

Abstract
Missense mutations as well as a null allele of the human glycine receptor α1 subunit gene GLRA1 result in the neurological disorder hyperekplexia [startle disease, stiff baby syndrome, Mendelian Inheritance in Man (MIM) #149400]. In a pedigree showing dominant transmission of hyperekplexia, we identified a novel point mutation C1128A of GLRA1. This mutation encodes an amino acid substitution (P250T) in the cytoplasmic loop linking transmembrane regions M1 and M2 of the mature α1 polypeptide. After recombinant expression, homomeric α1P250T subunit channels showed a strong reduction of maximum whole-cell chloride currents and an altered desensitization, consistent with a prolonged recovery from desensitization. Apparent glycine binding was less affected, yielding an approximately fivefold increase inKi values. Topological analysis predicts that the substitution of proline 250 leads to the loss of an angular polypeptide structure, thereby destabilizing open channel conformations. Thus, the novel GLRA1 mutant allele P250T defines an intracellular determinant of glycine receptor channel gating.
Keywords: glycine, hyperekplexia, inhibition, receptor, startle disease, stiff baby syndrome
Strychnine-sensitive glycine receptors (GlyRs) represent a family of ligand-gated chloride channels that exist as pentameric protein complexes. The GlyR isoform prevailing in brainstem and spinal cord of adult mammals is an assembly of ligand-binding α1 and structural β subunits (Betz, 1992; Becker, 1995; Becker and Langosch, 1998). In addition, α2, α3, and α4 subunit genes have been identified in the human and rodents (Grenningloh et al., 1990; Kuhse et al., 1990; Kingsmore et al., 1994;Matzenbach et al., 1994; Nikolic et al., 1998). Mature GlyR subunit polypeptides are thought to cross the postsynaptic membrane four times, with transmembrane segment M2 delineating the inner wall of the anion pore. Determinants of ligand binding have been assigned to the large extracellular N-terminal domain of the α subunit variants (Betz, 1992; Breitinger and Becker, 1998). Glycinergic agonist responses also depend on amino acid residues situated within the extracellular loop linking segments M2 and M3 (Becker and Langosch, 1998). The human genes encoding the α1 (GLRA1), α2 (GLRA2), α3 (GLRA3), and β subunits (GLRB) have been localized to the chromosomal regions 5q32, Xp21.2-p22.1, 4q33-q34, and 4q31.3, respectively (Grenningloh et al., 1990; Shiang et al., 1993; Baker et al., 1994; Shiang et al., 1995;Handford et al., 1996; Milani et al., 1998; Nikolic et al., 1998).
Glycine binding is efficiently antagonized by the plant alkaloid strychnine, which produces both increases in muscle tone and exaggerated startle responses to external stimuli (Becker, 1995). Symptoms of the human neurological disorder hyperekplexia [startle disease, stiff baby syndrome, STHE, Mendelian Inheritance in Man (MIM) #14940] are reminiscent of strychnine-induced GlyR dysfunction (Tijssen et al., 1995). Affected infants display exaggerated startle responses and severe muscle stiffness, which may result in fatal apnea. During the first year of life, muscle tone returns to normal whereas excessive startling, which may culminate in immediate, unprotected falling, persists into adulthood (Ryan et al., 1994; Tijssen et al., 1995; Brune et al., 1996). Dominant traits of hyperekplexia were found to correlate to GLRA1 missense mutations affecting segment M2 and the extracellular M2-M3 loop (Shiang et al., 1993, 1995; Elmslie et al., 1996; Milani et al., 1996). In two recessive traits, amino acid exchanges have been identified within segment M1 (Rees et al., 1994;Becker and Langosch, 1998). Moreover, homozygosity for a null allele demonstrated that the complete loss of GLRA1 gene function may be tolerated in the human (Brune et al., 1996). Homologous phenotypes shown by mouse lines carrying GlyR α1 and β mutant alleles further support the causative role of GlyR alterations in hypertonic motor disorders (Mülhardt et al., 1994; Ryan et al., 1994; Saul et al., 1994; Kling et al., 1997).
This study reports on a novel GLRA1 allele causing dominant hyperekplexia. A missense mutation results in the substitution of P250, which is located within the intracellular M1-M2 loop. Recombinant α1P250T receptors displayed moderate changes in agonist affinity yet dramatic alterations in chloride conductance, defining proline (α1)250 as an important intracellular determinant of GlyR channel gating.
MATERIALS AND METHODS
GLRA1 gene structure, single-strand conformation polymorphism analysis, and sequencing of genomic DNA
The numbering scheme for GLRA1 gene structure used here follows the designations given by Matzenbach et al. (1994) for the murine GlyR subunit genes. This is consistent with recent revisions of the GLRA1 gene structure (Shiang et al., 1993, 1995). Genomic DNA was obtained by phenol/chloroform extraction of peripheral blood leukocytes from participating family members. PCR amplification of GLRA1 exons and subsequent single-strand conformation polymorphism (SSCP) analysis at constant temperatures (10, 15, 20, and 25°C) was performed as described (Milani et al., 1998). After the detection of an informative polymorphism, amplimers of exon 7 were cloned into pBluescript II SK− (Stratagene, La Jolla, CA) and subjected to DNA sequencing. Direct sequencing of genomic PCR amplimers was performed on an Applied Biosystems Prism 377 automated DNA sequencer.
Generation and expression of GlyR α1 subunit constructs
GlyR α1 subunit cDNAs (Grenningloh et al., 1990) were cloned into a pSP64T-derived vector (Krieg and Melton, 1984). Employing the oligonucleotide-directed PCR mutagenesis method of Ho et al. (1993), the point mutation C1128A coding for the mutant subunit α1P250T was introduced to the cDNA construct. For functional expression in Xenopus laevis oocytes, recombinant full-length plasmids were used to generate synthetic capped and polyadenylated cRNA using SP6 RNA polymerase (Promega, Madison, WI). The cRNAs were purified by phenol/chloroform extraction, and ribonucleotides were eliminated using Chromaspin columns (Clontech, Palo Alto, CA). RNA contents were quantified photometrically. For expression in the human embryonic kidney cell line (HEK 293), the α1 and α1P250T cDNAs were cloned into the vector pCIS in which the human cDNA was expressed under the control of the cytomegalovirus promotor. The cells were transfected as described for 48 hr and subjected to biochemical and physiological analysis (Sontheimer et al., 1989).
