. 4B,C). Administration of 6 nmol of mPer1 antisense significantly reduced the expression of mPer1 mRNA (68 ± 8.7% of random ODN treatment; n = 4; p < 0.05; Student’s t test) but not that of mPer2 mRNA (99 ± 11% of random ODN treatment; n = 4;p > 0.05; Student’s t test). This result suggests that phenotypic effects of mPer1 antisense ODN treatment on light-induced phase delay are mediated by the specific inhibition of mPer1 expression in the SCN.
Fig. 4.

Effects of mPer1antisense ODN on the mPer1 expression in the SCN.A, Distribution of biotinylated ODN 2 hr after injection into the brain. mPer1 antisense ODN (5′-biotnylated; 6 nmol in 5 ul) was microinjected intracerebroventricularly. Mice were killed 2 hr later, followed by detection of biotinylated ODN. Anarrow on the top slice shows antisense ODN injection site. An arrow on thebottom slice shows the position of SCN. The ODNs were most extensively distributed around the third ventricle including the SCN. B, Inhibition of light induction ofmPer1 transcript in the SCN by in vivo mPer1 antisense ODN treatment. Total RNA was isolated 1.5 hr after light exposure from mPer1 antisense ODN-pretreated mice, and mPer1, mPer2, and GAPDH RNA were amplified by an RT-PCR method. Lane 1, Treated with vehicle; lane 2, treated with 2 nmol of antisense ODN;lane 3, treated with 4 nmol of ODN; lane 4, treated with 6 nmol of antisense ODN. The PCR products ofmPer1, mPer2, and GAPDH gene are indicated by arrows. C, Semiquantitative analysis of RT-PCR products shown in B. The band intensity of RT-PCR products of mPer1 andmPer2 mRNA was measured by one-dimensional analysis software (Eastman Kodak), and their amounts were normalized against GAPDH.
DISCUSSION
Administration of mPer1 antisense ODN at CT1, but not at other CTs, significantly delayed the locomotor rhythm of mice. Injection of vehicle, mPer1 sense ODN, or mPer1random ODN at CT1 had little effect. It is currently believed that circadian oscillators, including those in mammals, are comprised of transcription/translation-based negative feedback loops controlled by clock genes (Hall, 1998; Reppert, 1998; Young, 1998). Peripheral or intra-SCN injections of translation inhibitors such as anisomycin or cycloheximide at early subjective days (CT1–4) have been reported to produce a phase delay in wheel-running rhythms (Takahashi and Turek, 1987; Inouye et al., 1988). Transcript of mPer1 is endogenously rhythmic with a consistent peak of expression in the subjective day at CT4 (Tei et al., 1997). We observed that the injection of mPer1 antisense ODN 4 hr before the light pulse did not block the light-induced phase delay of locomotor rhythm (data not shown). Therefore, the largest reduction of mPer1expression by antisense ODN might occur when antisense ODN is injected 2–3 hr before the mPer1 peak and may be the reason why antisense ODN delayed the circadian rhythm only at CT1.
In this study, we demonstrated that blockade of acute mPer1induction after light exposure by antisense ODN prevents the light-induced phase shifts of the circadian activity rhythm. This block in light-induced phase shift was caused by selective inhibition ofmPer1 induction, because mPer1 antisense ODN alone did not interfere with the free-running rhythm at CT16. Moreover, it is interesting that both mPer1 mRNA expression and phase delay of locomotor activity induced by light at CT16 were reduced by 4 nmol of mPer1 antisense ODN but not by 2 nmol. Thus, we observed the parallel reduction of mPer1 expression and phase delay. In the present experiment, 4 and 6 nmol ofmPer1 antisense ODN reduced to 60–70% of mPer1RNA expression induced by light exposure. Although we do not detect the protein production of mPer1 after light exposure, we can estimate 30–40% reduction of mPer1 mRNA may affect the light-induced phase changes in mouse behavior. Present results suggest that the reduction of light-induced phase delay by antisense ODNin vivo is a result of the inhibition of light-induced acute induction of mPer1 gene in the SCN. Further experiments are needed to locate the specific region of antisense ODN action (for example, direct antisense ODN injection into the SCN or immunostaining of mPer1 antibody there).
