

Abstract
The ionotropic glutamate receptor GluR6 exhibits strongly and rapidly desensitizing current responses. Treatment of heterologically expressed GluR6 with the lectin concanavalin A (ConA) inXenopus oocytes as well as in human embryonic kidney-293 cells results in a considerable increase of the steady-state current, presumably by inhibiting receptor desensitization. In the present study, we investigated the molecular basis of this effect using a systematic mutagenesis approach. We found that although N-glycosylation is an absolute prerequisite for the lectin-mediated inhibition of desensitization, no single one of the nine extracellular consensus sites for N-glycosylation of GluR6 is required. Rather, each of the nine N-linked carbohydrate side chains is independently capable of modulatory interaction with the lectin. Moreover, even artificially introduced N-glycosylation sites can substitute for native sites. Thus, the specific site of the lectin binding does not appear to be important for its desensitization-inhibiting action. Furthermore, we show that the extent of the receptor’s ConA sensitivity depends on its state of activation, because the desensitized GluR6 exhibits significantly lower lectin sensitivity than the nondesensitized receptor. We conclude that binding of ConA “locks” the receptor in the activatable state, thereby inhibiting conformational changes required to shift the receptor to the desensitized state.
Keywords: GluR6, kainate receptor, N-glycosylation, lectin, concanavalin A, receptor desensitization, ectopic sites, mutagenesis
Ionotropic glutamate receptors (iGluRs) are found throughout the vertebrate brain (Monaghan et al., 1989), where they constitute the dominant excitatory neurotransmitter system. On the basis of pharmacological and electrophysiological properties as well as sequence homologies, GluRs have been divided into three major subfamilies: AMPA receptors, kainate (KA) receptors, and NMDA receptors.
As important mediators of cell-to-cell signaling, iGluRs are tightly regulated and functionally modulated by several post-transcriptional [alternative splicing (Sommer et al., 1992) and RNA editing (Seeburg, 1996)] and post-translational mechanisms [protein phosphorylation (Raymond et al., 1994), palmitoylation (Pickering et al., 1995), and N-glycosylation (Taverna et al., 1994; Everts et al., 1997)]. A characteristic feature of ionotropic glutamate receptors, especially of the kainate receptor subfamily, is the rapid and effective desensitization of the ionic current response that has a role in shaping the postsynaptic response. Treatment with the lectin concanavalin A (ConA) potentiates currents of native (Mathers and Usherwood, 1976; Mayer and Vyklicky, 1989; Thio et al., 1993; Wong and Mayer, 1993) and recombinant (Geoffroy et al., 1989; Partin et al., 1993, 1995; Yue et al., 1995; Everts et al., 1997) glutamate receptors. By far the largest increase of current responses can be observed with kainate receptors. A commonly suggested mechanism is the inhibition of receptor desensitization (Mayer and Vyklicky, 1989; Huettner, 1990). Potentiation factors for the kainate receptor GluR6 expressed inXenopus oocytes can be several thousand-fold (Egebjerg et al., 1991; Everts et al., 1997). Although lectin-mediated current potentiation has been known for a long time, its mechanism is not yet understood. In the present study we therefore set out to investigate the molecular basis of ConA-mediated modulation of GluR6 current responses.
Lectins such as ConA are proteins that bind specifically to certain carbohydrates. Therefore, carbohydrate side chains of glutamate receptor are obvious candidates to mediate the lectin-induced inhibition of desensitization. The amino acid sequence of GluR6 contains nine potential extracellular sites for N-glycosylation that conform to the universal consensus sequence N–X–S/T, with X ≠ P. These sites occur in the two large domains of the receptor that according to the currently recognized three-transmembrane domain model are located in the two extracellular domains: the N terminus and the loop between transmembrane domains B and C (Hollmann et al., 1994; Wo and Oswald, 1995). One additional consensus site appears to be localized intracellularly and should therefore not be glycosylated (see Fig. 3). We created two series of mutants with altered N-glycosylation properties. In the first series of mutants, the nine extracellular consensus N-glycosylation sites were removed one at a time. In the second series, nine mutants were made that each retained only a single N-glycosylation site. Three additional mutants were engineered to contain an ectopic N-glycosylation site after all native sites had been eliminated by mutation. For all mutants, the desensitization behavior and the desensitization-inhibiting effects of ConA were determined.
Fig. 3.

Putative transmembrane topology of GluR6.Black boxes represent transmembrane domains. The localization of native and ectopic N-glycosylation consensus sequences (N–X–S/T, X ≠ P) is indicated by • and ○, respectively.
MATERIALS AND METHODS
Concanavalin A (grade VI) and polymannosyl–bovine serum albumin [(BSA)-p-aminophenyl-α-d-mannopyranoside, 26 mol monosaccharide per mol albumin] were obtained from Sigma (Munich, Germany). Tunicamycin was purchased from Boehringer Mannheim (Mannheim, Germany). Other drugs were purchased from Sigma unless noted otherwise. Adult female frogs (Xenopus laevis) were obtained from Nasco (Fort Atkinson).
cRNA synthesis. Template was prepared from circular plasmid cDNA by linearizing each clone with a suitable restriction enzyme. The vector used throughout this study was pSGEM, a modified version of pGEMHE (Villmann et al., 1997). cRNA was prepared from 1 μg of linearized template using an in vitro transcription kit (Stratagene, La Jolla, CA) with a modified standard protocol that uses each of the nucleotides at 800 μm (except for GTP at 200 μm), 400 μmm7GpppG (Pharmacia, Piscataway, NJ) for capping, and an extended reaction time of 3 hr with T3 or T7 RNA polymerase. All cRNAs were trace-labeled with [32P]UTP (Amersham, Arlington Heights, IL) to allow for quality checks by gel electrophoresis and calculation of the yield.
Electrophysiological recordings from Xenopusoocytes. Frog oocytes of stages V–VI were obtained by surgically removing parts of the ovaries of Xenopus laevis anesthetized with tricaine (2 gm/l). The removed ovaries were chopped and incubated with 815 U/ml (= 2.8 mg/ml) collagenase type I (Worthington, Freehold, NJ) and 2200 U/ml (= 0.15 mg/ml) trypsin at 20°C for 2 hr in calcium-free Barth’s solution (see below) with slow agitation to remove the follicular cell layer and then washed extensively with Barth’s solution (88 mm NaCl, 1.1 mm KCl, 2.4 mm NaHCO3, 0.3 mmCa(NO3)2, 0.3 mmCaCl2, 0.8 mm MgCl2, 15 mm HEPES, pH 7.6 with NaOH). Oocytes were maintained in Barth’s solution supplemented with 100 μg/ml gentamycin, 40 μg/ml streptomycin, and 63 μg/ml penicillin. Oocytes were injected with 10 ng of wild-type (wt) or mutant GluR6(Q) cRNA 24 hr after collagenase treatment using a 10 μl Drummond microdispenser; 5–6 d after RNA injection, oocytes were recorded in amphibian Ringer’s solution (115 mm NaCl, 2.5 mm KCl, 1.8 mmCaCl2, 10 mm HEPES, pH 7.2 with NaOH) under voltage clamp at a holding potential of −70 mV, with a Turbo Tec-10CD amplifier (NPI). Voltage electrodes had resistances of 1–3 MΩ and were filled with 3 m KCl; current electrodes had resistances of ∼1 MΩ and were filled with 3 m CsCl. Agonist was applied by superfusion for 10 sec at a flow rate of 6 ml/min in a 50 μl recording chamber. For each oocyte, steady-state currents (after 10 sec of agonist superfusion) were recorded before and after ConA treatment. Lectin treatment was performed by pipetting 100 μl of 10 μm ConA in amphibian Ringer’s solution (calculated for the tetramer of 102 kDa) directly into the recording chamber while perfusion was stopped. Oocytes were incubated for 8 min in the lectin solution and then perfusion was restarted for 1 min before the next agonist application. To estimate EC50values, eight different agonist concentrations (A) were applied, and steady-state values of the evoked currents (I) were measured and fitted with the SigmaPlot program (Jandel Scientific, San Rafael, CA) to the equation I =Imax/[1 + EC50/AnH], whereImax is the maximal current, EC50 is the agonist concentration yielding half-maximal currents, and nH is the Hill coefficient.
Electrophysiological recordings from human embryonic kidney-293 cells. Human embryonic kidney (HEK)-293 cells were transiently transfected with plasmids (pcDNA3; Invitrogen, Carlsbad, CA) encoding wild-type (wt) or mutant GluR6(Q) by calcium phosphate precipitation for 6–8 hr (Chen and Okayama, 1987). HEK-293 cells were used for electrophysiology from 1 to 3 d later.
Recordings were made using an Axopatch 200 amplifier and pCLAMP 6 software (Axon Instruments, Foster City, CA). Data were sampled at 20 kHz and filtered at 2 kHz. Extracellular solution consisted of the following (in mm): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, pH 7.3. Intracellular solution consisted of the following (in mm): 110 cesium gluconate, 20 CsCl, 4 NaCl, 1 MgCl2, 5 EGTA, 0.5 CaCl2, and 10 HEPES, pH 7.3.
