

Abstract
Neuronal intranuclear inclusions are found in the brains of patients with Huntington’s disease and form from the polyglutamine-expanded N-terminal region of mutant huntingtin. To explore the properties of inclusions and their involvement in cell death, mouse clonal striatal cells were transiently transfected with truncated and full-length human wild-type and mutant huntingtin cDNAs. Both normal and mutant proteins localized in the cytoplasm, and infrequently, in dispersed and perinuclear vacuoles. Only mutant huntingtin formed nuclear and cytoplasmic inclusions, which increased with polyglutamine expansion and with time after transfection. Nuclear inclusions contained primarily cleaved N-terminal products, whereas cytoplasmic inclusions contained cleaved and larger intact proteins. Cells with wild-type or mutant protein had distinct apoptotic features (membrane blebbing, shrinkage, cellular fragmentation), but those with mutant huntingtin generated the most cell fragments (apoptotic bodies). The caspase inhibitor Z-VAD-FMK significantly increased cell survival but did not diminish nuclear and cytoplasmic inclusions. In contrast, Z-DEVD-FMK significantly reduced nuclear and cytoplasmic inclusions but did not increase survival. A series of N-terminal products was formed from truncated normal and mutant proteins and from full-length mutant huntingtin but not from full-length wild-type huntingtin. One prominent N-terminal product was blocked by Z-VAD-FMK. In summary, the formation of inclusions in clonal striatal cells corresponds to that seen in the HD brain and is separable from events that regulate cell death. N-terminal cleavage may be linked to mutant huntingtin’s role in cell death.
Keywords: NH2-terminal huntingtin fragments, nuclear inclusions, cytoplasmic inclusions, full-length huntingtin, apoptosis, apoptotic bodies, membrane blebbing, Z-VAD-FMK, Z-DEVD-FMK, striatal hybrid cells
The genetic mutation in Huntington’s disease (HD) is a polyglutamine expansion in the N-terminal region of huntingtin (Huntington’s Disease Collaborative Research Group, 1993). The mechanism of HD pathogenesis is unclear. Recent studies in HD postmortem brain tissue show that the N-terminal region of mutant huntingtin aggregates in nuclear inclusions (NI) in cortical and striatal neurons (DiFiglia et al., 1997; Becher et al., 1998). The appearance of NI in regions most affected in the HD brain implicates the aggregation of huntingtin in the pathogenesis of HD. In support of this idea, HD transgenic mice expressing exon 1 with a highly expanded CAG repeat domain develop NI before the onset of a clinical phenotype (Davies et al., 1997; Scherzinger et al., 1997) and before a reduction in mRNAs for neurotransmitter receptors in the striatum and cortex (Cha et al., 1998). Inclusions also appear in patients with other expanded CAG repeat disorders including spinocerebellar ataxia 1 (SCA-1; Skinner et al., 1997), spinocerebellar ataxia 3 (SCA-3; Machado-Joseph’s disease; Paulson et al., 1997), and dentatopallidoluysian atrophy (DRPLA; Becher et al., 1998), indicating that a common mechanism may contribute to the formation of these structures. Despite these interesting observations, the involvement of inclusions in cell death remains unclear.
Proteins with expanded polyglutamines, when introduced into non-neuronal cells in vitro, produce minimal or no NI, but form aggregates in the cytoplasm when expressed from truncated transcripts (Paulson et al., 1997; Skinner et al., 1997; Igarashi et al., 1998; Martindale et al., 1998). Nuclear and cytoplasmic aggregates have been observed in non-neuronal cells after the expression of small N-terminal mutant huntingtin fragments, but not after the expression of large fragments or full-length huntingtin (Cooper et al., 1998; Hackam et al., 1998). These results suggest that the intrinsic properties required to generate N-terminal products from larger proteins may be cell-specific. Activated caspases cleave normal and mutant huntingtins near the NH2 terminus (Goldberg et al., 1996; Wellington et al., 1998), and their presence may be needed to form inclusions from full-length mutant huntingtin.
Here we report the effects of transient expression in an immortalized striatal cell line of partial and full-length huntingtin with normal and expanded polyglutamine regions. Expression of normal or mutant proteins occurred mainly in the cytoplasm, produced apoptotic effects, and generated N-terminal huntingtin fragments in Western blots. Mutant huntingtin formed NI and CI, which were reduced by the caspase inhibitor Z-DEVD-FMK without altering survival. The inhibitor, Z-VAD-FMK, had no effect on the formation of inclusions but markedly increased survival and blocked the cleavage of a prominent N-terminal product. We speculate that inclusions formed by mutant huntingtin are independent of events involved with cell death.
MATERIALS AND METHODS
Construction of expression plasmids. cDNA transcripts containing the 5 prime one third (3221 bases for wild-type) of huntingtin or the full-length cDNA (9774 for wild-type) were constructed with 18, 46, and 100 CAG repeats. The 100 CAG repeat sequence was interrupted by a CGG between the 28th and 30th CAG triplets. The truncated transcripts included putative sites of caspase cleavage. Cloned cDNAs were verified by restriction digestion and sequencing. The cDNA for the FLAG epitope was placed 5 prime to that of huntingtin, and the transcripts were cloned into the mammalian expression vector pcDNA3 (Invitrogen, San Diego, CA) and identified as FH3221-18, -46, and -100, and FH9774-18, -46, and -100. The pcDNA3 vectors containing truncated huntingtin with normal and expanded (100) polyglutamines without the FLAG epitope (H3221-18 and -100) were also made. In another set of expression plasmids, the shorter huntingtin transcript (3221 bases) was cloned into pEGFP-N1 (Clontech, Palo Alto, CA) in frame with a 3 prime transcript for green fluorescent protein (GFP). These expression plasmids are identified as H3221GFP18, 46, and 100. The cytomegalovirus promoter drives protein expression in pcDNA3 and pEGFP-N1. The respective plasmids with FLAG or GFP only were used as controls in some experiments.
Cell culture. Clonal striatal cells (X57 cell line) derived by somatic cell fusion of embryonic day 18 mouse striatal neurons and neuroblastoma cells (N18TG2; Wainwright et al., 1995) were grown on untreated glass coverslips. A mouse-derived mesenchephalic dopaminergic cell line (MN9D; Choi et al., 1991) was also used.