Transcript of mPer1 is rapidly induced by light in a time-of-day-dependent manner (Shigeyoshi et al., 1997). The responsiveness of mPer1 mRNA to light is gated so that little or no increase was seen during the subjective day, whereas robust induction was seen during the subjective night. Gating is also present in light-induced phase shifts of behavioral rhythm. Their dose and threshold is closely correlated with mPer1 inducibility in the SCN. These results with our present results suggest thatmPer1 plays a central role in the circadian clock.mPer2 gene was also shown to be induced by light but in a delayed manner compared with mPer1 (Takumi et al., 1998b), possibly reflecting a different regulatory mechanism. Recently,mPer3 has been isolated and shown not to be light inducible (Takumi et al., 1998a; Zylka et al., 1998), suggesting thatmPer genes have different roles in the light-induced phase shift. Therefore, injection of mPer2 or mPer3antisense ODN or cocktails containing antisense ODNs of mPergenes may be a good strategy for determining the roles of these genes.
To exclude the possibility that other regions of the brain might have added to the effects of mPer1 antisense ODN treatment, we examined the neural rhythm of SCN using slice culture. Administration of mPer1 antisense ODN blocked the glutamate-induced phase delay of the SCN circadian firing rhythm. Thus, glutamate-induced phase shifts may be involved in the expression of mPer1 mRNA in the SCN. SCN is entrained to the environmental light/dark cycle via a retinal projection, the retinohypothalamic tract (RHT). Glutamate is a transmitter of the RHT (de Vries et al., 1993). Glutamate and NMDA application to rat SCN in vitro have been reported to cause phase delays in SCN firing rhythms when applied at early subjective night (Shibata et al., 1994; Shirakawa and Moore, 1994). Furthermore, glutamate receptor antagonists and inhibitors of nitric oxide synthase, calmodulin, or calcium calmodulin kinase II antagonize phase shifts in the SCN firing rhythm induced by glutamate or NMDA in vitro(Shibata et al., 1994; Watanabe et al., 1994; Fukushima et al., 1997). Therefore, we cannot rule out the possibility that mPer1antisense ODN interferes with these biochemical steps. However, the sequence specificity of the ODNs on light- or glutamate-induced phase delay strongly suggest this is not the case.
Light-induced phase shifts of circadian rhythms induce immediately early genes (IEGs) such as c-fos, junB, and NGFI-A mRNAs specifically in the SCN (Rusak et al., 1990; Morris et al., 1998). Blockade of expression of c-fos or Jun B expression in the SCN has been shown to inhibit light-induced phase shifts in mammalian circadian clocks (Wollnik et al., 1995). These proteins are believed to dimerize and bind to AP-1, which are CRE/CaRE consensus sequences that are present in the promoters of many genes (Takeuchi et al., 1993). The light-induced induction of IEGs is also gated asmPer1 and mPer2. The time courses of c-fos andmPer1 mRNA induction are similar, but it is unknown whether c-fos protein is involved in transcription of mPer1 (ormPer2) or the induction of the c-fos and mPer are simultaneous.