To mimic synaptic activation of glutamate receptors and to measure their rapid desensitization, a fast perfusion system was used to apply glutamate. Control and test solutions, under the control of solenoid valves, were gravity-fed through a triple-barrel glass pipette pulled to a diameter of 175 μm. This flow pipette was mounted on a piezoelectric bimorph to switch the barrel position. Solution exchange was ≤500 μsec. After breaking into the whole-cell configuration, the recording electrode was raised and the HEK-293 cell was lifted. Proper positioning of the flow pipette relative to the lifted cell was critical for obtaining rapid activation of kainate receptors.
Percentage desensitization was measured as 100 × [(peak − steady state)/peak], where the steady-state current was measured at the end of a 100 msec application of glutamate. The time constant for desensitization τ was taken from the fitted monoexponential decay of the desensitizing current, starting from 90% of the peak current using Clampfit software.
Inhibition of N-glycosylation by tunicamycin. To express nonglycosylated receptors, we used tunicamycin, a potent inhibitor of N-glycosylation (Duksin and Mahoney, 1982).
HEK-293 cells were bath-treated with 5 μg/ml tunicamycin immediately before transfection, and this concentration was maintained in the medium until the cells were recorded 24 hr later.
Oocytes were preinjected with 50 nl of 400 μg/ml tunicamycin (= 20 ng/oocyte or ∼22 μg/ml for an oocyte of 1.2 mm diameter) 1 d before injection with cRNA. Immediately before tunicamycin injection, a stock solution of 10 mg/ml tunicamycin in dimethylsulfoxide (DMSO) was diluted to 4% DMSO with amphibian Ringer’s solution; 4% DMSO did not adversely affect the oocytes. In contrast to cell cultures, bath treatment of oocytes with tunicamycin turned out to be largely ineffective in inhibiting N-glycosylation (data not shown).
Site-directed mutagenesis. Single nucleotide exchanges were introduced by PCR-mediated site-directed mutagenesis using mutagenetic primers as described previously (Hollmann et al., 1994). For all constructs we used the “Q” editing variant of GluR6, GluR6(Q), which from here on will be referred to as GluR6. To create N-glycosylation site mutants, we first deleted single N-glycosylation consensus sites (N–X–S/T, X ≠ P) in the GluR6 sequence by changing the S or T to an A. The 10 resulting mutants were numbered in the order of appearance of their respective consensus sites in the GluR6 cDNA (NG = N-glycosylation site): GluR6(Q)[T38A] = GluR6-ΔNG1; GluR6(Q)[T44A] = GluR6-ΔNG2; GluR6(Q)[T246A] = GluR6-ΔNG3; GluR6(Q)[T349A] = GluR6-ΔNG4; GluR6(Q)[T383A] = GluR6-ΔNG5; GluR6(Q)[T394A] = GluR6-ΔNG6; GluR6(Q)[S401A] = GluR6-ΔNG7; GluR6(Q)[T517A] = GluR6-ΔNG8; GluR6(Q)[T576A] = GluR6-ΔNG9; and GluR6(Q)[T722A] = GluR6-ΔNG10.
A second series of nine mutants had only one N-glycosylation site left, plus site NG9. The site NG9 was retained unmutated in all of these mutants because this consensus sequence for N-glycosylation is located intracellularly (see Fig. 3) and therefore cannot be glycosylated. The eight other sites were deleted. The resulting mutants were named after the one true N-glycosylation site that was retained. In an additional mutant, all nine extracellular consensus sequences for N-glycosylation were deleted: GluR6-NG1 contains all of the mutations mentioned except T38A and T576A; GluR6-NG2 contains all of the mutations except T44A and T576A; GluR6-NG3 contains all of the mutations except T246A and T576A; GluR6-NG4 contains all of the mutations except T349A and T576A; GluR6-NG5 contains all of the mutations except T383A and T576A; GluR6-NG6 contains all of the mutations except T394A and T576A; GluR6-NG7 contains all of the mutations except S401A and T576A; GluR6-NG8 contains all of the mutations except T517A and T576A; GluR6-NG9 contains all of the mutations except T576A; and GluR6-NG10 contains all of the mutations except T722A and T576A.
Several ectopic N-glycosylation sites were introduced in the mutant GluR6-NG9 by creating artificial N-glycosylation consensus sequences: GluR6-EG1 = based on GluR6-NG9 with F284N creating site EG1; GluR6-EG2 = based on GluR6-NG9 with S396N creating site EG2; and GluR6-EG3 = based on GluR6-NG9 with I637N creating site EG3.
All mutations were verified by chain-termination method sequencing using the Sequenase kit from USB.
Labeling of cell-surface glycoproteins with biotinylated ConA. To identify only that fraction of receptor protein that is inserted in the plasma membrane of the oocytes, surface proteins were tagged with biotin and isolated by streptavidin–Sepharose-mediated precipitation of the labeled protein 4–6 d after RNA injection. Briefly, intact oocytes were incubated in 1 mg/ml NHS-SS-Biotin (Pierce, Rockford, IL) solution for 2 hr at 4°C. After five washes for 10 min each in frog Ringer’s solution, 10 oocytes were homogenized with a Teflon pestle in 200 μl H-buffer (100 mm NaCl, 20 mm Tris-HCl, pH 7.4, 1% Triton X-100, and 1 mmphenylmethylsulfonyl fluoride (PMSF) plus a mixture of additional proteinase inhibitors: 2.5 μg/ml leupeptin, 20 μg/ml aprotinin, 2.5 μg/ml pepstatin, and 20 μg/ml benzamidine hydrochloride). The homogenate was kept on ice for 60 min. After centrifugation for 60 sec at 16,000 × g to remove yolk platelets and the melanin pigment granula, the supernatants were supplemented with 20 μl streptavidin–Sepharose beads (Sigma) and incubated for 3 hr at 4°C on a rotating rod. The streptavidin–Sepharose beads were pelleted by a 60 sec spin and washed three times with H-buffer, and the washed pellets were boiled in 40 μl/oocyte SDS polyacrylamide gel loading buffer (0.8 m β-mercaptoethanol, 6% SDS, 20% glycerol, 25 mm Tris-HCl, pH 6.8, and 0.1% bromphenol blue).
SDS gel electrophoresis and Western blotting. The solubilized membrane proteins were separated at 4°C on discontinuous SDS polyacrylamide gels with a 5% stacking gel and a 7.5% separating gel (Hollmann et al., 1994). The gel was blotted onto Amersham Hybond ECL nylon membranes (Hollmann et al., 1994). Membranes were blocked with 1× Roti-block (Roth) and were probed (4°C, overnight) with the primary polyclonal anti-GluR6 antibody (kindly provided by Dr. R. Wenthold) diluted in antibody incubation buffer (0.1× Roti-block, 0.1% Triton X-100, 20 mm Tris-HCl, pH 7.6, 140 mm NaCl). Immunoreactive bands were detected by peroxidase-labeled donkey anti-rabbit IgG antibodies (Jackson Laboratory, Bar Harbor, ME) in the case of polyclonal primary antibodies. All antibodies were diluted in antibody incubation buffer and visualized with the chemoluminescence method (ECL detection kit, Amersham).
[3H]KA binding to HEK-293 cell membranes. For the assay of [3H]KA binding to GluR6 and GluR6 mutants present on membranes of transfected cells, the following protocol was used. Adhesive HEK-293 (ATCC No. CRL 1573) cells transfected using the calcium phosphate precipitation technique were harvested in ice-cold 0.5 mm EDTA, 1 mm PMSF, and PBS. After centrifugation at 4000 × g, the pelleted cells (from 10–30 plates, 10 cm diameter) were homogenized with a Teflon–glass homogenizer in ice-cold 50 mmTris-acetate buffer containing 10 mm EDTA, 1 mmPMSF, 30 mg/ml leupeptin, and 0.15 u/ml aprotinin, and centrifuged at 8000 × g. The supernatant was collected and centrifuged at 600,000 × g. The resulting pellets were suspended and homogenized in ice-cold 150 mm NaCl, 50 mm Tris-acetate buffer, pH 7.0, and centrifuged at 600,000 × g. The latter step was repeated twice more in NaCl-free 50 mm Tris-acetate buffer at pH 7.3. After suspension and homogenization of the pellets in NaCl-free 50 mm Tris-acetate buffer at pH 7.3, membranes were frozen and kept in liquid nitrogen until use for the [3H]KA binding assay. Displacement curves were constructed by incubating on ice membranes (50–150 mg protein) with 80 nm[3H]KA (58 Ci/mmol) in a total volume of 250 ml of 50 mm Tris-acetate buffer at pH 7.3 in the presence of increasing concentrations of unlabeled kainate (10 nm, 30 nm, 100 nm, 300 nm, 1 μm, 3 μm, and 10 μm). After 60 min, the membranes were filtered through Whatman GF/C filters that were then washed twice with 5 ml ice-cold 50 mmTris-acetate buffer. The filters were dried in air and counted with scintillation fluid (Lumax-xylene). The specific binding of [3H]KA was defined as the total binding minus the binding obtained in the presence of 1 mm kainate. All experiments were performed in triplicate, and data are shown ± SEM. Binding data were analyzed using the Ligand program (Munson and Rodbard, 1980).