Membrane preparation and [3H]strychnine binding assay
Crude membrane fractions were prepared from transfected cells as described (Sontheimer et al., 1989). For radioligand displacement, membranes were incubated with 16.7 nm[3H]strychnine (DuPont NEN, Boston, MA; specific activity 30 Ci/mmol) and increasing concentrations of unlabeled ligands. Specific binding to membrane fractions was determined in triplicate by filtration assay using 50 μg of total protein (Kling et al., 1997). Binding data were analyzed by a nonlinear algorithm provided by the GraphPad program.
Electrophysiological recordings
Recording conditions and dose–response relationships. Whole-cell recordings (ambient temperature) fromXenopus laevis oocytes were performed on a two-microelectrode voltage-clamp system (Kuner and Schoepfer, 1996). Oocytes were perfused with Ringer’s solution containing (in mm) 115 NaCl, 2.5 KCl, 1.8 CaCl2, 1 MgCl2, and 10 HEPES, adjusted to pH 7.2 with NaOH. Glycine-induced currents were recorded from outside-out patches (Hamill et al., 1981), using an EPC-9 amplifier with Pulse software (Heka electronics GmbH, Lambrecht, Germany). Solutions were applied using a Piezo-driven double-barrel fast application system (Colquhoun et al., 1992). The solutions (pH 7.2) consisted of either Mg2+-free Ringer’s solution (external), or (in mm) 100 KCl, 2 MgCl2, and 10 HEPES (internal).
Dose–response curves were constructed from peak currents induced by seven appropriately spaced concentrations of glycine at a holding potential of −70 mV. Data were fitted to the Hill equation to derive the EC50 and Hill coefficient using the program Igor (WaveMetrics, Inc., Lake Oswego, OR). For homomeric α1 channels, EC50 values depended on the total current expression (Saul et al., 1994), whereas such a relation was not detectable for α1P250T channels. With whole-cell currents exceeding −10 μA, the EC50 for glycine was 0.08 ± 0.01 mm (n = 3) in α1 channels, displaying a slightly biphasic dose–response (data not shown). For current values more than −4 μA, the EC50 for glycine was 0.24, and the dose–response was monophasic (see Results), consistent with observations by Taleb and Betz (1994).
Current expression levels and ion selectivity. Oocytes were injected with cRNA solution (23 nl, 100 ng/μl) using a Nanoject Injector (Drummond Inc., Broomall, PA). For both α1 and α1P250T channels, the peak currents elicited by saturating glycine concentrations (α1, 1 mm; α1P250T, 10 mm) were quantified in 10 different oocytes. As current expression levels may vary among different batches of oocytes, the ratioIwt/Imut was calculated from the average currents determined for the same batch of oocytes. Ratios averaged from three different batches were taken as the mean difference in current expression between α1P250T and α1. The reversal potential of glycine-induced whole-cell currents was determined by changing the voltage rampwise from −60 mV to +40 mV within 2 sec. Ramps recorded in the absence of glycine were subtracted from ramps recorded in the presence of glycine. Two such glycine-activated ramps were recorded before, during, and after exposure to 50% diluted Ringer’s solution. Reversal potentials were corrected for liquid junction potentials. Assuming that cytoplasmic Cl− concentrations ofXenopus oocytes are in the range of 100–110 mm, the Nernst equation predicts shifts of 12.7–15.1 mV, respectively.
Kinetic parameters and current–voltage (I–V) curves. Whole-cell current signals were low-pass filtered at fc = 3.3 kHz and digitized at 10 kHz. The current traces (decaying part: 300 msec, starting at the peak) were fitted to single (Eq. 1: y = k0 +k1 * exp(−x/τ)) or double (Eq. 2:y = k0 +k1 * exp(−x/τ1) +k2 * exp(−x/τ2)) exponential functions to derive the decay time constants (τ). The rate of solution exchange (20–80% rise time, typically 3 msec) was determined after each experiment by application of 10% Ringer’s solution to the recording pipette (open-tip response). Voltage steps were repeatedly applied with increments of 10 mV from -100 to +100 mV. To ascertain recovery from desensitization, single steps were separated by 5 sec (α1) or 10 sec (α1P250T). Glycine was applied for 400 msec within a voltage step lasting for 600 msec.
Single-channel analysis. Single-channel currents were low-pass filtered at 10 kHz, digitized with a modified pulse-code modulation device (Sony, model ES 701), and recorded on videotape. For analysis, data were replayed from tape, low-pass filtered at fc = 2.5 kHz with the help of an eight-pole low-pass Bessel filter (Frequency Devices, Haverhill, MA), and digitized at 10 kHz using the analog-to-digital interface of the EPC-9 (Heka). Amplitudes were determined manually using MacTAC (Skalar Instruments, Inc., Seattle, WA). Three patches from different batches of oocytes were analyzed for α1 constructs, and for each patch 500–1000 events were considered. Three patches expressing α1P250T channels were analyzed with nonstationary variance analysis as described by Spruston et al. (1995). Briefly, the mean variance (ς2) of 10–40 current responses to pulses of 1 mm glycine was plotted as a function of the mean current of all responses analyzed and fitted to Equation 3: ς2 = iI − (1/N) * I2 + ςb2(I, total current; i single-channel current;N, number of channels in the patch; ςb2, mean background variance).popen was determined from the relationI/N * i.
RESULTS
Occurrence of hereditary hyperekplexia in family BS was diagnosed clinically, and the mode of inheritance was indicative of dominant transmission (Gabriel and Lenard, 1984). Of 17 family members participating in genetic examination, hyperekplexia was diagnosed in 10 subjects. In some of the cases, generalized stiffness was reported in early infancy, which largely disappeared within the first year of life. The spectrum of clinical symptoms varied from excessive startle reactions to a predominance of muscular hypertonia. Consistent with the guidelines of the local committee on ethics, informed consent was obtained from all individuals participating.
Genomic DNAs of members of family BS were subjected to SSCP screening for GLRA1 mutant alleles. After amplification of sequences corresponding to GLRA1 exon 7, a polymorphism linked to the disorder was identified (Fig.1A). Cloning of the corresponding DNA amplimer and sequencing of nine recombinants revealed a single nucleotide substitution, C1128A, encoding a threonine residue instead of a proline in position 250 of the mature α1 polypeptide (Fig. 1B). In addition, heterozygosity for the mutant allele was confirmed by direct sequencing of genomic PCR amplimers (data not shown). Presence of theGLRA1P250T allele was associated with hyperekplexia in family BS, with the exception of one individual showing mild startle reactions in addition to a pronounced fear syndrome, who was found to be homozygous for the normal alleleGLRA1. This patient was not available for further physiological exploration of reflex latencies indicative of startle disease (Brune et al., 1996). As a younger sibling to an affected individual, however, this patient may suffer from a behavioral disorder producing a phenocopy of hyperekplexia.