In this study, we used antisense ODN as pharmacological tools to inhibit mPer1 expression in vivo and in vitro. The mechanism of inhibition of physiological systems by antisense ODNs is believed to be the result of specific hybridization of the antisense ODN to its complementary mRNA, causing disruption of the translation of the mRNA into protein (Talamo, 1998). We have not determined the amounts of mPer1 protein expression, because we have not obtained anti-mPer1 antibody. Antisense ODN is also believed to bind to the genomic DNA region of the corresponding gene and inhibit binding of transcription factors and to bind to mRNA and accelerate degradation of targeted mRNA by RNaseH (Kashihara et al., 1998). Both of these mechanisms should lower the level of mRNA. These effects may be sequence-specific; arising from inhibition of imperfectly matched target genes, or sequence-independent effects on gene expression. Antisense ODNs may also effect nontargeted genes or even be toxic to physiological systems (Talamo, 1998). In the present study, we showed that mPer1 mRNA in the SCN was reduced by treatment withmPer1 antisense ODN, but treatment did not affectmPer2 and GAPDH mRNA levels, demonstrating that the antisense ODN used in this study specifically effects onlymPer1 gene expression. We used phosphotioate-substituted ODNs, which have longer biological half-lives than unsubstituted ODNs but may be toxic (Agrawal et al., 1991). In this study, some animals exhibited altered locomotor activity for the first several hours after injection. However, this effect was observed in both mPer1antisense ODN-injected animals and control ODNs-injected animals, suggesting this change is caused by a toxic effect of the administration of ODNs. In all cases, locomotor activity was restored to normal under constant darkness. In our previous experiments (Ono et al., 1996, Watanabe et al., 1996), methamphetamine and adenosine antagonists inhibited the light-induced phase shift, although these chemicals increase or decrease motor activity, respectively. Thus, the circadian oscillator may be unaffected by ODN injection. We also demonstrated that ODNs injected intracerebroventricularly were distributed in specific regions of the brain after 2 hr, especially around the third ventricle including the SCN. However, other regions of the brain might have added to the effects of mPer1 antisense ODN treatment.
In summary, the present results indicate that acute induction ofmPer1 mRNA after light exposure is necessary for light-induced phase shifting of the mouse locomotor rhythm. Further genetic dissection of mPer genes, possibly with knock-out mice is useful to identify the role of these genes in detail.
Footnotes
This study was partially supported by grants to S.S. from Research Project for Future Program (RFTF96L00310), from the Japan Society for the Promotion of Science, and from the Japanese Ministry of Education, Science, Sports and Culture (09470018).
Correspondence should be addressed to Shigenobu Shibata, Department of Pharmacology and Brain Science, School of Human Sciences, Waseda University, Tokorozawa, Saitama 359–1192, Japan. E-mail:shibata@human.waseda.ac.jp
REFERENCES
- 1.Agrawal S, Temsamani J, Tang JY. Pharmacokinetics, biodistribution, and stability of oligodeoxynucleotide phosphorothioates in mice. Proc Natl Acad Sci USA. 1991;88:7595–7599. doi: 10.1073/pnas.88.17.7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albrecht U, Sun ZS, Eichele G, Lee CC. A differential response of two putative mammalian circadian regulators, mPer1 and mPer2, to light. Cell. 1997;91:1055–1064. doi: 10.1016/s0092-8674(00)80495-x. [DOI] [PubMed] [Google Scholar]
- 3.Colwell CS, Ralph MR, Menaker M. Do NMDA receptors mediate the effects of light on circadian behavior? Brain Res. 1990;523:117–120. doi: 10.1016/0006-8993(90)91643-u. [DOI] [PubMed] [Google Scholar]