RESULTS
Time and dose dependence of the ConA-induced inhibition of GluR6 desensitization
To achieve optimal conditions for the ConA-mediated inhibition of receptor desensitization during our experiments, we investigated the time and concentration dependence of the lectin-induced effect. Current responses of GluR6-injected Xenopus oocytes were recorded (reference values), and the oocytes were then incubated with 0.1 μm, 1 μm, or 10 μm ConA (calculated for the tetramer) for varying periods of time. Current responses were then recorded for a second time, followed by treatment of all oocytes with 10 μm ConA for 10 min to induce maximal inhibition of desensitization. The current responses measured after the second ConA treatment were taken as 100%. For both kainate- and glutamate-induced currents, ConA-mediated potentiation depended on lectin concentration as well as incubation time (Fig.1A). We observed that for every concentration of ConA used, the kainate-induced current response increased faster than the glutamate-induced response. After a 20 min incubation with the lowest concentration of ConA (0.1 μm), the potentiation of the glutamate-induced currents reached only 0.04% of maximal potentiation, whereas the kainate-induced currents reached 0.3% (Fig. 1A). With a concentration of 1 μm ConA, current potentiation was much closer to the maximum but still significantly less than 100%. Incubation of GluR6 with 10 μm ConA resulted in the maximal increase of amplitudes after 8 min for both kainate- and glutamate-induced currents (Fig. 1A). Therefore, for our standard procedure to inhibit receptor desensitization we chose the incubation conditions to be 10 μm ConA for 8 min. Receptor potentiation was virtually irreversible. Even prolonged washing (several hours) did not return steady-state amplitudes to pre-ConA treatment levels. In fact, ConA-potentiated GluR6-expressing oocytes could be stored overnight without loss of potentiation.
Fig. 1.

A, Time and concentration dependence of the ConA-induced inhibition of GluR6-wt desensitization. GluR6-injected oocytes were incubated with ConA solutions of different concentrations for increasing periods of time. Kainate-evoked (■) and glutamate-evoked (•) current responses were recorded. All oocytes were then incubated with 10 μm ConA (calculated for a tetramer) for 10 min, followed by another recording of current responses. The amplitudes of the second recordings were defined as 100%. ConA solutions of 0.1 and 1 μm did not achieve maximal inhibition of desensitization and apparently are not yet at equilibrium after 20 min of incubation. Treatment with a 10 μm ConA solution resulted in maximal inhibition of desensitization after 8 min for both agonists kainate and glutamate. Values represent means ± SEM (n = 3).B, Decrease of current responses recorded from ConA-treated GluR6-wt during long agonist application to oocytes. Despite the ConA-induced inhibition of desensitization, a residual slow decay of the current can be observed.
We found that even after prolonged ConA treatment the GluR6 current responses still desensitized to a certain extent. After long-lasting post-ConA agonist applications (up to 15 min), the current amplitudes were reduced to 67 ± 3% and 54 ± 6% for kainate and glutamate, respectively, compared with the initial peak current amplitudes (Fig. 1B). This decay could not be prevented by additional ConA incubation (data not shown). Similar observations have been made with GluR6-transfected, ConA-treated HEK-293 cells, where glutamate-evoked currents decrease to 63 ± 11% of the initial peak values (see Fig. 7, top panel). Therefore, receptor desensitization does not seem to be inhibited completely by lectin treatment, but it is significantly reduced or considerably slowed down.
Fig. 7.

Current traces of GluR6-wt and some N-glycosylation mutants, expressed in HEK-293 cells. Inhibition of N-glycosylation by tunicamycin treatment did not abolish current responses of GluR6-wt. Except for tunicamycin-treated cells, current responses were recorded before and after ConA treatment. After lectin incubation, a reduced desensitization level can be observed. This effect is least distinct at the tested mutant, GluR6-NG2, which has only one N-glycosylation site left.
ConA-induced inhibition of GluR6 desensitization is mediated by N-linked carbohydrate side chains
All iGluRs are extensively N-glycosylated (Yue et al., 1995;Everts et al., 1997). This suggested that the desensitization-inhibiting effect of lectins, which are carbohydrate-binding proteins, may be mediated by the carbohydrate side chains attached to the N-glycosylaton sites of the receptors. To test this hypothesis, we analyzed whether the ConA-induced potentiation of current responses can be inhibited by an excess of free carbohydrates that compete with the glycoprotein for the carbohydrate-binding site of the lectin. ConA exhibits affinities for carbohydrate ligands in the order of glucose < mannose <p-nitrophenyl-α-mannopyranoside ≪ branched (α-1,2) oligomannose chains (Goldstein and Poretz, 1986). Therefore, we used glucose, p-nitrophenyl-α-mannopyranoside, and polymannosyl–BSA (an oligomannose ligand with 26 mol mannose per mol BSA) as competitors. The lectin solutions were preincubated with the respective carbohydrate for 10 min before the application to GluR6. We observed that neither glucose norp-nitrophenyl-α-mannopyranoside was able to significantly reduce the desensitization-inhibiting properties of ConA, not even when used at a 1000-fold excess. Presumably, these compounds exhibit a considerably lower affinity for the lectin than the N-linked carbohydrate side chains of the receptor protein (Fig.2A). The carbohydrate side chains of GluR6 expressed in oocytes are presumably high-affinity ligands for ConA, possibly N-linked carbohydrates of the high mannose type (Rogers et al., 1991; Hullebroeck and Hampson, 1992). Therefore, competitors with lower affinities such as glucose orp-nitrophenyl-α-mannopyranoside are unable to prevent binding of the lectin to the glycoprotein; however, they may slow the kinetics of the interaction between ConA and the protein.
Fig. 2.
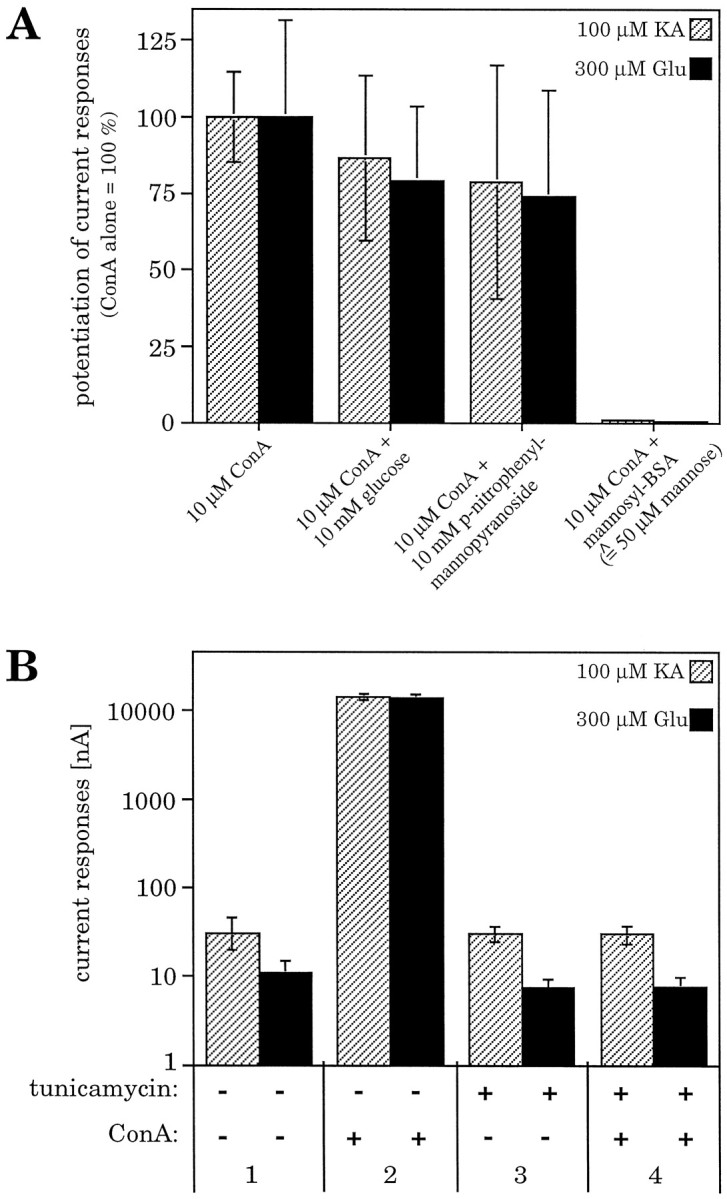
A, Potencies of various soluble carbohydrates for preventing the ConA-induced inhibition of GluR6 desensitization. The carbohydrates compete with the glycosylated receptor molecules for the carbohydrate-binding sites of the lectin. Even in a 1000-fold excess, the monosaccharide glucose orp-nitrophenyl-α-mannopyranoside was not able to significantly reduce the ConA-induced inhibition of receptor desensitization. In contrast, polymannosyl–BSA, equivalent to branched oligomannose residues, inhibits the ConA-induced effect when applied only at fivefold excess (calculated for mannose units). Values represent means ± SEM (n = 3 oocytes).B, The inhibition of GluR6 desensitization by ConA is N-glycosylation dependent. When N-glycosylation was prevented by tunicamycin pretreatment of the oocytes, steady-state current responses of non-N-glycosylated receptors were identical with responses at glycosylated receptors (1, 3). The ConA-induced increase of current amplitudes could only be observed at the glycosylated receptor (2) but was completely abolished at the tunicamycin-treated receptor (4). Values represent means ± SEM (n = 9–11 oocytes).