Fig. 1.

A, Hyperekplexia allele of theGLRA1 gene in family BS. A, Pedigree of family BS. Affected individuals are indicated by filled symbols and unaffected individuals by open symbols. Only individuals volunteering for participation are included, and birth order was altered to avoid identification of affected individuals. SSCP conformers of DNA samples are depicted beneath the symbols of the corresponding individuals. Theasterisk denotes an individual displaying mild startle reactions, in addition to a pronounced fear syndrome, who was found to be homozygous for the normal allele GLRA1.B, Analysis of a normal and the hyperekplexia allele ofGLRA1. The nucleotide substitution (C → A) corresponding to position 1128 of the cDNA predicts the amino acid exchange P250T in the hyperekplexia α1 subunit allele (coding strand, gel lanes: G, A, T, C). The amino acid sequences (single letter code) encoded by the two DNA ladders and reading frombottom to top are listed next to the gel patterns.
Within the transmembrane topology predicted for GlyR subunit polypeptides (Becker and Langosch, 1998), amino acid position 250 locates to the cytoplasmic loop linking transmembrane segments M1 and M2. As noted earlier (Galzi et al., 1992), sequence alignments show that all glycine and GABAA receptor α polypeptides known carry a proline residue in the homologous position (Fig.2). Analysis of the protein secondary structure with the Chou–Fasman algorithm (Chou and Fasman, 1974) predicted that the substitution of P250, which is likely to confer an angular conformation on a peptide sequence, by a threonine residue significantly increases the propensity to form a continuous α-helical structure (data not shown). Although the success of predictive methods is hard to assess in individual cases, this nevertheless suggests that the P250T mutation induces a major change in secondary structure.
Fig. 2.

Alignment of amino acid sequences of wild-type and mutant glycine, and GABAA receptor subunits. Sequences represent the cytoplasmic loop between transmembrane segments M1 and M2 including the flanking regions. The last rowindicates point mutations encoded by the GLRA1 mutant alleles. Positions of transmembrane regions M1 and M2 are marked. The amino acid exchange P250T is given in bold. Sequences were retrieved from the EMBL nucleotide sequence database (http://www.ebi.ac.uk/embl.html).
To characterize the functional properties of GlyRs comprising the α1P250T subunit, mutant constructs were generated by site-directed mutagenesis from wild-type α1 cDNAs. After transfection with wild-type and mutant receptor constructs, HEK 293 cells were subjected to Western blot analysis using monoclonal antibody (mAb) 4a, which defines an epitope common to all GlyR α subunits (Becker et al., 1988; Sontheimer et al., 1989). For both α1 and α1P250T constructs, an immunoreactive polypeptide band of 48 kDa was observed (Fig.3A). No differences in staining intensities were detectable, indicative of similar expression efficiencies of these cDNA constructs. The ligand-binding properties of recombinant α1 and α1P250T GlyRs were determined by [3H]strychnine binding to membrane fractions of transfected cells (Sontheimer et al., 1989). Equal numbers of binding sites (α1, 22.68 pmol/mg; α1P250T, 22.18 pmol/mg of membrane fraction) became apparent for both constructs confirming the conclusion that α1 and α1P250T subunit proteins are present in the eukaryotic expression system at roughly equal amounts. However, the apparent glycine-binding affinities derived from displacement assays were 5.5- to 6-fold lower for α1P250T than for α1 GlyRs (Ki values, 36 ± 2 μm for α1 vs 205 ± 33 μm for α1P250T) (Fig. 3B). Binding affinities for the agonists β-alanine and taurine were also reduced with α1P250T GlyRs (data not shown).
Fig. 3.

Properties of the recombinant α1P250T receptor protein. A, Western blot of membrane preparations from HEK 293. Immunostaining by monoclonal antibody mAb 4a, which specifically recognizes GlyR α subunits, produced no detectable differences between cells transfected with the wild-type (wt) or the mutated (P250T) α1 cDNA construct.B, Ligand-binding properties of recombinant GlyR α1 and α1P250T receptors. Values present displacement of [3H]strychnine binding by unlabeled strychnine and glycine.
To assess the influence of the P250T substitution on physiological properties of recombinant GlyR channels, α1 and α1P250T cRNAs were injected into Xenopus laevis oocytes. Whole-cell current responses were recorded from the oocytes, and dose–response characteristics (EC50) were established by application of various glycine concentrations (Fig.4A,B). For α1 channels, an EC50 value of 0.24 ± 0.02 mm glycine (mean ± SEM, n = 6; currents more than −4 μA, see Materials and Methods) was determined, whereas α1P250T channels produced half-maximal responses at 0.54 ± 0.03 mm glycine (n = 6). The corresponding Hill coefficients were 3.4 ± 0.1 in α1 channels and 1.9 ± 0.1 in α1P250T channels (Fig. 4C). The observed differences were statistically significant (p < 0.01, unpaired Student’s ttest). Furthermore, both constructs differed with respect to the maximum current amplitude elicited by application of saturating glycine concentrations (Fig. 4). Oocytes injected with identical amounts (2.3 ng) of either wild-type or mutant cRNA produced currents of 11.4 ± 7.3 μA (n = 29, mean ± SD) and 1.2 ± 0.8 μA (n = 28), respectively. To account for the large batch-specific variability of whole-cell current expression levels, the ratioIα1/Iα1(P250T) was separately assessed for each batch. The average ratio was 10 ± 2 (mean ± SEM) for three batches, indicating that mutant channels yielded approximately 10-fold smaller currents.
Fig. 4.