In vitro transfection and experimental procedures.Transfections were performed with Lipofectamine (2.5 μg/35 mm dish; Life Technologies, Gaithersburg, MD). Mock transfections omitted the plasmid DNA and were performed as controls. For treatment with caspase inhibitors or transglutaminase inhibitor, cells were grown in 35 mm culture dishes to 60–80% confluence and treated with Lipofectamine and FH3221-18 or -100 (2.5 μg) for 6 hr. Cells were harvested and plated onto glass coverslips and treated with the cell-permeable caspase inhibitors Z-VAD-FMK (5–100 μm) or Z-DEVD-FMK (50–200 μm) (obtained from Enzyme Systems Products, Livermore, CA) or the transglutaminase inhibitor cystamine (0.2–1 mm) continuously for 3 d. Fresh inhibitor was replaced with each feeding every 24 hr. Cells were washed in PBS and fixed and processed for FLAG immunofluorescence. The concentrations of the caspase inhibitors Z-VAD-FMK and Z-DEVD-FMK were in the range used by other investigators to examine effects on survival of neuronal and non-neuronal cells in culture (Eldadah et al., 1997; MacFarlane et al., 1997; McCarthy et al., 1997; D’Mello et al., 1998).
Immunohistochemistry. Transfected striatal hybrid cells grown on glass coverslips were rinsed briefly with PBS and fixed for 20 min with 4% paraformaldehyde in PBS at room temperature. After fixation, cells were rinsed multiple times and stored at 4°C in PBS. Cells were made permeable by a 60 min incubation in 0.2% Triton X-100 in PBS and were treated with 4% normal goat serum before incubation in primary antibodies against FLAG (M5 monoclonal antibody; Eastman Kodak, Rochester, NY), huntingtin (polyclonal Ab 1, Ab 585, Ab 2527; 1–4 μg/ml; DiFiglia et al., 1995; Velier et al., 1998), or bromodeoxyuridine (BrdU; monoclonal; Boehringer Mannheim, Indianapolis, IN). For studies of BrdU incorporation, cells were incubated in 0.2 NHCl at room temperature for 1 hr after fixation. All single or combined incubations of primary antisera were overnight at 4°C. Secondary antibody was used at a 1:400 dilution and produced no labeling in the absence of primary antibody. Plasmids containing the cDNAs for FLAG or GFP only produced no labeling in cells at the DNA concentrations used for transfection (2.5 μg/35 mm culture dish). For FLAG immunofluorescence, Bodipy FL anti-mouse IgG was used (Molecular Probes, Eugene, OR). For double labeling with anti-BrdU, sections were treated sequentially in Cy3 anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA) and anti-BrdU-fluorescein (Boehringer Mannheim). Sections were examined using conventional immunofluorescence microscopy and a Bio-Rad (Hercules, CA) 1024 laser confocal microscope. Image processing was performed with Adobe Photoshop.
Western blot assays. Total protein extracts from the transfected cells were detected in Western blot assay with anti-huntingtin antisera Ab 1 as previously described (DiFiglia et al., 1995).
RESULTS
Patterns of huntingtin localization
Wild-type or mutant huntingtin localized diffusely to the cytoplasm in the majority of cells (X57) studied 1–3 d after transfection, regardless of polyglutamine length or length of huntingtin (Fig. 1, Table1). Among cells expressing mutant huntingtin in the cytoplasm were some with labeling also in the nucleus (Figs. 1, 2d). In 1–10% of neurons, wild-type and mutant huntingtins were heavily concentrated in irregular or tubular-shaped cytoplasmic vacuoles independent of polyglutamine length in huntingtin. The vacuoles were distributed ubiquitously in the cell body (dispersed vacuoles) or as a complex of interconnected tubules in the perinuclear region (perinuclear vacuoles) and were more frequent with the truncated than the full-length proteins. Cells with perinuclear vacuoles were significantly smaller in cross-sectional area than nontransfected cells in the same culture dishes (p < 0.05; n = 50 labeled and unlabeled neurons in cultures expressing wild-type or mutant proteins), suggesting that they were undergoing apoptosis.
Fig. 1.

Patterns of localization of FLAG–huntingtin fusion proteins in clonal striatal neurons 1–3 d after transfection of FH3221 with 18, 46, or 100 glutamines. Diffuse cytoplasmic labeling, dispersed vacuoles, and condensed perinuclear vacuoles occur regardless of polyglutamine length. Neurons with mutant huntingtin show some diffuse nuclear labeling and nuclear and cytoplasmic (“ring-like”) inclusions (arrows).
Table 1.
Patterns of wild-type and mutant huntingtin localization
| Pattern of FLAG staining | #CAGs | 18 | 46 | 100 | |||
|---|---|---|---|---|---|---|---|
| Huntingtin | Truncateda | Full-lengthb | Truncatedc | Full-lengthd | Truncatede | Full-lengthf | |
| Diffuse cytoplasmic | Day 1 | 88.5 | 92.2 | 94.3 | 92.3 | 89.4 | 96.7 |
| Day 3 | 96.7 | 100 | 94.6 | 100 | 94.9 | 88.8 | |
| Dispersed vacuoles | Day 1 | 3.3 | 0.4 | 3.8 | 0.3 | 1.4 | 0 |
| Day 3 | 3.3 | 0 | 1.4 | 0 | 0.9 | 0 | |
| Perinuclear vacuoles | Day 1 | 8.2 | 7.4 | 1.3 | 0 | 7.2 | 2.2 |
| Day 3 | 0 | 0 | 2.7 | 0 | 0.2 | 0 | |
| Nuclear inclusions | Day 1 | 0 | 0 | 0.6 | 0 | 0.7 | 1.1 |
| Day 3 | 0 | 0 | 1.4 | 0 | 2.6 | 10.3 | |
| Cytoplasmic inclusions | Day 1 | 0 | 0 | 0 | 0 | 1.4 | 0 |
| Day 3 | 0 | 0 | 0 | 0 | 1.4 | 0.9 | |
Number of cells: a1481 (Day 1); 361 (Day 3) b213 (Day 1); 63 (Day 3)c2578 (Day 1); 370 (Day 3)d311 (Day 1); 66 (Day 3)e4032 (Day 1); 1312 (Day 3)f349 (Day 1); 95 (Day 3).