In line with this interpretation, polymannosyl–BSA almost completely prevented the ConA-induced increase of current responses at an excess as little as fivefold (calculated for mannose-residues) (Fig.2A). This is consistent with the fact that polymannosyl–BSA is a multivalent ligand with high affinity for ConA and therefore competes with the GluR6 subunit for ConA binding, thus abolishing the inhibition of receptor desensitization. This experiment suggests that it is indeed the carbohydrate-binding property of ConA that is responsible for its desensitization-inhibiting activity.
To confirm this result and to exclude that O-glycosylation (Hullebroeck and Hampson, 1992) plays a role in ConA-mediated receptor potentiation, we expressed the kainate receptor subunit GluR6 in N-glycosylation-incompetent Xenopus oocytes. N-glycosylation incompetence was achieved by preinjecting the oocytes with the antibiotic tunicamycin 1 d before injection of cRNA. This procedure completely blocks N-glycosylation of newly synthesized protein (Duksin and Mahoney, 1982; Hollmann et al., 1994; Everts et al., 1997) but leaves O-glycosylation unaffected (Elbein, 1987). The steady-state current amplitudes of GluR6 in tunicamycin-treated oocytes were the same as in untreated controls (Fig. 2B). We also were able to detect GluR6 current responses in tunicamycin-treated HEK-293 cells. However, peak amplitudes were drastically reduced (see Fig. 7, second panel from top), whereas steady-state currents were much less affected. Thus, N-glycosylation is not required for ion channel function at GluR6. In contrast, after ConA incubation, huge differences were observed between tunicamycin-treated and untreated oocytes. Although the receptors expressed in untreated oocytes exhibited an enormous potentiation of the steady-state amplitudes (5293 ± 1218-fold for glutamate-induced currents, 1607 ± 446-fold for kainate-induced currents), the tunicamycin-treated, non-N-glycosylated receptors showed no current increase at all (Fig. 2B). These results unequivocally demonstrate that N-glycosylation is essential for the lectin-induced inhibition of GluR desensitization. The effect is mediated by the N-linked carbohydrate side chains, whereas O-glycosylation does not contribute to the potentiation of the current responses.
GluR6 mutants lacking any single N-glycosylation site still show ConA-induced inhibition of desensitization
To test whether any specific N-glycosylation site is responsible for mediating the inhibition of GluR6 desensitization by ConA, we engineered a series of 10 N-glycosylation site mutants. In each mutant, one of the potential N-glycosylation sites (amino acid consensus sequence N–X–S/T, X ≠ P) (Fig.3) was rendered nonfunctional through site-directed mutagenesis. All mutants were expressed in oocytes, and current responses were recorded before and after treatment with ConA. To calculate the potentiation factors, steady-state amplitudes after ConA treatment were divided by the amplitudes recorded before lectin incubation. All mutants showed a clear increase of the current response (Fig. 4A,B). Thus, in all cases desensitization was still blocked, although one N-glycosylation site was missing. This demonstrates that no single N-linked carbohydrate side chain is the sole mediator of the lectin-induced effect. For most mutants, the potentiation factors were very close or equal to wild-type [GluR6-ΔNG1, GluR6-ΔNG2, GluR6-ΔNG3, GluR6-ΔNG4, GluR6-ΔNG8, GluR6-ΔNG10 (see Fig.4A)]. For GluR6-ΔNG9, however, current amplitudes were dramatically reduced, most likely because of the localization of the mutated amino acid within the putative ion pore-forming domain. Because of its localization, this site would appear unlikely to become glycosylated. In fact, absence of N-glycosylation at site NG9 had been reported earlier by others (Taverna et al., 1994) and was reconfirmed by us (data not shown). Because no current was observed before lectin treatment, the potentiation factors of this mutant could only be approximated, and they represent the minimal possible values. The mutants GluR6-ΔNG5, GluR6-ΔNG6, and GluR6-ΔNG7 exhibited significantly increased currents before ConA treatment compared with wild type (Fig. 4B). Because ConA-potentiated current responses of these three mutants were the same as wild type, their calculated potentiation factors are smaller than the factors found for wild type (Fig. 4A).
Fig. 4.

A, Inhibition of desensitization at GluR6 mutants lacking one N-glycosylation site. Inhibition of desensitization is expressed as potentiation of current responses after ConA treatment. Potentiation factors were calculated as ratio of amplitudes after ConA incubation compared with amplitudes before ConA. Potentiation factors found for kainate- and glutamate-induced current responses, respectively, were 1607 ± 446 and 5293 ± 1218 at GluR6-wt, 2154 ± 582 and 6996 ± 3656 at GluR6-ΔNG1, 2200 ± 973 and 5983 ± 3351 at GluR6-ΔNG2, 980 ± 331 and 3825 ± 2634 at GluR6-ΔNG3, 1814 ± 632 and 5859 ± 1910 at GluR6-ΔNG4, 28 ± 7 and 118 ± 33 at GluR6-ΔNG5, 24 ± 9 and 55 ± 24 at GluR6-ΔNG6, 56 ± 34 and 217 ± 99 at GluR6-ΔNG7, 754 ± 428 and 1930 ± 931 at GluR6-ΔNG8, >5.2▿ and >8.8▿ at GluR6-ΔNG9, and 3860 ± 1914 and 6159 ± 2402 at GluR6-ΔNG10. Values represent means ± SEM (n = 4–7 oocytes). ▿, Estimated minimal potentiation factors. An exact calculation was not possible because no responses could be observed before ConA treatment. Kainate-evoked currents before ConA treatment in these cases were assumed to be maximally 1 nA (which represents the detection limit), whereas glutamate-evoked responses were considered to be maximally 0.5 nA. B, Current traces of GluR6-wt and two representative glycosylation mutants expressed in oocytes. GluR6-ΔNG3 is shown as an example for wild-type-like current responses; GluR6-ΔNG6 exemplifies a mutant with increased current amplitudes before ConA-induced inhibition of desensitization (see Results). The sharp spike in GluR6-wt and GluR6-ΔNG3 represents rapidly inactivating channels, the peak amplitude of which cannot be resolved in oocytes. These spikes are different from those sometimes observed as a result of calcium-activated chloride channels endogenous to the oocyte, as is evidenced by their persistence in calcium-free Ringer’s solution in which CaCl2 has been substituted with 1.8 mmMgCl2 (data not shown).
To determine whether a change of the protein expression levels or of protein transport to the cell surface might contribute to the altered current amplitudes of any of the mutants, we compared the amount of total receptor protein as well as that of plasma membrane-inserted protein in oocytes. Immunological detection of the protein on Western blots revealed that no change could be observed in either total protein preparations or preparations of plasma membrane-inserted protein, except for the mutant GluR6-ΔNG9; the amount of protein of this mutant is significantly smaller than the wild-type level (Fig.5A). The reduced current amplitudes recorded for this mutant therefore can be partly explained by a reduction in protein expression. In contrast, the three mutants that exhibited increased current responses do not show increased protein expression levels (Fig. 5A). Thus in these cases the changed current amplitudes are not likely the result of the presence of more receptor protein but rather of a change in ion channel properties.
Fig. 5.

Western blots of plasma membrane protein preparations. A, GluR6-wt- and ΔNG mutant-injected oocytes. B, GluR6-wt- and NG mutant-injected oocytes.C, GluR6-wt- and EG mutant-injected oocytes.D, Negative control for the plasma membrane protein preparation method (same samples as in C but without incubation with biotinylated ConA; see Materials and Methods). Blots were probed with polyclonal, affinity-purified C-terminal antibodies against GluR6 (provided by Dr. R. Wenthold). Ten nanograms of cRNA had been injected per oocyte. Aliquots of 10 oocytes were loaded per lane. The position of prestained protein markers (83, 119, and 213 kDa) is indicated on the right. Arrows indicate the specific band of the receptor protein. *, Immunoreactive band not related to the heterologously expressed protein because it is recognized by the antibody even in uninjected oocytes. Expression of this band varies considerably even between oocytes from the same frog.
Some of the potential N-glycosylation sites of GluR6 (NG5, NG6, NG7, NG8, and NG10) are located in regions that contribute to the putative ligand-binding pocket (Stern-Bach et al., 1994; Mano et al., 1996). A change of an amino acid or the lack of a carbohydrate side chain in this region potentially could cause a change in the EC50values for agonists. We recorded dose–response curves for the agonists kainate and glutamate for all mutants and calculated EC50values, Hill coefficients (nH), and theoretical maximal current responses (Imax). Only for the mutant GluR6-ΔNG8 were the EC50 values altered significantly. They were increased ∼3.5-fold for glutamate and ∼13-fold for kainate (Table 1). The observation that glutamate and kainate show different increases in EC50 values can be explained by the finding that their binding sites are not identical but rather overlapping (Paas et al., 1996). None of the other mutations altered the ligand-binding properties. The Hill coefficients remain unchanged for all mutants (Table 1), indicating that the cooperativity of ligand binding is not modified.
Table 1.