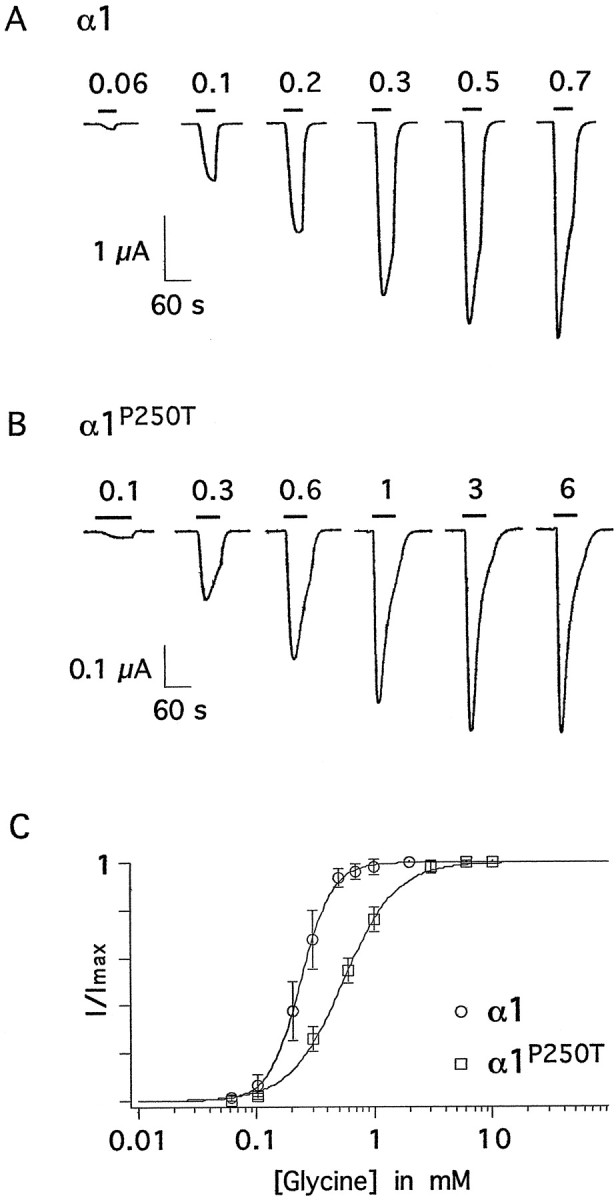
Whole-cell current responses of recombinant α1 and α1P250T receptor channels. A,B, Whole-cell current responses at a holding potential of −70 mV elicited by different concentrations of glycine applied to oocytes expressing homomeric α1 (A) or α1P250T (B) receptor channels. Bars indicate glycine applications; concentrations are millimolar. Note that the vertical scales for wild-type (A) and mutant (B) channels are different. C, Dose–response curves for wild-type (circles) and mutant (squares) receptor channels. EC50 values for glycine and Hill coefficients are presented in Results.
In ligand-gated ion channels of the nAChR type, mutations of an amino acid residue homologous to position GlyR α1(250) contribute to alterations of ion selectivity (Galzi et al., 1992). By analogy, we analyzed whether the anion selectivity of GlyR channels is affected by the P250T substitution. Replacing the external Ringer’s solution with a 50% diluted Ringer’s solution revealed no significant difference in the shift of the reversal potential between α1 channels (12.5 ± 0.3 mV; n = 4) and α1P250T mutant channels (14.1 ± 0.7 mV; n = 4). Indeed, these values are close to the reversal potential predicted from the Nernst equation (13–15 mV, data not shown). As expected for a Cl− selective conductance, the reversal potential of α1 and α1P250T mutant channels (−24 ± 2 mV; n = 7) was close to the Cl−equilibrium potential of Xenopus oocytes (−22 mV; Fraser et al., 1993). Hence, mutant channels remained anion-selective, but exhibited a moderate reduction in the apparent glycine affinity and a strong reduction of the maximum current amplitude.
Comparing the traces shown in Figure 4, A and B, reveals that α1P250T mutant channels desensitize more strongly than α1 channels. Indeed, rapid desensitization of mutant channels, eluding detection by means of whole-cell recordings caused by large oocytes and slow agonist application, may account for the decreases in whole-cell currents and increases in EC50for glycine that we observed. To further investigate GlyR desensitization, we determined the macroscopic kinetic parameters of the current response elicited by brief applications of saturating glycine concentrations to channels present in outside-out patches. Currents mediated by α1 channels showed a rapidly desensitizing component at negative potentials, whereas at positive potentials desensitization was only weak (Fig.5A). The desensitization, i.e., the decay of the inward current in the continued presence of glycine, could best be fitted with a double exponential function, yielding τ1 = 12 ± 3 msec and τ2 = 192 ± 78 msec (mean ± SEM, n = 5). The current–voltage (I–V) relation of the peak current was essentially linear, whereas the inward plateau current was reduced in a voltage-dependent manner (Fig. 5B). Currents mediated by α1P250T channels, in contrast, were strongly desensitizing at both positive and negative potentials, without reaching a discrete plateau (Fig. 5C). Consistent with the lack of the fast initial current component, the desensitization could be fitted with a single exponential function, giving rise to τ = 261 ± 52 msec (n = 5). Mutant α1P250T channels exhibited a slightly outwardly rectifying I–V relation of both peak and “plateau” current (Fig. 5D). In both types of channels, the peak current and the plateau current reverse direction at the same potential, indicating that the observed desensitization of the current in fact reflects true desensitization rather than a chloride shift (Akaike and Kaneda, 1989). Figure 5E directly compares normalized current responses, emphasizing the biphasic versus monophasic desensitization of α1 and α1P250Tchannels, respectively. Currents mediated by α1 channels reach a plateau accounting for ∼30–50% of the initial peak current, whereas most α1P250T channels desensitized within ∼1 sec, consistent with a prolonged phase of recovery from desensitization. Taken together, the 10-fold reduction in whole-cell current amplitudes observed does not reflect fast desensitization of the mutant channels relative to wild-type. Rather, the extent of desensitization in mutant channels, reflecting a slower rate of recovery from densitization, may most significantly contribute to this difference.
Fig. 5.

Kinetics of recombinant α1 and α1P250T receptor channels. A, Fast application of saturating concentrations of glycine (thick bar) to outside-out patches containing homomeric α1 channels. The 400 msec pulse of 1 mm glycine elicited outward currents at +70 mV (top trace) and inward currents at −70 mV (bottom trace). The dotted lineindicates the baseline, currents are corrected for the leak.B, Current–voltage (I–V) relation of wild-type channels. Filled symbols are theI–V relation of the peak current, andopen symbols show the I–Vrelation determined 400 msec after the peak (plateau). The current reverses at ∼0 mV. C, Same as in A for homomeric α1P250T channels, with thebar indicating the application of 10 mmglycine. Note the different dimension of the vertical scale bar in comparison with A. D,I–V relation of mutant channels, seeB: 400 msec after exposure to glycine. E, Comparison of the desensitizing component of the current mediated by wild-type and mutant channels. Both traces are normalized to their respective peak currents. The horizontal bar indicates the application of a 1.5 sec pulse of glycine. Note the different time scale as compared with A and C. The data were low-pass filtered at fc = 300 Hz and digitized at 1 kHz.