Fig. 2.

Features of NI and CI in clonal striatal and midbrain neurons. The inclusions were formed by FH3221-100 (a–c, e) or FH9774-100 (d). Inclusions in striatal cells appear more consolidated and intensely FLAG-positive (a–d) than in the midbrain neuron (e). Striatal cell ina has three small and two large NI. Cells inb and d have both a nuclear and a cytoplasmic inclusion. Cell in c shows retraction of cytoplasm away from the cytoplasmic inclusion. All cells have diffuse cytoplasmic labeling, and cell in d has diffuse nuclear labeling.
Nuclear inclusions (NI; 0.7–10% of neurons) or cytoplasmic inclusions (CI; 0.9–1.4% of neurons) formed from the shortened (Figs. 1,2a–c) or full-length huntingtin (Fig.2d) using FH3221-100 and FH9774-100, respectively. NI also developed in 0.6–1.4% of neurons using FH3221-46 (Fig. 1), but they were not found after 3 d with the full-length cDNA expressed by FH9774-46 (Table 1). Similar to HD brain, the NI were spherical or ovoid and occupied 10–30% of the cross-sectional area of the nucleus. NI developed in clonal midbrain dopaminergic neurons (Wainwright et al., 1995) with FH3221-100 (Fig. 2e), but the NI were less intensely labeled than those found in the striatal cells. Between days 3 and 4, the rate of cell proliferation and the loss of FLAG-positive cells varied for different transfections and affected the percent of cells with inclusions at these time points. Nevertheless, ∼4–6 d after transfection, the percent of NI and CI induced by FH3221-100 began to increase, and by day six constituted 43 and 27% of the total labeled cells, respectively (Fig.3). The rise in cells with NI and CI overlapped a period of marked loss in the total FLAG-positive neurons. NI and CI occurred together in only ∼10% of labeled cells. Thus, by day six, 60% of the FLAG- positive neurons that remained in the cultures had some form of inclusion. CI typically occurred singly in cells, frequently near blebs in the plasma membrane (Figs.2c, 5e). CI were spherical and uniformly dense (Fig. 2b,c) or had clear centers, which gave them a distinct ring-like appearance (Figs. 1,4g). CI were distinct from dispersed and condensed perinuclear vacuoles in a number of ways. CI occurred singly in neurons and were uniformly spherical, whereas vacuoles were numerous, interconnected, irregularly shaped, or tubular.
Fig. 3.

Effects of the number of days after transfection on the formation of NI and CI. Cells were transfected with FH3221-100. Percent of total FLAG+ neurons with inclusions progressively increases between 4 and 6 d. Note that the total number of FLAG+ neurons (indicated over each set of bars) decreases markedly by 5–6 d after transfection. Results are shown for one transfection experiment. Results were similar in two other experiments.
Fig. 5.
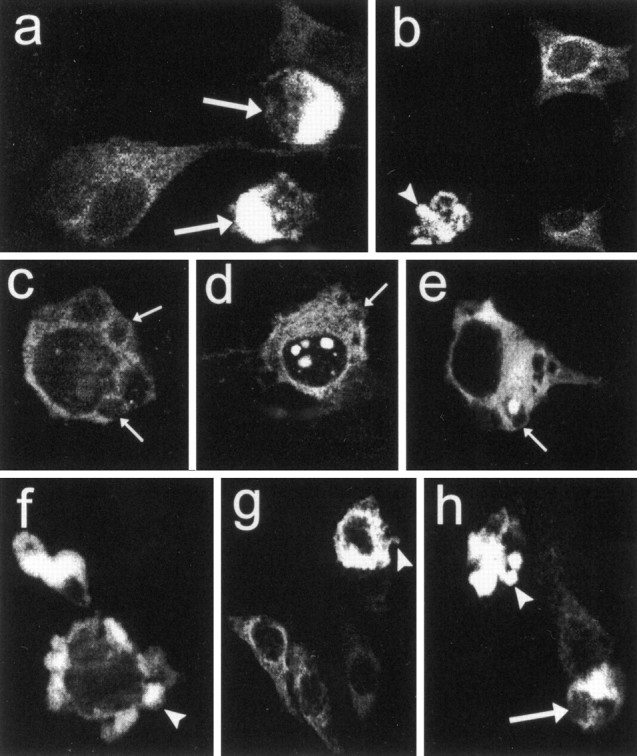
Features of apoptotic cell death in clonal striatal cells expressing wild-type (a, b) and mutant huntingtin (c–h). Expression plasmids were FH3221-18 in a and b, FH3221-100 in c–e, H3221GFP-100 in f, and FH9774-100 in g andh. Shrunken cells (a, h,large arrows), plasma membrane blebs (c–e, small arrows), and cellular fragmentation (b, f–h, arrowheads) were the most commonly seen apoptotic features. Note membrane blebs along the plasma membrane appear in neurons that have diffuse cytoplasmic FLAG labeling or in addition have NI (d) and CI (e).
Fig. 4.

Mutant huntingtin with 100 glutamines localized in clonal striatal cells. a–d show staining with anti-huntingtin antisera Ab 1 after transfection of H3221-100 (no epitope tag), ande–g show GFP after transfection of H3221GFP100 (tagged at the COOH terminus). Both constructs produce mainly diffuse labeling in the cytoplasm (a, e), some cells with perinuclear vacuoles (b, f), nuclear inclusions (c, e), and cytoplasmic inclusions (d, g). Note the ring-like structure of the cytoplasmic inclusion in g. The results indicate that the expression of mutant huntingtin alone or with a COOH-terminal GFP tag shares the same subcellular compartments as FLAG–huntingtin shown in Figures 1and 2.
The patterns of FLAG staining in striatal cells transfected with FH3221 constructs were also seen with Ab 1, which recognizes the NH2-terminus in huntingtin (results not shown). Ab 1 detected similar patterns of normal and mutant huntingtin localization after expression of H3221-18 and H3221-100 (Fig. 4a–d, shown for H3221-100), suggesting that the presence of the FLAG epitope at the NH2 terminus did not affect protein targeting. In neurons expressing FH3221, anti-huntingtin antisera Ab 585, which recognizes an epitope downstream of the polyglutamine tract in huntingtin, detected mainly diffuse cytoplasmic labeling in neurons. It also weakly recognized the cytoplasmic face of dispersed and perinuclear vacuoles that accumulated normal and mutant proteins. Ab 585 did not label NI or CI. Similarly, Ab 2527, which recognizes an epitope near the COOH-terminal, detected mainly diffuse cytoplasmic labeling in neurons and did not recognize vacuoles or inclusions after expression of FH9774. The limited recognition of vacuoles and inclusions by Ab 585 and Ab 2527 suggested that these compartments had accumulated mainly cleaved NH2 huntingtin products.