GluR6 mutants lacking single N-glycosylation sites
| Receptor | Glu | KA | ||||
|---|---|---|---|---|---|---|
| EC50(μm) | nH | Imax(nA) | EC50(μm) | nH | Imax(nA) | |
| GluR6-wt | 37.1 ± 8.4 | 1.0 ± 0.1 | 14378 ± 965 | 0.6 ± 0.04 | 1.3 ± 0.03 | 15263 ± 1112 |
| GluR6-ΔNG1 | 46.8 ± 1.4 | 1.4 ± 0.1 | 14912 ± 3180 | 1.3 ± 0.2 | 1.4 ± 0.1 | 15167 ± 2811 |
| GluR6-ΔNG2 | 34.3 ± 3.8 | 1.5 ± 0.1 | 14312 ± 2905 | 1.0 ± 0.04 | 1.5 ± 0.1 | 14621 ± 2924 |
| GluR6-ΔNG3 | 57.3 ± 2.5 | 1.3 ± 0.1 | 12437 ± 3679 | 2.0 ± 0.3 | 1.1 ± 0.1 | 13120 ± 4204 |
| GluR6-ΔNG4 | 44.3 ± 1.5 | 1.3 ± 0.1 | 4888 ± 916 | 1.7 ± 0.2 | 1.2 ± 0.1 | 5925 ± 735 |
| GluR6-ΔNG5 | 56.1 ± 1.5 | 1.3 ± 0.1 | 14800 ± 2343 | 2.1 ± 0.2 | 1.0 ± 0.1 | 14890 ± 2541 |
| GluR6-ΔNG6 | 29.9 ± 2.1 | 1.5 ± 0.1 | 13556 ± 2035 | 0.8 ± 0.1 | 1.5 ± 0.2 | 14849 ± 2121 |
| GluR6-ΔNG7 | 27.5 ± 2.5 | 1.2 ± 0.1 | 14027 ± 352 | 1.2 ± 0.1 | 1.1 ± 0.1 | 14303 ± 768 |
| GluR6-ΔNG8 | 123.6 ± 3.0 | 1.8 ± 0.2 | 5180 ± 825 | 11.2 ± 2.7 | 1.0 ± 0.1 | 8559 ± 1622 |
| GluR6-ΔNG9 | 60.3 ± 8.5 | 1.0 ± 0.2 | 4.2 ± 1.9 | 1.4 ± 0.2 | 1.0 ± 0.1 | 8.7 ± 2.5 |
| GluR6-ΔNG10 | 52.8 ± 16.1 | 1.3 ± 0.1 | 11100 ± 2378 | 2.8 ± 0.2 | 1.1 ± 0.1 | 11254 ± 2351 |
EC50 values, Hill coefficients (nH), and maximal current responses (Imax) for kainate (KA) and glutamate (Glu) of GluR6-wt and -ΔNG glycosylation mutants in which single native N-glycosylation sites have been rendered nonfunctional. Values represent means ± SEM (n = 3).
To test the assumption that the changed N-glycosylation patterns and not the mutated amino acids produced the observed alteration in receptor function of the mutants GluR6-ΔNG5, GluR6-ΔNG6, and GluR6-ΔNG7, we expressed those mutants in tunicamycin-treated, N-glycosylation-incompetent oocytes. This procedure results in receptors that differ only in the mutated amino acid but not in the extent of N-glycosylation that is lacking in all of them. Under these conditions, the three mutants showed the same current amplitudes as the nonglycosylated wild-type receptor (Fig.6). Thus, the increased current responses of the respective mutants were caused by their specific N-glycosylation pattern. We additionally compared [3H]kainate ligand-binding properties of wild-type GluR6 and mutants GluR6-ΔNG5 and GluR6-ΔNG6. Displacement curves with unlabeled kainate gave IC50 values (kainate concentration causing half-maximal inhibition) of 30 ± 12, 100 ± 17, and 30 ± 12 nm, respectively, indicating little if any detectable differences in ligand binding.
Fig. 6.

Current amplitudes of GluR6-wt and some N-glycosylation mutants, compared in the glycosylated and non-N-glycosylated state. When glycosylated, the mutants GluR6-ΔNG5, GluR6-ΔNG6, and GluR6-ΔNG7 show significantly larger current responses than the wild-type receptor (hatched bars). If N-glycosylation is prevented by tunicamycin treatment, current amplitudes are identical for wild type and mutants (black bars). Values represent means ± SEM (n = 4 oocytes).
One possible reason for the increased current amplitudes of the mutants GluR6-ΔNG5, GluR6-ΔNG6, and GluR6-ΔNG7 before ConA treatment could be a slowing of desensitization kinetics. This assumption is supported by the observation that it takes these mutants much longer than the wild-type receptor to reach a steady-state current plateau (data not shown). Therefore, the observed higher amplitudes might be a result of incomplete desensitization after the standard agonist application time of 10 sec. Indeed, when the agonist was applied for a longer period of time (up to 15 min), a further decrease of the current amplitude could be seen. However, the remaining steady-state current was still significantly higher than that of GluR6-wt. An analysis of the respective current traces led to the estimation that in oocytes the “apparent desensitization time constants” (defined as the half-time necessary to reach the steady-state current) is up to 10 times larger for the N-glycosylation mutants GluR6-ΔNG5, GluR6-ΔNG6, and GluR6-ΔNG7 than for GluR6-wt (data not shown).
To more accurately determine the desensitization properties of these mutants and wild-type GluR6, we transfected HEK-293 cells with the respective cDNAs and recorded current responses to rapidly applied glutamate in lifted whole cells (Fig. 7). Surprisingly, we were not able to detect significant differences in the extent or rate of desensitization between wild type and mutants. The extent of desensitization was 98.9 ± 0.2, 99.7 ± 0.1, 98.5 ± 0.3, and 98.8 ± 0.4% for GluR6-wt, GluR6-ΔNG5, GluR6-ΔNG6, and GluR6-ΔNG7, respectively. Desensitization time constants τ were 4.9 ± 0.2, 3.3 ± 0.3, 4.8 ± 0.4, and 6.6 ± 0.7 msec for GluR6-wt, GluR6-ΔNG5, GluR6-ΔNG6, and GluR6-ΔNG7, respectively. Thus, the altered desensitization properties of the mutants appear to be restricted to theXenopus oocyte expression system. Alternatively, the desensitization observed in oocytes may reflect a different type of desensitization not found in HEK-293 cells. This may be caused by differences in the carbohydrate composition of the N-linked side chains in the different cell types.
Any single native N-glycosylation site is able to mediate the ConA-induced inhibition of GluR6 desensitization
Because single N-glycosylation site deletion mutants behaved virtually like wild-type GluR6 with respect to ConA-mediated current potentiation, we next engineered double, triple, and quadruple deletion mutants. Because these mutations failed to abolish the effect of ConA (data not shown), we went on to investigate the properties of mutants with single N-glycosylation sites. We engineered a second series of nine GluR6-mutants that retained only one each of the nine extracellular N-glycosylation sites. In an additional mutant, all nine extracellular sites were removed. Western Blot experiments revealed that when total protein or plasma membrane protein was examined the expression levels of these mutants were not different from GluR6-wt (Fig. 5B). However, a notable shift to a lower molecular weight was observed because of the lack of most carbohydrate side chains.
The EC50 values of all nine mutants were significantly larger than those of the wild-type receptor. The smallest increase was observed for the mutant GluR6-NG8 (which retained only the N-glycosylation site NG8). In this case, EC50 values were doubled for glutamate and increased ∼4.5-fold for kainate as agonist (Table 2). This finding is in line with our results from the series of single-site mutants in which the mutation introduced to remove NG8 has the strongest impact on the EC50 value. The only mutant of the second series that does not carry this particular mutation is GluR6-NG8. Thus, its EC50 value is lower compared with any of the other mutants. For all other mutants, EC50 values went up distinctly (5- to 10-fold for glutamate and 11- to 27-fold for kainate) (Table 2). The observed changes of the EC50 values are likely caused by the alteration of several amino acids in the ligand-binding domain rather than by the lack of most of the carbohydrate side chains. This interpretation is supported by the fact that GluR6 wild-type receptors expressed in tunicamycin-treated, non-N-glycosylating oocytes exhibit EC50 values identical to those of properly glycosylated receptors (Everts et al., 1997). Competition ligand-binding experiments on one of the mutants, GluR6-NG2, produced IC50 values for kainate of 30 ± 12 nm, which is identical to wild-type GluR6. This indicates that the amino acid exchanges introduced in this mutant affect channel gating (and thus the EC50 value) rather than ligand binding.
Table 2.