To further elucidate whether a change in microscopic kinetic properties or a reduction of the single-channel conductance may account for the current reduction, we evaluated single-channel currents of mutant and wild-type channels in outside-out patches. Single-channel openings of α1 channels in the presence of 100 μm glycine (Fig.6A, top trace) exhibited a predominant conductance state of ∼80 pS as previously observed (Bormann et al., 1993). In contrast, application of 1 mm glycine to outside-out patches containing α1P250T channels elicited currents reminiscent of whole-cell current responses, but with a very small amplitude (Fig.6A, bottom trace). This was consistently observed in eight patches with currents ranging from 1 to 10 pA and might be explained by the presence of multiple small conductances in the patch. The current amplitudes were dependent on the glycine concentration and returned to baseline during the continued presence of glycine (data not shown). Although no distinct single-channel events could be detected for α1P250T channels, the increased noise after application of glycine is consistent with the presence of open channels (Fig. 6A, bottom trace). Indeed, nonstationary variance analysis (Fig.6B,C) predicted the presence of minute, short-lived channel conductances in outside-out patches containing α1P250T channels with a mean conductance of 1.3 ± 0.2 pS (n = 3) and an open probability of 0.02 ± 0.01. Given the very low open probability, the error associated with these estimates of kinetic parameters might be large. Nevertheless, the single-channel kinetics of α1P250T channels are strongly different from those of α1 channels (53.5 ± 12.8 pS; popen = 0.7 ± 0.4), consistent with an impairment of channel gating. Because native GlyRs exist as α/β heteromers, desensitization behavior and single-channel properties of heteromeric channels comprising either α1P250T or α1 subunits were analyzed after coexpression with the β subunit in HEK 293 cells. None of these properties was detectably different for α1P250T and α1P250T/β channels (H.-G. Breitinger, unpublished observations).
Fig. 6.
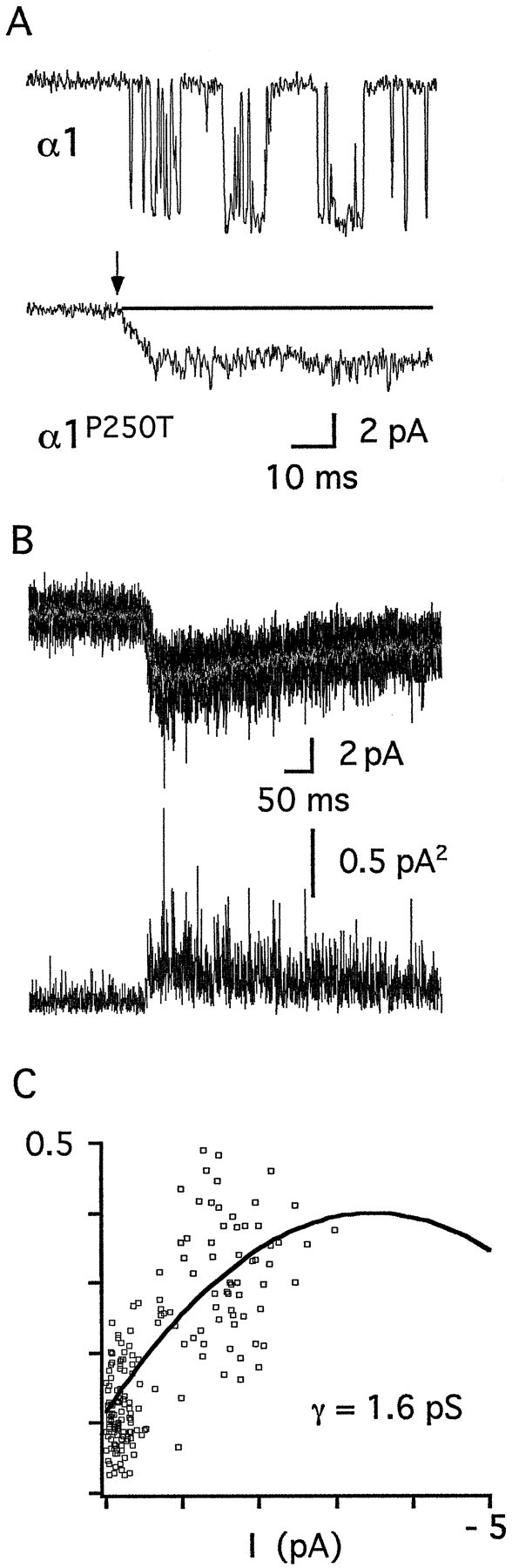
Single-channel properties of recombinant α1 and α1P250T receptor channels. A, Thetop trace shows single-channel currents of homomeric wild-type channels recorded from an outside-out patch in the presence of 100 μm glycine. The bottom trace shows an experiment with a patch containing multiple homomeric mutant receptor channels, with the arrow indicating the application of 1 mm glycine. In the continued presence of glycine, the baseline (dotted line) was reached within 1 sec. Note the increase in noise after the application of glycine. The holding potential was −100 mV. For display, data were refiltered at fc = 1 kHz. B, Nonstationary variance analysis of outside-out patches. The top panel shows 10 superimposed traces with the mean current printed ingray. The bottom panel shows the mean variance plotted versus time. C, Plot of the mean variance obtained from a total of 30 responses in this patch as a function of the mean current. The data were fitted with Equation 3 (see Materials and Methods), the obtained parameters are:i = 1.6 pS; popen = 0.01; N = 42.
DISCUSSION
Inhibitory GlyRs are ligand-gated chloride channels that represent pentameric assemblies of glycine-binding α1 polypeptides and structural β subunits, as analyzed in spinal cord of adult rodents (Betz, 1992). The human neurological disorder hyperekplexia has previously been attributed to various mutant alleles ofGLRA1, the gene encoding the GlyR α1 subunit. These hyperekplexia alleles of GLRA1 predict amino acid substitutions which, according to the generally accepted model of GlyR transmembrane topology, reside within a region ranging from M1 to the extracellular M2-M3 loop (Becker and Langosch, 1998). Here, we characterize a novel GLRA1 allele resulting in dominant hyperekplexia in which the codon encoding proline 250 of the normal allele is mutated into a threonine codon. After recombinant expression, the exchange of this proline residue located within the intracellular M1-M2 loop strongly diminished glycine-induced chloride conductances rather than agonist binding.