- 4.Daan S, Pittendrigh C. A functional analysis of circadian pacemakers in nocturnal rodents II. The variability of phase response curves. J Comp Physiol [A] 1976;106:253–266. [Google Scholar]
- 5.de Vries MJ, Nunes Cardozo B, van der Want J, de Wolf A, Meijer JH. Glutamate immunoreactivity in terminals of the retinohypothalamic tract of the brown Norwegian rat. Brain Res. 1993;612:231–237. doi: 10.1016/0006-8993(93)91665-f. [DOI] [PubMed] [Google Scholar]
- 6.Edmunds LN. Cellular and molecular basis of biological clocks. Springer; New York: 1988. [Google Scholar]
- 7.Fukushima T, Shimazoe T, Shibata S, Watanabe A, Ono M, Hamada T, Watanabe S. The involvement of calmodulin and Ca2+/calmodulin-dependent protein kinase II in the circadian rhythms controlled by the suprachiasmatic nucleus. Neurosci Lett. 1997;227:45–48. doi: 10.1016/s0304-3940(97)00297-8. [DOI] [PubMed] [Google Scholar]
- 8.Hall JC. Genetics of biological rhythms in Drosophila. Adv Genet. 1998;38:135–184. doi: 10.1016/s0065-2660(08)60143-1. [DOI] [PubMed] [Google Scholar]
- 9.Hastings MH. Central clocking. Trends Neurosci. 1997;20:459–464. doi: 10.1016/s0166-2236(97)01087-4. [DOI] [PubMed] [Google Scholar]
- 10.Inouye ST, Takahashi JS, Wollnik F, Turek FW. Inhibitor of protein synthesis phase shifts a circadian pacemaker in mammalian SCN. Am J Physiol. 1988;255:R1055–R1058. doi: 10.1152/ajpregu.1988.255.6.R1055. [DOI] [PubMed] [Google Scholar]
- 11.Kashihara N, Maeshima Y, Makino H. Antisense oligonucleotides. Exp Nephrol. 1998;6:84–88. doi: 10.1159/000020509. [DOI] [PubMed] [Google Scholar]
- 12.Konopka RJ, Benzer S. Clock mutants of Drosophila melanogaster. Proc Natl Acad Sci USA. 1971;68:2112–2116. doi: 10.1073/pnas.68.9.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morris ME, Viswanathan N, Kuhlman S, Davis FC, Weitz CJ. A screen for genes induced in the suprachiasmatic nucleus by light. Science. 1998;379:1544–1547. doi: 10.1126/science.279.5356.1544. [DOI] [PubMed] [Google Scholar]
- 14.Ono M, Watanabe A, Matsumoto Y, Fukushima T, Nishikawa Y, Moriya T, Shibata S, Watanabe S. Methamphetamine modifies the photic entraining responses in the rodent suprachiasmatic nucleus via serotonin release. Neuroscience. 1996;72:213–224. doi: 10.1016/0306-4522(95)00500-5. [DOI] [PubMed] [Google Scholar]
- 15.Reppert SM. A clockwork explosion! Neuron. 1998;21:1–4. doi: 10.1016/s0896-6273(00)80234-2. [DOI] [PubMed] [Google Scholar]
- 16.Rusak B, Robertson HA, Wisden W, Hunt SP. Light pulses that shift rhythms induce gene expression in the suprachiasmatic nucleus. Science. 1990;248:1237–1240. doi: 10.1126/science.2112267. [DOI] [PubMed] [Google Scholar]
- 17.Shearman LP, Zylka MJ, Weaver DR, Kolakowski LF, Jr, Reppert SM. Two period homologs: circadian expression and photic regulation in the suprachiasmatic nuclei. Neuron. 1997;19:1261–1269. doi: 10.1016/s0896-6273(00)80417-1. [DOI] [PubMed] [Google Scholar]
- 18.Shibata S, Moore RY. Neuropeptide Y and optic chiasm stimulation affect suprachiasmatic nucleus circadian function in vitro. Brain Res. 1993;615:95–100. doi: 10.1016/0006-8993(93)91118-c. [DOI] [PubMed] [Google Scholar]
- 19.Shibata S, Tsuneyoshi A, Hamada T, Tominaga K, Watanabe S. Effect of substance P on circadian rhythms of firing activity and the 2-deoxyglucose uptake in the rat suprachiasmatic nucleus in vitro. Brain Res. 1992;597:257–263. doi: 10.1016/0006-8993(92)91482-t. [DOI] [PubMed] [Google Scholar]
- 20.Shibata S, Watanabe A, Hamada T, Ono M, Watanabe S. N-methyl-d-aspartate induces phase shifts in circadian rhythm of neuronal activity of rat SCN in vitro. Am J Physiol. 1994;267:R360–R364. doi: 10.1152/ajpregu.1994.267.2.R360. [DOI] [PubMed] [Google Scholar]