Neurons transfected with H3221 GFP-100 showed huntingtin–GFP localization comparable to FLAG–huntingtin 1 d after transfection (Fig. 4e,g). Among GFP-positive neurons (n = 7096 cells), 95% had diffuse cytoplasmic labeling 0.14% had NI, and 1.8% had CI. The presence of huntingtin–GFP within both nuclear and cytoplasmic inclusions suggested that the large, uncleaved form of mutant huntingtin was incorporated into aggregates. CI were identified about as frequently with the FLAG and GFP-tagged cDNAs. However, NI were labeled more frequently (5–18×) with the FLAG-tagged construct than the GFP-tagged cDNA. This suggested that NI incorporated mainly cleaved N-terminal fragments of mutant huntingtin. In an analysis of double-labeled cells (n = 161 cells) cotransfected with FLAG and GFP-tagged cDNAs, the NI (n = 7) detected had FLAG labeling but not GFP, indicating that in this small sampling of cells only a cleaved N-terminal fragment of mutant huntingtin had translocated to the nucleus. Vacuoles were also less frequently detected with the GFP-tagged construct (0.23% dispersed vacuoles and 2.4% perinuclear vacuoles) than with the FLAG-tagged cDNA (Table 1; range, 1.4–8.2%). These results provided additional support that the vacuoles accumulated primarily NH2 products of the normal and mutant proteins.
Apoptotic features in neurons expressing wild-type and mutant huntingtins
Apoptosis occurs in affected neurons of the HD brain (Portera-Cailliau et al., 1995). Morphological characteristics of apoptotic cell death including cell shrinkage, membrane blebs, and cellular fragmentation (Kerr et al., 1972) were evident in cells expressing partial and full-length wild-type or mutant huntingtins (Fig. 5). Shrunken cells had a marked accumulation of wild-type or mutant huntingtin in the cytoplasm (Fig.5a,h) and were seen under basal conditions and in cultures differentiated with forskolin or sodium butyrate. Severe membrane blebs (blisters) of the plasma membrane, a feature of early apoptotic cell death, occurred after normal or mutant protein expression. Membrane blebs were found in cells where mutant huntingtin appeared diffusely in the cytoplasm (Fig. 5c) or additionally formed NI and CI (Fig. 5c,d). An early stage of cellular fragmentation, indicated by protrusions from the cell surface, was seen with FLAG- (Fig.5b,g,h) or GFP-tagged (Fig. 5f) transcripts. Isolated cell fragments that contained vacuoles appeared when FH3221-18 or 46 were expressed (Fig. 6a,left). Cell fragments with ring-like inclusions were prevalent when FH3221-100 was transfected (Fig.6a, right) and were increased in proportion to total cell fragments 1–6 d after transfection (Fig. 6b) in parallel with the rise in CI in neurons (see Fig. 3). The ratio of cell fragments to neurons was significantly greater in cultures transfected with FH3221-100 than in cultures treated with FH3221-18 or -46 (Fig. 6c).
Fig. 6.
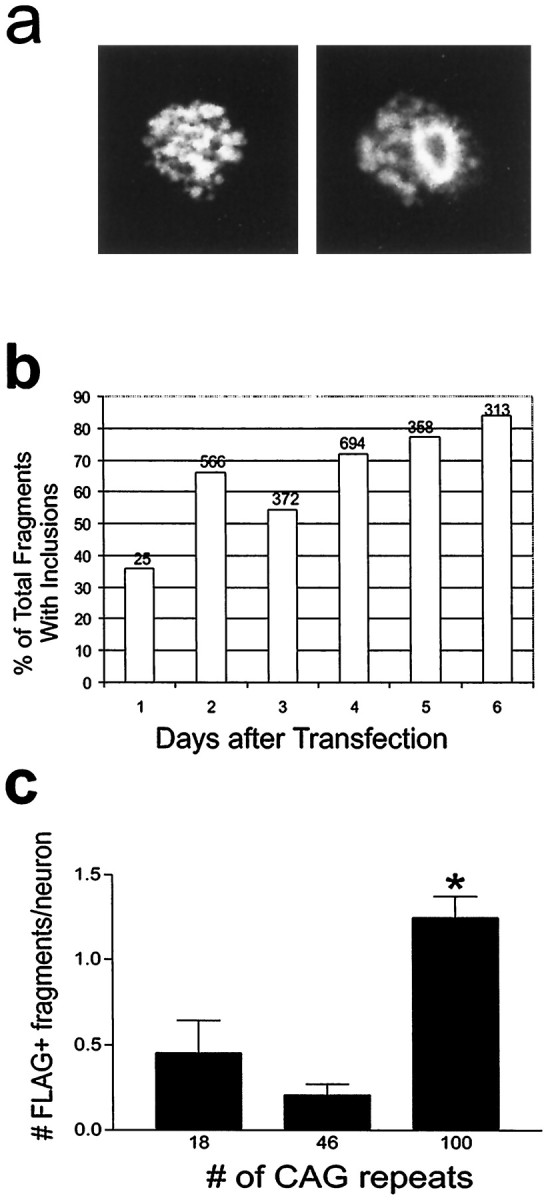
Analysis of apoptotic cell fragments.a, Cell fragments with vacuoles (left) and ring-like inclusions (right). b, Cell fragments with ring-like inclusions increase in proportion to total cell fragments 1–6 d after transfection of FH3221-100. Number of cells is shown at the top of each bar for one time course experiment. c, Ratio of cell fragments per neuron is significantly greater in cultures expressing huntingtin with 100 glutamines than with 18 or 46 glutamines (p < 0.01; n = 6 per group).