Screening for GLRA1 mutations of a large pedigree with dominant hyperekplexia resulted in the identification of the novel allele GLRA1P250T. Although heterozygosity for this allele was associated with hyperekplexia, developmental as well as interindividual variations of clinical phenotypes became apparent between affected individuals. Indeed, the spectrum of symptoms varied from excessive startle reactions triggered by unexpected sounds to a predominance of muscular hypertonia. Distinction has been made between a “major” and “minor” form of this disease in which the latter could not be assigned to GLRA1 mutant alleles (Tijssen et al., 1995). In family BS, however, variations in phenotype are most likely explained by differences in genetic penetrance of theGLRA1P250T allele because of as yet unidentified background genes modulating the clinical presentation ofGLRA1 mutations. The allele GLRA1P250Tfurther adds to the genetic heterogeneity of hyperekplexia. Although the number of hyperekplexia alleles as yet identified precludes any definitive conclusions, an interesting relationship emerges between transmembrane topologies and modes of inheritance of α1 subunit mutations. The amino acid substitutions Q266H (Milani et al., 1996), R271Q/l (Shiang et al., 1993), K276E (Elmslie et al., 1996), and Y279C (Shiang et al., 1995) associated with dominant hyperekplexia cluster within or adjacent to the channel-lining M2 segment of the predicted α1 polypeptide. Heterologous expression shows that these mutations impair agonist binding and/or channel gating of mutant receptors, suggestive of a negative dominant effect resulting in a partial loss of function (Langosch et al., 1994; Rajendra et al., 1994;Laube et al., 1995; Lynch et al., 1997). In contrast, the recessive missense mutations predict exchanges located within M1 or the large N-terminal domain, i.e., S231R (Becker and Langosch, 1998), I244N (Rees et al., 1994), and, in the spasmodic mouse, A52S (Ryan et al., 1994; Saul et al., 1994). By both criteria, its dominant mode of inheritance and a close proximity of the site of amino acid exchange to M2, the allele GLRA1P250T would be assigned to the first group of missense mutations.
The proline residue affected by this mutation is conserved in the homologous position of all GlyR and GABAA receptor α polypeptides known (Fig. 2), suggesting a functional selection pressure on this site for ligand-gated anion channels. On the other hand, insertion of a proline plus an additional amino acid residue into the corresponding region of recombinant α7 subunits of the nicotinic acetylcholine receptor changes the channel selectivity from cationic to anionic (Galzi et al., 1992). However, the loss of this proline residue from recombinant GlyR α1P250T receptor channels did not detectably alter ionic selectivity, but strongly affected glycine-mediated current responses. Which mechanism underlies the reduction in whole-cell current amplitudes? Based on Western blot analysis and an equal number of ligand-binding sites, membrane insertion of the receptor protein appears to be undisturbed. Consistent with the assignment of determinants of agonist binding to the large N-terminal domain (Betz, 1992), apparent glycine binding was only weakly affected by the intracellular amino acid substitution P250T. However, reductions in apparent agonist-binding affinities may also be secondary to changes in receptor conformations associated with gating (Colquhoun and Farrant, 1993). Indeed, the observation that affinities for the antagonist strychnine were not altered in α1P250T receptors supports the notion that the ligand-binding domain remained unaffected by the mutation. Moreover, dose–response analysis of whole-cell currents revealed a pattern characteristic for a gating-deficient channel (Spivak, 1995). Although the EC50 value was only slightly shifted, the Hill coefficient was decreased, and the maximal current amplitude was strongly reduced in α1P250T channels. Mutant channels differed from the wild-type in their desensitization and resensitization properties, consistent with an alteration in channel gating. Single-channel analysis suggested a pronounced change in microscopic gating kinetics, combined with a decrease in single-channel conductance. Taken together, the novel hyperekplexia alleleGLRA1P250T defines an intracellular determinant of GlyR channel gating. This is consistent with previous observations on the recombinant α1 subunit mutants W243A, I244N, and I244A, which exhibit increased desensitization rates, implying that the M1-M2 loop in toto contributes to GlyR desensitization properties (Lynch et al., 1997). At present, it is not entirely clear how these changes in functional properties relate to GlyR protein architecture. Sterical analysis (Chou and Fasman, 1974) predicted the substitution of proline 250 by threonine to change an angular into a helical polypeptide structure. However, the mutant α1P250A has been shown to display only slightly altered whole-cell currents (Lynch et al., 1997). Considering the statistical nature of structural predictions and the limited effect of the P250A mutation on channel properties (Lynch et al., 1997), the amino acid substitution P250T may nevertheless be speculated to disturb a hinge function of the M1-M2 loop, which positions the adjacent segment M2, thereby destabilizing open-channel conformations (Fig.7). This conclusion fits into a topological model of the nicotinic acetylcholine receptor superfamily, positioning the gate of these channels close to the cytoplasmic end of segment M2, near the M1-M2 loop (Wilson and Karlin, 1998). Conversely, the M2-M3 loop has been proposed to serve as the extracellular hinge of segment M2, thought to mediate the interaction of the ligand-binding and channel activation site (Lynch et al., 1997).
Fig. 7.

Topological predictions for the cytoplasmic M1-M2 loop of recombinant α1 and α1P250T receptor channels. Location of transmembrane segments M1 and M2 are indicated.
Using recombinant analysis of the GLRA1 mutant alleles known, it has become possible to attribute the neurological disorder hyperekplexia to disturbances in GlyR physiology. At present, however, the diverse clinical phenotypes of hyperekplexia cannot be correlated with the distinct parameters affected in GlyR function, such as ligand binding, intramolecular signal transduction (activation/gating), channel conductance, and their implications for neuronal signal processing (Jones and Westbrook, 1996). This suggests an all-or-none mechanism of GlyR dysfunction to generate the symptoms of hyperekplexia. This conclusion implies that the function of glycinergic inhibition in the human is in obvious need of further investigation.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft, Bundesministerium für Bildung und Forschung, the European Union, and the Fonds der Chemischen Industrie. We thank the members of family BS for participation in this study. Generous support by P. H. Seeburg, help with Western blotting by C. Kling, invaluable discussions with H.-G. Breitinger, and a critical reading of this manuscript by T. Bonk are gratefully acknowledged. We thank N. Spruston for providing Igor noise analysis macros.
Dr. Saul and Dr. Kuner contributed equally to this work.
Correspondence should be addressed to Dr. Cord-Michael Becker, Institut für Biochemie, Universität Erlangen-Nürnberg, Fahrstrasse 17, D-91054 Erlangen, Germany.