GluR6 mutants with single N-glycosylation sites
| Receptor | Glu | KA | ||||
|---|---|---|---|---|---|---|
| EC50(μm) | nH | Imax(nA) | EC50(μm) | nH | Imax(nA) | |
| GluR6-wt | 37.1 ± 8.4 | 1.0 ± 0.1 | 14378 ± 965 | 0.6 ± 0.04 | 1.3 ± 0.03 | 15263 ± 1112 |
| GluR6-NG1 | 271 ± 6 | 1.0 ± 0.1 | 12 ± 2 | 9.3 ± 0.9 | 0.9 ± 0.1 | 34 ± 5 |
| GluR6-NG2 | 394 ± 5 | 1.0 ± 0.1 | 38 ± 6 | 8.1 ± 0.1 | 1.1 ± 0.1 | 100 ± 14 |
| GluR6-NG3 | 389 ± 21 | 1.1 ± 0.2 | 2.6 ± 0.9 | 9.6 ± 0.8 | 1.0 ± 0.1 | 11 ± 6 |
| GluR6-NG4 | 388 ± 27 | 1.0 ± 0.1 | 15 ± 3 | 16 ± 5 | 0.9 ± 0.1 | 29 ± 8 |
| GluR6-NG5 | 328 ± 15 | 1.0 ± 0.1 | 6.0 ± 1.4 | 8.9 ± 0.1 | 1.1 ± 0.1 | 19 ± 5 |
| GluR6-NG6 | 296 ± 25 | 1.1 ± 0.1 | 33 ± 9 | 8.8 ± 1.5 | 0.9 ± 0.12 | 43 ± 19 |
| GluR6-NG7 | 279 ± 2 | 1.2 ± 0.2 | 25 ± 8 | 15 ± 5 | 0.7 ± 0.2 | 56 ± 17 |
| GluR6-NG8 | 91 ± 7 | 1.1 ± 0.1 | 100 ± 18 | 2.7 ± 0.1 | 1.1 ± 0.1 | 219 ± 42 |
| GluR6-NG9 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| GluR6-NG10 | 179 ± 15 | 1.0 ± 0.1 | 5.5 ± 1.3 | 6.8 ± 2.1 | 0.8 ± 0.2 | 36 ± 14 |
| GluR6-EG1 | 314 ± 86 | 1.3 ± 0.1 | 52 ± 17 | 13 ± 3 | 1.3 ± 0.1 | 84 ± 11 |
| GluR6-EG2 | 390 ± 37 | 1.1 ± 0.1 | 4.8 ± 1.2 | 17 ± 4 | 1.1 ± 0.1 | 8.6 ± 2.5 |
| GluR6-EG3 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
EC50 values, Hill coefficients (nH), and maximal current responses (Imax) for kainate (KA) and glutamate (Glu) of GluR6-wt and -NG glycosylation mutants that retain only one of the native N-glycosylation sites or only one ectopic N-glycosylation site. n.d., Not detectable. Values represent means ± SEM (n = 3).
To analyze the lectin-induced modulation of the desensitization properties at mutants with only a single N-glycosylation site, current responses were recorded before and after treatment with ConA. Surprisingly, mutants carrying any single site still showed increased steady-state currents after lectin incubation, although only one N-glycosylation site was left (Fig. 8). Thus, the presence of a single N-glycosylation site is sufficient for lectin interaction. Furthermore, any single carbohydrate side chain is able to mediate the inhibition of desensitization, regardless of its localization in the receptor protein. However, the potentiation factors and maximal current amplitudes are significantly reduced compared with the GluR6 wild type (Fig. 8A,B). Reduced but still significant potentiation of steady-state currents was also observed in HEK-293 cells, as shown in the case of GluR6-NG2 (Fig. 7).
Fig. 8.

A, Inhibition of desensitization at GluR6 mutants retaining only one native N-glycosylation site. Inhibition of desensitization is expressed as potentiation of current responses after ConA treatment. Potentiation factors were calculated as in Figure 4. Except for GluR6-NG9 (see Results), all mutants showed an increase in current responses after lectin incubation. Potentiation factors found for kainate- and glutamate-induced current responses, respectively, were 1607 ± 446 and 5293 ± 1218 at GluR6-wt, 23 ± 8 and 43 ± 17 at GluR6-NG1, 21 ± 7 and 50 ± 22 at GluR6-NG2, 33 ± 17 and >18▿ at GluR6-NG3, 78 ± 42 and 65 ± 35 at GluR6-NG4, 34 ± 17 and >17▿ at GluR6-NG5, 38 ± 13 and 62 ± 29 at GluR6-NG6, 24 ± 8 and 24 ± 6 at GluR6-NG7, 70 ± 19 and 129 ± 31 at GluR6-NG8, 1.2 ± 0.9 and not detectable at GluR6-NG9, and 35 ± 17 and 20 ± 9 at GluR6-NG10. Values represent means ± SEM (n = 5–7 oocytes).▿, Estimated minimal potentiation factors. An exact calculation was not possible because no responses could be observed before ConA treatment. Glutamate-evoked currents before ConA were assumed to be maximally 0.5 nA in these cases. B, Current traces of GluR6-wt and some glycosylation mutants expressed in oocytes. GluR6-NG8 is shown as an example of a mutant with increased current responses after lectin treatment despite the absence of eight of nine N-glycosylation sites. GluR6-NG9 shows no ConA-induced effect because no N-linked carbohydrate side chains are present in this mutant (see Results and Fig. 3).
The only mutant that showed no detectable increase in current responses after ConA treatment was GluR6-NG9, the mutant that according to the now widely accepted three-transmembrane domain topology model has no extracellular N-glycosylation site left because site NG9 is probably located within the membrane (Fig. 3). This result confirms the observations made for tunicamycin-treated, non-N-glycosylated GluR6-wt (Fig. 2B) and strengthens the evidence that the lectin-induced inhibition of desensitization is solely mediated by N-linked carbohydrate side chains.
Even ectopic N-glycosylation sites can mediate ConA-induced inhibition of GluR6 desensitization
Our data are consistent with the conclusion that although N-glycosylation is critical for the action of ConA in inhibiting desensitization, the exact location of the N-glycosylation is not critical. To test this idea further, three more mutants were engineered. At three different positions we introduced ectopic N-glycosylation sites into the N-glycosylation site-deficient mutant GluR6-NG9 (Fig. 3). We chose positions that are located at three distinct positions in the protein: at the N-terminal end of the receptor protein outside the putative ligand-binding domain and far away from any native site (EG1), in the N-terminal part of the ligand-binding domain and in close vicinity to native sites NG6 and NG7 (EG2), and in the extracellular loop between TMD B and TMD C (EG3) (Fig. 3). Because the parent construct GluR6-NG9 has no remaining native extracellular N-glycosylation sites, the ectopic sites were the only sites present in these three mutants. One of these mutants, GluR6-EG3, did not show any measurable current responses before or after lectin incubation, although surface expression of the protein could easily be detected (Fig. 5C). The two other mutants, GluR6-EG1 and GluR6-EG2, formed functional ion channels, and in every respect examined behaved identically to the mutants with a single native N-glycosylation site (Fig. 9). Protein expression levels were not different from wild-type receptor, and the EC50 values were in the same range as for the GluR6-NG mutants (Table 2). The two functional mutants with ectopic N-glycosylation responded to ConA treatment and exhibited inhibition of desensitization, confirming our hypothesis that the exact location of N-glycosylation is not critical. All that is necessary for ConA inhibition of desensitization is that there is at least one N-glycosylation site somewhere in the extracellular domain.
Fig. 9.

A, Inhibition of desensitization at GluR6 mutants carrying a single ectopic N-glycosylation site. Inhibition of desensitization is expressed as potentiation of current responses after ConA treatment. Potentiation factors were calculated as in Figure 4. GluR6-EG1 and GluR6-EG2 are sensitive to ConA treatment, although no native N-glycosylation site is left. GluR6-EG3 did not show detectable current responses under any condition. Potentiation factors found for kainate- and glutamate-induced current responses, respectively, were 1607 ± 446 and 5293 ± 1218 at GluR6-wt, 26 ± 12 and 50 ± 29 at GluR6-EG1, and 9.5 ± 3.4 and >7▿ at GluR6-EG2. Values represent means ± SEM (n = 6–7 oocytes). ▿, Estimated minimal potentiation factor. An exact calculation was not possible because no responses could be observed before ConA treatment. Glutamate-evoked currents were assumed to be maximally 0.5 nA in these cases. B, Current traces of GluR6-EG1 and GluR6-EG2 expressed in oocytes, before and after inhibition of desensitization by ConA. Current amplitudes are significantly increased after lectin treatment.
Intrasubunit cross-linking is not involved in the ConA effect
A widely accepted mechanism for many of the documented lectin-induced biological effects is the cross-linking of proteins or cells caused by binding of the multivalent lectins. To test whether cross-linking might play a crucial role in inhibiting GluR6 desensitization, we compared the effects of the dimeric derivative succinyl-ConA (Gunther et al., 1973) and of the nonderivatized, tetrameric ConA, which is thought to efficiently mediate cross-linking. Treatment of the GluR6-wt subunit with succinyl-ConA resulted in slightly decreased but still significant potentiation factors amounting to ∼70% of ConA-induced potentiation. The same was observed with mutants with only a single native or ectopic N-glycosylation site. Our data exclude the possibility that intrasubunit cross-linking, which in any case is impossible with “monovalent” receptor subunits, is the key mechanism of lectin-induced GluR6 potentiation. Because the dimeric succinyl-ConA has been shown to be considerably less effective in intermolecular cross-linking than the tetrameric form (Gunther et al., 1973), the above findings also suggest that clustering of receptor subunits or receptor complexes does not contribute substantially to the lectin-induced inhibition of GluR6 desensitization.