Effects of caspase inhibitors Z-VAD-FMK and Z-DEVD-FMK
Striatal cells expressing FH3221-18 or 100 and treated with the caspase inhibitor Z-VAD-FMK showed marked increases in cell number on day 3 (Fig. 7) compared with neurons without the caspase inhibitor. Increased survival with Z-VAD-FMK was dose-dependent and maximal at 100 μm (Fig.7). The proportion of FLAG–huntingtin-negative cells in the cultures was modestly increased by Z-VAD-FMK (mean increase was 49%) compared with FLAG–huntingtin-positive cells (240–310%). Moreover, the mean percent of FLAG+ cells incorporating BrdU, a marker of S-phase activity, was not significantly changed by Z-VAD-FMK (Student’st test; p > 0.05) either for the wild-type (−Z-VAD-FMK, 4.8%; +Z-VAD-FMK, 4.3%) or mutant proteins (100 CAGs, −Z-VAD-FMK, 25.5%; +Z-VAD-FMK, 16.5%). This showed that the inhibitor had increased viability and not cell proliferation. Similar protective effects of Z-VAD-FMK on cell survival were seen when the full-length wild-type and mutant huntingtins were expressed (mean increases were 397% for FH9774-18 and 198% for FH9774-100; n = 6 experiments). In contrast to its effects on survival, Z-VAD-FMK did not change the proportion of total neurons that developed NI and CI (Fig. 7).
Fig. 7.

Effects of caspase inhibitors Z-VAD-FMK (100 μm) and Z-DEVD-FMK (200 μm) on the number of FLAG-positive neurons and the proportion of neurons with NI and CI present 3 d after transfection. Striatal neurons were transfected with FH3221-18 or -100. Bar graphs show a significant increase in neuron survival (*p < 0.05; n = 6) after treatment with Z-VAD-FMK and a significant reduction (*p < 0.05;n = 6) in the proportion of neurons that develop nuclear and cytoplasmic inclusions after treatment with Z-DEVD-FMK. Mean values based on total FLAG-positive neurons counted per coverslip in six coverslips. Line graphs show effects of concentration of Z-VAD-FMK on the number of FLAG-positive neurons and of Z-DEVD-FMK on the percent of FLAG-positive neurons with nuclear inclusions in cultures treated with FH3221-100. Mean values are based on analysis of 20 microscopic fields per coverslip in three coverslips. *Signifies a difference from 0 concentration atp < 0.05, Student’s t test.
Treatment with the caspase 3 inhibitor, Z-DEVD-FMK, caused a dose-dependent reduction in the proportion of neurons with NI and CI formed by FH3221-100 (Fig. 7). This was offset by a significant rise in the proportion of neurons with diffuse cytoplasmic labeling (results not shown). Z-DEVD-FMK had no effect on the survival of cells expressing FH3221-18, -46 (data not shown), or -100 (Fig. 7).
Effects of transglutaminase inhibitors
Transglutaminase is a substrate for polyglutamine-enriched proteins (Cooper et al., 1997) and causes mutant huntingtin to aggregate (Kahlem et al., 1998). Treatment over 3 d with a 0.2 mm concentration of the transglutaminase inhibitor cystamine did not reduce NI or CI formed by mutant huntingtin in our striatal cells, and 0.5 and 1 mm cystamine were toxic to striatal cells.
Western blot assay of protein extracts from the transfected striatal cells
Total protein extracts from cells transfected with FH3221-18, 46, and 100 were examined by Western blot with anti-huntingtin antisera Ab 1. The fully expressed proteins migrated at ∼140 kDa (Fig. 8). A larger fragment of ∼175 kDa also appeared when mutant huntingtin contained 100 glutamines. A series of three N-terminal fragments were generated by the normal and mutant proteins. The size of the products seen with FH3221-18 was ∼60, 70, and 80 kDa. FH3221-46 generated products of ∼70, 80, and 90 kDa, and FH3221-100 produced bands of ∼80, 90, and 100 kDa. The variation in size of the N-terminal products of the normal and mutant proteins was consistent with the variable polyglutamine regions in these proteins. The two smaller N-terminal products generated by the normal (60 and 70 kDa) and mutant (70 and 80 kDa for FH3221-46 and 80 and 90 kDa for FH3221-100) proteins were more prominent than the larger product. In some blots these smaller fragments resolved as doublets, indicating that each arose from cleavage at two nearby sites in huntingtin. These results demonstrated that three to five cleavage products were produced by the large truncated normal and mutant proteins.
Fig. 8.

Western blots of huntingtin expression in protein extracts of transfected striatal cells and the effects of caspase inhibitors. a, Human huntingtin is seen at the expected size of ∼140 kDa (top arrowhead) in cells exposed to the FH3221 expression plasmids. A slightly larger band at ∼175 kDa is also seen with FH3221-100. N-terminal products (bottom arrowheads) occur with wild-type (18 CAGs) and mutant (46 and 100 CAGs) huntingtins and show variable size consistent with polyglutamine expansion. Native huntingtin appears at the top of first lane. Z-VAD-FMK (100 μm) markedly inhibits a prominent intermediate N-terminal product in wild-type huntingtin (70 kDa) and mutant huntingtins (80 kDa for 46 CAGs and 90 kDa for 100 CAGs). b, Expression of full-length wild-type huntingtin (18 CAGs) and mutant huntingtin (100 CAGs) with FH9774 constructs generates N-terminal products (arrowheads) only from mutant huntingtin. Lane c was nontransfected and shows native mouse huntingtin at thetop of the blot. Full-length human mutant huntingtin migrates above native mouse protein. c,d, Time course of appearance of N-terminal products after expression of FH3221-100. Results in cand d are from different transfections. The blot at theright in c is a longer exposure of the 7 and 9 hr time points. The 140 kDa band appears at 5 and 6 hr. The 100 kDa product is seen at 9 hr (c, right blot). All three N-terminal products are present at 15 hr (d). The 90 kDa band is the last to appear at 15 hr (d) and the most prominent at 45 hr (c). Native huntingtin appears at thetop of most lanes (c).e, Effects of Z-VAD-FMK concentration (0–100 μm) on mutant huntingtin expressed from FH3221-100. The 90 kDa N-terminal product is attenuated at all concentrations and maximally at 100 μm. Increased expression of the 140, 80, and 100 kDa proteins are seen and may be related to increased viability. f, Effects of concentration of Z-DEVD-FMK (0–200 μm) on mutant huntingtin expression by FH3221-100. No inhibitory effects on cleavage are seen at any concentration. N-terminal products are slightly elevated, but the 140 kDa protein is unchanged in cells treated with the inhibitor. Protein extracts (20 μg/lane) were taken 24 hr after transfection in a, b,e, and f. Antibody Ab 1 was used to detect huntingtin. Molecular mass markers are shown to theleft of each blot.