REFERENCES
- 1.Akaike N, Kaneda M. Glycine-gated chloride current in acutely isolated rat hypothalamic neurons. J Neurophysiol. 1989;62:1400–1409. doi: 10.1152/jn.1989.62.6.1400. [DOI] [PubMed] [Google Scholar]
- 2.Baker E, Sutherland GR, Schofield PR. Localization of the glycine receptor alpha 1 subunit gene (GLRA1) to chromosome 5q32 by FISH. Genomics. 1994;22:491–493. doi: 10.1006/geno.1994.1419. [DOI] [PubMed] [Google Scholar]
- 3.Becker C-M. Glycine receptors: molecular heterogeneity and implications for disease. The Neuroscientist. 1995;1:130–141. [Google Scholar]
- 4.Becker C-M, Langosch D. Glycine receptors. In: Stephenson FA, Turner AJ, editors. Amino acid neurotransmission. Portland; London: 1998. pp. 93–112. [Google Scholar]
- 5.Becker C-M, Hoch W, Betz H. Glycine receptor heterogeneity in rat spinal cord during postnatal development. EMBO J. 1988;7:3717–3726. doi: 10.1002/j.1460-2075.1988.tb03255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betz H. Structure and function of inhibitory glycine receptors. Q Rev Biophys. 1992;25:381–394. doi: 10.1017/s0033583500004340. [DOI] [PubMed] [Google Scholar]
- 7.Bormann J, Rundström N, Betz H, Langosch D. Residues within transmembrane segment M2 determine chloride conductance of glycine receptor homo- and heterooligomers. EMBO J. 1993;12:3729–3737. doi: 10.1002/j.1460-2075.1993.tb06050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breitinger HG, Becker CM. The inhibitory glycine receptor: prospects for a therapeutic orphan? Curr Pharm Design. 1998;4:315–334. [PubMed] [Google Scholar]
- 9.Brune W, Weber RG, Saul B, von Knebel Doeberitz M, Grond Ginsbach C, Kellermann K, Meinck HM, Becker CM. A GLRA1 null mutation in recessive hyperekplexia challenges the functional role of glycine receptors. Am J Hum Genet. 1996;58:989–997. [PMC free article] [PubMed] [Google Scholar]
- 10.Colquhoun D, Farrant M. Molecular pharmacology. The binding issue. Nature. 1993;366:510–511. doi: 10.1038/366510b0. [DOI] [PubMed] [Google Scholar]
- 11.Colquhoun D, Jonas P, Sakmann B. Action of brief pulses of glutamate on AMPA/kainate receptors in patches from different neurones of rat hippocampal slices. J Physiol (Lond) 1992;458:261–287. doi: 10.1113/jphysiol.1992.sp019417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chou PY, Fasman GD. Prediction of protein conformation. Biochemistry. 1974;13:222–245. doi: 10.1021/bi00699a002. [DOI] [PubMed] [Google Scholar]
- 13.Elmslie FV, Hutchings SM, Spencer V, Curtis A, Covanis T, Gardiner RM, Rees M. Analysis of GLRA1 in hereditary and sporadic hyperekplexia: a novel mutation in a family cosegregating for hyperekplexia and spastic paraparesis. J Med Genet. 1996;33:435–436. doi: 10.1136/jmg.33.5.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fraser SP, Moon C, Djamgoz MBA. Electrophysiology of Xenopus oocytes: an expression system in molecular neurobiology. In: Wallis DI, editor. The practical approach series. IRL; Oxford: 1993. pp. 65–88. [Google Scholar]
- 15.Gabriel N, Lenard HG. Das congenitale “stiff baby”-Syndrom. Fortschr Myologie. 1984;7:194–201. [Google Scholar]
- 16.Galzi JL, Devillers TA, Hussy N, Bertrand S, Changeux JP, Bertrand D. Mutations in the channel domain of a neuronal nicotinic receptor convert ion selectivity from cationic to anionic. Nature. 1992;359:500–505. doi: 10.1038/359500a0. [DOI] [PubMed] [Google Scholar]
- 17.Grenningloh G, Schmieden V, Schofield PR, Seeburg PH, Siddique T, Mohandas TK, Becker CM, Betz H. Alpha subunit variants of the human glycine receptor: primary structures, functional expression and chromosomal localization of the corresponding genes. EMBO J. 1990;9:771–776. doi: 10.1002/j.1460-2075.1990.tb08172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 19.Handford CA, Lynch JW, Baker E, Webb GC, Ford JH, Sutherland GR, Schofield PR. The human glycine receptor beta subunit: primary structure, functional characterisation and chromosomal localisation of the human and murine genes. Mol Brain Res. 1996;35:211–219. [PubMed] [Google Scholar]
- 20.Ho C, Kulaeva OI, Levine AS, Woodgate R. A rapid method for cloning mutagenic DNA repair genes: isolation of umu-complementing genes from multidrug resistance plasmids R391, R446b, and R471a. J Bacteriol. 1993;175:5411–5419. doi: 10.1128/jb.175.17.5411-5419.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones MV, Westbrook GL. The impact of receptor desensitization on fast synaptic transmission. Trends Neurosci. 1996;19:96–101. doi: 10.1016/s0166-2236(96)80037-3. [DOI] [PubMed] [Google Scholar]
- 22.Kingsmore SF, Suh D, Seldin MF. Genetic mapping of the glycine receptor alpha 3 subunit on mouse chromosome 8. Mamm Genome. 1994;5:831–832. doi: 10.1007/BF00292030. [DOI] [PubMed] [Google Scholar]
- 23.Kling C, Koch M, Saul B, Becker CM. The frameshift mutation oscillator (Glra1spd-ot) produces a complete loss of glycine receptor alpha1-polypeptide in mouse central nervous system. Neuroscience. 1997;78:411–417. doi: 10.1016/s0306-4522(96)00567-2. [DOI] [PubMed] [Google Scholar]
- 24.Krieg PA, Melton DA. Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucleic Acids Res. 1984;12:7057–7070. doi: 10.1093/nar/12.18.7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuhse J, Schmieden V, Betz H. Identification and functional expression of a novel ligand binding subunit of the inhibitory glycine receptor. J Biol Chem. 1990;265:22317–22320. [PubMed] [Google Scholar]