The activation state of the receptor determines the extent of ConA-induced inhibition of GluR6 desensitization
We further investigated whether the activation state of the GluR6 receptor complex during lectin incubation has any influence on the modulation of desensitization behavior. Our standard lectin incubation was performed on the nonactivated receptor (not agonist bound, not desensitized). To test the ConA effect on the activated, desensitized state, we applied saturating concentrations of agonist to GluR6-injected oocytes for a period long enough to allow the channel to desensitize to a constant steady-state current level. ConA was then added with the agonist continuously present. After the standard incubation time of 8 min, the oocytes were washed, and agonist-induced responses were elicited as usual. Under these conditions, desensitization of GluR6 was inhibited to a much smaller extent as compared with our standard procedure (Fig.10). When the receptors had been desensitized with kainate before lectin incubation, the potentiation factors decreased to 19 ± 4 and 18 ± 6% for kainate- and glutamate-evoked responses, respectively, compared with control values produced by a standard lectin incubation of the nonactivated, nondesensitized receptor in a parallel batch of oocytes. Using glutamate as the desensitizing agonist before ConA treatment, ConA potentiation factors decreased even more (11 ± 6 and 6 ± 3% of the control values for kainate- and glutamate-evoked responses, respectively). Because glutamate is a more strongly desensitizing agonist at GluR6 than kainate, the efficiency of the ConA-induced inhibition of GluR6 desensitization appears to be inversely related to the desensitization state of the receptor during lectin treatment.
Fig. 10.

The extent of lectin-induced inhibition of GluR6 desensitization depends on the activation state of the receptor. Lectin effect on nondesensitized receptors (standard lectin incubation conditions, hatched bars), on receptors desensitized by kainate application (open bars), and on receptors desensitized by glutamate application (black bars). ConA-mediated potentiation in the desensitized state is significantly smaller than in the nondesensitized state. Values represent means ± SEM (n = 3–6 oocytes).
In summary, we conclude that lectin-induced inhibition of desensitization of GluR6 is caused by an irreversible block of the conformational change leading from the activated state to the desensitized state and that any single N-linked carbohydrate side chain on the GluR6 receptor can bind the lectin to mediate this modulatory action.
DISCUSSION
Molecular requirements for ConA-induced inhibition of GluR6 desensitization
The results of the present study shed some light on the molecular mechanism of the ConA-induced inhibition of GluR6 desensitization. We show that there is no single N-linked carbohydrate side chain that by itself is necessary for mediating the lectin effect, because the deletion of any one of the nine N-glycosylation sites does not abolish the lectin-induced receptor potentiation. Rather, the presence of any single N-glycosylation site is sufficient to lead to effective inhibition of desensitization by ConA, no matter whether this site is a native one or has been artificially introduced. Furthermore, the precise location of the lectin-binding site in the extracellular domains of the receptor protein does not seem to be important for the induction of receptor potentiation. This is evident from our observation that every GluR6 mutant with only a single remaining N-glycosylation site, regardless of position, or even with solely an ectopic N-glycosylation site, can still be potentiated by ConA treatment.
The lack of one carbohydrate side chain in mutants GluR6-ΔNG5, GluR6-ΔNG6, and GluR6-ΔNG7 appeared to cause a reduced level of desensitization even in the absence of lectin, resulting in an increased level of steady-state currents. Thus, certain carbohydrate side chains might play a role in the mechanism of desensitization. All electrophysiological measurements were performed with two frequently used GluR agonists: kainate and glutamate. For each mutant tested, glutamate- and kainate-evoked responses are potentiated to a similar extent, indicating that ConA does not discriminate between the slightly different binding sites and/or gating mechanisms of the two agonists. The same appears to be true for other agonists such as domoic acid and methyl glutamate (Everts, 1998). Mutant GluR6-ΔNG8 was the only single-site mutant that showed altered EC50 values for glutamatergic agonists. Because this site is localized within the proposed lobe 1 of the S2 domain of the ligand-binding domain (Mano et al., 1996), the mutation presumably affects the ligand-binding pocket. The lack of any major shift in EC50 values confirms our previous finding (Everts et al., 1997) that carbohydrate side chains are not involved in the formation of the ligand-binding site.
Inhibition of GluR6 desensitization can be achieved by ConA treatment of the receptors in the nonactivated, nondesensitized state or, albeit to a much lesser extent, in the activated, desensitized state (Fig.10). Therefore, the binding site(s) for ConA on the GluR6 protein is obviously not only rendered accessible on ligand binding but is accessible even in the absence of ligand. However, after desensitization on prolonged agonist application, ConA, although definitely still binding, might bind to a different site(s), or maybe to a different conformation of the same site(s). This is evident from the fact that the extent of receptor potentiation by the lectin decreases significantly with progressive desensitization. Similar observations have been made on native locust muscle glutamate receptors (Evans and Usherwood, 1985), the subunit composition of which, however, remains unknown.
All GluR6 mutants with only a single remaining N-glycosylation site can still be activated by agonist application, before as well as after ConA treatment. Thus, whichever of the nine extracellular N-glycosylation consensus sites is retained in the GluR6 mutants, the irreversible binding of ConA does not interfere with ligand binding. In particular, the presence of ConA does not appear to disturb the conformational change(s) that the two lobes of the ligand-binding site (Stern-Bach et al., 1994) have to go through to bind ligand and trigger channel opening. However, it should be possible to find positions in the protein at which, on binding of ConA, ectopic N-glycosylation sites will indeed disturb ligand binding itself or the channel-gating mechanism. Presumably, such sites are in close vicinity to or identical with the residues identified as being involved in ligand binding (Stern-Bach et al., 1994). A possible example for such a site is seen in our mutant GluR6-EG3, which is nonfunctional.
A conformational “lock” may explain the action of ConA
Ligand binding induces a conformational change of the receptor protein that first leads to the opening of the ion pore and then, probably by pulling the two lobes of the ligand-binding site closer together, to the desensitization of the ion channel (Mano et al., 1996). The gating mechanism itself obviously is not hindered by the binding of ConA because large currents can easily be elicited. Quite to the contrary, current responses are potentiated dramatically because channel desensitization is inhibited by the lectin treatment (Mayer and Vyklicky, 1989). Therefore, we postulate that the conformational rearrangement leading from ligand binding to channel desensitization can be divided into two distinct stages: a first step that is ConA insensitive and opens (activates) the channel, and a second step that is ConA sensitive and leads to desensitization.
ConA appears to inhibit the conformational change leading from the active, open state to the desensitized state, and thereby increases the steady-state current of the receptor. The lectin may act to lock, i.e., immobilize, the activatable, nondesensitized receptor conformation. This locking might be caused by the bulky ConA (∼102 kDa) simply hanging on to a domain that needs to make a crucial move when undergoing a conformational change. The bulky mass of the lectin attached to the domain in question may slow down or sterically hinder to some extent (in an extreme scenario even to a stand-still) the movement of such a domain. This model is in line with the finding that even after ConA treatment a very slow desensitization of agonist-evoked current responses at GluR6 can still be observed (Fig.1B) (see Results). The observation that the dimeric derivative succinyl-ConA is somewhat less effective in inhibiting desensitization than the unmodified tetrameric ConA might thus be explained by the smaller molecular weight of the derivative, resulting in incomplete inactivation domain immobilization.
This suggested locking mechanism would not require cross-linking and is therefore consistent with the observation that even “monovalent” GluR6 mutants with only a single N-glycosylation site can be potentiated by ConA. This latter finding effectively rules out intrasubunit cross-linking of N-linked carbohydrates as a possible mechanism of inhibition of desensitization. The observation that even the lectin derivative succinyl-ConA is able to inhibit receptor desensitization (Yue et al., 1995; Everts, 1998) (see Results) also supports a mechanism other than cross-linking. The dimeric succinyl-ConA is frequently used to determine whether a ConA-mediated effect is caused by cross-linking events. Succinyl-ConA cannot assume the usual, tetrameric form of ConA that is thought to mediate cross-linking, whereas its binding properties are identical (Gunther et al., 1973).
The hypothesis of an immobilization of extracellular receptor domains by bound ConA is strengthened by the observation that the extent of lectin-mediated GluR6 potentiation at different glycosylation mutants depends on the number of retained extracellular N-glycosylation consensus sites. The nine mutants that lack only one of nine N-glycosylation sites (i.e., ConA-binding sites) exhibit an average potentiation factor of ∼3450 and ∼1320 for glutamate- and kainate-induced responses, respectively. In contrast, the nine mutants that retain only one single N-glycosylation site show an average potentiation factor of only ∼50 and ∼40 for glutamate and kainate, respectively. Mutants with intermediate numbers of N-glycosylation sites consistently show intermediate potentiation factors (data not shown). It is easy to imagine that immobilization would be more complete if several bulky ConA molecules bind to the receptor.
Although the exact molecular rearrangements that GluRs undergo during ConA binding still have to be worked out, this study demonstrates that lectin-induced modulation is dependent on N-glycosylation and is closely coupled to the mechanism of receptor desensitization but not gating. ConA turns out to be a valuable tool that allows uncoupling of the channel activation step of the kainate receptor GluR6 from the desensitization step. Thus, investigation of lectin-induced modulation of GluRs may help in understanding the mechanism of receptor desensitization that is of crucial importance for synaptic physiology.