The NH2-terminal products produced by FH3221-100 were also detected after expression of full-length mutant huntingtin (Fig. 8b). The 90 kDa product was the most prominent of the three products seen. N-terminal products were not produced by full-length wild-type huntingtin (Fig.8b). Cells split from the same transfections were used to verify by FLAG staining that the transfections were successful for both the full-length normal and mutant proteins. The results suggested that polyglutamine expansion in full-length huntingtin facilitated production of N-terminal fragments.
The generation of N-terminal products was examined 5–45 hr after transfection of FH3221-100. The 140 kDa protein was detected at 5–6 hr, was maximal by 24 hr, and was reduced at 45 hr. The 100 kDa fragment was seen at 9 hr, peaked between 15 and 24 hr, and was lower by 45 hr. The 90 kDa band was first seen at 15 hr and was prominent at 24 and 45 hr. The 80 kDa band was low at 9 hr, maximal at 15 hr, reduced by 24 hr, and nearly absent by 45 hr (Fig.8c,d). After long film exposures of the chemiluminescence reaction (∼1 hr), multiple smaller molecular mass products (in the range of 20–50 kDa for FH3221-100) were detected (data not shown). This suggested that the products were being processed to smaller fragments. The results indicated that the generation of N-terminal products from FH3221-100 was time-dependent but coincided with the appearance of inclusions in the striatal cells. The emergence and duration of the products also appeared to differ, with the 90 kDa species being the most long-lived.
Z-VAD-FMK blocked formation of the intermediate 90 kDa fragment seen with FH3221-100, and the corresponding 70 and 80 kDa products formed from FH3221-18 and FH3221-46, respectively (Fig. 8a,e). The inhibitory effect was correlated in a dose-dependent manner (Fig. 8e, shown for FH3221-100) with increased survival (Fig. 7). Z-VAD-FMK increased the fully expressed 140 kDa protein and enhanced expression of the 80 and 100 kDa products generated by FH3221-100 (Fig. 8a,f). The rise in the 140 kDa protein may have resulted from increased cell survival and/or the inhibition of the 90 kDa from the 140 kDa protein. Z-VAD-FMK also blocked the 90 kDa fragment generated by full-length mutant huntingtin with FH9774-100 (results not shown). Z-DEVD-FMK had no inhibitory effects on N-terminal products of normal or mutant huntingtin. Treatment with the inhibitor at all doses tested had no effect on the 140 kDa band but slightly increased the N-terminal products formed by FH3221-100 (Fig. 8f) compared with the N-terminal products in untreated cells.
DISCUSSION
The recent identification in HD brain of nuclear inclusions derived from N-terminal fragments of mutant huntingtin (DiFiglia et al., 1997) has heightened interest about their formation and potential role in the pathogenesis of the disease. We have shown that the predominant cytoplasmic distribution of huntingtin localization seen in immortalized mouse striatal cells transfected with human huntingtin cDNAs fits with that of neurons in normal and HD brain (DiFiglia et al., 1995, 1997; Sapp et al., 1997). The development and appearance of NI in the transfected cells also showed a remarkable correspondence with HD neurons. The formation of NI in culture, as in the HD brain, was polyglutamine and time-dependent. The CI formed in culture evolved similarly to the NI and most likely corresponds to the mutant huntingtin aggregates identified in dystrophic neurites in the HD brain (DiFiglia et al., 1997), although the ring-like architecture of some CI has not been seen in HD neurons.
Inclusions may contain huntingtin proteins of various sizes. NI contained mostly cleaved N-terminal products consistent with other recent findings (Cooper et al., 1998; Hackam et al., 1998), whereas CI had a mixture of cleaved and larger complete proteins. Immunohistochemical and biochemical studies in HD brain suggest that mutant huntingtin may be present in the nucleus as a full-length protein (DiFiglia et al., 1997, their Fig. 3; Sapp et al., 1997). N-terminal products also accumulated in dispersed and perinuclear vacuoles. In contrast to inclusions, which contained only mutant huntingtin, vacuoles incorporated normal or mutant proteins. The subcellular compartment to which vacuoles belong is unclear, but recent observations suggest that they are part of the endoplasmic reticulum (K. Kegel and M. DiFiglia, unpublished observations).
Features of apoptosis occurred in striatal neurons expressing normal and mutant proteins. The characteristics of apoptotic cell death included membrane blebbing, cellular fragmentation, and shrinkage (Kerr et al., 1972). The striatal cells most reduced in size were those that accumulated high levels of N-terminal normal or mutant huntingtin into dispersed and perinuclear vacuoles, indicating that accumulation into these compartments could be toxic. The generation of apoptotic fragments was polyglutamine-dependent and occurred significantly more frequently in the striatal cells expressing a highly expanded mutant protein than in cells with the moderately expanded or wild-type huntingtin. The apoptotic morphology induced by wild-type huntingtin was somewhat surprising because it has not been reported in studies with other cell lines. The immortalized striatal cells may be especially sensitive to huntingtin-induced apoptosis or may be less able to handle the accumulation of large amounts of huntingtin produced by transient transfection than other cells. Native huntingtin exists at low levels in striatal hybrid cells cultured under basal conditions (Kim et al., 1999) and is also low in medium-sized striatal neurons compared with other neurons in mouse and human brain (Bhide et al., 1996; Sapp et al., 1997).