- 26.Kuner T, Schoepfer R. Multiple structural elements determine subunit specificity of Mg2+ block in NMDA receptor channels. J Neurosci. 1996;16:3549–3558. doi: 10.1523/JNEUROSCI.16-11-03549.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langosch D, Laube B, Rundstrom N, Schmieden V, Bormann J, Betz H. Decreased agonist affinity and chloride conductance of mutant glycine receptors associated with human hereditary hyperekplexia. EMBO J. 1994;13:4223–4228. doi: 10.1002/j.1460-2075.1994.tb06742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laube B, Langosch D, Betz H, Schmieden V. Hyperekplexia mutations of the glycine receptor unmask the inhibitory subsite for beta-amino-acids. NeuroReport. 1995;6:897–900. doi: 10.1097/00001756-199504190-00018. [DOI] [PubMed] [Google Scholar]
- 29.Lynch JW, Rajendra S, Pierce KD, Handford CA, Barry PH, Schofield PR. Identification of intracellular and extracellular domains mediating signal transduction in the inhibitory glycine receptor chloride channel. EMBO J. 1997;16:110–120. doi: 10.1093/emboj/16.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matzenbach B, Maulet Y, Sefton L, Courtier B, Avner P, Guenet JL, Betz H. Structural analysis of mouse glycine receptor alpha subunit genes. Identification and chromosomal localization of a novel variant. J Biol Chem. 1994;269:2607–2612. [PubMed] [Google Scholar]
- 31.Milani N, Dalpra L, del Prete A, Zanini R, Larizza L. A novel mutation (Gln266–>His) in the alpha 1 subunit of the inhibitory glycine-receptor gene (GLRA1) in hereditary hyperekplexia. Am J Hum Genet. 1996;58:420–422. [PMC free article] [PubMed] [Google Scholar]
- 32.Milani N, Mülhardt C, Weber RG, Lichter P, Kioschis P, Poustka A, Becker C-M. The human glycine receptor β subunit gene (GLRB): structure, refined chromosomal localization and population polymorphism. Genomics. 1998;50:341–345. doi: 10.1006/geno.1998.5324. [DOI] [PubMed] [Google Scholar]
- 33.Mülhardt C, Fischer M, Gass P, Simon-Chazottes D, Guenet JL, Kuhse J, Betz H, Becker CM. The spastic mouse: Aberrant splicing of glycine receptor beta subunit mRNA caused by intronic insertion of L1 element. Neuron. 1994;13:1003–1015. doi: 10.1016/0896-6273(94)90265-8. [DOI] [PubMed] [Google Scholar]
- 34.Nikolic Z, Laube B, Mülhardt C, Weber RG, Lichter P, Kioschis P, Poustka A, Becker CM. The human glycine receptor subunit α3: Gene structure, chromosomal location, and functional characterization of alternative transcripts. J Biol Chem. 1998;273:19708–19714. doi: 10.1074/jbc.273.31.19708. [DOI] [PubMed] [Google Scholar]
- 35.Rajendra S, Lynch JW, Pierce KD, French CR, Barry PH, Schofield PR. Startle disease mutations reduce the agonist sensitivity of the human inhibitory glycine receptor. J Biol Chem. 1994;269:18739–18742. [PubMed] [Google Scholar]
- 36.Rees MI, Andrew M, Jawad S, Owen MJ. Evidence for recessive as well as dominant forms of startle disease (hyperekplexia) caused by mutations in the alpha 1 subunit of the inhibitory glycine receptor. Hum Mol Genet. 1994;3:2175–2179. doi: 10.1093/hmg/3.12.2175. [DOI] [PubMed] [Google Scholar]
- 37.Ryan SG, Buckwalter MS, Lynch JW, Handford CA, Segura L, Shiang R, Wasmuth JJ, Camper SA, Schofield P, O’Connell P. A missense mutation in the gene encoding the alpha 1 subunit of the inhibitory glycine receptor in the spasmodic mouse. Nat Genet. 1994;7:131–135. doi: 10.1038/ng0694-131. [DOI] [PubMed] [Google Scholar]
- 38.Saul B, Schmieden V, Kling C, Mulhardt C, Gass P, Kuhse J, Becker CM. Point mutation of glycine receptor alpha 1 subunit in the spasmodic mouse affects agonist responses. FEBS Lett. 1994;350:71–76. doi: 10.1016/0014-5793(94)00736-5. [DOI] [PubMed] [Google Scholar]
- 39.Shiang R, Ryan SG, Zhu YZ, Hahn AF, O’Connell P, Wasmuth JJ. Mutations in the alpha 1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat Genet. 1993;5:351–358. doi: 10.1038/ng1293-351. [DOI] [PubMed] [Google Scholar]
- 40.Shiang R, Ryan SG, Zhu YZ, Fielder TJ, Allen RJ, Fryer A, Yamashita S, O’Connell P, Wasmuth JJ. Mutational analysis of familial and sporadic hyperekplexia. Ann Neurol. 1995;38:85–91. doi: 10.1002/ana.410380115. [DOI] [PubMed] [Google Scholar]
- 41.Sontheimer H, Becker CM, Pritchett DB, Schofield PR, Grenningloh G, Kettenmann H, Betz H, Seeburg PH. Functional chloride channels by mammalian cell expression of rat glycine receptor subunit. Neuron. 1989;2:1491–1497. doi: 10.1016/0896-6273(89)90195-5. [DOI] [PubMed] [Google Scholar]
- 42.Spivak CE. Correlations among Hill parameters reflect models of activating ligand-gated ion channels. Trends Pharmacol Sci. 1995;16:39–42. doi: 10.1016/s0165-6147(00)88976-2. [DOI] [PubMed] [Google Scholar]
- 43.Spruston N, Jonas P, Sakmann B. Dendritic glutamate receptor channels in rat hippocampal CA3 and CA1 pyramidal neurons. J Physiol (Lond) 1995;482:325–352. doi: 10.1113/jphysiol.1995.sp020521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taleb O, Betz H. Expression of the human glycine receptor alpha 1 subunit in Xenopus oocytes: apparent affinities of agonists increase at high receptor density. EMBO J. 1994;13:1318–1324. doi: 10.1002/j.1460-2075.1994.tb06384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tijssen MA, Shiang R, van Deutekom J, Boerman RH, Wasmuth JJ, Sandkuijl LA, Frants RR, Padberg GW. Molecular genetic reevaluation of the Dutch hyperekplexia family. Arch Neurol. 1995;52:578–582. doi: 10.1001/archneur.1995.00540300052012. [DOI] [PubMed] [Google Scholar]
- 46.Wilson GG, Karlin A. The location of the gate in the acetylcholine receptor channel. Neuron. 1998;20:1269–1281. doi: 10.1016/s0896-6273(00)80506-1. [DOI] [PubMed] [Google Scholar]