Footnotes
This work was supported by a Deutsche Forschungsgemeinschaft Heisenberg fellowship to M.H., a German–Israeli Foundation for Scientific Research and Development grant to M.H. and V.T., and Sonderforschungsbereich 406. We thank Dr. Robert Wenthold (National Institute on Deafness and Other Communication Disorders–National Institutes of Health, Bethesda, MD) for the generous donation of the affinity-purified anti-GluR6 antiserum used in this study.
Correspondence should be addressed to Dr. M. Hollmann, Glutamate Receptor Laboratory, Max-Planck-Institute for Experimental Medicine, Hermann-Rein-Strasse 3, D-37075 Göttingen, Germany.
REFERENCES
- 1.Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duksin D, Mahoney WC. Relationship of the structure and biological activity of the natural homologues of tunicamycin. J Biol Chem. 1982;257:3105–3109. [PubMed] [Google Scholar]
- 3.Egebjerg J, Bettler B, Hermans-Borgmeyer I, Heinemann S. Cloning of a cDNA for a glutamate receptor subunit activated by kainate but not AMPA. Nature. 1991;351:745–748. doi: 10.1038/351745a0. [DOI] [PubMed] [Google Scholar]
- 4.Elbein AD. Inhibitors of the biosynthesis and processing of N-linked oligosaccharide chains. Annu Rev Biochem. 1987;56:497–534. doi: 10.1146/annurev.bi.56.070187.002433. [DOI] [PubMed] [Google Scholar]
- 5.Evans ML, Usherwood PNR. The effect of lectins on desensitisation of Locust muscle glutamate receptors. Brain Res. 1985;358:34–39. doi: 10.1016/0006-8993(85)90945-x. [DOI] [PubMed] [Google Scholar]
- 6.Everts I. PhD thesis. University of Kiel; 1998. N-Glykosylierung an ionotropen Glutamatrezeptoren: Ihre Bedeutung für die Rezeptorfunktion und deren Modulation durch Lektine. . [Google Scholar]
- 7.Everts I, Villmann C, Hollmann M. N-glycosylation is not a prerequisite for glutamate receptor function but is essential for lectin modulation. Mol Pharmacol. 1997;52:861–873. doi: 10.1124/mol.52.5.861. [DOI] [PubMed] [Google Scholar]
- 8.Geoffroy M, Lambolez B, Calvalho LP, Rossier J, Stinnakre J. Concanavalin A potentiates NMDA-evoked responses in the Xenopus oocyte expression system. Eur J Pharmacol. 1989;166:355–356. doi: 10.1016/0014-2999(89)90084-8. [DOI] [PubMed] [Google Scholar]
- 9.Goldstein IJ, Poretz RD. Isolation, physicochemical characterization, and carbohydrate-binding specificity of lectins. In: Liener IE, Sharon N, Goldstein IJ, editors. The lectins: properties, functions, and applications in biology and medicine. Academic; New York: 1986. pp. 33–248. [Google Scholar]
- 10.Gunther GR, Wang JL, Yahara I, Cunningham BA, Edelman GM. Concanavalin A derivates with altered biological activities. Proc Natl Acad Sci USA. 1973;70:1012–1016. doi: 10.1073/pnas.70.4.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hollmann M, Maron C, Heinemann S. N-glycosylation site tagging suggests a three transmembrane domain topology for the glutamate receptor GluR1. Neuron. 1994;13:1331–1343. doi: 10.1016/0896-6273(94)90419-7. [DOI] [PubMed] [Google Scholar]
- 12.Huettner JE. Glutamate receptor channels in rat DRG neurons: activation by kainate and quisqualate and blockade of desensitization by Con A. Neuron. 1990;5:255–266. doi: 10.1016/0896-6273(90)90163-a. [DOI] [PubMed] [Google Scholar]
- 13.Hullebroeck MF, Hampson DR. Characterization of the oligosaccharide side chains on kainate binding proteins and AMPA receptors. Brain Res. 1992;590:187–192. doi: 10.1016/0006-8993(92)91094-u. [DOI] [PubMed] [Google Scholar]
- 14.Mano I, Lamed Y, Teichberg VI. A venus flytrap mechanism for activation and desensitization of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors. J Biol Chem. 1996;271:15299–15302. doi: 10.1074/jbc.271.26.15299. [DOI] [PubMed] [Google Scholar]
- 15.Mathers DD, Usherwood PNR. Concanavalin A blocks desensitization of glutamate receptors on insect muscle fibers. Nature. 1976;259:409–411. doi: 10.1038/259409a0. [DOI] [PubMed] [Google Scholar]
- 16.Mayer ML, Vyklicky L. Concanavalin A selectively reduces desensitization of mammalian neuronal quisqualate receptors. Proc Natl Acad Sci USA. 1989;86:1411–1415. doi: 10.1073/pnas.86.4.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monaghan DT, Bridges RJ, Cotman CW. The excitatory amino acid receptors: their classes, pharmacology, and distinct properties in the function of the central nervous system. Annu Rev Pharmacol Toxicol. 1989;29:365–402. doi: 10.1146/annurev.pa.29.040189.002053. [DOI] [PubMed] [Google Scholar]
- 18.Munson PJ, Rodbard D. LIGAND: a versatile computerized approach for characterization of ligand binding systems. Anal Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- 19.Paas Y, Eisenstein M, Medevielle F, Teichberg VI, Devillers-Thiery A. Identification of the amino-acid subsets accounting for the ligand binding specificity of a glutamate receptor. Neuron. 1996;17:979–990. doi: 10.1016/s0896-6273(00)80228-7. [DOI] [PubMed] [Google Scholar]
- 20.Partin KM, Patneau DK, Winters CA, Mayer ML, Buonanno A. Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin A. Neuron. 1993;11:1069–1082. doi: 10.1016/0896-6273(93)90220-l. [DOI] [PubMed] [Google Scholar]
- 21.Partin KM, Bowie D, Mayer ML. Structural determinants of allosteric regulation in alternatively spliced AMPA receptors. Neuron. 1995;14:833–843. doi: 10.1016/0896-6273(95)90227-9. [DOI] [PubMed] [Google Scholar]
- 22.Pickering DS, Taverna FA, Salter MW, Hampson DR. Palmitoylation of the GluR6 kainate receptor. Proc Natl Acad Sci USA. 1995;92:12090–12094. doi: 10.1073/pnas.92.26.12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raymond LA, Tingley WG, Blackstone CD, Roche KW, Huganir RL. Glutamate receptor modulation by protein phosphorylation. J Physiol (Paris) 1994;88:181–192. doi: 10.1016/0928-4257(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 24.Rogers SW, Hughes TE, Hollmann M, Gasic GP, Deneris ES, Heinemann S. The characterization and localization of the glutamate receptor subunit GluR1 in the rat brain. J Neurosci. 1991;11:2713–2724. doi: 10.1523/JNEUROSCI.11-09-02713.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seeburg PH. The role of RNA editing in controlling glutamate receptor channel properties. J Neurochem. 1996;66:1–5. doi: 10.1046/j.1471-4159.1996.66010001.x. [DOI] [PubMed] [Google Scholar]
- 26.Sommer B, Monyer H, Wisden W, Verdoorn TA, Burnashev N, Sprengel R, Sakmann B, Seeburg PH. Glutamate-gated ion channels in the brain: genetic mechanism for generating molecular and functional diversity. Drug Res. 1992;42:209–210. [PubMed] [Google Scholar]
- 27.Stern-Bach Y, Bettler B, Hartley M, Sheppard PO, O’Hara PJ, Heinemann SF. Agonist selectivity of glutamate receptors is specified by 2 domains structurally related to bacterial amino acid-binding proteins. Neuron. 1994;13:1345–1357. doi: 10.1016/0896-6273(94)90420-0. [DOI] [PubMed] [Google Scholar]
- 28.Taverna FA, Wang LY, Macdonald JF, Hampson DR. A transmembrane model for an ionotropic glutamate receptor predicted on the basis of the location of asparagine-linked oligosaccharides. J Biol Chem. 1994;269:14159–14164. [PubMed] [Google Scholar]
- 29.Thio LL, Clifford DB, Zorumski CF. Concanavalin A enhances excitatory synaptic transmission in cultured rat hippocampal neurons. Synapse. 1993;13:94–97. doi: 10.1002/syn.890130111. [DOI] [PubMed] [Google Scholar]
- 30.Villmann C, Bull L, Hollmann M. Kainate binding proteins possess functional ion channel domains. J Neurosci. 1997;17:7634–7643. doi: 10.1523/JNEUROSCI.17-20-07634.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wo ZGL, Oswald RE. A topological analysis of goldfish kainate receptors predicts three transmembrane segments. J Biol Chem. 1995;270:2000–2009. doi: 10.1074/jbc.270.5.2000. [DOI] [PubMed] [Google Scholar]
- 32.Wong LA, Mayer ML. Differential modulation by cyclothiazide and concanavalin A of desensitization at native α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-preferring and kainate-preferring glutamate receptors. Mol Pharmacol. 1993;44:504–510. [PubMed] [Google Scholar]
- 33.Yue KT, Macdonald JF, Pekhletski R, Hampson DR. Differential effects of lectins on recombinant glutamate receptors. Eur J Pharmacol Mol Pharmacol. 1995;291:229–235. doi: 10.1016/0922-4106(95)90062-4. [DOI] [PubMed] [Google Scholar]