A pathogenic mechanism for HD has been proposed, whereby polyglutamine expansion induces the production of an altered protein that causes cell death possibly through the formation of inclusions (Goldberg et al., 1996; Davies et al., 1997; Hackam et al., 1998; Martindale et al., 1998). The caspase inhibitor Z-VAD-FMK increased cell viability without changing the formation of inclusions. Treatment with the caspase inhibitors Z-DEVD-FMK (this study) and Z-IETD-FMK (M. Kim and M. DiFiglia, unpublished observations) significantly reduced the proportion of neurons that formed NI and CI but was not associated with increased survival. Because caspase inhibitors are not selective and may affect cell survival and morphological features of apoptosis through multiple ICE-like proteases and other substrates (Eldadah et al., 1997; MacFarlane et al., 1997; D’Mello et al., 1998), the mechanisms by which these inhibitors acted in our study are unclear. Nevertheless, the results of experiments with Z-VAD-FMK and DEVD-FMK suggest that the formation of inclusions is not essential to cause cell death. In accord with this idea, results in transfected 293T cells exposed to tamoxifen showed that even in the absence of inclusions, the mutant protein was more toxic than wild-type (Hackam et al., 1998). During the review of this manuscript, another study reported that the formation of nuclear inclusions in cultured embryonic rat striatal neurons by N-terminal fragments of mutant huntingtin did not correlate with the extent of apoptotic cell death (Saudou et al., 1998).
Huntingtin expression in clonal striatal cells produced a series of three to five N-terminal fragments, consistent with the presence of endogenous proteolytic activity. These products are in the range of sizes consistent with cleavage at several putative caspase cleavage sites near the NH2 terminus of huntingtin (Wellington et al., 1998). Apoptotic extracts (Goldberg et al., 1996) and purified caspases 1 and 3 produce N-terminal products (two for each caspase) from in vitro translated full-length normal and mutant huntingtin, and deletion mutation has verified at least one caspase 3-sensitive site (Wellington et al., 1998). Thus, some of the N-terminal products of huntingtin seen in the striatal cells may result from caspase 1 and/or caspase 3 activity. The marked attenuation by Z-VAD-FMK of a prominent N-terminal product in huntingtin (90 kDa product generated from FH3221-100) is compatible with the interruption of a caspase 1 and/or 3 consensus site in huntingtin. In the range of concentrations used in our study, Z-VAD-FMK was shown to inhibit the activity of caspases 1 and 3 in apoptotic cells (Eldadah et al., 1997; MacFarlane et al., 1997). It is not clear whether the N-terminal products generated by mutant huntingtin expression in the striatal cells contributed directly to the formation of nuclear inclusions or involved further proteolytic digestion as suggested by biochemical assay in the HD brain (DiFiglia et al., 1997).
Although both normal and mutant proteins expressed from truncated cDNAs were susceptible to cleavage, only mutant huntingtin was cleaved from the full-length protein. While our paper was being reviewed, Lunkes and Mandel (1998) also reported cleavage of full-length mutant but not wild-type huntingtin using stable and inducible neuroblastoma cell lines. The findings in both studies support previous evidence that polyglutamine expansion enhances N-terminal cleavage (Goldberg et al., 1996). Z-VAD-FMK, which increased survival, blocked a prominent N-terminal product generated by truncated normal and mutant huntingtin proteins. The same product was formed by mutant huntingtin but not wild-type huntingtin when the full-length protein was expressed. These observations strongly implicate an N-terminal product of mutant huntingtin in apoptosis but do not prove that this product functions as an apoptotic initiator or mediator.
Despite evidence that transglutaminase induces polymerization of mutant huntingtin (Kahlem et al., 1998), the inhibition of transglutaminase was ineffective in reducing NI and CI or in changing survival. However, concentrations of the transglutaminase inhibitor cystamine, which were found effective in reducing cytoplasmic aggregates formed by mutant DRPLA in COS-7 cells (Igarashi et al., 1998), were toxic to the striatal cells. Therefore, our negative results do not exclude the possibility that transglutaminase contributes to the aggregation of mutant huntingtin in the HD brain.
We used an immortalized cell line derived from a fusion of embryonic mouse cells of striatal origin and neuroblastoma cells to investigate huntingtin expression. This cell line shows characteristics of striatal cells, including an enrichment in dopamine receptors, and forms neurites when differentiated (Wainright et al., 1995). Also, native huntingtin in the clonal striatal cells is known to have the same cytoplasmic localization and vesicle membrane associations found in mouse striatal cells in vivo (Kim et al., 1999; Bhide et al., 1996). Because transformed cells have altered growth characteristics, they may have some limitations for the study of neuronal function compared with primary cultured neurons. Among the significant advantages of immortalized neurons, however, is their potential use for high through-put assays in drug screening.
In summary, our findings show that clonal mouse striatal cells transiently expressing human huntingtin in vitro exhibit patterns of mutant huntingtin expression seen in the HD brain, including the polyglutamine-dependent and time-dependent formation of NI. Experiments using caspase inhibitors revealed that the formation of inclusions could be separated from events that control cell survival. Immortalized striatal cells provide a more accurate model of the HD cellular phenotype than that achieved with other types of transformed cells recently studied and should afford a rapid way of dissecting the signal transduction events involved with HD pathology.
Footnotes
This work was supported by National Institutes of Health Grants NS16367 to M.D., NS31579 to M.D. and N.A., the Hereditary Disease Foundation to M.D. and N.A., the Huntington’s Disease Society of America to M.D., the Howard Hughes Medical Institute to G.L., and MH28942 to A.H. and L.W.
Correspondence should be addressed to Marian DiFiglia, Laboratory of Cellular Neurobiology, Massachusetts General Hospital, MGH-East, 149 13th Street, Charlestown, MA 02129.