- 21.Shigeyoshi Y, Taguchi K, Yamamoto S, Takekida S, Yan L, Tei H, Moriya T, Shibata S, Loros JJ, Dunlap JC, Okamura H. Light-induced resetting of a mammalian circadian clock is associated with rapid induction of the mPer1 transcript. Cell. 1997;91:1043–1053. doi: 10.1016/s0092-8674(00)80494-8. [DOI] [PubMed] [Google Scholar]
- 22.Shirakawa T, Moore RY. Glutamate shifts the phase of the circadian neuronal firing rhythm in the rat suprachiasmatic nucleus in vitro. Neurosci Lett. 1994;178:47–50. doi: 10.1016/0304-3940(94)90286-0. [DOI] [PubMed] [Google Scholar]
- 23.Sun ZS, Albrecht U, Zhuchenko O, Bailey J, Eichele G, Lee CC. RIGUI, a putative mammalian ortholog of the Drosophila period gene. Cell. 1997;19:1003–1011. doi: 10.1016/s0092-8674(00)80366-9. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi JS, Turek FW. Anisomycin, an inhibitor of protein synthesis, perturbs the phase of a mammalian circadian pacemaker. Brain Res. 1987;405:199–203. doi: 10.1016/0006-8993(87)91010-9. [DOI] [PubMed] [Google Scholar]
- 25.Takeuchi J, Shannon W, Aronin N, Schwartz WJ. Compositional changes of AP-1 DNA-binding proteins are regulated by light in a mammalian circadian clock. Neuron. 1993;11:825–836. doi: 10.1016/0896-6273(93)90112-5. [DOI] [PubMed] [Google Scholar]
- 26.Takumi T, Taguchi K, Miyake S, Sakakida Y, Takashima N, Matsubara C, Maebayashi Y, Okumura K, Takekida S, Yamamoto S, Yagita K, Yan L, Young MW, Okamura H. A light-independent oscillatory gene mPer3 in mouse SCN and OVLT. EMBO J. 1998a;17:4753–4759. doi: 10.1093/emboj/17.16.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takumi T, Matsubara C, Shigeyoshi Y, Taguchi K, Yagita K, Maebayashi Y, Sakakida Y, Okumura K, Takashima N, Okamura H. A new mammalian period gene predominantly expressed in the suprachiasmatic nucleus. Genes Cells. 1998b;3:167–176. doi: 10.1046/j.1365-2443.1998.00178.x. [DOI] [PubMed] [Google Scholar]
- 28.Talamo BR. Introduction: antisense oligonucleotides: strategies and successes. Chem Senses. 1998;23:243–247. doi: 10.1093/chemse/23.2.243. [DOI] [PubMed] [Google Scholar]
- 29.Tei H, Okamura H, Shigeyoshi Y, Fukuhara C, Ozawa R, Hirose M, Sakaki Y. Circadian oscillation of a mammalian homologue of the Drosophila period gene. Nature. 1997;389:512–516. doi: 10.1038/39086. [DOI] [PubMed] [Google Scholar]
- 30.Watanabe A, Hamada T, Shibata S, Watanabe S. Effects of nitric oxide synthase inhibitors on N-methyl-d-aspartate-induced phase delay of circadian rhythm of neuronal activity in the rat suprachiasmatic nucleus in vitro. Brain Res. 1994;646:161–164. doi: 10.1016/0006-8993(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 31.Watanabe A, Moriya T, Nishikawa Y, Arakito, Hamada T, Shibata S, Watanabe S. Adenosine A1-receptor agonist attenuates the light-induced phase shifts and fos expression in vivo and optic nerve stimulation-evoked field potentials in the suprachiasmatic nucleus in vitro. Brain Res. 1996;740:329–336. doi: 10.1016/s0006-8993(96)00881-5. [DOI] [PubMed] [Google Scholar]
- 32.Wollnik F, Brysch W, Uhlmann E, Gillardon F, Bravo R, Zimmermann M, Schlingensiepen KH, Herdegen T. Block of c-Fos and JunB expression by antisense oligonucleotides inhibits light-induced phase shifts of the mammalian circadian clock. Eur J Neurosci. 1995;7:388–393. doi: 10.1111/j.1460-9568.1995.tb00334.x. [DOI] [PubMed] [Google Scholar]
- 33.Young MW. The molecular control of circadian behavioral rhythms and their entrainment in Drosophila. Annu Rev Biochem. 1998;67:135–152. doi: 10.1146/annurev.biochem.67.1.135. [DOI] [PubMed] [Google Scholar]
- 34.Zylka MJ, Shearman LP, Weaver DR, Reppert SM. Three period homologs in mammals: differential light responses in the suprachiasmatic circadian clock and oscillating transcripts outside of brain. Neuron. 1998;20:1103–1110. doi: 10.1016/s0896-6273(00)80492-4. [DOI] [PubMed] [Google Scholar]