REFERENCES
- 1.Becher MW, Kotzuk JA, Sharp AH, Davies SW, Bates GP, Price DL, Ross CA. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis. 1998;4:387–397. doi: 10.1006/nbdi.1998.0168. [DOI] [PubMed] [Google Scholar]
- 2.Bhide PG, Day M, Sapp E, Schwarz C, Sheth A, Kim J, Young AB, Penney J, Golden J, Aronin N, DiFiglia M. Expression of normal and mutant huntingtin in the developing brain. J Neurosci. 1996;16:5523–5535. doi: 10.1523/JNEUROSCI.16-17-05523.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington’s disease gene. Proc Natl Acad Sci USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi HK, Won LA, Kontur PJ, Hammond DN, Fox AP, Wainer B, Hoffman PC, Heller A. Immortalization of embryonic mesencephalic dopaminergic neurons by somatic cell fusion. Brain Res. 1991;552:67–76. doi: 10.1016/0006-8993(91)90661-e. [DOI] [PubMed] [Google Scholar]
- 5.Cooper AJ, Sheu KF, Burke JR, Onodera O, Strittmatter WJ, Roses AD, Blass JP. Polyglutamine domains are substrates of tissue transglutaminase: does transglutaminase play a role in expanded CAG/poly-Q neurodegenerative diseases? J Neurochem. 1997;69:431–434. doi: 10.1046/j.1471-4159.1997.69010431.x. [DOI] [PubMed] [Google Scholar]
- 6.Cooper JK, Schilling G, Peters MF, Herring WJ, Sharp AH, Kaminsky Z, Masone J, Khan FA, Borchelt DM, Dawson VL, Dawson TM, Ross CA. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum Mol Genet. 1998;7:783–790. doi: 10.1093/hmg/7.5.783. [DOI] [PubMed] [Google Scholar]
- 7.Davies SW, Turmain M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;9:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 8.DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel J-P, Reeves S, Carraway R, Boyce FM, Aronin N. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 9.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 10.D’Mello SR, Aglieco F, Roberts MR, Borodezt, Haycock JW. A DEVD-inhibited caspase other than CPP32 is involved in the commitment of cerebellar granule neurons to apoptosis induced by K+ deprivation. J Neurochem. 1998;70:1809–1818. doi: 10.1046/j.1471-4159.1998.70051809.x. [DOI] [PubMed] [Google Scholar]
- 11.Eldadah BA, Yakovlev AG, Faden AI. The role of CED-3-related cystein proteases in apoptosis of cerebellar granule cells. J Neurosci. 1997;17:6105–6113. doi: 10.1523/JNEUROSCI.17-16-06105.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldberg YP, Nicholson DW, Rasper DM, Kalchman MA, Doide HB, Graham RK, Bromm M, Kazemi-Esfarjani P, Thornberry NA, Vaillancourt JP, Hayden MR. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet. 1996;13:442–449. doi: 10.1038/ng0896-442. [DOI] [PubMed] [Google Scholar]
- 13.Hackam A, Singaraja T, Wellington CL, Metzler M, McCutcheon K, Zhang T, Kalchaman M, Hayden MR. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol. 1998;141:1097–1105. doi: 10.1083/jcb.141.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 15.Igarashi S, Koide R, Shimohata T, Yamada M, Hayashi Y, Takano H, Data H, Oyake M, Sato T, Sato A, Egawa S, Ikeuchi T, Tanaka H, Nakano R, Tanaka K, Hozumi I, Inuzuka T, Takahashi H, Tsuji S. Suppression of aggregate formation and apoptosis by transglutaminase inhibitors in cells expressing truncated DRPLA protein with an expanded polyglutamine stretch. Nat Genet. 1998;18:111–117. doi: 10.1038/ng0298-111. [DOI] [PubMed] [Google Scholar]
- 16.Kahlem P, Green H, Djian P. Transglutaminase action imitates Huntington’s disease: selective polymerization of huntingtin containing expanded polyglutamine. Mol Cell. 1998;1:595–601. doi: 10.1016/s1097-2765(00)80059-3. [DOI] [PubMed] [Google Scholar]
- 17.Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim M, Velier J, Chase K, LaForet G, Kalchman MA, Hayden MR, Won L, Heller A, Aronin N, DiFiglia M (1999) Forskolin and dopamine D1 receptor activation increase huntingtin’s association with endosomes in immortalized neuronal cells of striatal origin. Neuroscience, in press. [DOI] [PubMed]
- 19.Lunkes A, Mandel J-L. A cellular model that recapitulates major pathogenic steps of Huntington’s disease. Hum Mol Genet. 1998;7:1355–1361. doi: 10.1093/hmg/7.9.1355. [DOI] [PubMed] [Google Scholar]
- 20.MacFarlane M, Cain K, Sun X-M, Alnemri ES, Cohen GM. Processing/activation of at least four interleukin-1β converting enzyme-like proteases occurs during the execution phase of apoptosis in human monocytic tumor cells. J Cell Biol. 1997;137:469–479. doi: 10.1083/jcb.137.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, Bredesen DE, Tufaro F, Hayden MR. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet. 1998;18:150–154. doi: 10.1038/ng0298-150. [DOI] [PubMed] [Google Scholar]
- 22.McCarthy NJ, Whyte MKB, Gilbert CS, Evan GI. Inhibition of CED-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J Cell Biol. 1997;136:215–227. doi: 10.1083/jcb.136.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, Pittman RN. Intraneuronal nuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333–344. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 24.Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos VE. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sapp E, Schwarz C, Chase K, Bhide PG, Young AB, Penney J, Vonsattel JP, Aronin N, DiFiglia M. Huntingtin localization in brains of normal and Huntington’s disease patients. Ann Neurol. 1997;42:604–612. doi: 10.1002/ana.410420411. [DOI] [PubMed] [Google Scholar]
- 26.Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 27.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 28.Skinner PJ, Koshy BT, Cummings CJ, Klement IA, Helin K, Servadio A, Zoghbi HY, Orr HT. Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures. Nature. 1997;389:971–974. doi: 10.1038/40153. [DOI] [PubMed] [Google Scholar]
- 29.Velier J, Kim M, Schwarz C, Kim TW, Sapp E, Chase K, Aronin N, DiFiglia M. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol. 1998;152:34–40. doi: 10.1006/exnr.1998.6832. [DOI] [PubMed] [Google Scholar]
- 30.Wainwright MS, Perry BD, Won L, O’Malley KL, Wang WY, Ehrlich ME, Heller A. Immortalized murine striatal neuronal cell lines expressing dopamine receptors and cholinergic properties. J Neurosci. 1995;15:676–688. doi: 10.1523/JNEUROSCI.15-01-00676.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wellington CL, Ellerby LM, Hackam AS, Margolis RL, Trifiro MA, Singaraja R, McCutcheon K, Salvesen GS, Propp SS, Bromm M, Rowland KJ, Zhang T, Rasper D, Roy S, Thornberry N, Pinsky L, Kakizuka A, Ross CA, Nicholson DW, Bredesen DE, Hayden MR. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem. 1998;273:9158–9167. doi: 10.1074/jbc.273.15.9158. [DOI] [PubMed] [Google Scholar]